
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermodynamic consequences of Tyr to Trp mutations in the cation–π-mediated binding of trimethyllysine by the HP1 chromodomain†
Mackenzie W.
Krone‡
 a,
Katherine I.
Albanese‡
a,
Gage O.
Leighton
b,
Cyndi Qixin
He
c,
Ga Young
Lee
c,
Marc
Garcia-Borràs
c,
Alex J.
Guseman
a,
David C.
Williams
Jr
de,
K. N.
Houk
c,
Eric M.
Brustad
a and
Marcey L.
Waters
*a
a,
Katherine I.
Albanese‡
a,
Gage O.
Leighton
b,
Cyndi Qixin
He
c,
Ga Young
Lee
c,
Marc
Garcia-Borràs
c,
Alex J.
Guseman
a,
David C.
Williams
Jr
de,
K. N.
Houk
c,
Eric M.
Brustad
a and
Marcey L.
Waters
*a
aUniversity of North Carolina at Chapel Hill, 131 South Road, Campus Box 3290, Chapel Hill, NC 27599, USA. E-mail: mlwaters@email.unc.edu
bDepartment of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, 120 Mason Farm Rd, Campus Box 7260, Chapel Hill, NC 27599, USA
cDepartment of Chemistry and Biochemistry, University of California at Los Angeles, 607 Charles E. Young Drive East, Box 951569, Los Angeles, CA 90095-1569, USA
dDepartment of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, Campus Box 7525, Brinkhous-Bullitt Building, Chapel Hill, NC 27599, USA
eLineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, 450 West Drive, Chapel Hill, NC 27599, USA
First published on 4th March 2020
Abstract
Evolution has converged on cation–π interactions for recognition of quaternary alkyl ammonium groups such as trimethyllysine (Kme3). While computational modelling indicates that Trp provides the strongest cation–π interaction of the native aromatic amino acids, there is limited corroborative data from measurements within proteins. Herein we investigate a Tyr to Trp mutation in the binding pocket of the HP1 chromodomain, a reader protein that recognizes Kme3. Binding studies demonstrate that the Trp-mediated cation–π interaction is about −5 kcal mol−1 stronger, and the Y24W crystal structure shows that the mutation is not perturbing. Quantum mechanical calculations indicate that greater enthalpic binding is predominantly due to increased cation–π interactions. NMR studies indicate that differences in the unbound state of the Y24W mutation lead to enthalpy–entropy compensation. These results provide direct experimental quantification of Trp versus Tyr in a cation–π interaction and afford insight into the conservation of aromatic cage residues in Kme3 reader domains.
Introduction
In the last 20 years, the cation–π interaction, defined as the favorable interaction between a cationic species and the electron-rich face of an aromatic ring (or other π system), has entered into the standard lexicon of noncovalent interactions that contribute to biomolecular structure and function.1–3 These interactions are of particular importance in proteins, invoking both cationic and aromatic amino acid side chains, and accordingly mediate critical biological processes including protein folding and enzymatic catalysis as well as protein–ligand and protein–protein interactions (PPIs).1,2,4 Statistical analysis of protein structures, gas phase measurements, and computational studies all indicate that Trp provides the most favorable cation–π interaction among the naturally occurring aromatic amino acids due to its larger surface area and greater electrostatic potential, which provides more extensive electrostatic stabilization of a neighboring cation (Fig. 1A).5,6 Nonetheless, there is a dearth of direct experimental data in proteins demonstrating that Trp provides a greater driving force for cation–π interactions than Phe or Tyr.7–13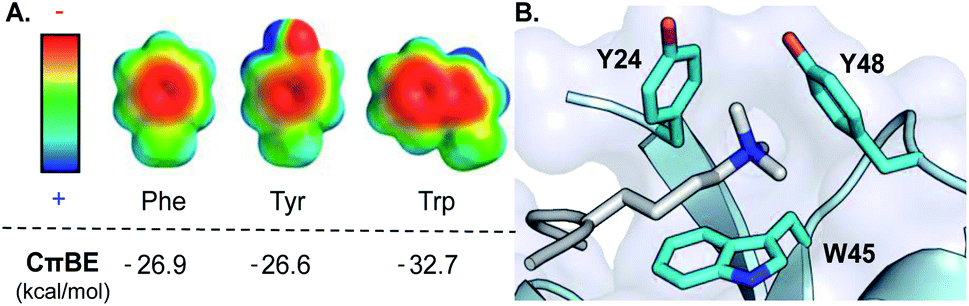 | ||
| Fig. 1 (A) Electrostatic potential maps of Phe, Tyr, and Trp and the calculated cation–π binding energy (CπBE) of the aromatic side chain. Electrostatic potentials were calculated in Spartan at the B3LYP/6-31G* level of theory (+100 to −100 kcal mol−1). CπBE are based on gas phase interactions of the aromatic side chain with Na+. (B) Aromatic cage (cyan) of the HP1 chromodomain bound to Kme3 (silver). PDB ID: 1KNE. | ||
