
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intra- and intermolecular Fe-catalyzed dicarbofunctionalization of vinyl cyclopropanes†
Lei
Liu
 ,
Wes
Lee
,
Mingbin
Yuan
,
Chris
Acha
,
Michael B.
Geherty
,
Brandon
Williams
and
Osvaldo
Gutierrez
*
,
Wes
Lee
,
Mingbin
Yuan
,
Chris
Acha
,
Michael B.
Geherty
,
Brandon
Williams
and
Osvaldo
Gutierrez
*
Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, USA. E-mail: ogs@umd.edu
First published on 27th February 2020
Abstract
Design and implementation of the first (asymmetric) Fe-catalyzed intra- and intermolecular difunctionalization of vinyl cyclopropanes (VCPs) with alkyl halides and aryl Grignard reagents has been realized via a mechanistically driven approach. Mechanistic studies support the diffusion of alkyl radical intermediates out of the solvent cage to participate in an intra- or intermolecular radical cascade with a range of VCPs followed by re-entering the Fe radical cross-coupling cycle to undergo (stereo)selective C(sp2)–C(sp3) bond formation. This work provides a proof-of-concept of the use of vinyl cyclopropanes as synthetically useful 1,5-synthons in Fe-catalyzed conjunctive cross-couplings with alkyl halides and aryl/vinyl Grignard reagents. Overall, we provide new design principles for Fe-mediated radical processes and underscore the potential of using combined computations and experiments to accelerate the development of challenging transformations.
Introduction
Iron-catalyzed C–C cross-coupling reactions have attracted much attention due to the higher abundance, cost-effectiveness, and lower toxicity of iron in comparison to precious transition metals.1 Methods for ligand-supported (e.g., N-heterocycles, bisphosphines, and diamines) and ligand-free systems for iron-catalyzed C–C cross-coupling reactions using C(sp), C(sp2), and C(sp3) partners have been developed.2 In particular, bisphosphine-iron systems have emerged as highly versatile and promising candidates for the formation of new C–C bonds with a range of organometallic nucleophiles including Mg- (Kumada),3 Zn- (Negishi),4 B- (Suzuki–Miyaura),5 and Al6 reagents with alkyl halides and redox active esters.4a Electron-poor vinyl cyclopropanes have also been used as π-coupling partners in Fe-catalyzed C(sp2)–C(sp3) bond formation (Scheme 1A). In particular, Fürstner used low valent iron ferrates to promote tandem ring-opening/monoarylation of electron-poor vinyl cyclopropanes (VCPs) using aryl Grignard reagents.7 In a related study, Plietker used a low valent, electron rich ferrate complex (Bu4N[Fe(CO)3(NO)]) to promote ring-opening/monoarylation of electron-deficient VCPs with acidic pronucleophiles.8 Despite these advancements, to the best of our knowledge, there are only two reports of asymmetric iron-catalyzed cross-couplings: between aryl Grignard reagents or lithium aryl borates as nucleophiles and α-halo esters as electrophiles (Scheme 1A).9,10 Further, despite the use of vinylcyclopropanes as useful reagents in organic synthesis, the application of VCPs in Fe-catalyzed conjunctive cross-couplings is not known. Thus, development of new (asymmetric) iron-catalyzed radical cascade/C(sp2)–C(sp3) cross-coupling reactions will expand the synthetic toolbox and lead to an increase in diversification of carbocycles.11 Herein, we used a mechanistically guided approach to design and develop a new and selective Fe-catalyzed intra- and inter-molecular dicarbofunctionalization of synthetically versatile vinyl cyclopropanes (Scheme 1B).12,13 Conceptually, this work establishes the use of readily accessible vinyl cyclopropanes in Fe-catalyzed conjunctive cross-coupling reactions.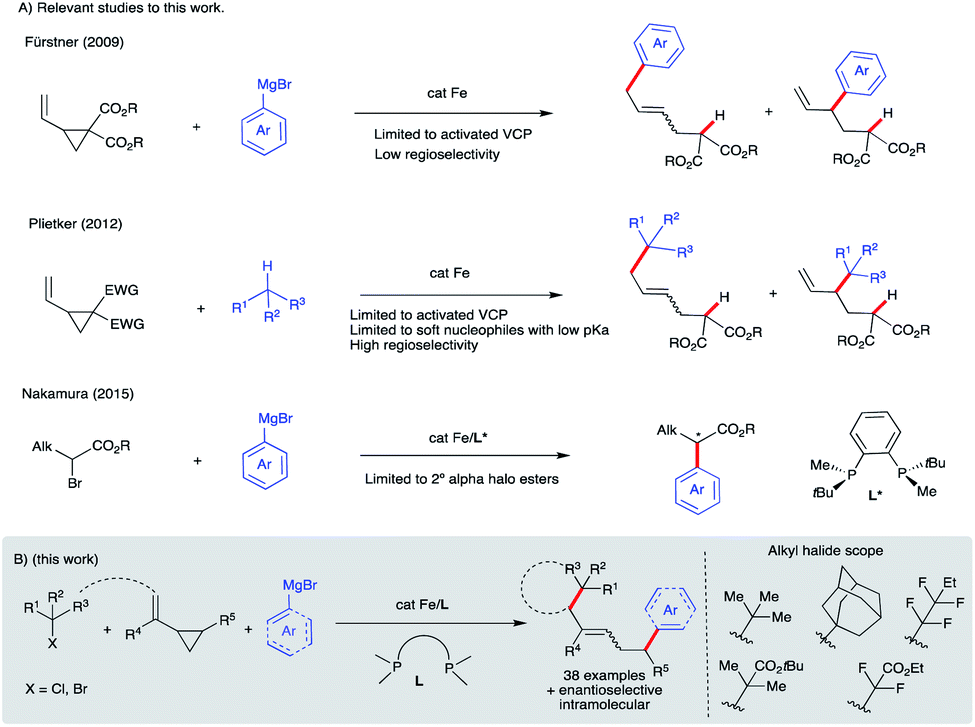 | ||
| Scheme 1 Design and development of Fe-catalyzed intra- and inter-molecular difunctionalization of vinyl cyclopropanes via a new radical cascade reaction. | ||
Contributing to the scarcity of iron-catalyzed asymmetric reactions is likely the fact that mechanistic details of iron-catalyzed cross-coupling are not well understood, in comparison to palladium systems.14 Pioneering mechanistic studies by Kochi15 in the 1970s and more recent reports by Bedford,16 Nakamura,17 Norrby,18 Fürstner,19 Tonzetich,20 Koszinowski,21 and Neidig22 have led to a greater understanding of these transformations. In 2017, parallel quantum mechanical studies in our lab23 and by Morokuma24 were reported on the mechanism of chiral bisphosphine cross-coupling reactions between α-chloro esters and aryl Grignard reagents. These studies revealed a mechanism involving halogen abstraction by an aryl Fe(I) complex, leading to an alkyl radical and halo aryl Fe(II) species (Scheme 2; circled). In turn, these two species could combine, leading to an Fe(III) intermediate which will then undergo reductive elimination, leading to the desired cross-coupled product. More experimental studies (i.e., spectroscopic and kinetic) are needed to assess the validity of the computational models, and the mechanism likely depends on subtle changes to the alkyl halide, Grignard, and ligand structures. Nonetheless, based on these mechanistic studies, we envisage diverting the reactivity from the Fe radical cross-coupling catalytic cycle (black) to a programmed intra- and intermolecular radical cascade (red) with vinyl cyclopropanes, leading to a new alkyl radical that could then re-enter the catalytic cycle and undergo stereoselective C(sp2)–C(sp3) bond formation. Given that most of the transition-metal catalyzed cascade reactions terminate with C–H bond formation25 and fail to control the stereoselectivity at the termination step, if successful, this approach could lead to a rapid increase in molecular diversity.
