
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New reductive rearrangement of N-arylindoles triggered by the Grubbs–Stoltz reagent Et3SiH/KOtBu†
Andrew J.
Smith
a,
Daniela
Dimitrova
a,
Jude N.
Arokianathar
a,
Krystian
Kolodziejczak
 a,
Allan
Young
a,
Mark
Allison
a,
Darren L.
Poole
b,
Stuart G.
Leach
b,
John A.
Parkinson
a,
Tell
Tuttle
*a and
John A.
Murphy
*a
a,
Allan
Young
a,
Mark
Allison
a,
Darren L.
Poole
b,
Stuart G.
Leach
b,
John A.
Parkinson
a,
Tell
Tuttle
*a and
John A.
Murphy
*a
aDepartment of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: john.murphy@strath.ac.uk; tell.tuttle@strath.ac.uk
bGlaxoSmithKline Medicines Research Centre, Gunnels Wood Road, Stevenage, SG1 2NY, UK
First published on 11th March 2020
Abstract
N-Arylindoles are transformed into dihydroacridines in a new type of rearrangement, through heating with triethylsilane and potassium tert-butoxide. Studies indicate that the pathway involves (i) the formation of indole radical anions followed by fragmentation of the indole C2–N bond, and (ii) a ring-closing reaction that follows a potassium-ion dependent hydrogen atom transfer step. Unexpected behaviors of ‘radical-trap’ substrates prove very helpful in framing the proposed mechanism.
Introduction
Indoles are an important class of compounds that feature widely in medicinal chemistry. Accordingly, the synthesis and reactivity of indoles have both been well explored. The indole nucleus is electron-rich, and indoles routinely act as aromatic nucleophiles, principally undergoing substitution by electrophiles or addition of electrophiles at the 2, 3-double-bond.In view of their position as electron-rich substrates, reductive transformations of indoles are very rare,1 but we now report a new radical-based rearrangement of N-arylindoles that arises from reductive activation. The reaction is brought about by the Grubbs–Stoltz reagent,2–9 resulting from heating triethylsilane with potassium tert-butoxide. Since its discovery in 2013, this reagent has been shown to drive a range of useful transformations, including cleavage of Ar–O (Scheme 1, 1 → 2)2,5 and Ar–S bonds (3 → 4),5 regioselective C-silylation of indoles (5 → 6),3,4,6,7 reductive debenzylation of N-benzylindoles (7 → 8)8 and reduction of unsaturated hydrocarbons (9 → 10).8 A wide range of mechanisms has been proposed for these transformations featuring diverse intermediates, including (i) silyl radicals 14,2,5,8,9 (ii) silyl radical anions, e.g.15 (ref. 6, 8 and 9) and (iii) silanate anions, e.g.16;2,6,7,9 leading to extensive discussion in the literature. This is illustrated by both open-shell and closed-shell mechanisms being proposed by the same authors in back-to-back papers, as possible pathways for converting 5 → 6.6,7
 | ||
| Scheme 1 Selected transformations with both (i) triethylsilane or diethylsilane and (ii) potassium tert-butoxide. | ||
Most recently, in 2019, a unique role for the closely related KOtBu + Et2SiH2 (as well as KOtBu + Et3SiH) in hydrosilylation of styrenes (11 → 12) was proposed by Jeon et al., and supported with good evidence in a reaction that critically depends on K+–arene interactions. (In the absence of potassium ions, polymer 13 is formed instead of 12).10
Thus, this apparently simple mixture of base and silane provides great diversity in its reactions. Its applications already include methods for the desulfurisation of fuels5 and for the silylation of amines,11 together with proposed applications to the controlled cleavage of lignins.2,5 To harness its chemistry, further understanding is clearly needed of the mechanisms of its reactions. In this paper, we report a new rearrangement that transforms N-arylindoles to dihydroacridines, and we provide evidence supporting (i) an initial fragmentation of the C2–N bond of indole radical anions, followed by (ii) K+ ion-assisted hydrogen atom transfer chemistry for the conversion of the ring-opened intermediates to dihydroacridines.