The recognition of trimethyllysine (Kme3) on histone proteins by Kme3 reader proteins is a biologically and medicinally relevant group of PPIs mediated by cation–π interactions. These epigenetic PPIs, which mediate gene expression, have generally been shown to be driven by cation–π interactions between the trimethylammonium group of Kme3 in the histone tail and an aromatic cage within the reader protein,14–18 in which the tetra-alkyl ammonium ion in Kme3 preferentially interacts with the face of an aromatic ring through charge–quadrupole interactions.2,19,20 Given the significant number of reader proteins for Kme3 with diverse aromatic cages varying in compositions of 2–4 Phe, Tyr, and Trp residues, this family of proteins provides an ideal model system for studying cation–π interactions in PPIs.14
Herein we describe Tyr-to-Trp mutational studies of a conserved Kme3 reader protein, heterochromatin protein 1 (HP1) from Drosophila melanogaster, to experimentally determine the relative contributions of these cation–π interactions in a protein binding event. Our combined thermodynamic, crystallographic, and computational studies show that the cation–π interaction is indeed stronger with Trp, as predicted, but the mutations cause changes to the unbound state that counterbalance enthalpic gains in binding affinity. These results provide the first direct measure in a protein of the difference in magnitude of Trp versus Tyr in a cation–π interaction, as well as insight into the exclusive evolutionary selection of Tyr and Phe at certain positions in the aromatic binding pocket of this family of Kme3 reader proteins.
Results and discussion
HP1 chromodomain is a model reader protein for studying cation–π interactions
We chose the chromodomain of HP1 from Drosophila melanogaster, which binds Kme3 at position 9 of histone H3 (H3K9me3), as our model reader protein, because its structure and mechanism of binding have been extensively characterized.21,22 Recognition of Kme3 is mediated by a conserved binding pocket comprised of two Tyr residues (Y24 and Y48) and a Trp residue (W45, Fig. 1B). Previous studies from our groups have demonstrated that binding is driven by cation–π interactions by establishing the significance of the positive charge on Kme3 through replacement with a neutral isostere and by tuning the contribution of each tyrosine residue using unnatural amino acid mutagenesis.15,16The aromatic amino acids present in the binding pocket for Kme3 are highly conserved in the HP1 and polycomb chromodomain families.18 For example, the composition of the pocket shows only small variations between D. melanogaster (HP1, dPC) and mammalian (CBX 1–7) chromodomain homologs (Fig. S1†). W45 (HP1 numbering) is invariant in both HP1 and polycomb proteins. Y48 has converged to Tyr or Phe residues in HP1 proteins but is a conserved Trp at the homologous position of polycomb proteins. Interestingly, despite the fact that tryptophan should provide an improved cation–π interaction, the phenyl ring at position 24 appears to be important, as only Tyr or Phe are observed at this site. In a previous report, we demonstrated that replacement of Tyr24 and Tyr48 in HP1 with Phe results in similar binding affinity when compared to wild type, suggesting some plasticity in the pocket.16 Tyr24 and Tyr48 are both surface exposed, and preliminary mutational modeling in PyMOL suggested that the predominant Trp rotamers at each position could occupy similar geometries to Tyr without significant structural perturbation (Fig. S2†). Accordingly, we sought to investigate the effect of introducing tryptophan mutations to this conserved aromatic cage.
