
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing the selectivity of prolinamide organocatalysts using the mechanical bond in [2]rotaxanes†
María
Calles
a,
Julio
Puigcerver
a,
Diego A.
Alonso
 b,
Mateo
Alajarin
a,
Alberto
Martinez-Cuezva
*a and
Jose
Berna
*a
b,
Mateo
Alajarin
a,
Alberto
Martinez-Cuezva
*a and
Jose
Berna
*a
aDepartamento de Química Orgánica, Facultad de Química, Regional Campus of International Excellence “Campus Mare Nostrum”, Universidad de Murcia, E-30100, Murcia, Spain. E-mail: ppberna@um.es; amcuezva@um.es
bDepartamento de Química Orgánica, Facultad de Ciencias, Universidad de Alicante, E-03080, Alicante, Spain
First published on 11th March 2020
Abstract
The synthesis of a pair of switchable interlocked prolinamides and their use as organocatalysts in three different enamine-activated processes are reported. A diacylaminopyridine moiety was incorporated into the thread for directing [2]rotaxane formation further allowing the association of complementary small molecules. The rotaxane-based systems were tested as organocatalysts in asymmetric enamine-mediated processes, revealing a significantly improved catalytic ability if compared with the non-interlocked thread. The presence of an electron-withdrawing nitro group at the macrocycle helps to achieve high conversions and enantioselectivities. These systems are able to interact with N-hexylthymine as a cofactor to form supramolecular catalysts displaying a divergent catalytic behaviour. The presence or absence of the cofactor controls the chemoselectivity in competitive reactions.
1. Introduction
Asymmetric organocatalysis has become a powerful tool for the synthesis of sophisticated molecules from easily available starting materials.1 In this area, (S)-proline was found to be a promising catalyst for asymmetric aldol transformations,2,3 and consequently the development of catalytic scaffolds bearing this privileged moiety is still a hot topic.During the last few decades the interest of the scientific community in the synthesis and study of mechanically interlocked molecules (MIMs) has undergone a huge evolution.4 Within the range of applications of rotaxanes, the most frequently employed MIMs, those related to the study of their chemical reactivity, for example their use as organocatalysts5 or ligands in metal catalysed transformations,6 are lately capturing the attention of chemists. In this regard, switchable rotaxane-based catalysts, which are able to change their catalytic activity (ON/OFF, enantio- or diastereo-selectivity alterations and diverse activation modes), are of high interest.7 In general, the macrocycle inhibits catalysis when located over the catalytically active site.8 As a result, non-interlocked threads are more reactive than the corresponding interlocked systems, although displaying poorer selectivities. In contrast, the more rigid and confined interlocked catalysts9 oftentimes afford higher diastereo- or enantio-selectivities.10 In this line, we have demonstrated that the location of a polyamide ring close to a pyrrolidine active site of an interlocked catalyst switches the enantioselective course of a process, generating both possible enantiomers of the final products via an enantiodivergent approach.11 Recently, Leigh and coworkers reported controlled dynamic switching allowing the enantioselectivity of a rotaxane-catalyzed reaction to be reversed, thus highlighting the importance of the position of the ring with regard to the catalytically active site.12
Keeping in mind the potential of mechanically interlocked architectures for acting as catalysts,13 herein we have designed a series of chiral prolinamide-based rotaxanes bearing a diacylaminopyridine (DAP) moiety as the template,14 ready to be used as organocatalysts (Fig. 1).15 The selection of the DAP functionality as a binding site might enable these systems to modulate their activity by interaction with complementary molecules, such as N-hexylthymine acting as a supramolecular co-catalyst.16,17 Furthermore, we postulated that the proximity of the macrocycle to the catalytically active site (pyrrolidine core, Fig. 1, green circle) and its restricted translational motion between the bulky groups (Fig. 1, grey circle) could influence the course of the studied processes with unforeseeable trends. The position of the ring near the active site would generate a restricted dynamic chiral pocket leading to an increase of the selectivity of the catalytic process. In this context, the effect of the strength of the interaction between the interlocked components on the catalytic behaviour has been also studied by varying the acidity the NH of the isophthalamide units through the incorporation of different substituents18 on the macrocycle (Fig. 1, blue circle).