Our recent report8 used Et3SiH/KOtBu to transform N-benzylindoles e.g.7 to indoles (8), and we proposed that electron transfer from radical anion 15 to the substrate could induce the fragmentation of the N-benzyl bond. In the same paper, N-allyl-3-methylindole 17 (Scheme 2) was converted to 3-methylindole 19 (35%); unexpectedly, the reaction also afforded 2-iso-propylaniline 18 (18%). Reductive ring-opening reactions of indoles, as seen in the formation of 18, were unknown at the time and inspired the investigation reported in this paper; however, very recently, elegant studies by Yorimitsu et al.1 have reductively opened indoles 20 with excess Li powder, trapping the intermediate vinyl and amidyl dianions with boron electrophiles to afford benzazaborin products 21 in high yield.1,12
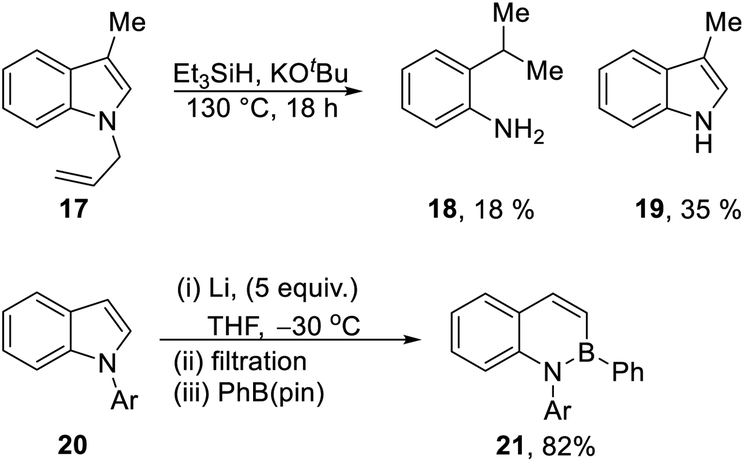
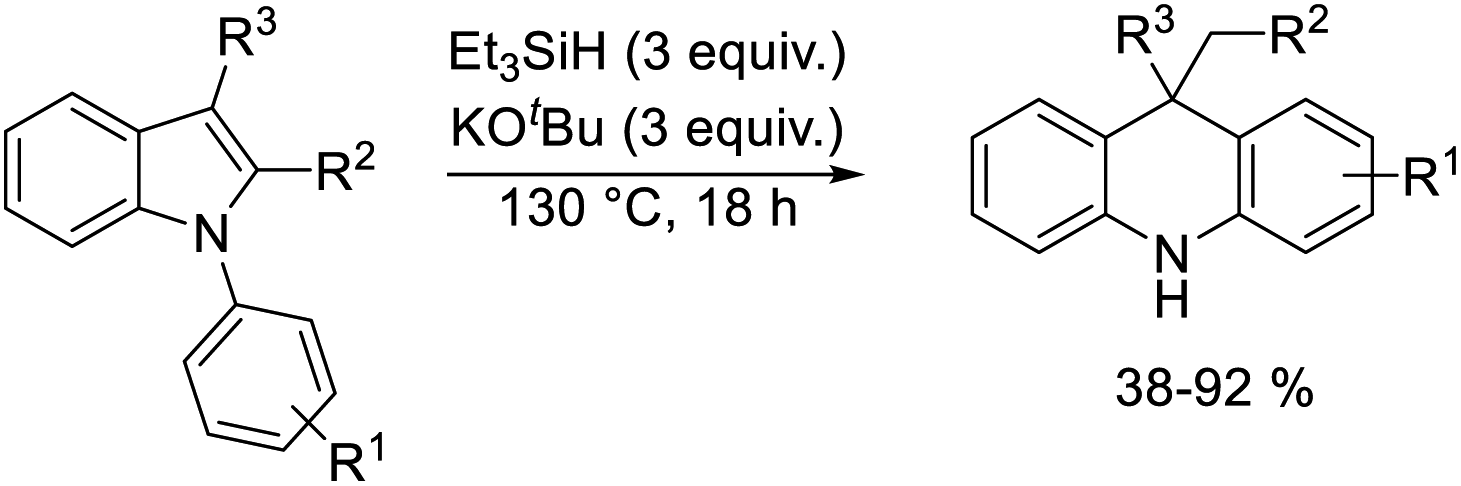
A selection of N-arylindoles 22–29 was prepared (see ESI†) and tested under the conditions shown in Table 1. Indole heterocyclic rings were cleaved, as the substrates underwent unexpected and facile conversion to 9,10-dihydroacridines 30–36 in moderate to excellent yields. Substituents were tolerated in the 2- and 3-positions of the indole (entries 1–7), as well as in the N-aryl group (entry 8). To rationalise the rearrangements, we adopt a working hypothesis for the mechanism, shown in Scheme 3. A key intermediate is radical anion 37. This could form by electron transfer from donor 15 to the indole or, alternatively, by H-atom addition (H-atom addition is discussed later in this paper) to the indole ring system, to form 39 or 40 (R = H) or an isomer, followed by deprotonation under the basic conditions of the reaction. This radical anion then fragments to afford the distal radical anion 38. Abstraction of a hydrogen atom by the vinyl radical (likely donors would include triethylsilane or silanate 16 or intermediates 39 or 40) affords the styrene/amide anion 41. This styrene now undergoes regioselective hydrogen atom addition as proposed by Jeon et al.10 to give benzyl radical anion 42, and instead of forming a C–Si bond as in Jeon's example, a C–C bond is formed as cyclisation gives radical anion 43. This could function directly as an H-atom donor to form 46 or, alternatively, deprotonation of the cyclohexadienyl radical then affords the electron-rich species 44 (or, following silylation, 45 (ref. 11)) which behaves as an electron donor.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yield% |
| a Reactions were carried out neat (i.e. without added solvent). | |||
| 1 | 22, R1 = R2 = R3 = H | 30, R1 = R2 = R3 = H | 58 |
| 2 | 23, R1 = R2 = H, R3 = Me | 31, R1 = R2 = H, R3 = Me | 77 |
| 3 | 24, R1 = R2 = H, R3 = Et | 32, R1 = H, R2 = R3 = Me | 66 |
| 4 | 25, R1 = H, R2 = R3 = Me | 32, R1 = H, R2 = R3 = Me | 53 |
| 5 | 26, R1 = R2 = H, R3 = Ph | 33, R1 = R2 = H, R3 = Ph | 92 |
| 6 | 27, R1 = R2 = H, R3 = C8H17 | 34, R1 = R2 = H, R3 = C8H17 | 55 |
| 7 | 28, R1 = R2 = H, R3 = C4H9 | 35, R1 = R2 = H, R3 = C4H9 | 71 |
| 8 | 29, R1 = 4-Me, R2 = R3 = H | 36, R1 = 2-Me, R2 = R3 = H | 38 |
 | ||
| Scheme 3 | ||
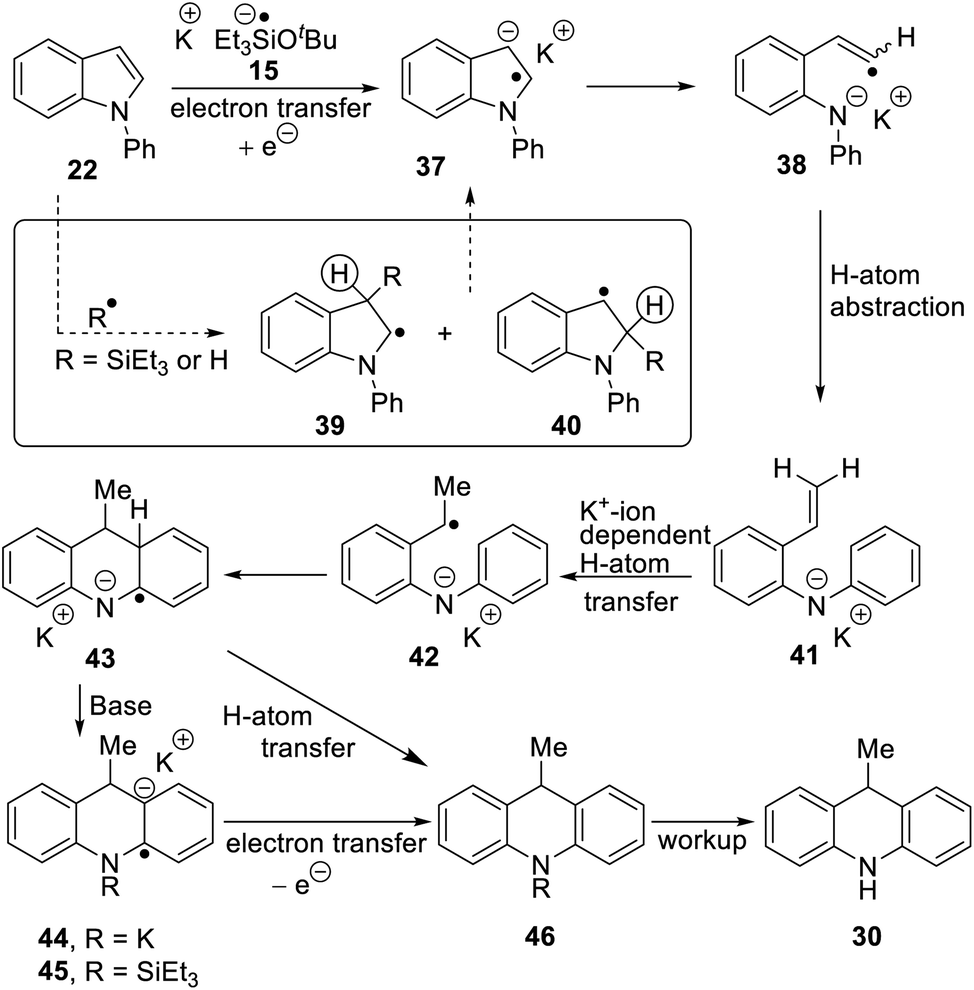
Considering the behavior of radical anion 37, fragmentation of the 5-membered ring could depend on the excess electron of the radical anion being localised extensively in the indole ring system, rather than in the N-phenyl group (Fig. 1a, Case A). Computational studies performed on the 3-methyl analogue 23 are consistent with this (Fig. 1b). We rationalise that the greater delocalisation associated with the bicyclic π-system of the indole results in this preferential localisation of the excess electron density of the radical anion.