Thermodynamics of binding in HP1 tryptophan mutants
We cloned and expressed the corresponding single and double Trp mutations at the 24 and 48 locations in HP1, denoted HP1–Y24W, HP1–Y48W, and HP1–Y24W/Y48W (Fig. S3†). Binding constants and thermodynamic parameters (Kd, ΔH°, and ΔS°) of association between an H3K9me3 peptide (residues 1–15) and the Trp variants were measured by isothermal titration calorimetry (ITC, Table 1 and Fig. S5†). Based on calculated cation–π energies for Trp and Tyr (Fig. 1A), we hypothesized that noncovalent interactions between Kme3 and the aromatic cage would be more favorable with Trp in place of Y24 and Y48. However, the affinity of HP1–Y24W for H3K9me3 remained unchanged with respect to the wild type and HP1–Y48W binding was slightly attenuated (Table 1). Binding to the HP1–Y24W/Y48W double mutant was significantly weaker.| Proteina | K d (μM) | ΔH° (kcal mol−1) | −TΔS° (kcal mol−1) |
|---|---|---|---|
| a Experiments were performed by titrating H3K9me3 peptide (3–3.5 mM) into HP1 mutants (180–230 μM) in 50 mM sodium phosphate, pH 7.4, 150 mM NaCl, 2 mM TCEP at 25 °C. Values are average of three independent experiments, and calculated error is standard deviation of measurements. | |||
| HP1 wild type | 15 ± 2 | −5.8 ± 0.3 | −0.9 ± 0.3 |
| HP1–Y24W | 14 ± 3 | −10.6 ± 0.7 | 4.0 ± 0.8 |
| HP1–Y48W | 23 ± 3 | −10.4 ± 0.6 | 4.1 ± 0.6 |
| HP1–Y24W/Y48W | 55 ± 16 | −9 ± 2 | 3 ± 2 |
Interestingly, the thermodynamic binding parameters of all three Trp variants showed significant differences when compared to the wild type protein. While the mutations did not result in improved binding affinities, the enthalpic values for binding indicate enhanced noncovalent interactions consistent with more favorable cation–π interactions. For example, the enthalpy of association of the H3K9me3 peptide is more favorable by almost two-fold for HP1–Y24W and HP1–Y48W when compared to wild type protein (Table 1). This is consistent with the hypothesis that the larger Trp indole provides a stronger cation–π interaction relative to Phe or Tyr. However, in both cases, the improved enthalpy of binding is counterbalanced by an entropic cost that reduces the overall affinity of each mutant.
Structural investigation of HP1–Y24W bound to H3K9me3
To investigate the structural basis of measured differences in binding, we determined the crystal structure of HP1–Y24W in complex with H3K9me3 peptide at 1.52 Å (see ESI Methods and Table S1†). The overall fold of the chromodomain bound to the native ligand was not perturbed (RMSD = 0.19 Å) and clear electron density was observed for the Y24W mutation (Fig. S6 and S7†). Consistent between the wild type and HP1–Y24W structures, the backbone of the H3 peptide forms a characteristic three-stranded β-sheet with the chromodomain while K9me3 interacts with the surface exposed aromatic cage. Importantly, the general topology of the aromatic cage remains intact (Fig. 2A).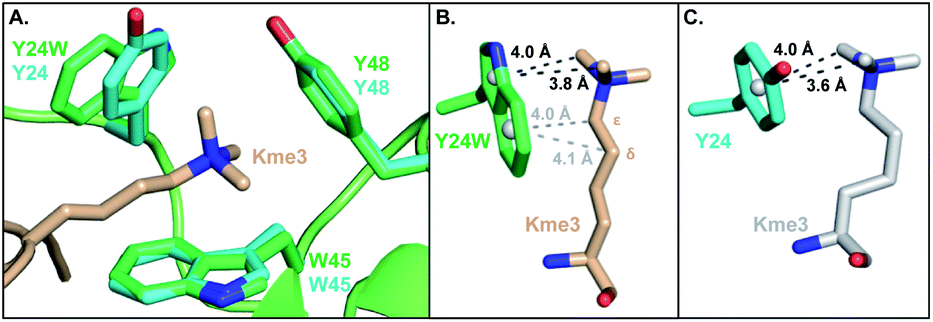 | ||
| Fig. 2 (A) Overlay of the HP1–Y24W crystal structure (PDB: 6MHA) with wild type HP1 (PDB: 1KNE). The wild type cage is shown in cyan and the HP1–Y24W cage is shown in green. Both chromodomains were crystallized in the presence of H3K9me3, but only the Kme3 peptide of HP1–Y24W (wheat) is shown for clarity. (B and C) Rotated perspective from (A) highlighting the differences in ligand contacts and conformation between (B) Y24W and (C) Y24 in HP1. | ||
The backbone and Cβ atoms of Y24W superpose with the wild type tyrosine. The 5-membered ring of Y24W contacts the Kme3 trimethyl ammonium cation in a comparable position and orientation to the phenol moiety of native Y24. The six-membered ring of Y24W adopts an orientation that permits additional packing interactions with the alkyl chain of the Kme3. Distinct conformational changes are observed in the alkyl chain of Kme3, which adopts a relaxed anti conformation (Fig. 2B). All other reported HP1–ligand structures show Kme3 in a strained gauche conformation (Fig. 2C).16,21 This change in side chain geometry tunes the distance and angle of interactions between Kme3 and aromatic cage and allows Kme3 to make additional contacts with Y24W.