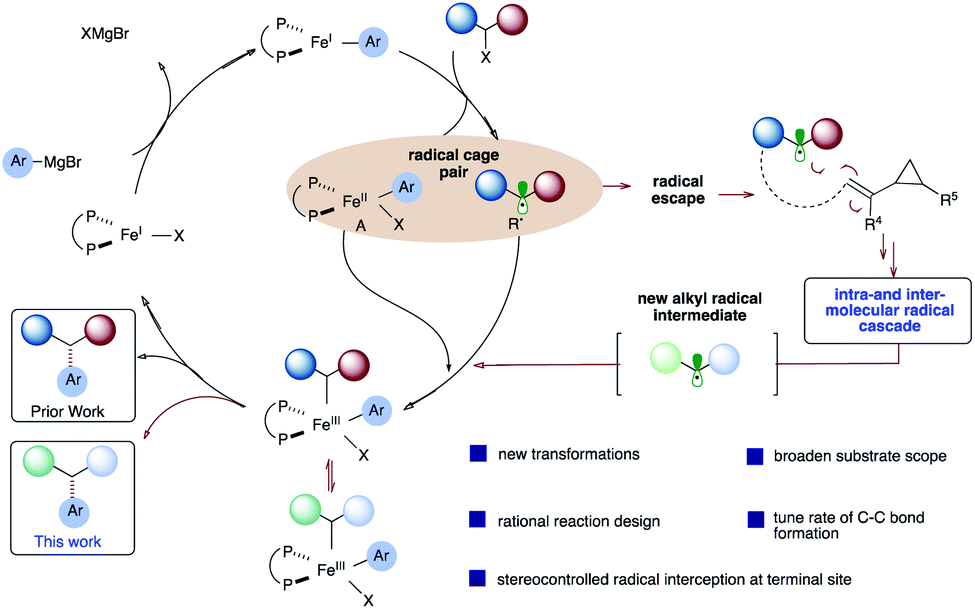 | ||
| Scheme 2 A proposed mechanism of iron-catalyzed cross-coupling reactions. | ||
Results and discussion
Stereoselective intra-molecular dicarbofunctionalization of VCPs
Li's group demonstrated the use of cyclopropyl olefins to promote a radical alkylation, ring-opening, intramolecular arylation cascade reaction under photoredox conditions.26 Fu and co-workers reported that chiral nickel catalysts could achieve enantioselective cross-coupling of a wide range of racemic alkyl halides (as radical precursors) with organometallic nucleophiles27 including those involving stereoconvergent radical cyclization/arylation.28 We hypothesize that upon radical formation and in the presence of a pendant vinyl cyclopropane (Scheme 2; right), we could divert reactivity from the cross-coupling cycle to promote an intra-molecular radical cascade reaction. Specifically, we envisage a 5-exo-trig cyclization outcompeting radical rebound to an aryl Fe species and divert reactivity towards Fe-catalyzed 1,5-dicarbofunctionalization of vinyl cyclopropanes.Indeed, as shown in Scheme 3, quantum mechanical calculations support our hypothesis. Specifically, the barrier for radical 5-exo-cyclization (viaTSA-B) to form B˙ is 6.0 kcal mol−1 lower in energy than the barrier for the radical rebound transition state TSA-P1 that will lead to cross-coupling product P1 (7.1 kcal mol−1vs. 13.1 kcal mol−1, respectively). In turn, the kinetically favored cyclic alkyl radical B˙ will then undergo radical ring-opening (the barrier is only 1.6 kcal mol−1) to form the thermodynamically favored alkyl radical C˙. Finally, C˙ could then re-enter the iron cross-coupling cycle and undergo C(sp2)–C(sp3) bond formation (viaTSC-P2; the barrier is 14.8 kcal mol−1 from C˙) leading, after reductive elimination (not shown), to the radical cascade product P2. Overall, these calculations suggest that the radical cyclization/ring-opening/cross-coupling pathway (leading to P2) is kinetically favored over cross-coupling (leading to P1). Moreover, we anticipate that the chiral aryl iron species could control the final radical coupling step (with C˙) and permit high-levels of stereocontrol.29
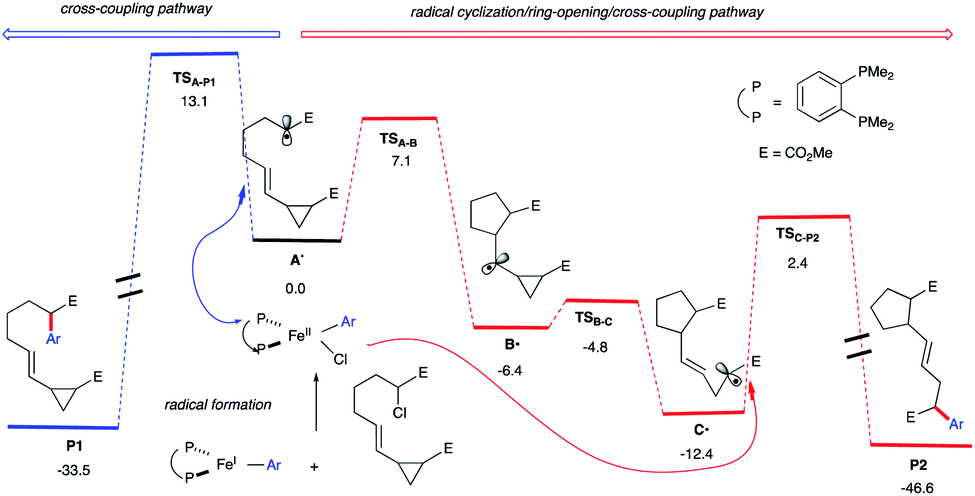 | ||
| Scheme 3 Energetics for the in-cage (blue) and out-of-cage (red) arylation computed at the UPBEPBE/6-311+G(d,p)-SDD(Fe)-THF(SMD)//UB3LYP/6-31G(d) levels of theory. | ||
Gratifyingly, we found that we can divert reactivity towards the asymmetric intra-molecular 1,5-difunctionalization of vinyl cyclopropanes using 1 as the substrate (Scheme 4). Specifically, using the standard conditions for Fe-catalyzed α-arylation,30 the reaction of 1 with 2-napthylmagnesium bromide gave a mixture of diastereomeric radical cascade products 2a (see ESI†). Using column chromatography, we separated and identified the trans, E stereoisomer as the major diastereomer (see ESI†). At this moment, noting that most of the transition-metal catalyzed cascade reactions terminate with C–H bond formation25 and fail to control stereoselectivity at the termination step, we were primarily concerned with determining if the chiral iron species controlled the stereochemistry of the terminating C(sp2)–C(sp3) arylation. As a proof-of-concept, to determine stereochemistry at the terminating C–C bond forming step, we subjected 2a to a one-pot procedure of ozonolysis followed by reduction with NaBH4 that led to the corresponding product 3a in good yields and enantioselectivities. Notably, as