 | ||
| Fig. 1 (a) Case A, when the unpaired electron in radical anion of N-arylindoles localises on the indole group in the radical anion of 23 (i.e.23ra), then fragmentation of the indole nucleus results, while with the excess electron is housed in the N-aryl group in 47ra, then cleavage of the indole N-aryl bond results (Case B); (b) radical anion 23ra showing localisation of spin in the indole nucleus and (c) radical anion 47ra, showing localisation of spin in the naphthyl group; (d) substrates 47 → 49 undergo indole N-aryl bond cleavage (for computational methods, see ESI†). | ||
We investigated further substrates by computation and experiment, starting with the N-naphthyl substrate 47. In this case, the naphthyl π-system is the preferred site for the radical anion (Fig. 1c). In the laboratory, that should result in cleavage of the N-naphthyl bond to form an indolyl anion and a naphthyl radical, rather than in cleavage of the indole nucleus (Fig. 1a, Case B). Accordingly, the α-naphthyl substrate 47 (Fig. 1d) was prepared. Under the standard reaction conditions, the predicted cleavage occurred to form 3-methylindole 19 (55%), and none of the corresponding dihydroacridine was detected.
Two additional substrates, 48 and 49, were prepared, featuring pyridine rings. Computation (see ESI†) suggested that, due to the electron-deficient nature of these rings, the radical anions of these substrates would house the added electron in the pyridine ring, leading again to [indole N]–pyridine bond cleavage, rather than cleavage of the indole nucleus. This was indeed the case, with indole being isolated in 48% and 39% from 48 and 49 respectively following workup, and no evidence of cleavage of the indole nucleus. Thus, the cleavages of substrates 22–29, and 47–49 follow the regioselectivity that is predicted from a study of the respective radical anions, providing strong evidence that radical anions are intermediates in these reactions.
To investigate the proposed mechanism involving SET, several other reaction conditions were investigated (Table 2) with substrate 22. The standard conditions for Et3SiH + KOtBu afforded 30 in 58% yield (entry 1). With a view to forming the radical anion of Me3SiOtBu, analogous to 15, without using triethylsilane or any other compound that contains a Si–H group, hexamethyldisilane was treated with the radical anion, potassium di-tert-butylbiphenylide (KDTBB) as reducing agent and KOtBu as base. This system afforded product 30 (33%, entry 2). This successful outcome would result if the disilane were reduced to a silyl radical and a silyl anion by KDTBB. Addition of a silyl radical to N-phenylindole7 could give 39, 40 (R = SiEt3, Scheme 3) or an isomeric radical. Both 39 and 40 have a labile hydrogen atom (circled) that can be removed either as a proton, e.g. by trimethylsilyl anion, to form Me3SiH, or as a hydrogen atom, e.g. by trimethylsilyl radical to again form Me3SiH, putting in place all the components for Scheme 3.
|
|
||
|---|---|---|
| Entry | Reagents (3 equiv. of each) | Yield 30 (%) |
| 1 | Et3SiH, KOtBu | 58 |
| 2 | KDTBB, Me6Si2, KOtBu | 33 |
| 3 | KDTBB, KOtBu | 0 |
| 4 | Me6Si2, KOtBu | 0 |
| 5 | KOtBu | 0 |
In the presence of KTDBB, but in the absence of any silane or disilane, no product 30 was formed (entry 3). When KTDBB was omitted (entry 4), no cleavage of the Si–Si bond could occur and so the reaction gave no rearranged product. Entry 5 shows that KOtBu alone could not bring about the rearrangement. From these results, it was clear that (i) an appropriate silyl compound, (ii) KOtBu and (iii) reducing power were all needed to bring about the rearrangement efficiently.
The requirement for all three components above was also shown when potassium metal was tested as a reducing agent in the presence of KOtBu (see ESI†). This led to consumption of the indole substrates. Acridine products, rather than dihydroacridines, were produced, reinforcing that reductive activation1 of the indole nucleus leads to fragmentation of the indole 5-membered ring, but the absence of H-atom sources meant that the reaction of Scheme 3 was not supported; the acridine products were formed in very low yield (5%, see ESI file†). However, from this result and from the results of Yorimitsu,1 it is clear that reductive activation of the indole nucleus of N-arylindoles does lead to fragmentation of the 5-membered ring.13
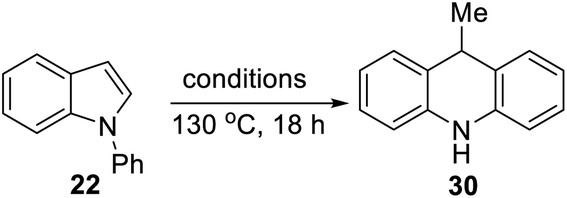
We now looked for evidence of reactive intermediates on the reaction pathway shown in Scheme 3, using substrates that incorporated radical clocks. These substrates provided most surprising outcomes and important mechanistic information. Substrates 50, 51, (Scheme 4), 61 (Scheme 6) and 73 (Scheme 7) were prepared and reacted under the standard conditions; the products were then analyzed to provide evidence of reaction intermediates.