Both conformations and distances are important determinants of the strength of cation–π interactions, with the preferred geometry placing the trimethylammonium directly over the center of an aromatic ring at a distance of ∼3.6 Å.1,6 To gain insight into how noncovalent interactions were altered in the binding pocket of HP1–Y24W, we measured distances from the centroids of aromatic rings to the N-methyl and methylene atoms of Kme3 (Fig. 2B and S8, Table S2†). We also generated structural views that allow the geometry of cation–π contacts to be observed normal to the plane of the ring (Fig. 3). In wild type HP1, the aromatic ring of Y24 makes close contacts (3.6 and 4.0 Å, Fig. 2C) with two N-methyl groups of Kme3 pointing towards the centroid of the phenol (Fig. 3). In HP1–Y24W (Fig. 2B), the 5-membered pyrrole ring of the HP1–Y24W indole interacts with the same two N-methyl groups at distances of 3.8 and 4.0 Å from the centroid of the pyrrole (Fig. 3), suggesting that the pyrrole may function in a similar manner to tyrosine at this position. Although not in an optimal position to participate in cation–π interactions, the 6-membered ring of Y24W makes van der Waals contacts with the ε and δ methylene groups of Kme3, resulting in additional favorable dispersion interactions (4.0 and 4.1 Å, Fig. 2B and 3).
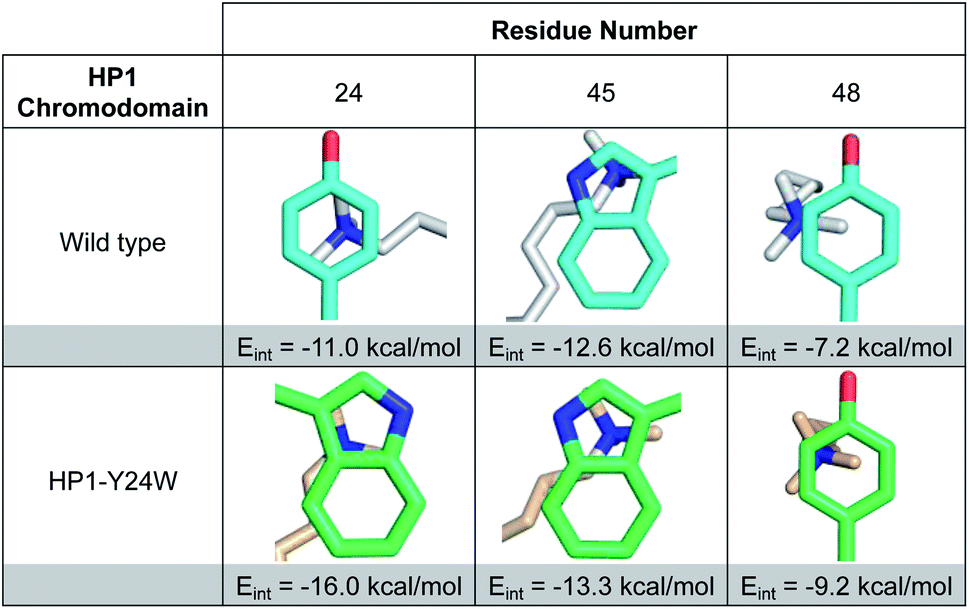 | ||
| Fig. 3 Images of Kme3 contacts with residues 24, 45, and 48 viewed normal to the plane of the ring for HP1 wild type and HP1–Y24W. Interaction energies (Eint) for Kme3 and each residue were calculated with the M06-2X/6-311+G(d,p) basis set (see ESI for computational protocol description†). | ||
Although Y48 and W45 were not mutated, the subtle rotation of the Kme3 ligand leads to additional contacts with both amino acids that may further enhance binding. In the case of Y48, a second N-methyl group is positioned closer to the centroid of Y48 in the HP1–Y24W structure (Fig. 3). Previous gas-phase computational studies by Dougherty predicted that a 2-point contact of NMe4+ with benzene is ∼1.3-fold stronger than a single-point contact, suggesting the added contact between Y48 and Kme3 improves binding.6 With respect to W45, the anti conformation of Kme3 appears to improve both cation–π contacts and VDW interactions (Fig. 3 and S8, Table S2†). In summary, the Y24W mutation and consequent structural changes enhance ligand interactions with each residue of the aromatic cage.