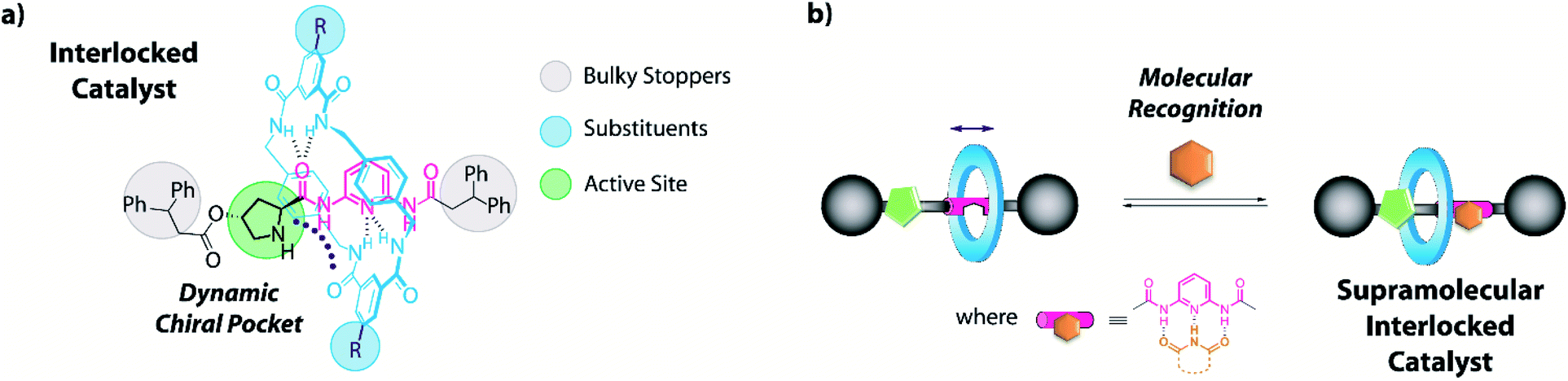 | ||
| Fig. 1 Design of (a) an interlocked DAP-based organocatalyst and (b) the formation of its hydrogen-bonding supramolecular complex. | ||
2. Results and discussion
2.1 Synthesis of mechanically interlocked DAP-based prolinamides
We started the synthesis from the commercially available Boc protected trans-4-hydroxy-L-proline 1 (Scheme 1). The amidation reaction between 1 and N-(6-aminopyridin-2-yl)-3,3-diphenylpropanamide (S1),14a in the presence of ethyl chloroformate and Et3N, yielded compound 2 in 59% yield. Then, the esterification of 2 with 3,3-diphenylpropanoyl chloride provided the protected thread 3 in moderate yield. The corresponding Leigh-type [2]rotaxanes 5a,b were readily obtained by carrying out a five-component reaction with p-xylylenediamine and a suitable isophthaloyl chloride (R2 = H or NO2) (see the ESI† for further details).19 Boc deprotection of the resulting prolinamides afforded the thread 4 and the rotaxanes 6a,b (in only 3 and 4 synthetic steps, respectively), which can be directly tested as organocatalysts.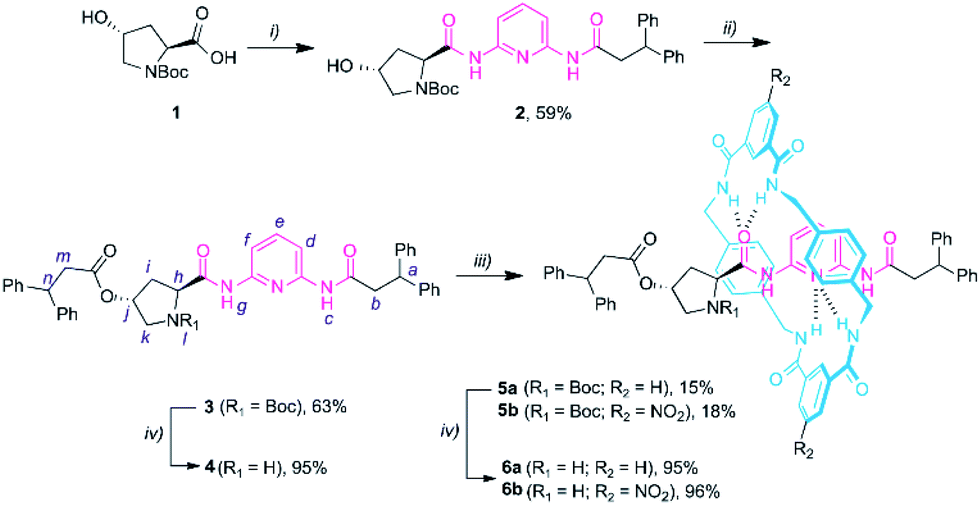 | ||
| Scheme 1 Synthesis of DAP-based non-interlocked and interlocked catalysts 4 and 6a-b. Reaction conditions: (i) ethyl chloroformate, Et3N, THF, 0 °C; then addition of N-(6-aminopyridin-2-yl)-3,3-diphenylpropanamide S1; 25 °C, overnight; then reflux for 3 h; (ii) 3,3-diphenylpropanoyl chloride, Et3N, CH2Cl2, 25 °C, overnight; (iii) p-xylylenediamine, isophthaloyl dichloride, Et3N, CHCl3, 25 °C, 4 h; (iv) TFA, CHCl3, overnight. Experimental procedures can be found in the ESI.† | ||
We investigated the ring location in the thread in rotaxane 6a. The comparison of the 1H NMR spectra of thread 4 and rotaxane 6a recorded in CDCl3 showed that the signals of the pyridine ring (Hd, He and Hf, see lettering in Scheme 1) are shifted to a higher field for 6a as a result of binding with the benzylic amide macrocycle. However, the magnitude of this shifting is slightly smaller (see the ESI, Table S1†) than that in other DAP-based rotaxanes14 pointing out that the ring could be also interacting with other hydrogen bonding motifs of the thread. Indeed, the proximity of the five-membered ring to the encircled DAP unit of 6a causes concomitant shifting of the 1H NMR signals of the pyrrolidine core indicating that the macrocycle also remains close to the active site, structurally defining a dynamic chiral pocket.
The DAP functionality is able to interact with neutral molecules, such as barbiturates, flavins or thymine derivatives, via a recognition process by forming a complementary DAD–ADA hydrogen bonding network.20 At this point, we reasoned that the DAP unit in 6 could enable the formation of a supramolecular complex with a suitable guest inducing the translation of the ring to the proline ester frame (see Fig. 2). In this regard, we next explored the ability of rotaxanes 6 to interact with N-hexylthymine (T) by calculating the association constant of the formed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex.14a We found that this cofactor is able to compete with the ring for the DAP unit in the thread. Titration 1H NMR experiments (CD2Cl2, 298 K) showed that 6a,b are able to bind to T with similar association constants of ∼20 M−1 through the DAP unit (see the ESI, Fig. S2–S5†). To further confirm this weak association we carried out 1H PGSE (Pulsed Gradient Spin Echo) diffusion measurements on solutions of T and 6b in CDCl3 at 298 K, revealing an 8% decrease of the D coefficient of the thymine derivative as a consequence of its complexation with the rotaxane (see the ESI, Tables S7 and S8†).