 | ||
| Scheme 4 | ||
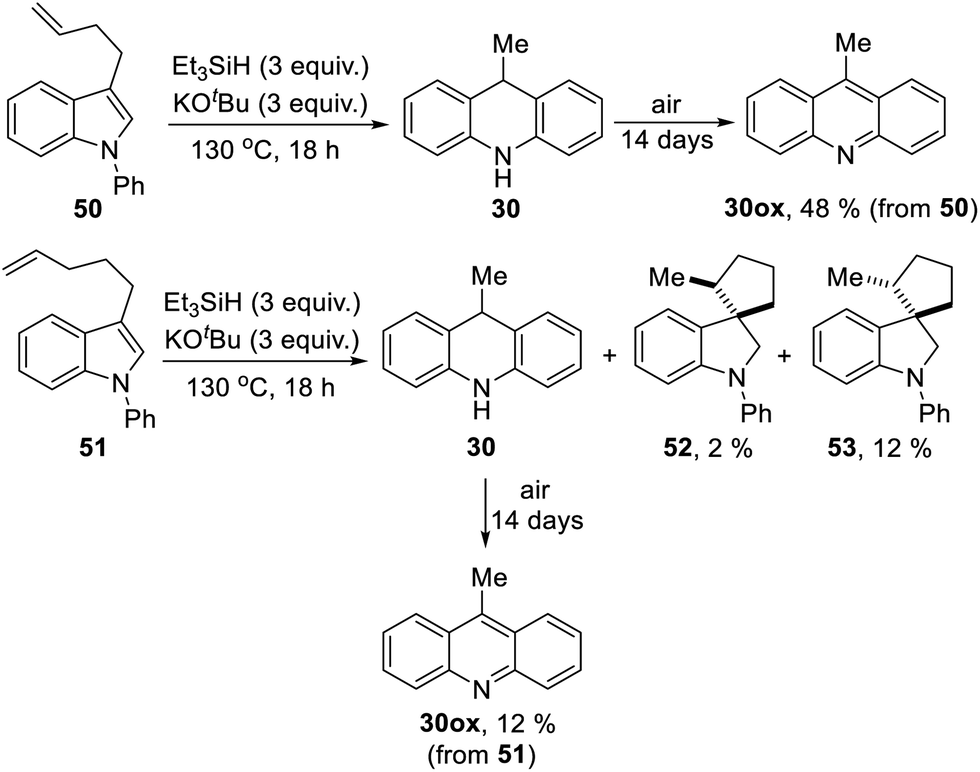
Substrate 50 gave an unexpected result when it afforded dihydroacridine 30 in good yield, confirmed by NMR. For ease of isolation, the product was exposed to oxidation in air, affording 30ox in 48% yield from 50. For this to happen, the butenyl side-chain must be severed during the reaction. Substrate 51 also afforded dihydroacridine 30 as well as the diastereomeric spiroindolines 52 and 53. The loss of the side-chains, seen in the formation of 30, is clearly associated with the alkene group in the side-chain, since Table 1 shows that substrates with saturated side-chains, 27 and 28, suffered no loss of their saturated alkyl side-chains.
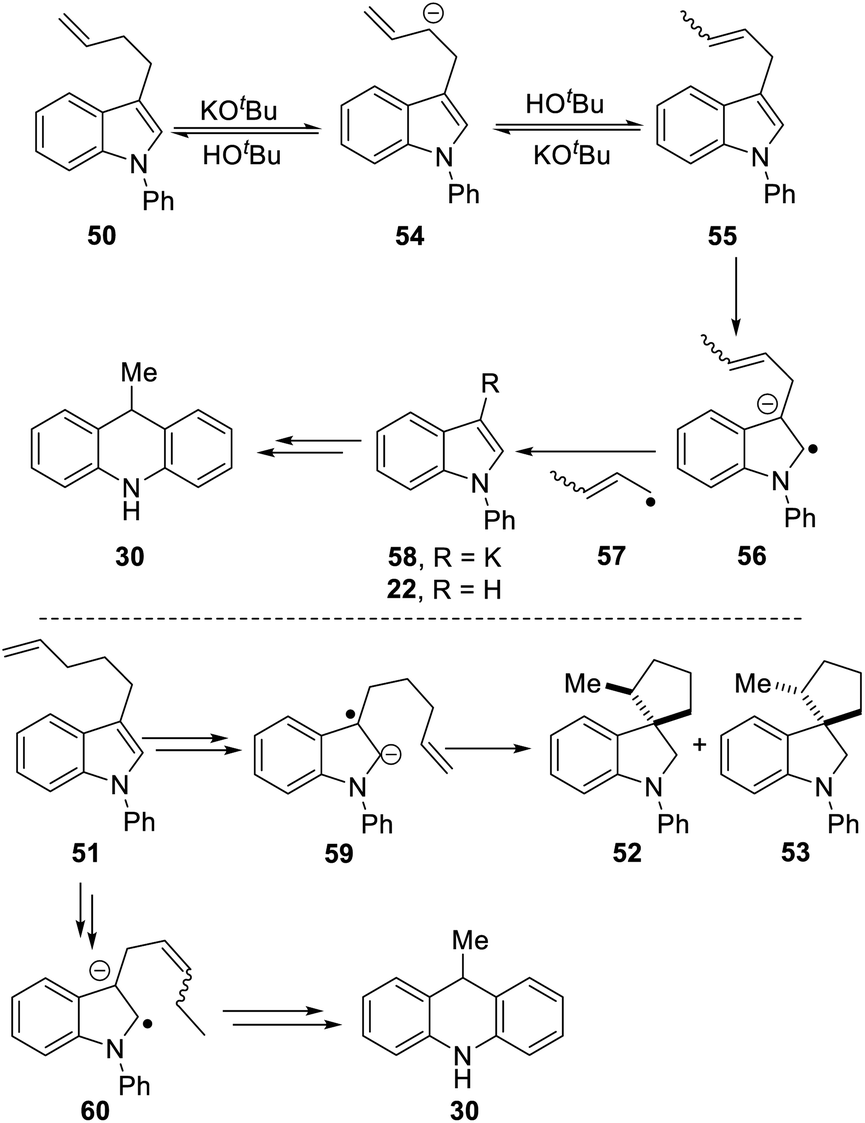
Scheme 5 proposes an explanation for both substrates 50 and 51. Substrate 50 can suffer reversible deprotonation by tert-butoxide in the allylic position to afford anion 54. Reprotonation of the anion can afford 55 which is converted to radical anion 56. (We draw the formation of the radical anion occurring following the regioisomerisation of the alkene, but the opposite sequence may well occur). Radical anion 56 then fragments to the 2-butenyl radical 57, leaving indolylpotassium 58; following proton abstraction, this gives N-phenylindole 22, which is then converted, as in Scheme 3, into 9-methyldihydroacridine 30. (For NMR evidence in favor of the alkene migration in the side-chain of 50, see ESI file†).
 | ||
| Scheme 5 | ||
The outcome of reaction with substrate 51 can be explained in like manner. In this case, conversion to its radical anion 59 leads to cyclisation to diastereomeric radicals that abstract hydrogen atoms to afford 52 and 53. On the other hand, if KOtBu-induced regioisomerisation migrates the alkene double-bond sequentially along the side-chain, the substrate then is converted to radical anion 60. This species can expel a delocalised pent-2-enyl radical, again forming indolylpotassium 58, which, following proton abstraction, is ultimately transformed to 9-methyldihydroacridine 30.