Computational investigations of Y24W bound to H3K9me3
To further support our observations, interaction energies between Kme3 and the aromatic cages of wild type HP1 and HP1–Y24W were calculated using truncated computational models generated from their corresponding crystal structures (Fig. S9, see computational protocol description in the ESI†).6,23 Computed electronic interaction energies (Eint) for the intact aromatic cage predict a stronger interaction for Kme3 and HP1–Y24W (Eint = −37.9 kcal mol−1) than for Kme3 and the wild type HP1 (Eint = −30.4 kcal mol−1). The estimated difference in Eint (ΔEint = −7.5 kcal mol−1) agrees well with experimentally observed binding enthalpies determined by ITC, which show a 5 kcal mol−1 difference between the HP1–Y24W mutant and HP1. Interaction energies for each individual residue in the aromatic cage were also calculated. Computations showed that the main improvement to the total Eint in HP1–Y24W mutant comes indeed from the mutated Y24W residue, which has an improved interaction of ΔEint = −5 kcal mol−1 with respect to the Y24 in the wild type (Fig. 3).To determine the significance of the additional CH–π interactions between the Y24W tryptophan and the Kme3 side chain, we decomposed the total Eint between Y24W and Kme3 using different truncated Kme3 models generated from the X-ray structure: Me(CH2)nNMe3+; where n = 0–3 (Fig. S9†). The computed total Eint for the native sidechain (n = 3) was −14.6 kcal mol; while the computed Eint for truncated models n = 2, 1 and 0 were −14.4, −14.1, and −13.3 kcal mol−1, respectively. Based on the estimated Eint for the truncated models, the contributions of γ-CH2 (n = 3) and δ-CH2 (n = 2) methylene units to the total interaction energy can be estimated to be about −1.2 kcal mol−1 due to van der Waals contacts and dispersion interactions, whereas interactions between Y24W and ε-CH2-NMe3+ provide the bulk of the favorable binding, amounting to −13.3 kcal mol−1 due to the cation–π interaction. The anti-conformation of the Kme3 sidechain adopted in the mutant (Fig. 2B), in contrast to the gauche sidechain conformation observed in wild type HP1 (Fig. 2C) allows these additional CH–π interactions to take place (Fig. S9†). Taken together, computations suggest that the observed improvement in the interaction energy in HP1–Y24W arises primarily from stronger cation–π interactions as well as additional van der Waals contacts attributed to Y24W mutation.
Potential source of entropic compensation in Y24W