expected from the radical cyclization event in the absence of chiral iron species (Scheme 3), the corresponding racemic cyclopentane fragment 3b′ was isolated in 77% yield in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. Having established, as a proof-of-concept, that we could control the enantioselectivity at the terminating step in the cascade reaction, we examine the aryl Grignard reagent scope in this transformation and its effect on enantioselectivity. As shown in Scheme 4B, the aryl Grignard reagent scope is broad. Both electron-poor and electron-rich aryl Grignard reagents participate in the diverted Fe-catalyzed intra-molecular difunctionalization of vinyl cyclopropanes to give the desired products in good yields (up to 82% over three steps) and enantioselectivities (up to 90:10 er). We established the absolute stereochemistry as (S) from the cyclized lactone (S)-13 from 3c. Notably, Grignard reagents that failed in the asymmetric Fe-catalyzed cross-coupling reactions were compatible reagents in these radical cascade/cross-coupling transformations. Specifically, highly electron-deficient (i.e., 3,4,5-trifluorophenyl 3h) and even sterically congested (e.g., ortho-methyl aryl 3i and 1-naphthyl 3k) aryl Grignard reagents formed the desired products with good enantioselectivities (77:23 to 90:10 er) albeit in lower yields (8–50%). However, at this moment, this method is limited to 5-exo-trig radical cyclization. Lengthening the tether with an extra methylene group leads to the formation of the Fe radical cross-coupled product and no products from diverted 6-exo-trig radical cyclization are observed (see ESI†). Presumably, the much higher energy barrier to undergo radical 6-exo-trig cyclization in comparison to 5-exo-trig radical cyclization prevents the formation of radical cascade/arylation (see Fig. S9 in the ESI† for energetics).
:1 dr. Having established, as a proof-of-concept, that we could control the enantioselectivity at the terminating step in the cascade reaction, we examine the aryl Grignard reagent scope in this transformation and its effect on enantioselectivity. As shown in Scheme 4B, the aryl Grignard reagent scope is broad. Both electron-poor and electron-rich aryl Grignard reagents participate in the diverted Fe-catalyzed intra-molecular difunctionalization of vinyl cyclopropanes to give the desired products in good yields (up to 82% over three steps) and enantioselectivities (up to 90:10 er). We established the absolute stereochemistry as (S) from the cyclized lactone (S)-13 from 3c. Notably, Grignard reagents that failed in the asymmetric Fe-catalyzed cross-coupling reactions were compatible reagents in these radical cascade/cross-coupling transformations. Specifically, highly electron-deficient (i.e., 3,4,5-trifluorophenyl 3h) and even sterically congested (e.g., ortho-methyl aryl 3i and 1-naphthyl 3k) aryl Grignard reagents formed the desired products with good enantioselectivities (77:23 to 90:10 er) albeit in lower yields (8–50%). However, at this moment, this method is limited to 5-exo-trig radical cyclization. Lengthening the tether with an extra methylene group leads to the formation of the Fe radical cross-coupled product and no products from diverted 6-exo-trig radical cyclization are observed (see ESI†). Presumably, the much higher energy barrier to undergo radical 6-exo-trig cyclization in comparison to 5-exo-trig radical cyclization prevents the formation of radical cascade/arylation (see Fig. S9 in the ESI† for energetics).