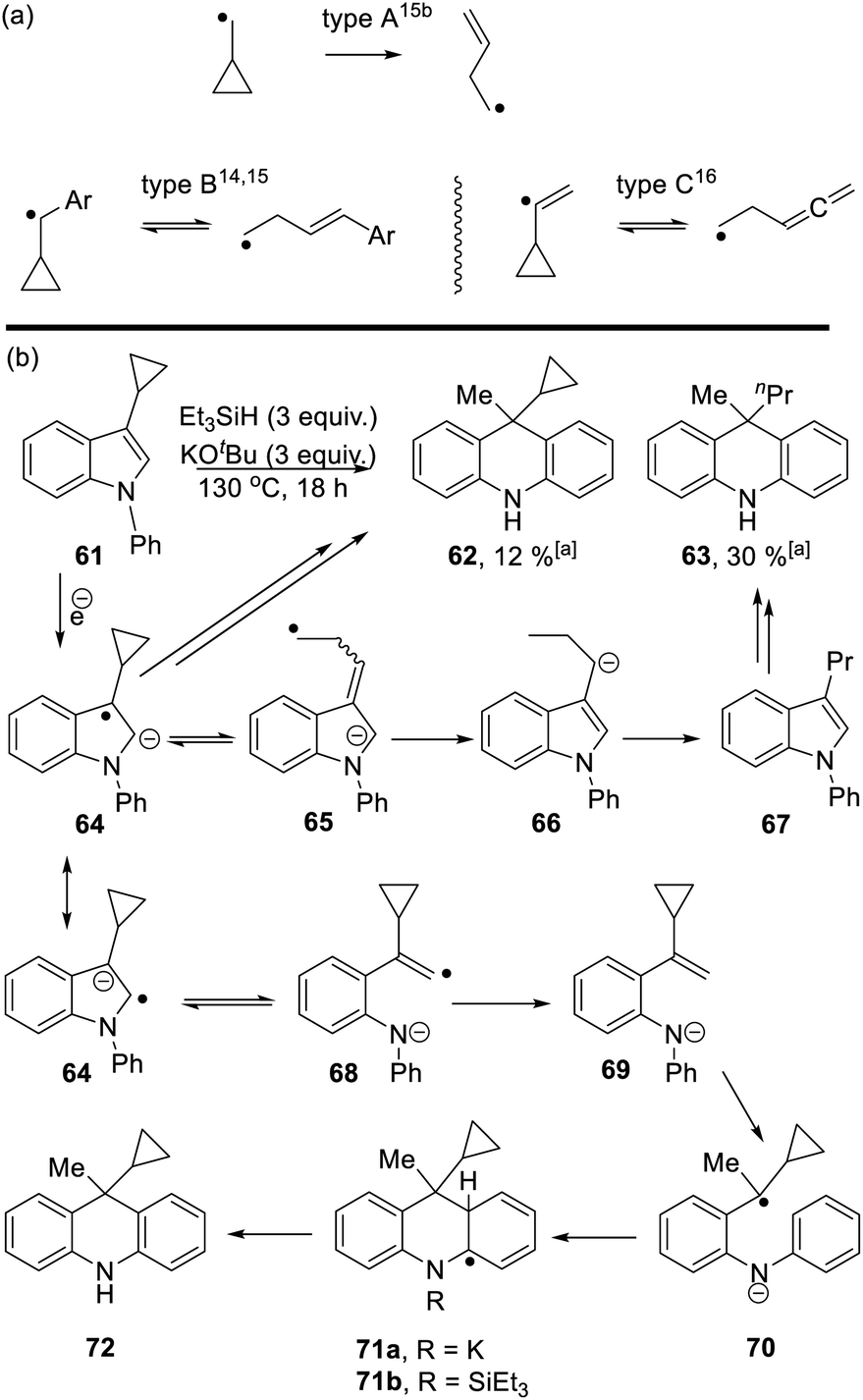
Studies were next carried out with substrate 61 and 73 bearing a cyclopropyl substituent as a radical probe6,10 (Scheme 6).
 | ||
| Scheme 6 | ||
Cyclopropyl groups are routinely used as indicators of radical intermediates. Simple cyclopropylcarbinyl radicals (type A, Scheme 6a) ring-open very rapidly to form butenyl radicals.14,15 The rate of the reverse reaction is comparatively very slow, so that these reactions routinely afford ring-opened products as the sole products from reactions. However, when the initial radical carbon is benzylic (type B)14,15 or is a vinyl radical (type C),16 then the cyclopropyl ring-opening is readily reversible and cyclic and/or open-chain products can result. As seen below, these subtleties of cyclopropanes as probes of radicals can be important in understanding mechanisms.
Starting with substrate 61, reaction with KOtBu and Et3SiH afforded a mixture of cyclopropyl product 62 (12%) and n-propyl analogue 63 (30%). Following formation of radical anion 64,8 fragmentation of the cyclopropyl ring would afford radical anion 65. As radical anion 64 incorporates a benzylic cyclopropylcarbinyl radical, this type B reaction will likely be reversible and thus both 64 and 65 can be considered as likely intermediates. For 65, abstraction of (i) a hydrogen e.g. from Et3SiH and (ii) a proton leads to 3-propyl-N-phenylindole 67. This compound can then be processed, according to Scheme 3, to afford dihydroacridine 63 following workup.
On the other hand, fragmentation of the 5-membered ring in 64 affords radical anion 68. Hydrogen atom abstraction leads to 69. Regioselective hydrogen atom transfer10 to styrene 69 gives benzylic radical 70. This is a cyclopropylbenzyl radical (type B) and so can open reversibly. Cyclisation affords radical anion 71a. Aromaticity is restored as 72 is formed; this could proceed with the amide anion still in place, as in 71a, or following silylation of the nitrogen as in 71b, as mentioned in Scheme 3.11
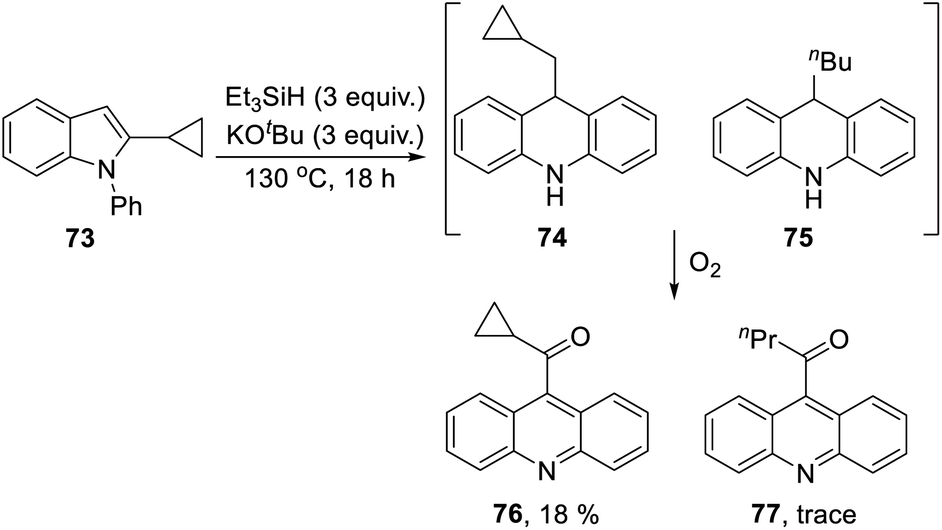
The 2-cyclopropyl substrate 73 (Scheme 7) was transformed into products from which 77 (trace amount) was separated and characterised. Exposure of the residue to air allowed isolation of acridine 76 (18%).