:1 complex.14a We found that this cofactor is able to compete with the ring for the DAP unit in the thread. Titration 1H NMR experiments (CD2Cl2, 298 K) showed that 6a,b are able to bind to T with similar association constants of ∼20 M−1 through the DAP unit (see the ESI, Fig. S2–S5†). To further confirm this weak association we carried out 1H PGSE (Pulsed Gradient Spin Echo) diffusion measurements on solutions of T and 6b in CDCl3 at 298 K, revealing an 8% decrease of the D coefficient of the thymine derivative as a consequence of its complexation with the rotaxane (see the ESI, Tables S7 and S8†).
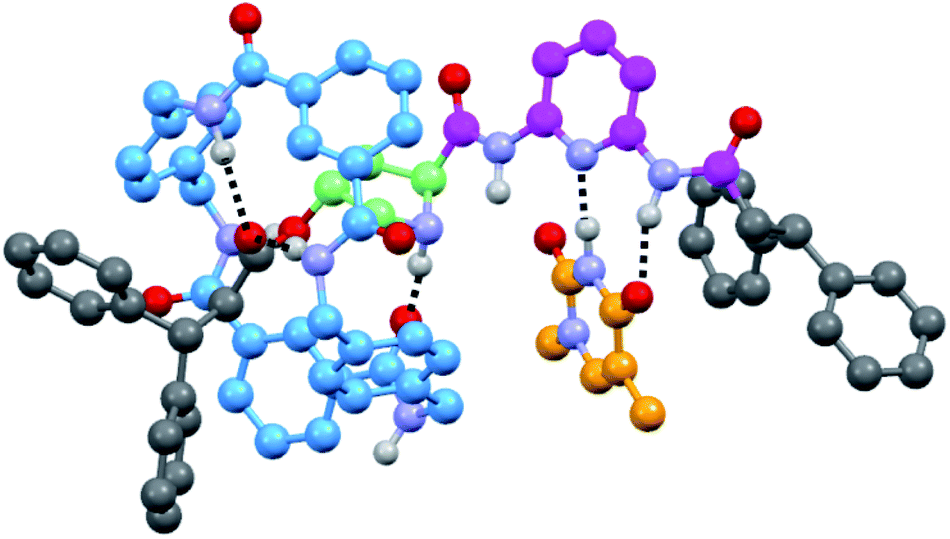 | ||
| Fig. 2 Computed minimum-energy co-conformer of the supramolecular complex 6a·T′ (with T′ = N1-methylthymine) at the M06/cc-pVDZ theoretical level. | ||
Note that the association constants of 6·T are lower than those of other reported similar complexes,14 the proline fragment being the only differential structural fragment. For further information, we computed the 6a·T′ (with T′ = N1-methylthymine) supramolecular aggregate at the DFT level (Fig. 2) in which the ring simultaneously interacts with the ester and the amino group of the pyrrolidine core (see the ESI†). The formation of only two out of three possible H-bonds between the DAP and thymine units, probably to favour aromatic interactions with the nearby diphenylmethyl group, accounts for the moderate strength of this interaction. It should be noted that the estimated magnitude of the association constants of 6·T are in the same order as that obtained for the free thread 4 (22 M−1) (see the ESI, Fig S6 and S7†), reinforcing the idea of the disturbing effect of the proximal five-membered ring on the H-bonding DAD array.
2.2 Background reactivity of the thread 4 and rotaxane 6a in the presence of acetone
A characteristic scenario found in enamine-type transformations catalysed by pyrrolidine-based systems is catalyst deactivation due to undesirable side reactions. The formation of cyclic species obtained by the intramolecular attack of the corresponding enamine on various side groups of the catalyst, such as secondary amides or carboxyl groups, is often reported.21 The presence of such relatively stable species generally results in a decrease of the reaction rate (low conversion) and selectivity (low e. r.). This is why we monitored the stability of thread 4 and rotaxane 6a in the presence of an excess of acetone (Scheme 2). We found that after 48 h, around 50% of the thread 4 was consumed, affording the cyclic imidazolidone 7 (Scheme 2a and Fig. S8†). Importantly, compound 7, easily isolable and characterised by HRMS (ESI+), showed a moderate stability in solution. After 72 h in CDCl3 at room temperature, 70% conversion of 7 into the initial thread 4 was observed, releasing free acetone (ESI, Fig. S9†). In stark contrast, rotaxane 6a remained completely unaffected when subjected to the same reaction conditions as the mechanical bond precludes the formation of the undesired cyclic byproduct 8 (Scheme 2b). | ||
| Scheme 2 Formation of the imidazolidone derivative of (a) thread 4 and (b) rotaxane 6a. Reaction conditions: (i) acetone (20 equiv.), CDCl3 (0.025 M), 25 °C, 2 days. The full experimental procedures can be found in the ESI.† | ||
2.3 Catalytic activity of the interlocked prolinamides
Once the DAP-based rotaxanes 6 were assembled, we decided to explore their aptitude as organocatalysts by comparing their activity with that of the free thread 4. For this study we chose three enamine-type transformations: aldol reactions between acetone and p-nitrobenzaldehyde22 or phenylglyoxylic acid23 and the Michael addition of acetone to β-nitrostyrene (Fig. 3).24 Testing the activity of N-hexylthymine as a cofactor was also planned.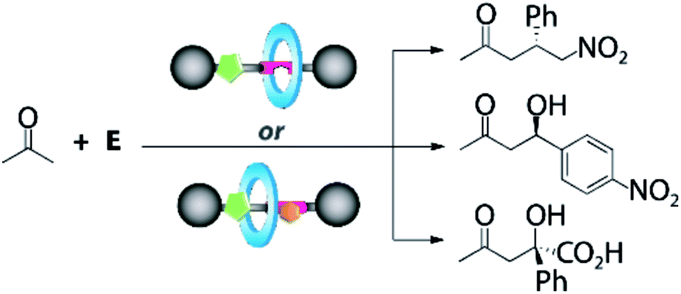 | ||
| Fig. 3 Selected enamine-type processes studied in this work. | ||
:4, 100:1). In contrast moderate conversions were achieved when rotaxanes 6a,b were used, although affording adduct 9 in a poor e. r. (Table 1, entries 2 and 3).