Perturbation of complex biological systems often leads to enthalpy–entropy compensation that may reflect either constraints on the observable binding free energy (ΔG ∼ constant) or extra thermodynamic sources.24 The origin of this compensation is often difficult to identify, but can provide useful structure–function information if determined. Thus, we evaluated the unbound and bound states of wild type and HP1–Y24W by NMR and circular dichroism (CD) to determine if we could detect any structural or dynamic differences that correlate with the change in the entropic cost of binding.Relaxation dispersion (RD) experiments, which measure conformational dynamics across the protein backbone, showed no significant differences in dynamics between the unbound wild type and mutant chromodomain (Fig. S11†), indicating that the biomolecular motion of Y24W is not substantially different from wild type on the micro to millisecond timescale. HSQC spectra for both the unbound and bound states of wild type and Y24W align well overall, indicating that the mutation does not substantially perturb the global protein fold (Fig. 4 and S10†). However, residues near the Kme3 binding pocket, including residues 45–48 and 51–53, show larger chemical shift perturbations (CSP) as a result of the Y24W mutation in the unbound (apo) state than the bound (holo) state (Fig. 4B and C). This suggests conformational differences between the unbound states of HP1 wild type and HP1–Y24W that extend beyond proximal residue effects due to the Tyr to Trp mutation, and are mainly focused at the binding pocket. Furthermore, the sidechain Nε of Y24W also showed a large change in chemical shift between the peptide bound and unbound states (Fig. 4A, inset), suggesting a change in conformation of the Trp sidechain upon binding.
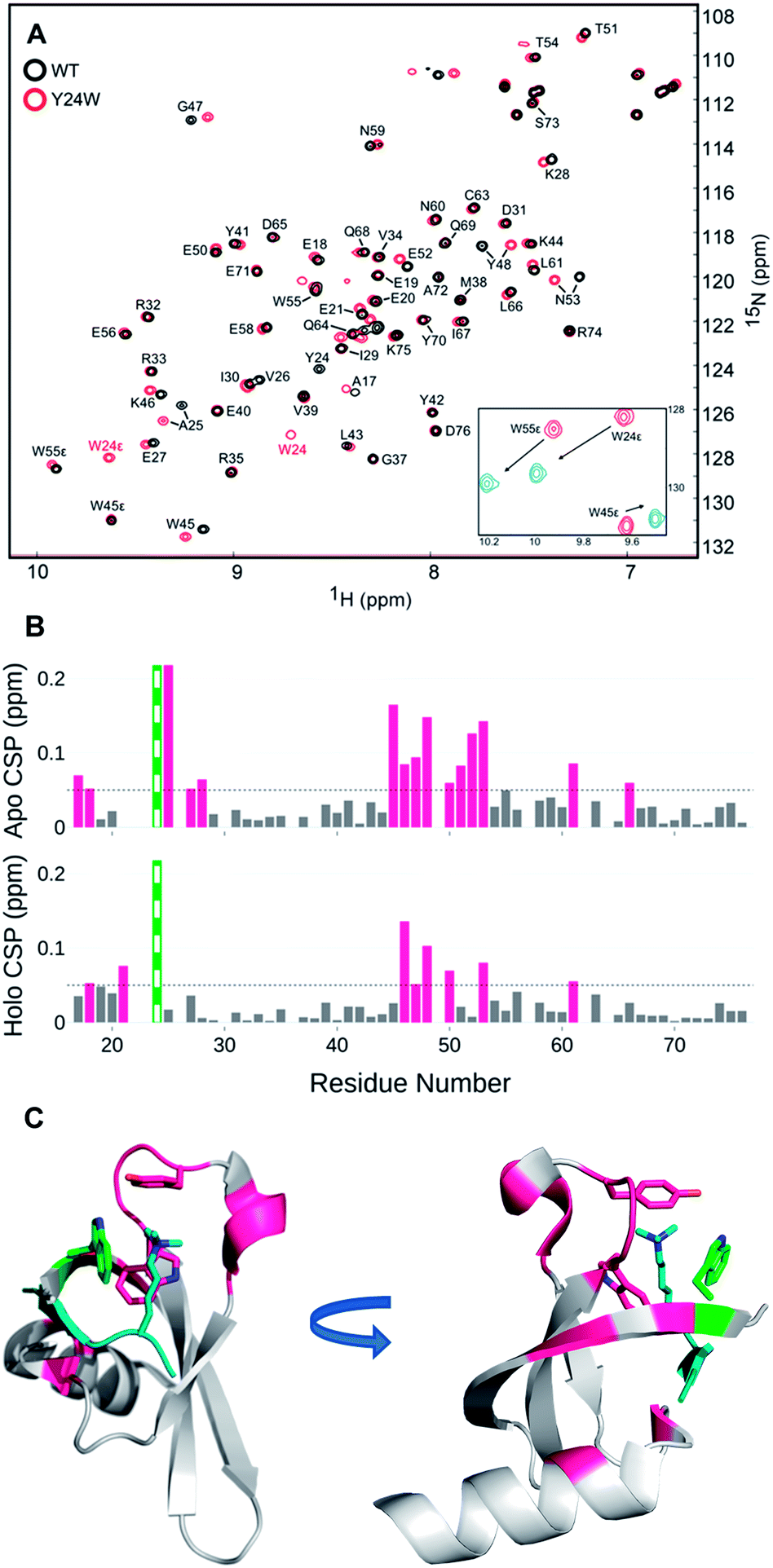 | ||
| Fig. 4 (A) [1H-15N] HSQC overlay of apo WT HP1 (black) and Y24W mutant (red). Y24W titration with the H3K9me3 peptide, stoichiometry from 0 (red) to 1.5 (blue), causes substantial chemical shift perturbations to the Nε of W24. (B) Several chemical shift perturbations (CSPs) in the apo (top) state are larger than the holo (bottom). CSP larger than 0.05 ppm are shown in pink. The green and white bar is the site of mutation. (C) The residues with CSP larger than 0.05 (pink) are mapped to the H3K9me3 (teal) bound Y24W HP1 structure (PDB: 6MHA) with W24 (green), W45, Y48, and K9me3 residues shown as sticks. | ||
Analogous to the HSQC data, the CD spectra of the unbound wild type HP1 and HP1–Y24W are generally similar, as indicated by the ratio of the mean residue ellipticity (MRE) value at 220 to 206 nm (Fig. S4†). Thermal CD melts showed only small changes in stability, further supporting that the overall structure of HP1 remains intact (Fig. S4†). In the unbound state, there are differences in the positive peak at 232 nm, which we primarily attribute to exciton coupling among residues in the aromatic cage (Fig. 5). Compared to wild-type, the exciton peak for HP1–Y24W is lower, suggesting a difference in the relative orientation of the aromatic residues in the aromatic cage. This local change in the aromatic cage region correlates well with the chemical shift perturbations present in the unbound HSQC data.