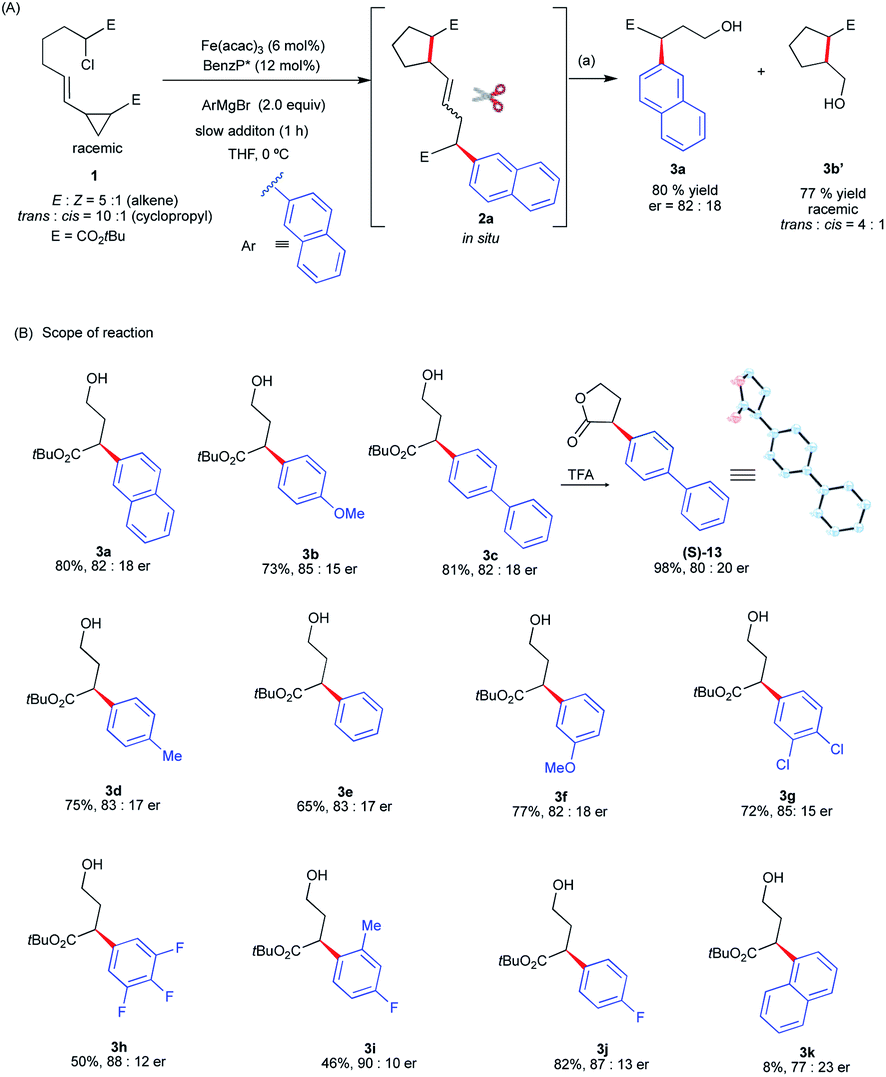 | ||
| Scheme 4 Design and application of asymmetric Fe-catalyzed intra-molecular dicarbofunctionalization of vinyl cyclopropanes. (a) O3, CH2Cl2/MeOH −78 °C, 5 min and then NaBH4 (10 equiv.) −78 °C to rt, 1 h. | ||
Design and application of inter-molecular dicarbo-functionalization of vinyl cyclopropanes
Given the scarcity of stereoconvergent transition metal-catalyzed radical cyclization-arylation cascades and underutilization of vinyl cyclopropanes in transition metal-catalyzed conjunctive cross-couplings, our proof-of-principle results with intramolecular functionalization of VCPs (Scheme 4) represent an attractive strategy towards this unmet need.31,32 Specifically, we envisage that by tuning the properties of alkyl halides, we could divert reactivity towards regioselective intermolecular ring-opening/dicarbofunctionalization of VCPs. Specifically, we hypothesize that sterically hindered tertiary alkyl halides will lead to higher barriers for Fe radical coupling and, instead, favor intermolecular radical addition to the vinyl cyclopropane (Scheme 5A). In turn, the incipient radical could then undergo cyclopropyl ring-opening and re-enter the Fe radical cross-coupling cycle to undergo (stereoselective) C(sp2)–C(sp3) arylation at the least sterically hindered site. Previous work by Fürstner and Plietker using iron as the catalyst with vinyl cyclopropanes (VCPs) resulted in ring-opening/monocarbofunctionalization, required electron-deficient VCPs, and terminated in protonation (Scheme 1A).7,8 Thus, if successful, this approach will expand the range of Fe-catalyzed transformations using vinyl cyclopropanes as valuable synthons in chemical synthesis. Also, if realized, this will represent the first example of (asymmetric) Fe-catalyzed 3-component cross-coupling.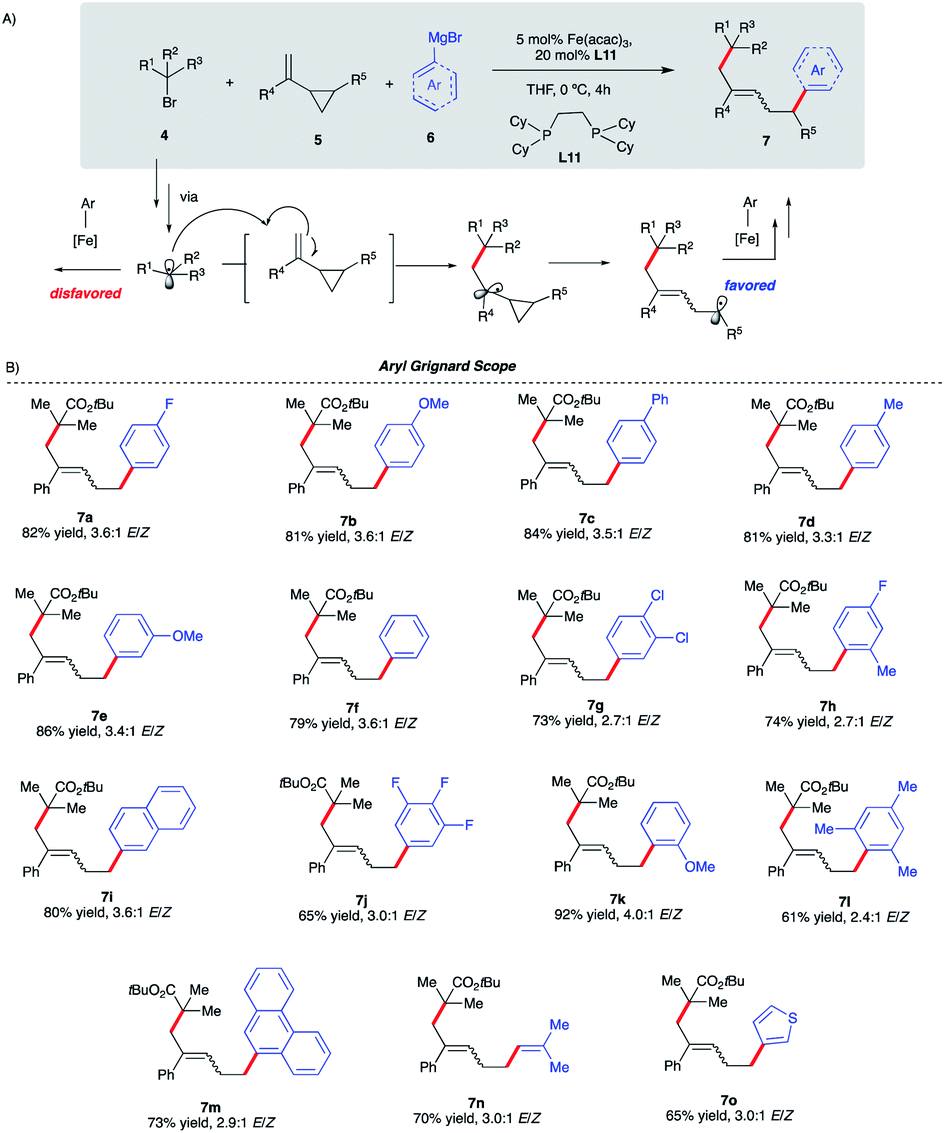 | ||
| Scheme 5 Reaction scope of the aryl Grignard reagent in Fe-catalyzed intermolecular dicarbofunctionalization of vinyl cyclopropanes. (a) Reactions were performed on a 0.20 mmol scale. (b) ArMgBr was added dropwise via a syringe pump over 4 h. (c) Isolated yield. | ||
We initiated our optimization studies using sterically hindered tert-butyl 2-bromo-2-methylpropanoate 4, (1-cyclopropylvinyl)-benzene 5 and aryl 4-fluorophenylmagnesium bromide 6 as model substrates (Scheme 5). Gratifyingly, after extensive ligand screening using bisphosphine, monophosphine, and diamine ligands (see Table S2 in the ESI†) and optimization studies, we identified acyclic 1,2-bis(dicyclohexylphosphino)ethane ligand L11 to be suitable to promote the desired and highly selective ring-opening/1,5-alkylarylation of vinyl cyclopropanes, forming compound 7a in 82% isolated yield and 3.6:1 E/Z ratio (Scheme 5B). With optimized conditions in hand, we next investigated the reaction scope of this transformation. As shown in Scheme 5B, the scope of this Fe-catalyzed 3-component dicarbofunctionalization is broad with respect to the Grignard nucleophile. Specifically, the reaction tolerated both withdrawing and electron-donating aryl nucleophiles, forming the desired 1,5-alkylaryl products