|
|
||||
|---|---|---|---|---|
| Entry | Cat | T | % convb | e. r.c |
| a Reaction conditions: acetone (0.25 mmol), β-nitrostyrene (0.025 mmol), catalyst (10 mol%), N-hexylthymine (0 or 0.125 mmol), CH2Cl2 (100 μL), 25 °C, 5 days. b Determined by 1H NMR from the crude reaction. c e. r. determined by HPLC using a Daicel ChiralPak AS-H column. | ||||
| 1 | 4 | NO | <5 | — |
| 2 | 6a | NO | 55 | 57:43 |
| 3 | 6b | NO | 28 | 54:46 |
| 4 | 4 | YES | 17 | 57:43 |
| 5 | 6a | YES | 85 | 78:22 |
| 6 | 6b | YES | 95 | 91:9 |
| 7 | — | YES | 0 | 0 |

Interestingly, the presence of up to 5 equivalents of N-hexylthymine (T) in the reaction media positively altered the course of the process (Table 1, entries 4–6; see the ESI for further details, Table S5†) whereas other complementary cofactors (barbital and non-alkylated thymine) did not cause similar pronounced changes (see the ESI, Table S4†). It should be noted that the reaction carried out in the presence of T without the catalyst was fully unproductive (Table 1, entry 7). In the case of thread 4, a slight increase of the conversion towards adduct 9 was detected (Table 1, compare entries 1 and 4), although it was practically unselective (57:43 e. r.). It is probable that the presence of T in the reaction media precludes the formation of 7 increasing the productivity of 4 for the formation of 9. In contrast, the more constrained rotaxanes 6 showed both better activity and selectivity. It is important to highlight that the enhancement of the catalytic behaviour of the interlocked systems compared with the free thread is a rather uncommon effect, as the macrocycle usually inhibits or reduces the activity of the functionalities settled inside or near its cavity.25 Note that the major reason for this overall outcome could lie in the inhibition of 4 by the substrate (see Scheme 2). Remarkably, rotaxane 6b, with nitro groups at the macrocycle, was observed to be the best catalyst, achieving nearly full conversion and remarkably increasing the enantiomeric ratio to 91:9 e. r. of the adduct 9 instead of the poor e. r. in the absence of T (Table 1, compare entries 3 and 6). Note that the acidity of the amide NH protons of the macrocycle in 6b is slightly increased by the electron-withdrawing NO2 groups. In this scenario it seems reasonable that an intermolecular hydrogen bonding interaction macrocycle-electrophile could be established, where the ring acts as a second activation site, similar to a bifunctional catalyst.26 This performance is noteworthy: catalyst 6b, which initially is a poorly active and completely unselective system, is converted to an enhanced supramolecular catalyst 6b·T upon addition of a complementary cofactor. Additional control experiments (see the ESI, Table S6†) carried out using the non-interlocked macrocycle as the catalyst, alone or in combination with 4 and/or T, demonstrated the need for the mechanical bond to obtain good activities and selectivities during the considered Michael addition.
:11, 50:50). Remarkably, higher conversions were obtained when the rotaxanes 6a,b were used and, more importantly, the formation of the enone 11 was almost avoided (Table 2, entries 2 and 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat | T | % convb |
10:11c |
e. r.d |
|
a Reaction conditions: acetone (0.25 mmol), p-nitrobenzaldehyde (0.025 mmol), catalyst (10 mol%), N-hexylthymine (0 or 0.125 mmol), CH2Cl2 (100 μL), 25 °C, 4 days.
b Determined by 1H NMR from the crude reaction.
c Ratio of 10:11 was determined by 1H NMR analysis.
d e. r. determined by HPLC using a Daicel ChiralPak AS-H column.
|
|||||
| 1 | 4 | NO | 14 | 50:50 |
76:24 |
| 2 | 6a | NO | 58 | 91:9 |
76:24 |
| 3 | 6b | NO | 60 | 100:0 |
88:12 |
| 4 | 4 | YES | 76 | 8317 |
62:38 |
| 6 | 6b | YES | 78 | 91:9 |
70:30 |
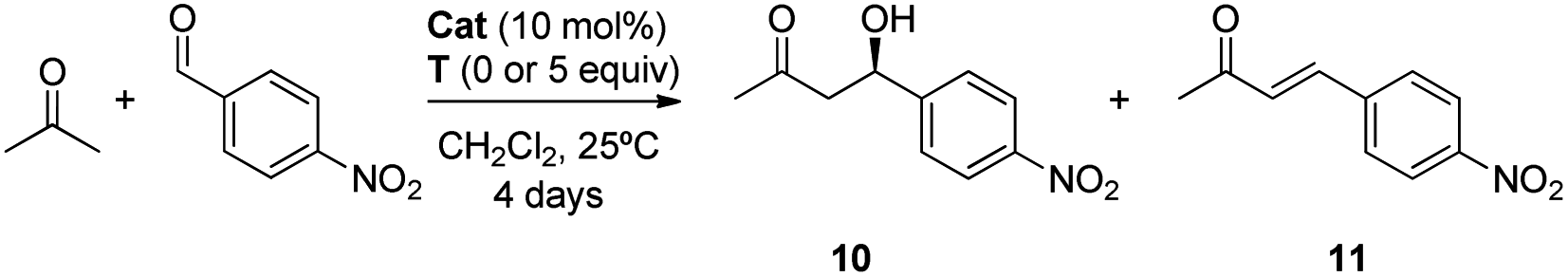
As in prior results, the nitro-containing rotaxane 6b was the best catalyst, improving the conversion to adduct 10 (60%), the chemoselectivity (enone 11 was not detected) and the enantioselectivity (88:12 e. r.) of the process (Table 2, entry 3). The presence of the mechanical bond apparently creates a well-defined chiral environment where the enamine intermediate is located, inside which the new C–C bond forming reaction occurs, generating enantioenriched products with higher selectivities.