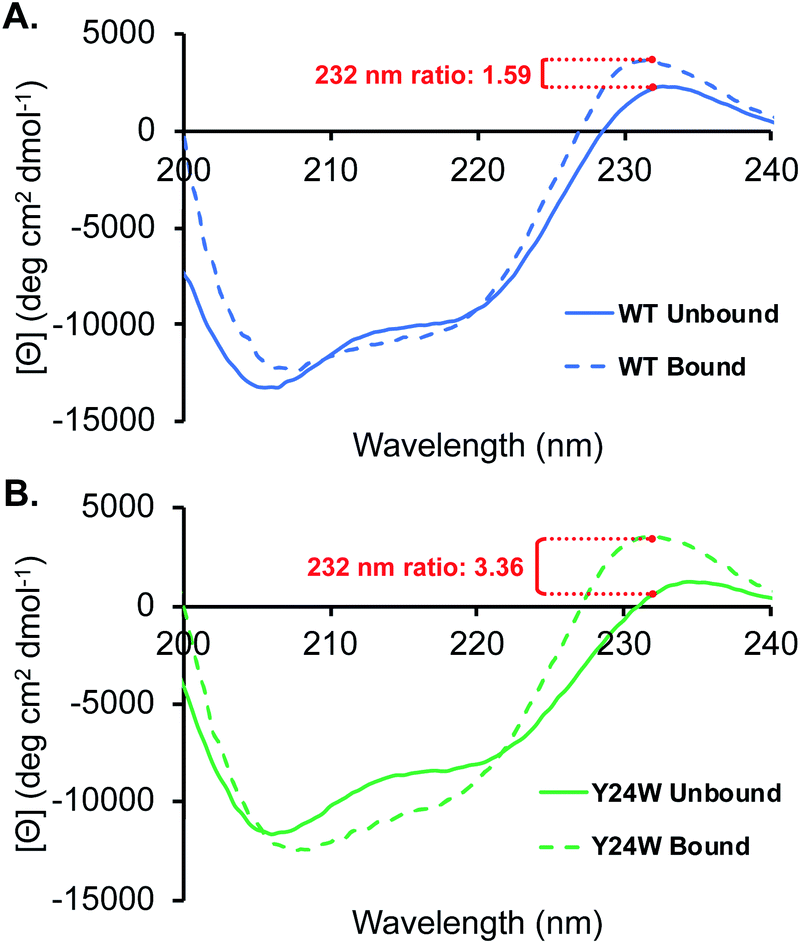 | ||
| Fig. 5 CD spectra of HP1 (A), and HP1–Y24W (B) in the presence and absence of H3K9me3. Ratio of the bound to unbound exciton peak are highlighted in red for each chromodomain. Spectra were obtained with 25 μM chromodomain and 100 μM H3K9me3 in 10 mM sodium phosphate buffer, pH 7.4 at 20 °C. | ||
In the presence of ligand, deeper minima from 210 to 220 nm in the bound state are observed for both proteins, indicating an increase in folding attributable to the formation of a three stranded beta-sheet between the chromodomain and the H3K9me3 peptide backbone upon binding (Fig. 5). The most significant difference between the HP1-Y24W and wild type CD spectra is the relative change in the positive exciton peak upon binding H3K9me3, which is reflected in the ratio of bound to unbound MRE values at 232 nm (Fig. 5). In Y24W, the change in exciton coupling is greater than wild type, while the magnitude in the bound states of both Y24W and wild type are similar. This is consistent with the NMR data, suggesting a more significant rearrangement of the aromatic cage in going from the unbound to bound state for Y24W. Taken together, these experiments suggest that differences in unbound states as well as changes in the conformation of W24 upon binding may contribute to the increased entropic cost of binding in the Y24W mutant.
Conclusions
These studies experimentally demonstrate for the first time that Trp provides a sizable increase in cation–π binding relative to Tyr in the HP1 chromodomain. Calculations yielded a more favorable interaction energy of −7.5 kcal mol−1 for HP1–Y24W, which is in reasonable agreement with the −5 kcal mol−1 enthalpic difference between wild type and HP1–Y24W measured by ITC. Calculations further suggest that the majority of this energetic gain arises from more favorable cation–π interactions, whereas increased van der Waals interactions with the Kme3 side chain provide minor stabilization amounting to about −2 kcal mol−1. Despite the enhanced interaction energy, binding affinity in this case is counterbalanced by an entropic penalty which appears to arise in part due to changes in the W24 conformation and surrounding binding pocket in the bound versus unbound states. Furthermore, these findings provide insight into the evolutionary basis of aromatic amino acids in a native Kme3 reader protein. While Trp, for example, provides the strongest cation–π interaction, natural selection is inherently governed by constraints that are rooted in structure and function.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of Health, National Institute of General Medical Sciences (GM 118499 to E. M. B. and M. L. W. and GM 109078 to K. N. H.). K. I. A. acknowledges funding from a Burroughs Wellcome Fellowship. G. O. L acknowledges the Molecular and Cellular Biophysics Training Program funded by the NIH (T32 GM008570). Crystallography was performed in the UNC Macromolecular Crystallography core facility that is supported by the National Cancer Institute of the National Institutes of Health under award number P30CA016086. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID (or 22-BM) beamline at the Advanced Photon Source, Argonne National Laboratory. SER-CAT is supported by its member institutions (see http://www.ser-cat.org/members.html), and equipment grants (S10_RR25528 and S10_RR028976) from the National Institutes of Health. We thank Dr Ashutosh Tripathy for assistance with calorimetry and circular dichroism, and Dr Christopher Snow (Colorado State University) for assistance with PyMOL scripts.References
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS PubMed.