in 61–92% yield. Notably, the reaction tolerated sterically hindered ortho-substituted aryl Grignard reagents (7h, 7k, 7l, and 7m) that proved to be problematic in previous direct Fe-catalyzed cross-coupling reactions.33 To highlight the versatility of this method, we also used both vinyl and heteroaryl Grignard reagents and, gratifyingly, obtained the desired products 7n and 7o, in good yields, 70% and 65% yield, respectively. Next, we explored the scope of vinyl cyclopropane and alkyl radical precursors (Scheme 6). Installing electron-rich or electron-poor aryl groups in the vinyl cyclopropane moiety (R4) allows the formation of the desired products in good to excellent yields (Scheme 6A). Notably, the reaction tolerates medicinally relevant 2- and 3-pyridyl moieties (7s, 7t). Moreover, the substituted cyclopropanes (R5) were suitable for this catalytic system, affording 7u and 7v in 67% and 34% yield, respectively. Unfortunately, sterically hindered tert-butyl 2-bromo-2-methylpropanoate 4 with alkyl VCPs such as 7w are beyond the reach of the present Fe-based system. In contrast to previously reported Fe-catalyzed cross-couplings, we found a wide range of alkyl bromides to be suitable radical precursors in this Fe-catalyzed conjunctive cross-coupling (Scheme 6B). Specifically, we found that alkyl fluorinated radical precursors are effective partners in the Fe-catalyzed intermolecular dicarbofunctionalization of VCPs, yielding the desired products 7x and 7y in 42% and 75% yield, respectively. Further, we observed a much higher E/Z ratio for 7y (12:1 E/Z), although the origin of this high selectivity is currently unknown. Finally, this method tolerated unactivated 3˙ alkyl radical precursors which led to the desired 3-component products 7z and 7za. We also performed quantum mechanical calculations on the intermolecular dicarbofunctionalization, and the calculated results are consistent with our hypothesis and the experimental result (see Fig. S10 in the ESI† for energetics). Specifically, tertiary alkyl radicals preferably undergo inter-molecular radical addition (with a barrier of 14.0 kcal mol−1) to the vinyl cyclopropane over radical coupling with Fe (with a barrier of 18.2 kcal mol−1).
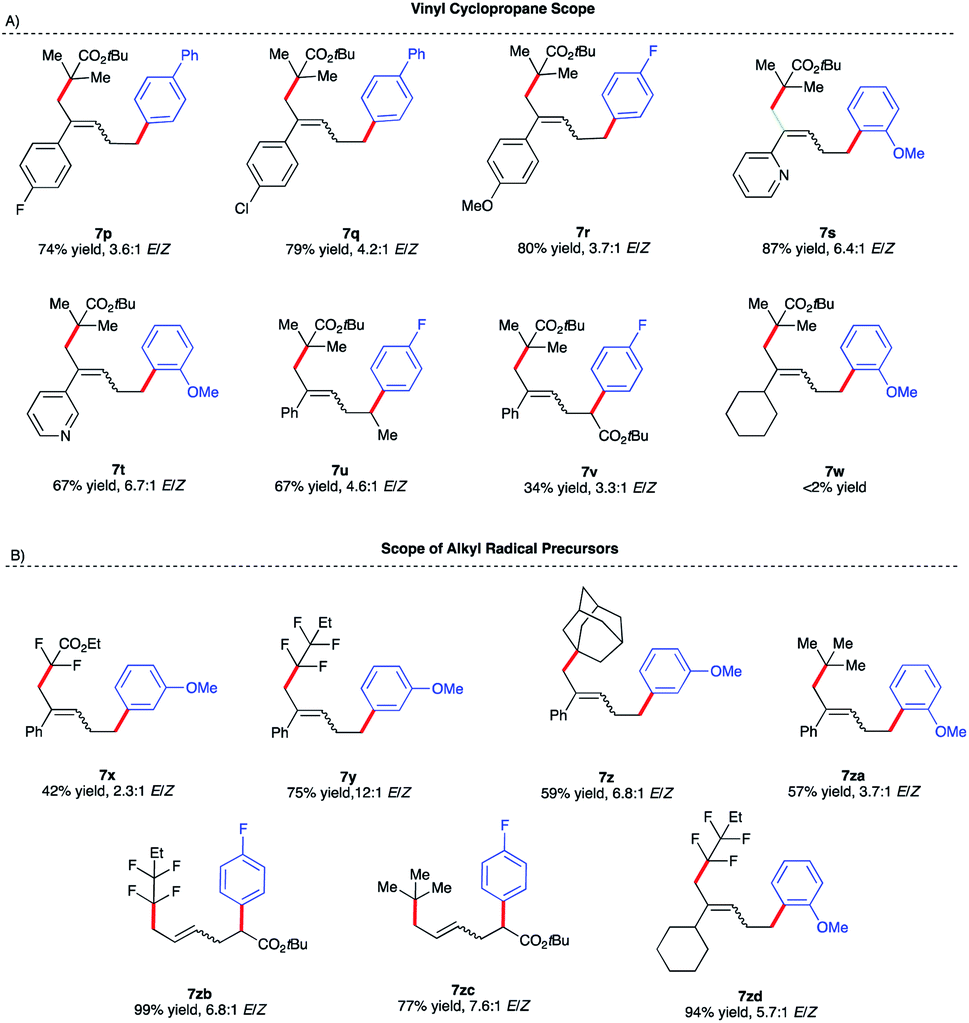 | ||
| Scheme 6 Vinyl cyclopropane and alkyl halide reaction scope. (a) Reactions were performed on a 0.20 mmol scale. (b) ArMgBr was added dropwise via a syringe pump over 4 h. (c) Isolated yield. | ||
Interestingly, fluorinated and tert-butyl radical precursors in combination with unactivated vinyl cyclopropanes led to the desired products 7zb, 7zc, and 7zd in excellent yields (77–99%). As observed for intramolecular Fe-catalyzed difunctionalization of VCPs (vide supra), preliminary results (Scheme 7) demonstrate that the chiral iron species controls the enantioselectivity at the terminal site of C–C bond formation in the presence of an ester moiety to form 8a and 8b in 35% and 75% yield, respectively. These results represent the first examples of an asymmetric Fe-catalyzed conjunctive cross-coupling. Ongoing work is geared towards expanding this transformation.