 | ||
| Scheme 7 | ||
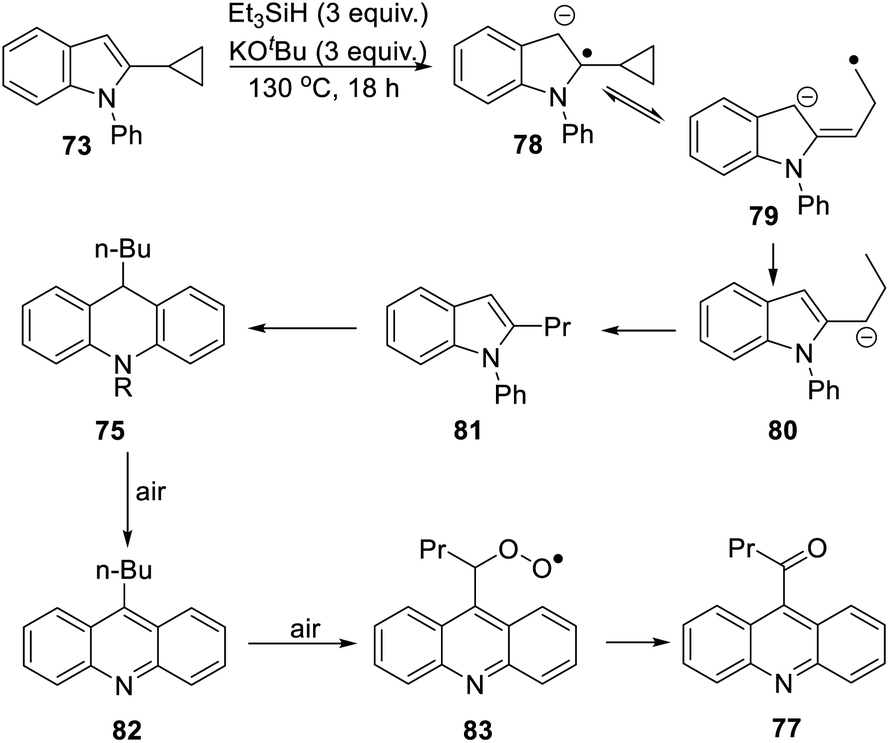
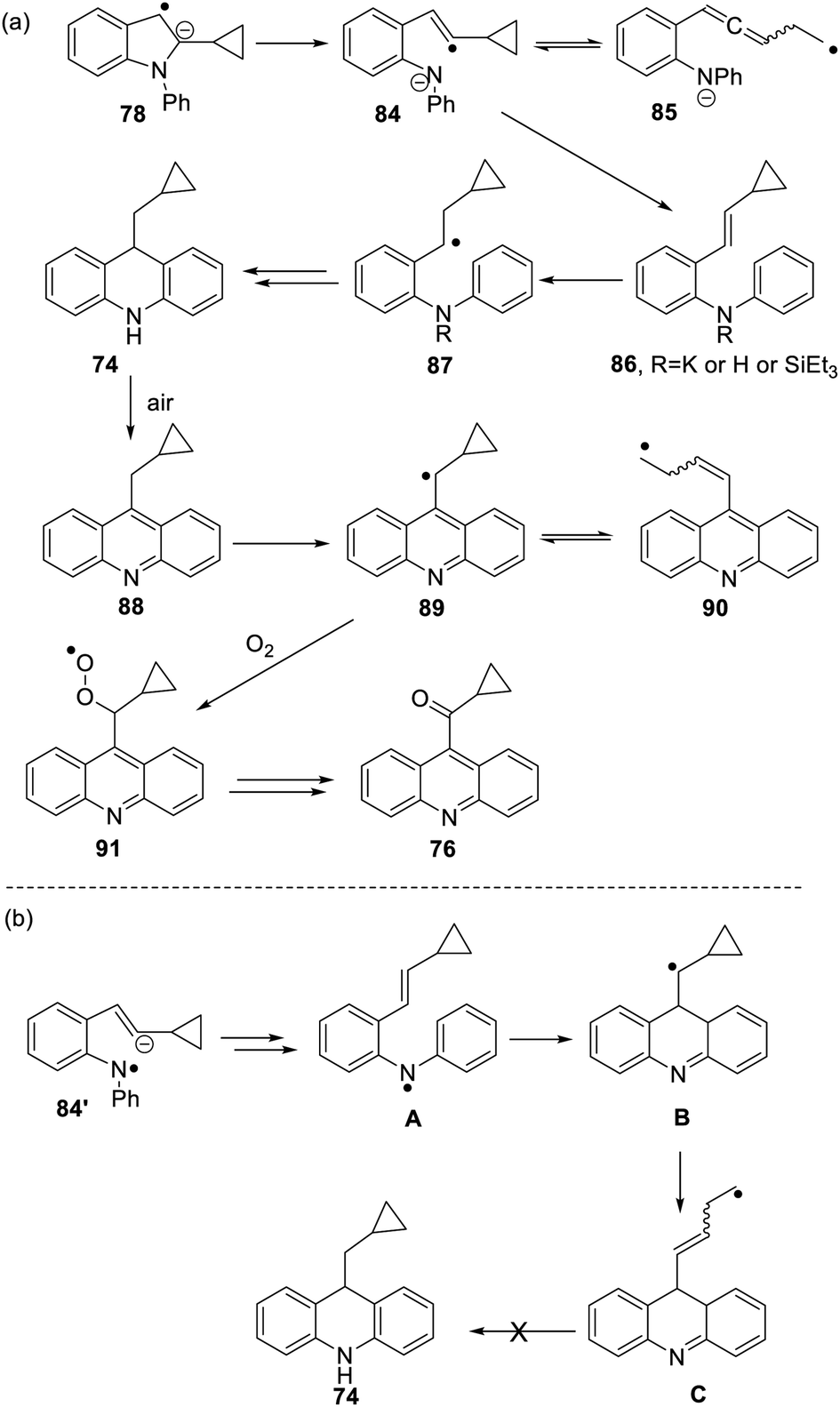
The formation of 77 follows pathways that are familiar from other examples in this paper, starting with the fragmentation of the 3-membered ring in radical anion 78; (Scheme 8); the oxidation of 82 by air likely involves abstraction of a benzylic hydrogen atom, with the peroxyl radical 83 as key intermediate on the pathway to ketone 77. The formation of compound 76 (Scheme 7) was also informative. Fragmentation of the 5-membered ring in 78 (see Scheme 9) leads to radical anion 84 which is a type C cyclopropylvinyl radical, likely to be in equilibrium with its ring-opened form 85. No product derived from 85 was detected, but 84 did lead to observed product.
 | ||
| Scheme 8 | ||
 | ||
| Scheme 9 | ||
Hydrogen abstraction by radical 84 leads to vinyl cyclopropane 86. Following Jeon,10 regioselective hydrogen atom addition gives radical 87, which then proceeds to the cyclopropylmethyl-substituted dihydroacridine 74. On deliberate exposure to air, aromatisation slowly occurs to yield 88, followed by activation of the benzylic C–H, ultimately leading to ketone 76.