In this transformation the addition of thymine as the cofactor was unfruitful, lowering the enantiomeric ratios, although displaying a slight increase of the conversion (Table 2, entries 4–6). Apparently, in this reaction the activating interaction of macrocycle-electrophile is better established in the initial rotaxane than in the thymine-cofactoring state.
:8 e. r. for aldol 12 after 48 hours). When thymine T is present the e. r. decreased (Table 3, entry 5).
|
|
||||
|---|---|---|---|---|
| Entry | Cat | T | % convb | e. r.c |
| a Reaction conditions: acetone (0.25 mmol), phenylglyoxylic acid (0.025 mmol), catalyst (10 mol%), N-hexylthymine (0 or 0.125 mmol), CH2Cl2 (100 μL), 25 °C, 2 days. b Determined by 1H NMR from the crude reaction. c e. r. determined by HPLC using a Daicel ChiralPak AS-H column. For analytical reasons, the adduct 12 was derivatized to its methyl ester (see the ESI). | ||||
| 1 | 4 | NO | 93 | 75:25 |
| 2 | 6a | NO | 54 | 89:11 |
| 3 | 6b | NO | 85 | 92:8 |
| 4 | 4 | YES | 100 | 60:40 |
| 5 | 6b | YES | 38 | 72:28 |
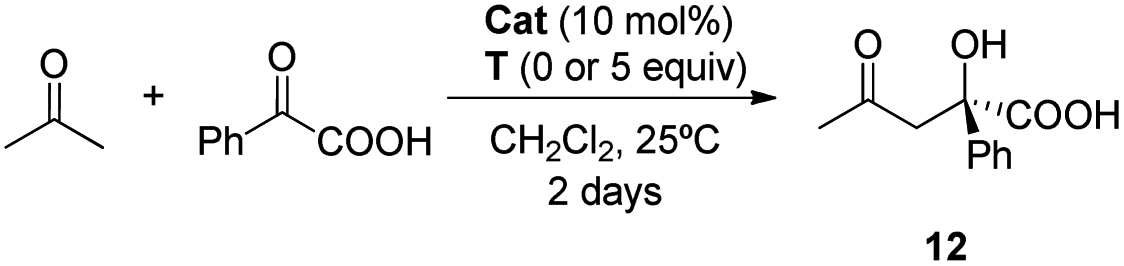
2.4 Exploring the activity of the catalyst 6b in competitive experiments
Catalyst 6b clearly works under two different regimes in the studied processes, either uncomplexed or complexed with N-hexylthymine forming the supramolecular interlocked catalyst 6b·T. Thus, we envisioned that the addition or not of the cofactor T could influence the distribution rate between the final adducts when two electrophiles are simultaneously present. Therefore, we carried out competitive experiments by adding p-nitrobenzaldehyde (1 equiv.) and β-nitrostyrene (1 equiv.) to acetone (1.5 equiv.) in the presence of catalyst 6b (10 mol%), with or without T (Scheme 3).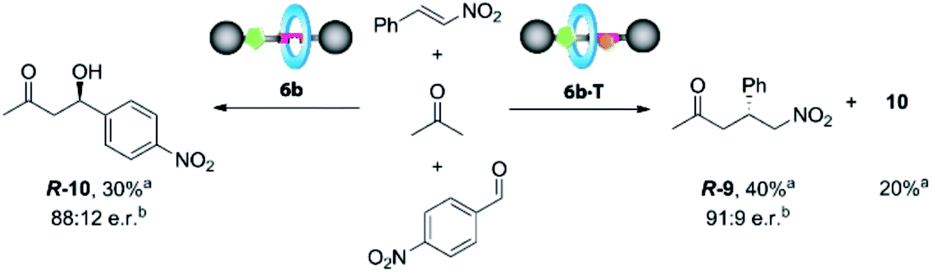 | ||
| Scheme 3 Michael versus aldol addition of acetone using rotaxane 6b as the catalyst in the presence or absence of N-hexylthymine (T). Reaction conditions: p-nitrobenzaldehyde (1 equiv.), trans-β-nitrostyrene (1 equiv.), acetone (1.5 equiv.), catalyst 6b (10 mol%), N-hexylthymine (5 equiv., if required), CH2Cl2 (0.25 M), 25 °C, 5 days.a Determined by 1H NMR from the crude reaction. b e. r. determined by HPLC using a Daicel ChiralPak AS-H column. | ||
After 5 days, we observed the formation of the aldol adduct 10 (30%), with only traces of the Michael adduct 9 (<5%) when T was not added. In contrast, the chemoselectivity became reversed in the presence of T, preferentially forming the Michael adduct 9 (40%), (ratio of 9:10, 2:1) (see the ESI, Fig. S10†). Importantly, the isolated adducts 9 and 10 maintained the enantiomeric ratios previously obtained in the individual experiments.