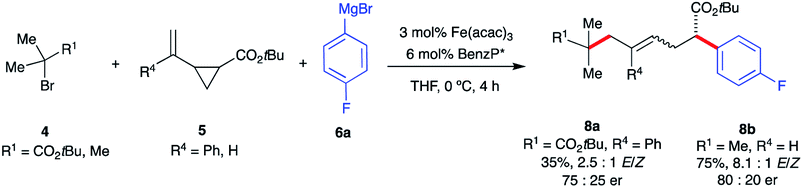 | ||
| Scheme 7 Preliminary results of asymmetric Fe-catalyzed intermolecular dicarbofunctionalization of vinyl cyclopropanes. (a) The enantiomeric ratio (er) values of (E)-8a and 8b were determined using chiral HPLC analysis. | ||
To demonstrate the synthetic versatility of the five-carbon unsaturated chain – potentially useful transformation, we performed the gram-scale synthesis of 7k (1.37 g, 87% yield) and performed several diversifications (Scheme 8). First, reduction of the alkene (Pd/C) provided the alkyl ester (9). Next, we performed hydrolysis of the ester to carboxylic acid (10) which could be a useful substrate in, inter alia, subsequent decarboxylative radical cross-couplings.34 Notably, derivatization of 7k to the carboxylic acid analog 10 permitted facile separation and purification of the major isomer (E). Finally, reduction of the ester formed the corresponding primary alcohol (11) which could undergo bromocyclization with N-bromosuccinimide (NBS) leading to the substituted tetrahydropyran with two stereogenic centers (12).
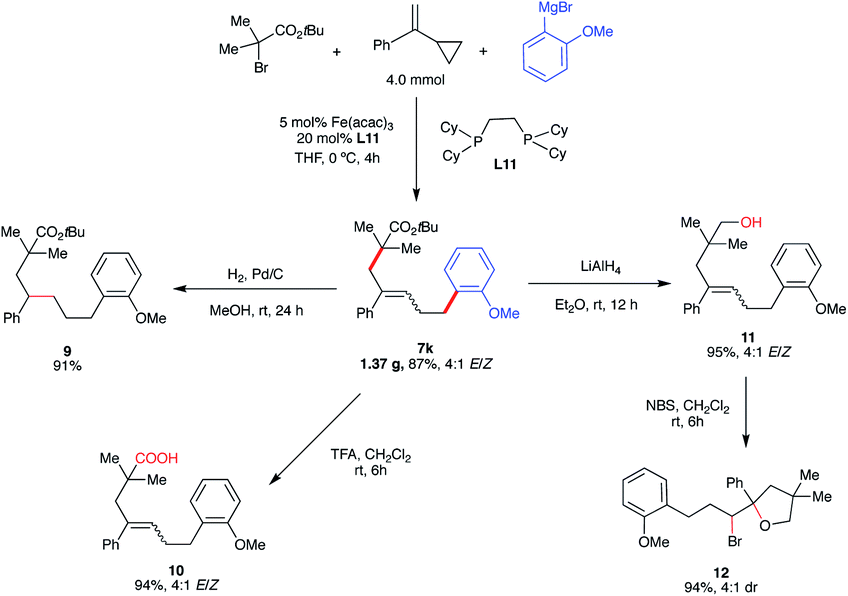 | ||
| Scheme 8 Synthetic applications of five-carbon unsaturated chain ester. (a) Isolated yield. (b) Yield of all isomers. | ||
Conclusions
In summary, Fe-catalyzed intra- and inter-molecular dicarbofunctionalizations of vinyl cyclopropanes have now been realized. In particular, we have used a mechanistically driven (computational and experimental) approach to divert the reactivity of the alkyl radical from the Fe radical cross-coupling cycle to undergo intra- and inter-molecular radical cascade reactions with activated and unactivated vinyl cyclopropanes. In turn, the incipient alkyl radical then re-enters the Fe cross-coupling cycle and undergoes (stereoselective) C(sp2)–C(sp3) bond formation. Finally, for the first time, as a proof-of-concept we show that VCPs can be used as effective conjunctive partners in asymmetric Fe-catalyzed 3-component cross-coupling reactions. We anticipate that, in particular, the intermolecular 3-component Fe-catalyzed dicarbofunctionalization reaction will impact the synthesis of medicinally relevant molecules. However, at present, some drawbacks of this method are the use of Grignard reagents that limits functional group incorporation into the nucleophile/radical precursors along with E/Z mixtures. Ongoing work is focused on expanding this strategy to a wide range of alkyl radical precursors, π-coupling partners, and nucleophilic partners participating in diverted asymmetric Fe-catalyzed radical cascade/cross-coupling reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support by the NSF (CAREER 1751568). O. G. is grateful to the University of Maryland College Park for start-up funds and computational resources from the UMD Deepthought2 and MARCC/BlueCrab HPC clusters and XSEDE (CHE160082 and CHE160053). We are grateful to Prof. Lai-Xi Wang (UMD) and Prof. Donald Watson (U. Delaware) for the use of the prep HPLC and polarimeter, respectively. Finally, we are grateful to Prof. Gary Molander (UPenn), Christopher Kelly (VCU), Prof. Martinez-Solorio (Drexel), Prof. Phillip DeShong (UMD), Prof. Davis (UMD), and Prof. Andrei Vedernikov (UMD) for helpful suggestions. We also acknowledge the Analytical NMR Service & Research Center (UMD) for NMR service/help provided by Dr Yinde Wang and Dr Fu Chen. We also thank Dr Yue Li (UMD) for help with the HRMS and Dr Peter Y. Zavalij for help with X-ray diffraction.References
- For a recent review, see: A. Piontek, E. Bisz and M. Szostak, Angew. Chem., Int. Ed., 2018, 57, 11116 CrossRef CAS PubMed.