Substrate 73 played an important role in our understanding of mechanism. An alternative route to the dihydroacridine products was initially considered [Scheme 9(b)] where fragmentation of the indole radical anion 78 would afford distal radical anion 84′. Proton abstraction, giving A and radical cyclisation would yield radical B. However, this cyclopropylcarbinyl radical of type A would open to give radical C, and no cyclopropyl product would be detected from this intermediate. Accordingly, H-atom addition to the styrene 86 is the favored route.
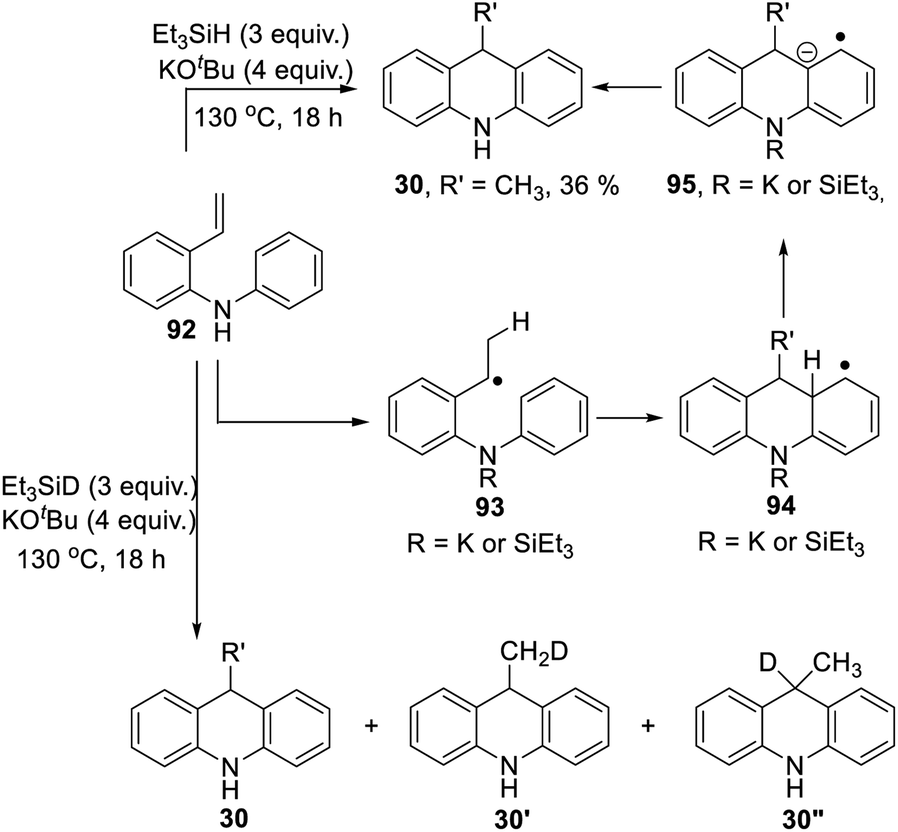
Scheme 3 proposed styrene 41 as a possible intermediate in the rearrangement reaction. To check its viability, substrate 92 was prepared (Scheme 10). On treatment with triethylsilane and KOtBu, this afforded 9-methyldihydroacridine 30 (36%). The upper part of Scheme 10 shows the proposed pathway. Hydrogen atom addition would form radical 93; notably, just a single H atom in 30 is proposed to derive from Et3SiH in this mechanism, and to be located in the methyl group of the product. To check this, the reaction was repeated using Et3SiD instead of Et3SiH. The 2H-NMR spectrum clearly showed labelling in the methyl group. The 13C {1H}-spectrum showed two resonances for the methyl carbon, one a 1:1:1 triplet, due to a CH2D group in 30′, and the other a singlet, due to an undeuterated methyl group in 30.
 | ||
| Scheme 10 | ||
Additionally, we detected 30′′. No signals were seen corresponding to more than one deuterium atom in the methyl group. Deuteration was also seen in the aryl groups. This process will have exchanged original Ar–H hydrogen atoms for D atoms and correspondingly produced Si–H bonds in place of Si–D bonds, and these H atoms will have led to some unlabeled methyl groups seen in the product.
To check that this conversion of 92 to 30 was dependent on potassium ions,10 the reaction was repeated with Et3SiH + NaOtBu; this gave no reaction.
We also subjected 3-methyl-N-phenylindole 23 to reaction with Et3SiD/KOtBu, focusing on deuterium incorporation into the methyl group of 31 (Scheme 11). In this case, we observed CH3, CH2D and CHD2 groups from the 13C{1H} and 13C{1H,2H} NMR spectra. This is in line with the proposed mechanism. The formation of some unlabelled methyl group and some monodeuterated CH2D group supports the exchange reactions mentioned above, where Ar–H hydrogen atoms on the substrate undergo exchange with R3Si–D bonds, and the resulting R3Si–H allows the formation of some unlabeled and monodeuterated methyl group in the dihydroacridine product. Consistent with the exchange reactions, deuteration of Ar–H positions was indeed seen in these reactions.