3. Conclusions
In summary, we have synthesized a series of chiral mechanically interlocked diacylaminopyridine-based prolinamides in a straightforward manner (only 4 synthetic steps). Their catalytic activity was modulated by complexation with a complementary DAD array (N-hexylthymine), forming a supramolecular catalyst. Importantly, the presence of the flexible and, at the same time, bulky isophthalamide ring improves the ability of the interlocked systems as catalysts when compared with the free thread, by creating a dynamic chiral pocket. As a result, the mechanical bond precludes the formation of inactive species that inhibit the catalysis, thus enhancing the overall catalytic activity of the interlocked systems. This remarkable behaviour differs from the considerably lower catalytic activity shown by previously reported rotaxane-based catalysts when compared with their non-interlocked threads. In our interlocked catalysts the benzylic amide macrocycle has not only a shielding effect but also an activating role. The presence of electron-withdrawing groups attached to the macrocycle, which increase the acidity of the NH amide groups, is beneficial for the outcomes of the assayed reactions, usually increasing the conversion and enantioselectivities. The addition or not of an external cofactor to the reaction media allows switching of the chemoselectivity in competitive experiments. All these facts allow this versatile system to work in two dissimilar regimes, being an effective catalyst in three different enamine-type processes.These results highlight that the employment of mechanically interlocked molecules as catalysts could open exciting paths in the field of asymmetric catalysis thus making a contribution to the design of new systems that could tackle challenging transformations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the MINECO and MICINN (CTQ2017-87231-P, RYC-2017-22700 and PGC2018-096616-B-I00) with joint financing by FEDER Funds and Fundación Seneca-CARM (Project 20811/PI/18). D. A. A. also thanks the University of Alicante for the financial support (VIGROB-173FI). We gratefully acknowledge Dr Marta Marin-Luna and Dr Aurelia Pastor for insightful discussions. The authors gratefully acknowledge the computer resources at Cibeles and the technical support provided by the Centro de Computación Científica-UAM (RES-QSB-2019-3-0012).Notes and references
- (a) B. List, Chem. Rev., 2007, 107, 5413–5415 CrossRef CAS; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed; (c) D. W. C. McMillan, Nature, 2008, 304–308 CrossRef PubMed.
- (a) Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615–1621 CrossRef CAS; (b) U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed. Engl., 1971, 10, 496–497 CrossRef CAS.
- P. I. Dalko, Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications, Wiley, Weinheim, 2013 Search PubMed.
- (a) J.-P. Sauvage and P. Garpard, From Non-Covalent Assemblies to Molecular Machines, Wiley, Weinheim, 2011 Search PubMed; (b) C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, Wiley, New York, 2016 CrossRef.
- (a) Y. Tachibana, N. Kihara and T. Takata, J. Am. Chem. Soc., 2004, 126, 3438–3439 CrossRef CAS PubMed; (b) J. Berna, M. Alajarin and R.-A. Orenes, J. Am. Chem. Soc., 2010, 132, 10741–10747 CrossRef CAS PubMed; (c) B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed; (d) J. Y. C. Lim, N. Yuntawattana, P. D. Beer and C. K. Williams, Angew. Chem., Int. Ed., 2019, 58, 6007–6011 CrossRef CAS PubMed; (e) N. Pairault, H. Zhu, D. Jansen, A. Huber, C. G. Daniliuc, S. Grimme and J. Niemeyer, Angew. Chem., Int. Ed., 2020, 59, DOI:10.1002/anie.201913781.
- (a) P. Thordarson, E. J. A. Bijsterveld, A. E. Rowan and R. J. M. Nolte, Nature, 2003, 424, 915–918 CrossRef CAS PubMed; (b) G. Hattori, T. Hori, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2007, 129, 12930–12931 CrossRef CAS PubMed; (c) Y. Suzaki, K. Shimada, E. Chihara, T. Saito, Y. Tsuchido and K. Osakada, Org. Lett., 2011, 13, 3774–3777 CrossRef CAS PubMed; (d) A. W. Heard and S. M. Goldup, Chem, 2020 DOI:10.1016/j.chempr.2020.02.006.
- (a) V. Blanco, A. Carlone, K. D. Hänni, D. A. Leigh and B. Lewandowski, Angew. Chem., Int. Ed., 2012, 51, 5166–5169 CrossRef CAS PubMed; (b) V. Blanco, D. A. Leigh, V. Marcos, J. A. Morales-Serna and A. L. Nussbaumer, J. Am. Chem. Soc., 2014, 136, 4905–4908 CrossRef CAS PubMed; (c) V. Blanco, D. A. Leigh, U. Lewandowska, B. Lewandowski and V. Marcos, J. Am. Chem. Soc., 2014, 136, 15775–15780 CrossRef CAS PubMed; (d) C. M. Álvarez, H. Barbero and D. Miguel, Eur. J. Org. Chem., 2015, 6631–6640 CrossRef; (e) A. Martinez-Cuezva, A. Saura-Sanmartin, T. Nicolas-Garcia, C. Navarro, R.-A. Orenes, M. Alajarin and J. Berna, Chem. Sci., 2017, 8, 3775–3780 RSC; (f) K. Eichstaedt, J. Jaramillo-Garcia, D. A. Leigh, V. Marcos, S. Pisano and T. A. Singleton, J. Am. Chem. Soc., 2017, 139, 9376–9381 CrossRef CAS PubMed; (g) Y.-J. Lee, K.-S. Liu, C.-C. Lai, Y.-H. Liu, S.-M. Peng, R. P. Cheng and S.-H. Chiu, Chem.–Eur. J., 2017, 23, 9756–9760 CrossRef CAS PubMed; (h) C. Biagini, S. D. P. Fielden, D. A. Leigh, F. Schaufelberger, S. Di Stefano and D. Thomas, Angew. Chem., Int. Ed., 2019, 58, 9876–9880 CrossRef CAS PubMed.