- For selected reviews, see: (a) C. Cassani, G. Bergonzini and C.-J. Wallentin, ACS Catal., 2016, 6, 1640 CrossRef CAS; (b) R. B. Bedford, Acc. Chem. Res., 2015, 48, 1485 CrossRef CAS PubMed; (c) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed; (d) W. M. Czaplik, M. Mayer, J. Cvengros and A. J. von Wangelin, ChemSusChem, 2009, 2, 396 CrossRef CAS PubMed; (e) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500 CrossRef CAS PubMed.
- (a) C.-L. Sun, H. Krause and A. Fürstner, Adv. Synth. Catal., 2014, 356, 1281 CrossRef CAS; (b) S. Kawamura and M. Nakamura, Chem. Lett., 2013, 42, 183 CrossRef CAS; (c) T. Hatakeyama, Y. Fujiwara, Y. Okada, T. Itoh, T. Hashimoto, S. Kawamura, K. Ogata, H. Takaya and M. Nakamura, Chem. Lett., 2011, 40, 1030 CrossRef CAS; (d) T. Hatakeyama, Y. Okada, Y. Yoshimoto and M. Nakamura, Angew. Chem., Int. Ed., 2011, 50, 10973 CrossRef CAS PubMed; (e) K. G. Dongol, H. Koh, M. Sau and C. L. L. Chai, Adv. Synth. Catal., 2007, 349, 1015 CrossRef CAS.
- (a) F. Toriyama, J. Cornella, L. Wimmer, T.-G. Chen, D. D. Dixon, G. Creech and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 11132 CrossRef CAS PubMed; (b) R. B. Bedford, E. Carter, P. M. Cogswell, N. J. Gower, M. F. Haddow, J. N. Harvey, D. M. Murphy, E. C. Neeve and J. Nunn, Angew. Chem., Int. Ed., 2013, 52, 1285 CrossRef CAS PubMed; (c) C. J. Adams, R. B. Bedford, E. Carter, N. J. Gower, M. F. Haddow, J. N. Harvey, M. Huwe, M. A. Cartes, S. M. Mansell, C. Mendoza, D. M. Murphy, E. C. Neeve and J. Nunn, J. Am. Chem. Soc., 2012, 134, 10333 CrossRef CAS PubMed; (d) T. Hatakeyama, Y. Kondo, Y. Fujiwara, H. Takaya, S. Ito, E. Nakamura and M. Nakamura, Chem. Commun., 2009, 1216 RSC; (e) R. B. Bedford, M. Huwe and M. C. Wilkinson, Chem. Commun., 2009, 600 RSC.
- (a) N. Nakagawa, T. Hatakeyama and M. Nakamura, Chem. Lett., 2015, 44, 486 CrossRef CAS; (b) T. Hatakeyama, T. Hashimoto, K. K. A. D. S. Kathriarachchi, T. Zenmyo, H. Seike and M. Nakamura, Angew. Chem., Int. Ed., 2012, 51, 8834 CrossRef CAS PubMed; (c) T. Hashimoto, T. Hatakeyama and M. Nakamura, J. Org. Chem., 2011, 77, 1168 CrossRef PubMed; (d) T. Hatakeyama, T. Hashimoto, Y. Kondo, Y. Fujiwara, H. Seike, H. Takaya, Y. Tamad, T. Ono and M. Nakamura, J. Am. Chem. Soc., 2010, 132, 10674 CrossRef CAS PubMed.
- (a) S. Kawamura, T. Kawabata, K. Ishizuka and M. Nakamura, Chem. Commun., 2012, 48, 9376 RSC; (b) S. Kawamura, K. Ishizuka, H. Takaya and M. Nakamura, Chem. Commun., 2010, 46, 6054 RSC.
- B. D. Sherry and A. Fürstner, Chem. Commun., 2009, 7116 RSC.
- A. P. Dieskau, M. S. Holzwarth and B. Plietker, J. Am. Chem. Soc., 2012, 134, 5048 CrossRef CAS PubMed.
- (a) T. Iwamoto, C. Okuzono, L. Adak, M. Jin and M. Nakamura, Chem. Commun., 2019, 55, 1128 RSC; (b) M. Jin, L. Adak and M. Nakamura, J. Am. Chem. Soc., 2015, 137, 7128 CrossRef CAS PubMed.
- For a recent review, see: H. Pellissier, Coord. Chem. Rev., 2019, 386, 1 CrossRef CAS.
- For recent reviews, see: (a) J. Choi and G. C. Fu, Science, 2017, 356, 6334 CrossRef; (b) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587 CrossRef CAS PubMed.
- For a recent review on vinyl cylcopropanes in synthesis, see: M. Meazza, H. Guo and R. Rios, Org. Biomol. Chem., 2017, 15, 2479 RSC.
- For iron-catalyzed functionalization of VCPs, see: C. Chen, X. Shen, J. Chen, X. Hong and Z. Lu, Org. Lett., 2017, 19, 5422 CrossRef CAS PubMed.
- (a) M. L. Neidig, S. H. Carpenter, D. J. Curran, J. C. DeMuth, V. E. Fleischaeur, T. E. Iannuzzi, P. G. N. Neate, J. D. Sears and N. J. Wolford, Acc. Chem. Res., 2019, 52, 140 CrossRef CAS PubMed; (b) J. D. Sears, P. G. N. Neate and M. L. Neidig, J. Am. Chem. Soc., 2018, 140, 11872 CrossRef CAS PubMed; (c) L. Liu, W. Lee, M. Yuan and O. Gutierrez, Comments Inorg. Chem., 2018, 38, 210 CrossRef CAS; (d) T. L. Mako and J. A. Byers, Inorg. Chem. Front., 2016, 3, 766 RSC.
- R. S. Smith and J. K. Kochi, J. Org. Chem., 1976, 41, 502 CrossRef CAS.
- P. B. Brenner, E. Carter, P. M. Cogswell, M. F. Haddow, J. N. Harvey, D. M. Murphy, J. Nunn and C. H. Woodall, Angew. Chem., Int. Ed., 2014, 53, 1804 CrossRef PubMed.
- (a) H. Takaya, S. Nakajima, N. Nakagawa, K. Isozaki, T. Iwamoto, R. Imayoshi, N. J. Gower, L. Adak, T. Hatakeyama, T. Honma, M. Takagaki, Y. Sunada, H. Nagashima, D. Hashizume, O. Takahashi and M. Nakamura, Bull. Chem. Soc. Jpn., 2015, 88, 410 CrossRef CAS; (b) D. Noda, Y. Sunada, T. Hatakeyama, M. Nakamura and H. Nagashima, J. Am. Chem. Soc., 2009, 131, 6078 CrossRef CAS PubMed.
- (a) A. Bekhradnia and P.-O. Norrby, Dalton Trans., 2015, 44, 3959 RSC; (b) A. Hedström, Z. Izakian, I. Vreto, C.-J. Wallentin and P.-O. Norrby, Chem.–Eur. J., 2015, 21, 5946 CrossRef PubMed; (c) J. Kleimark, A. Hedström, P.-F. Larsson, C. Johansson and P.-O. Norrby, ChemCatChem, 2009, 1, 152 CrossRef CAS; (d) A. Hedström, U. Bollmann, J. Bravidor and P.-O. Norrby, Chem.–Eur. J., 2011, 17, 11991 CrossRef PubMed.
- (a) A. Casitas, J. A. Rees, R. Goddard, E. Bill, S. DeBeer and A. Fürstner, Angew. Chem., Int. Ed., 2017, 56, 10108 CrossRef CAS PubMed; (b) A. Fürstner, H. Krause and C. W. Lehmann, Angew. Chem., Int. Ed., 2006, 45, 440 CrossRef PubMed.
- J. A. Przyojski, K. P. Veggeberg, H. D. Arman and Z. J. Tonzetich, ACS Catal., 2015, 5, 5938 CrossRef CAS.
- (a) T. Parchomyk, S. Demeshko, F. Meyer and K. Koszinowski, J. Am. Chem. Soc., 2018, 140, 9709 CrossRef CAS PubMed; (b) T. Parchomyk and K. Koszinowski, Chem.–Eur. J., 2016, 22, 15609 CrossRef CAS PubMed.
- (a) S. H. Carpenter, T. M. Baker, S. B. Muñoz III, W. W. Brennessel and M. L. Neidig, Chem. Sci., 2018, 9, 7931 RSC; (b) S. B. Muñoz III, S. L. Daifuku, J. D. Sears, T. M. Baker, S. H. Carpenter, W. W. Brennessel and M. L. Neidig, Angew. Chem., Int. Ed., 2018, 57, 6496 CrossRef; (c) V. E. Fleischauer, S. B. Muñoz III, P. G. N. Neate, W. W. Brennessel and M. L. Neidig, Chem. Sci., 2018, 9, 1878 RSC; (d) J. L. Kneebone, W. B. Brennessel and M. L. Neidig, J. Am. Chem. Soc., 2017, 139, 6988 CrossRef CAS PubMed; (e) S. B. Muñoz III, S. L. Daifuku, W. W. Brennessel and M. L. Neidig, J. Am. Chem. Soc., 2016, 138, 7492 CrossRef; (f) S. L. Daifuku, J. L. Kneebone, B. E. R. Snyder and M. L. Neidig, J. Am. Chem. Soc., 2015, 137, 11432 CrossRef CAS PubMed; (g) M. H. Al-Afyouni, K. L. Fillman, W. W. Brennessel and M. L. Neidig, J. Am. Chem. Soc., 2014, 136, 15457 CrossRef CAS PubMed; (h) S. L. Daifuku, M. H. Al-Afyouni, B. E. R. Snyder, J. L. Kneebone and M. L. Neidig, J. Am. Chem. Soc., 2014, 136, 9132 CrossRef CAS PubMed.
- W. Lee, J. Zhou and O. Gutierrez, J. Am. Chem. Soc., 2017, 139, 16126 CrossRef CAS PubMed.
- A. K. Sharma, W. M. C. Sameera, M. Jin, L. Adak, C. Okuzono, T. Iwamoto, M. Kato, M. Nakamura and K. Morokuma, J. Am. Chem. Soc., 2017, 139, 16117 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. P. Plesniak, H.-M. Huang and D. J. Procter, Nat. Rev. Chem., 2017, 1, 0077 CrossRef; (b) B. Quiclet-Sire and S. Z. Zard, Proc. R. Soc. A, 2017, 473, 20160859 CrossRef PubMed; (c) For a recent example of the radical cascade in total synthesis, see: Z. G. Brill, H. K. Grover and T. J. Maimone, Science, 2016, 352, 1078 CrossRef CAS PubMed; (d) U. Wille, Chem. Rev., 2013, 113, 813 CrossRef CAS; (e) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef CAS PubMed.
- J. Li, J. Chen, W. Jiao, G. Wang, Y. Li, X. Cheng and G. Li, J. Org. Chem., 2016, 81, 9992 CrossRef CAS PubMed.
- G. C. Fu, ACS Cent. Sci., 2017, 3, 692 CrossRef CAS.
- H. Cong and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 3788 CrossRef CAS PubMed.
- For computational details, see ESI†.
- L. Liu, W. Lee, J. Zhou, S. Bandyopadhyay and O. Gutierrez, Tetrahedron, 2019, 75, 129 CrossRef CAS.
- Potential challenges include: (a) diffusion-control rates of radical/radical recombination; (b) H-atom abstraction from solvent, non-stereoselective arylation with ferric species; (c) single electron transfer (SET) leading to carbocationic (or anionic) pathways, and, inter alia; (d) arylation in the outer-sphere of the chiral iron species that prevent high-levels of stereocontrol in the fragment-coupling step..
- (a) D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 74 CrossRef CAS PubMed; (b) H. Jiang, G. Seidler and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 16528 CrossRef CAS.
- M. Jin and M. Nakamura, Chem. Lett., 2011, 40, 1012 CrossRef CAS.
- (a) T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. Novak, J. Z. Zhong, R. B. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 260 CrossRef CAS; (b) S. Arshadi, S. Ebrahimiasl, A. Hosseinian, A. Monfraed and E. Vessally, RSC Adv., 2019, 9, 8964 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and full characterization of all starting materials and products, spectroscopic data, and computational details. CCDC 1957217. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00467g |
| This journal is © The Royal Society of Chemistry 2020 |