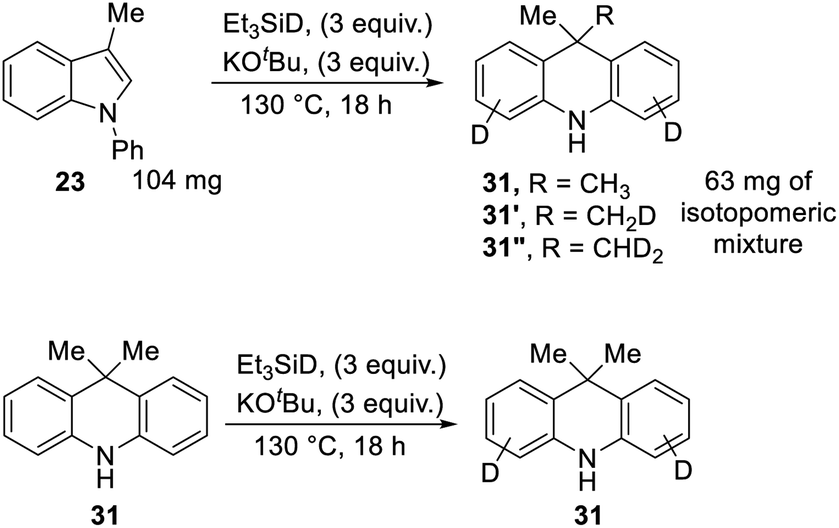 | ||
| Scheme 11 | ||
In a further experiment, unlabeled product 31 was subjected to the reaction conditions, to check whether the methyl groups were stable to the conditions or would undergo exchange. No exchange was seen in the methyl groups, although deuteration was seen on the aryl rings.
Additionally, and mindful of reports that H2 gas is produced on reaction of Et3SiH with KOtBu,2–7 those two reagents were heated for 1 hour, and then the headspace in the vessel, containing hydrogen, was replaced by deuterium gas and substrate 23 was introduced. This led to formation of the dihydroacridine 31. Examination of the 2H NMR spectrum showed deuterium incorporation into the methyl group of the product [and a much lesser degree of deuterium labelling of the Ar–H positions ortho to the NH group]. The methyl group carbon appeared as a singlet plus a 1:1:1 triplet, resulting from the presence of CH3 and CH2D (the combined yield of the products was 72%). Therefore, the gas in the headspace is not inert to the reaction. We further examined what happened when the hydrogen gas was removed and replaced by inert gas (argon). In this case, the transformation from indole substrate to dihydroacridine products was completely suppressed to < 1% yield.
As a final point, we investigated whether the reaction was specific to triethylsilane and to potassium tert-butoxide. Other silanes, notably Me2PhSiH, MePh2SiH and Ph3SiH were also successful in the conversion of 23 to 31. However, upon replacement of KOtBu with NaOtBu (Table S1,† entry 5), no reaction took place.
Conclusions
In summary, the Stoltz–Grubbs reducing system transforms N-arylindoles into 9,10-dihydroacridines in moderate to excellent yields. The ring-opening of the indole is triggered by fragmentation of intermediate indole radical anions; the styrenes formed in this fragmentation are then activated by potassium-ion-dependent hydrogen atom transfer to afford the dihydroacridine products. The transformation provides important information about the nature of chemistry that is undertaken by the Et3SiH + KOtBu reagent.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the following sources for financial support: (i) GSK and EPSRC for i–CASE awards (to AJS and DD) (ii) University of Strathclyde for studentship funding (iii) the EPSRC-funded ARCHIE-WeSt High Performance Computer (www.archie-west.ac.uk) for computational resource via EPSRC grant no. EP/K000586/1.Notes and references
-
(a) S. Tsuchiya, H. Saito, K. Nogi and H. Yorimitsu, Org. Lett., 2019, 21, 3855–3860 CrossRef CAS PubMed
; (b) H. Saito and H. Yorimitsu, Chem. Lett., 2019, 48, 1019–1028 CrossRef CAS
- A. Fedorov, A. A. Toutov, N. A. Swisher and R. H. Grubbs, Chem. Sci., 2013, 4, 1640–1645 RSC
- A. A. Toutov, W.-B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80–84 CrossRef CAS PubMed
- A. Toutov, W.-B. Liu, K. N. Betz, B. M. Stoltz and R. H. Grubbs, Nat. Protoc., 2016, 10, 1897–1903 CrossRef PubMed
- A. A. Toutov, M. Salata, A. Fedorov, Y.-F. Yang, Y. Liang, R. Cariou, K. N. Betz, E. P. A. Couzijn, J. W. Shabaker, K. N. Houk and R. H. Grubbs, Nat. Energy, 2017, 2, 17008 CrossRef CAS
- W.-B. Liu, D. P. Schuman, Y.-F. Yang, A. A. Toutov, Y. Liang, H. F. T. Klare, N. Nesnas, M. Oestreich, D. G. Blackmond, S. C. Virgil, S. Banerjee, R. N. Zare, R. H. Grubbs, K. N. Houk and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 6867–6879 CrossRef CAS PubMed
- S. Banerjee, Y.-F. Yang, I. D. Jenkins, Y. Liang, A. A. Toutov, W.-B. Liu, D. P. Schuman, R. H. Grubbs, B. M. Stoltz, E. H. Krenske, K. N. Houk and R. N. Zare, J. Am. Chem. Soc., 2017, 139, 6880–6887 CrossRef CAS PubMed
- A. J. Smith, A. Young, S. Rohrbach, E. F. O'Connor, M. Allison, H.-S. Wang, D. L. Poole, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2017, 56, 13747–13751 CrossRef CAS PubMed
- W. Xie, S.-W. Park, H. Jung, D. Kim, M.-H. Baik and S. Chang, J. Am. Chem. Soc., 2018, 140, 9659–9668 CrossRef CAS PubMed
- P. Asgari, Y. Hua, C. Thiamsiri, W. Prasitwatcharakorn, A. Karedath, X. Chen, S. Sardar, K. Yum, G. Leem, B. S. Pierce, K. Nam, J. Gao and J. Jeon, Nat. Catal., 2019, 2, 164–173 CrossRef CAS PubMed
-
(a)
A. A. Toutov, K. N. Betz, A. M. Romine and R. H. Grubbs, US Pat. Application US 2019/0218232A1, 2019
- In a different approach, that also adds electron density to the indole, but not involving radicals or radical anions, nucleophilic ring-opening of N-arylindoles by silyl anions, R3SiLi, was reported: P. Xu, E.-U. Würthwein, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2017, 56, 13872–13875 CrossRef CAS PubMed
- We did consider the possibility of the indole fragmentation in Scheme 3 arising, instead, from fragmentation of radical 40 rather than a radical anion 37. Radical 40 could arise by hydrogen atom addition to the 2-position of the indole 22. However, the literature features no reports of fragmentations of the indole nucleus arising from radicals like 40, in the presence of the Grubbs–Stoltz reagent or under other conditions.
- V. W. Bowry, J. Lusztyk and K. U. Ingold, J. Chem. Soc., Chem. Commun., 1990, 923–925 RSC
-
(a) T. A. Halgren, J. D. Roberts, J. H. Horner, F. N. Martinez, C. Tronche and M. Newcomb, J. Am. Chem. Soc., 2000, 122, 2988–2994 CrossRef CAS
- J. K. Crandall, G. L. Tindell and A. Manmade, Tetrahedron Lett., 1982, 23, 3769–3772 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data are provided as well as details of computational investigations. See DOI: 10.1039/d0sc00361a |
| This journal is © The Royal Society of Chemistry 2020 |