- For examples of the reduced reactivity of the functionalities located inside the macrocycle cavity see: (a) A. H. Parham, B. B. Windisch and F. Vögtle, Eur. J. Org. Chem., 1999, 1999, 1233–1238 CrossRef; (b) T. Oku, Y. Furusho and T. Takata, Org. Lett., 2003, 5, 4923–4925 CrossRef CAS PubMed; (c) D. A. Leigh and E. M. Perez, Chem. Commun., 2004, 2262–2263 RSC; (d) J. Winn, A. Pinczewska and S. M. Goldup, J. Am. Chem. Soc., 2013, 135, 13318–13321 CrossRef CAS PubMed.
- For examples of highly confined catalysts see: (a) S. R. Shenoy, F. R. P. Crisóstomo, T. Iwasawa and J. Rebek, J. Am. Chem. Soc., 2008, 130, 5658–5659 CrossRef CAS PubMed; (b) I. Čorić and B. List, Nature, 2012, 483, 315–319 CrossRef PubMed.
- (a) M. Galli, J. E. M. Lewis and S. M. Goldup, Angew. Chem., Int. Ed., 2015, 54, 13545–13549 CrossRef CAS PubMed; (b) S. Hoekman, M. O. Kitching, D. A. Leigh, M. Papmeyer and D. Roke, J. Am. Chem. Soc., 2015, 137, 7656–7659 CrossRef CAS PubMed.
- A. Martinez-Cuezva, M. Marin-Luna, D. A. Alonso, D. Ros-Ñiguez, M. Alajarin and J. Berna, Org. Lett., 2019, 21, 5192–5196 CrossRef CAS PubMed.
- (a) Y. Cakmak, S. Erbas-Cakmak and D. A. Leigh, J. Am. Chem. Soc., 2016, 138, 1749–1751 CrossRef CAS PubMed; (b) M. Dommaschk, J. Echavarren, D. A. Leigh, V. Marcos and T. A. Singleton, Angew. Chem., Int. Ed., 2019, 58, 14955–14958 CrossRef CAS PubMed.
- For a catenane-based catalyst, see: (a) R. Mitra, H. Zhu, S. Grimme and J. Niemeyer, Angew. Chem., Int. Ed., 2017, 56, 11456–11459 CrossRef CAS PubMed. For molecular knot-based catalysts, see: (b) V. Marcos, A. J. Stephens, J. Jaramillo-Garcia, A. L. Nussbaumer, S. L. Woltering, A. Valero, J.-F. Lemonnier, I. J. Vitorica-Yrezabal and D. A. Leigh, Science, 2016, 352, 1555–1559 CrossRef CAS PubMed; (c) G. Gil-Ramírez, S. Hoekman, M. O. Kitching, D. A. Leigh, I. Vitorica-Yrezabal and G. Zhang, J. Am. Chem. Soc., 2016, 138, 13159–13162 CrossRef PubMed.
- (a) A. Martinez-Cuezva, J. Berna, R.-A. Orenes, A. Pastor and M. Alajarin, Angew. Chem., Int. Ed., 2014, 53, 6762–6767 CrossRef CAS PubMed; (b) A. Martinez-Cuezva, A. Pastor, G. Cioncoloni, R.-A. Orenes, M. Alajarin, M. D. Symes and J. Berna, Chem. Sci., 2015, 6, 3087–3094 RSC; (c) A. Martinez-Cuezva, F. Carro-Guillen, A. Pastor, M. Marin-Luna, R.-A. Orenes, M. Alajarin and J. Berna, ChemPhysChem, 2016, 17, 1920–1926 CrossRef CAS PubMed; (d) A. Tron, I. Pianet, A. Martinez-Cuezva, J. H. R. Tucker, L. Pisciottani, M. Alajarin, J. Berna and N. D. McClenaghan, Org. Lett., 2017, 19, 154–157 CrossRef CAS PubMed.
- (a) B. List, Chem. Commun., 2006, 819–824 RSC; (b) P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138–6171 CrossRef CAS PubMed; (c) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600–1632 RSC.
- For selected examples, see: (a) D.-Q. Xu, H.-D. Yue, S.-P. Luo, A.-B. Xia, S. Zhang and Z.-Y. Xu, Org. Biomol. Chem., 2008, 6, 2054–2057 RSC; (b) Ö. Reis, S. Eymur, B. Reis and A. S. Demir, Chem. Commun., 2009, 9, 1088–1090 RSC; (c) D. Uraguchi, Y. Ueki and T. Ooi, Science, 2009, 326, 120–123 CrossRef CAS PubMed; (d) M. Bella, D. M. S. Schietroma, P. P. Cusella, T. Gasperi and V. Visca, Chem. Commun., 2009, 597–599 RSC; (e) N. El-Hamdouni, X. R. R. Companyo and A. Moyano, Chem.–Eur. J., 2010, 16, 1142–1148 CrossRef CAS PubMed; (f) A. S. Demir and S. Eymur, Tetrahedron: Asymmetry, 2010, 21, 112–115 CrossRef CAS; (g) K. Oh, J.-Y. Li and J. Ryu, Org. Biomol. Chem., 2010, 8, 3015–3024 RSC, For a review, see: (h) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed.
- (a) M. L. Clarke and J. A. Fuentes, Angew. Chem., Int. Ed., 2007, 46, 930–933 CrossRef CAS PubMed; (b) J. A. Fuentes, T. Lebl, A. M. Z. Slawin and M. L. Clarke, Chem. Sci., 2011, 2, 1997–2005 RSC.
- (a) S.-Y. Chang, H. Sop Kim, K.-J. Chang and K.-S. Jeong, Org. Lett., 2004, 62, 181–184 CrossRef PubMed; (b) A. Martinez-Cuezva, L. V. Rodrigues, C. Navarro, F. Carro-Guillen, L. Buriol, C. P. Frizzo, M. A. P. Martins, M. Alajarin and J. Berna, J. Org. Chem., 2015, 80, 10049–10059 CrossRef CAS PubMed.
- J. Berna, G. Bottari, D. A. Leigh and E. M. Perez, Pure Appl. Chem., 2007, 79, 39–54 CAS.
- (a) A. D. Hamilton and D. Van Engen, J. Am. Chem. Soc., 1987, 109, 5035–5036 CrossRef CAS; (b) S.-K. Chang, D. Van Engen, E. Fan and A. D. Hamilton, J. Am. Chem. Soc., 1991, 113, 7640–7645 CrossRef CAS; (c) N. Tamura, K. Mitsui, T. Nabeshima and Y. Yano, J. Chem. Soc., Perkin Trans. 2, 1994, 5, 2229–2237 RSC.
- (a) B. List, L. Hoang and H. J. Martin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5839–5842 CrossRef CAS PubMed; (b) D. Gryko and R. Lipiński, Adv. Synth. Catal., 2005, 347, 1948–1952 CrossRef CAS; (c) T. Kano, M. Ueda, J. Takai and K. Maruoka, J. Am. Chem. Soc., 2006, 128, 6046–6047 CrossRef CAS PubMed; (d) A. L. Fuentes de Arriba, L. Simon, C. Raposo, V. Alcázar, F. Sanz, F. M. Muñiz and J. R. Morán, Org. Biomol. Chem., 2010, 8, 2979–2985 RSC.
- (a) B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS; (b) B. List, P. Pojarliev and C. Castello, Org. Lett., 2001, 3, 573–575 CrossRef CAS PubMed; (c) A. Martinez, M. van Gemmeren and B. List, Synlett, 2014, 25, 961–964 CrossRef CAS; (d) A. Martinez, C. Zumbansen, A. Döhring, M. van Gemmeren and B. List, Synlett, 2014, 25, 932–934 CrossRef CAS.
- (a) Z. Tang, L.-F. Cun, X. Cui, A.-Q. Mi, Y.-Z. Jiang and L.-Z. Gong, Org. Lett., 2006, 8, 1263–1266 CrossRef CAS PubMed; (b) X.-Y. Xu, Z. Tang, Y.-Z. Wang, S.-W. Luo, L.-F. Cun and L.-Z. Gong, J. Org. Chem., 2007, 72, 9905–9913 CrossRef CAS PubMed.
- (a) B. List, P. Pojarliev and H. J. Martin, Org. Lett., 2001, 3, 2423–2425 CrossRef CAS PubMed; (b) N. Mase, K. Watanabe, H. Yoda, K. Takabe, F. Tanaka and C. F. Barbas, J. Am. Chem. Soc., 2006, 128, 4966–4967 CrossRef CAS PubMed; (c) C.-L. Cao, M.-C. Ye, X.-L. Sun and Y. Tang, Org. Lett., 2006, 8, 2901–2904 CrossRef CAS PubMed; (d) D. A. Alonso, A. Baeza, R. Chinchilla, C. Gómez, G. Guillena, I. M. Pastor and D. J. Ramón, Molecules, 2017, 22, 895–946 CrossRef PubMed.
- For previous studies reporting the activation of the reactivity due to the presence of the mechanical bond, see: (a) A. Martinez-Cuezva, C. Lopez-Leonardo, D. Bautista, M. Alajarin and J. Berna, J. Am. Chem. Soc., 2016, 138, 8726–8729 CrossRef CAS PubMed; (b) A. Martinez-Cuezva, D. Bautista, M. Alajarin and J. Berna, Angew. Chem., Int. Ed., 2018, 57, 6563–6567 CrossRef CAS PubMed; (c) F. Modicom, E. M. G. Jamieson, E. Rochette and S. M. Goldup, Angew. Chem., Int. Ed., 2019, 58, 3875–3879 CrossRef CAS PubMed.
- The interaction between an interlocked macrocycle and the same electrophile, supported by DFT calculations, was also considered a contributing factor for improving the final outcome of the same Michael reaction using a benzylic amide rotaxane as the catalyst (see ref 11).
- The formation of the corresponding enones as byproducts has also been observed in aldol reactions catalysed by L-proline and derivatives: (a) D. Gryko, M. Zimnicka and R. Lipiński, J. Org. Chem., 2007, 72, 964–970 CrossRef CAS PubMed; (b) Q.-Z. Liu, X.-L. Wang, S.-W. Luo, B.-L. Zheng and D.-B. Qin, Tetrahedron Lett., 2008, 49, 7434–7437 CrossRef CAS; (c) ref 22c .
- G. T. Fernando, S. Goswami, S. K. Chang, S. J. Geib and A. D. Hamilton, J. Am. Chem. Soc., 1990, 112, 7393–7394 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic and mass spectrometry data for all new compounds. See DOI: 10.1039/d0sc00444h |
| This journal is © The Royal Society of Chemistry 2020 |