- M. W. Nowak, P. C. Kearney, J. R. Sampson, M. E. Saks, C. G. Labarca, S. K. Silverman, W. Zhong, J. Thorson, J. N. Abelson, N. Davidson, P. G. Schultz, D. A. Dougherty and H. A. Lester, Science, 1995, 268, 439–442 CrossRef CAS PubMed.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS PubMed.
- M. R. Davis and D. A. Dougherty, Phys. Chem. Chem. Phys., 2015, 17, 29262–29270 RSC.
- J. M. Myslinski, J. H. Clements and S. F. Martin, Bioorg. Med. Chem. Lett., 2014, 24, 3164–3167 CrossRef CAS PubMed.
- G. N. Nagy, L. Marton, A. Contet, O. Ozohanics, L.-M. Ardelean, Á. Révész, K. Vékey, F. D. Irimie, H. Vial, R. Cerdan and B. G. Vértessy, Angew. Chem., Int. Ed., 2014, 53, 13471–13476 CrossRef CAS PubMed.
- W. Zhong, J. P. Gallivan, Y. Zhang, L. Li, H. A. Lester and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12088–12093 CrossRef CAS PubMed.
- C. Grauffel, B. Yang, T. He, M. F. Roberts, A. Gershenson and N. Reuter, J. Am. Chem. Soc., 2013, 135, 5740–5750 CrossRef CAS PubMed.
- D. L. Beene, G. S. Brandt, W. Zhong, N. M. Zacharias, H. A. Lester and D. A. Dougherty, Biochemistry, 2002, 41, 10262–10269 CrossRef CAS PubMed.
- S. A. Pless, J. D. Galpin, A. Frankel and C. A. Ahern, Nat. Commun., 2011, 2, 351 CrossRef PubMed.
- C.-Y. Li, X.-L. Chen, X. Shao, T.-D. Wei, P. Wang, B.-B. Xie, Q.-L. Qin, X.-Y. Zhang, H.-N. Su, X.-Y. Song, M. Shi, B.-C. Zhou and Y.-Z. Zhang, J. Bacteriol., 2015, 197, 3378–3387 CrossRef CAS PubMed.
- S. D. Taverna, H. Li, A. J. Ruthenburg, C. D. Allis and D. J. Patel, Nat. Struct. Mol. Biol., 2007, 14, 1025–1040 CrossRef CAS PubMed.
- R. M. Hughes, K. R. Wiggins, S. Khorasanizadeh and M. L. Waters, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11184–11188 CrossRef CAS PubMed.
- S. A. Baril, A. L. Koenig, M. W. Krone, K. I. Albanese, C. Q. He, G. Y. Lee, K. N. Houk, M. L. Waters and E. M. Brustad, J. Am. Chem. Soc., 2017, 139, 17253–17256 CrossRef CAS PubMed.
- J. J. A. G. Kamps, J. Huang, J. Poater, C. Xu, B. J. G. E. Pieters, A. Dong, J. Min, W. Sherman, T. Beuming, F. Matthias Bickelhaupt, H. Li and J. Mecinović, Nat. Commun., 2015, 6, 8911 CrossRef CAS PubMed.
- L. Kaustov, H. Ouyang, M. Amaya, A. Lemak, N. Nady, S. Duan, G. A. Wasney, Z. Li, M. Vedadi, M. Schapira, J. Min and C. H. Arrowsmith, J. Biol. Chem., 2011, 286, 521–529 CrossRef CAS PubMed.
- R. M. Hughes and M. L. Waters, J. Am. Chem. Soc., 2005, 127, 6518–6519 CrossRef CAS PubMed.
- S. E. Wheeler and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 3126–3127 CrossRef CAS PubMed.
- S. A. Jacobs and S. Khorasanizadeh, Science, 2002, 295, 2080–2083 CrossRef CAS PubMed.
- S. A. Jacobs, S. D. Taverna, Y. Zhang, S. D. Briggs, J. Li, J. C. Eissenberg, C. D. Allis and S. Khorasanizadeh, EMBO J., 2001, 20, 5232–5241 CrossRef CAS PubMed.
- Because cation–pi interactions have previously been shown to have a shallow distance dependence, this single-point approach based on the crystal structures is reasonable. See ref. 6 for additional details.
- J. M. Fox, M. Zhao, M. J. Fink, K. Kang and G. M. Whitesides, Annu. Rev. Biophys., 2018, 47, 223–250 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00227e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |