DOI:
10.1039/D0SC00197J
(Edge Article)
Chem. Sci., 2020,
11, 3829-3835
Interfering in apoptosis and DNA repair of cancer cells to conquer cisplatin resistance by platinum(IV) prodrugs†
Received
13th January 2020
, Accepted 20th February 2020
First published on 9th March 2020
Abstract
The dysregulation of apoptosis and DNA damage repair are two leading mechanisms of cisplatin resistance. Two anticancer PtIV prodrugs with the formulas [Pt(NH3)2Cl2(L1)2] (1, L1 = 3-chloro-benzo[b]thiophene-2-carboxylic acid) and [Pt(NH3)2Cl2(L2)2] (2, L2 = 3-chloro-6-methylbenzo[b]thiophene-2-carboxylic acid) were designed to target myeloid cell leukemia-1 (Mcl-1), a protein responsible for inhibiting apoptosis and promoting DNA damage repair. Complexes 1 and 2 exhibited high cytotoxicity against various cancer cell lines, especially cisplatin-resistant non-small-cell lung and ovarian cancer cells. The resistance factors of both complexes for cisplatin-resistant cancer cells also decreased markedly as compared with that of cisplatin. Both 1 and 2 could enter cancer cells effectively and cause DNA damage while simultaneously downregulating Mcl-1 to prompt a conspicuous apoptotic response. Complex 2 also downregulated the DNA damage repair proteins RAD51 and BRCA2 as well as inhibited the formation of RAD51 foci, which is regarded as a critical step and functional biomarker in homologous recombination. The acute toxicity of 1 and 2 to mice is lower than that of cisplatin, and more importantly, they show much stronger inhibition towards the growth of non-small-cell lung cancer in nude mice than cisplatin. Complexes 1 and 2 are the first Mcl-1-targeted PtIV prodrugs, and the latter could synchronously inhibit apoptosis and DNA repair related proteins in cisplatin-resistant cancer cells. The strategy of tuning both apoptosis and DNA repair pathways opens a promising window to overcoming resistance to cisplatin in anticancer chemotherapy, and is also a breakthrough in the design of multitalented platinum-based anticancer drugs.
Introduction
Cisplatin (CDDP) is one of the most effective anticancer drugs for treating various malignancies.1,2 It kills cancer cells primarily through damaging nuclear DNA to trigger apoptosis;3 unfortunately, its therapeutic efficacy is severely hindered by inherent and acquired drug resistance.4,5 In general, CDDP resistance is multifactorial, that is, it relies on the activation of multiple molecular or cellular responses.6,7 In these events, impeded apoptosis and enhanced DNA damage repair are two vital factors because some important proteins are dysregulated in CDDP-resistant tumors.6–9 For example, myeloid cell leukemia-1 (Mcl-1) is an antiapoptotic protein in the Bcl-2 family, which inhibits the progression of apoptosis via sequestering the proapoptotic proteins.10 Overexpression of Mcl-1 is the hallmark of several cancers and is associated with drug resistance and tumor relapse.11,12 Cancer cells can avoid apoptosis elicited by CDDP-mediated DNA damage through upregulating Mcl-1, leading to drug resistance;13,14 whereas the depletion of Mcl-1 can impair homologous recombination (HR),15 which is a major way to repair the DNA double-strand breaks (DSBs) triggered by CDDP.16,17 Therefore, impaired HR can block the repair of DNA DSBs and increase the curative effect of CDDP. We suppose that targeting Mcl-1 may not only promote apoptosis via mediating apoptosis-related pathways, but also interfere with DNA damage repair and ultimately potentiate CDDP towards CDDP-resistant tumors.
Octahedral PtIV prodrugs are kinetically inert in blood plasma and can be activated by biological reductants in tumor cells, such as ascorbic acid (AsA) and glutathione (GSH), producing PtII species to damage DNA and kill cancer cells.18,19 In addition to avoiding side reactions with biomolecules prior to DNA binding, PtIV complexes have two axial ligands to finely tune the pharmacological effects.20,21 This prodrug strategy has been widely used for the construction of multifunctional Pt complexes as illustrated in many studies.22–38
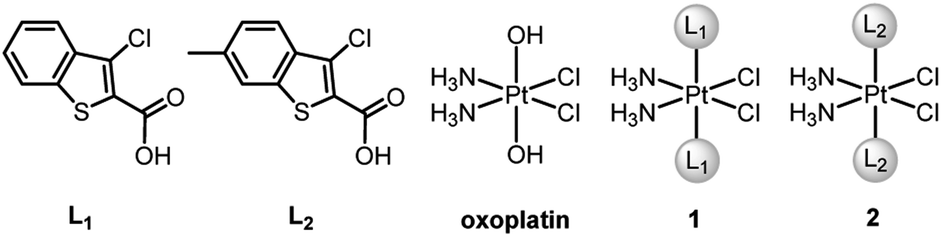
BRCA and RAD51 play essential roles in HR, and BRCA-proficient tumors are usually refractory to CDDP due to effective HR. Recently, we designed two PtIV–artesunate prodrugs, which inhibited HR via downregulating RAD51 and exhibited higher cytotoxicity against BRCA-proficient cancer cells than CDDP.39 However, platinum complexes simultaneously interfering in HR and apoptosis are rarely seen in the literature. Herein we report the design and properties of two PtIV prodrugs (1 and 2, Fig. 1) that target Mcl-1 to reverse CDDP resistance. The ligands L1 and L2, that is, Cl- and Cl-/Me-substituted benzothiophene-2-carboxylic acids, were identified as potential Mcl-1 inhibitors through preventing BH3-containing peptides from binding to Mcl-1.40,41 The incorporation of L1 and L2 into 1 and 2 respectively made these complexes exhibit potent antitumor potential and strong inhibition of Mcl-1 and HR proteins. Particularly, complex 2 is highly cytotoxic to CDDP-resistant non-small-cell lung cancer (NSCLC) and ovarian cancer cells and shows significant antitumor activity in vivo. The unique mechanism of action and remarkable activity of these complexes provide a new train of thought for designing PtIV prodrugs to overcome cisplatin resistance.
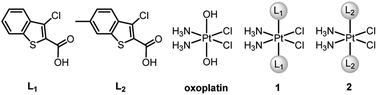 |
| | Fig. 1 Chemical structures of ligands L1 and L2, oxoplatin and complexes 1 and 2. | |
Results and discussion
Synthesis and characterization
cis-Diamminedichloro-trans-dihydroxyplatinum(IV) (oxoplatin) was prepared by oxidizing CDDP with hydrogen peroxide (30%).42 Complexes 1 and 2 were synthesized by directly reacting ligand L1 or L2 with oxoplatin using uronium salts as a coupling agent (Fig. 1 and Scheme S1†). The structures and purity of 1 and 2 were fully characterized by 1H, 13C and 195Pt NMR spectroscopy, HPLC and elemental analysis (see Fig. S1–S3†), which supported the proposed structures for the complexes. The proton resonances of the amine coordinated to the PtIV centre appear over a wide range from 6.0 to 7.0 ppm, which are similar to other PtIV complexes.43 The 195Pt NMR spectra show single resonance peaks at 1227.05 and 1226.80 ppm for 1 and 2, respectively, which are also similar to related PtIV complexes.44
Reduction, stability and lipophilicity
Prior to biological evaluation, the reduction potential, stability and lipophilicity of 1 and 2 were measured because they are crucial for antitumor activity.45 The reduction potentials of 1 and 2 determined by cyclic voltammetry are −0.66 and −0.72 V, respectively (see Fig. S4†). The relatively low values are indicative of less propensity for reduction. In fact, both 1 and 2 are stable in cell culture media and phosphate buffered saline (PBS) at pH 7.4 and 37 °C for 24 h (see Fig. S5 and S6†). The reduction process was further studied through the reactions between the complexes and AsA in PBS following the reported procedure,43 and the data indicate that 1 and 2 can be reduced by AsA (see Fig. S7†). The lipophilicities (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Po/w) of 1 and 2 determined in an n-octanol/buffer system via the shake-flask method were 0.60 and 0.84, respectively, which are more lipophilic than CDDP (−2.15), thus suggesting that complexes 1 and 2 enter cancer cells more readily than CDDP.
Po/w) of 1 and 2 determined in an n-octanol/buffer system via the shake-flask method were 0.60 and 0.84, respectively, which are more lipophilic than CDDP (−2.15), thus suggesting that complexes 1 and 2 enter cancer cells more readily than CDDP.
Cytotoxicity
The cytotoxicities of 1, 2, L1, L2, CDDP, and oxoplatin, as well as a mixture of CDDP and L1 or L2 at a molar ratio of 1:2 were tested against human NSCLC A549, human ovarian cancer A2780, and their CDDP-resistant counterparts A549/DDP and A2780/DDP cells using the MTT assay. Meanwhile, the breast cancer MCF-7, the human ovarian cancer Caov3, and the human normal liver L-02 cell lines were also subjected to the test. The median growth inhibitory concentrations (IC50) of the compounds at 72 h are listed in Tables 1 and S2.† Complexes 1 and 2 showed potent cytotoxicity towards all the tested cancer cell lines, particularly the A549/DDP and A2780/DDP cells. The IC50 values of 1 and 2 for A549/DDP cells are 18- and 32-fold less than that of CDDP, respectively; and those for A2780/DDP cells are 35- and 61-fold less than that of CDDP, respectively. The resistance factors (RFs), which are defined as the ratios of IC50 for resistant cells to that for sensitive cells, of CDDP, 1, and 2 for A549 and A549/DDP cells are 5.0, 0.9, and 0.9, respectively; and those for A2780 and A2780/DDP cells are 7.2, 0.9, and 0.7, respectively. These results indicate that 1 and 2 possess great potential to overcome CDDP resistance in NSCLC and ovarian cancer cells. All the control compounds or mixtures, including CDDP plus ligand, oxoplatin, and the ligands alone, only showed moderate or no cytotoxicity against these cell lines. Since Mcl-1 is overexpressed in many tumours, especially CDDP-resistant cancers,11 we tested the levels of Mcl-1 in these cells by western blot (WB). As expected, the expression of Mcl-1 in A549/DDP, A2780/DDP and CDDP-insensitive Caov3 cells is significantly higher than that in other cells (see Fig. S8†). Complexes 1 and 2 exhibited strong cytotoxicity towards all the tested cancer cells regardless of the Mcl-1 expression; whereas CDDP only showed high cytotoxicity against cancer cells expressing a low level of Mcl-1. The results highlighted the effectiveness of targeting Mcl-1 as a strategy for overcoming CDDP resistance.
Table 1 IC50 (μM) of the compounds against different cancer cell lines at 72 h
| Compounds |
A549 |
A549/DDP |
A2780 |
A2780/DDP |
|
1
|
1.5 ± 0.3 |
1.4 ± 0.4 |
0.8 ± 0.2 |
0.7 ± 0.2 |
|
2
|
0.9 ± 0.1 |
0.8 ± 0.1 |
0.6 ± 0.3 |
0.4 ± 0.1 |
| CDDP |
5.1 ± 0.3 |
25.7 ± 2.9 |
3.4 ± 1.0 |
24.4 ± 3.7 |
| CDDP + 2L1 |
6.0 ± 1.2 |
21.0 ± 2.1 |
3.6 ± 0.6 |
25.3 ± 2.6 |
| CDDP + 2L2 |
5.2 ± 1.3 |
18.0 ± 3.5 |
4.2 ± 0.3 |
23.1 ± 1.9 |
| Oxoplatin |
>50 |
>50 |
30.0 ± 5.6 |
>50 |
| L1 |
>100 |
>100 |
>100 |
>100 |
| L2 |
>100 |
>100 |
>100 |
>100 |
Cellular uptake
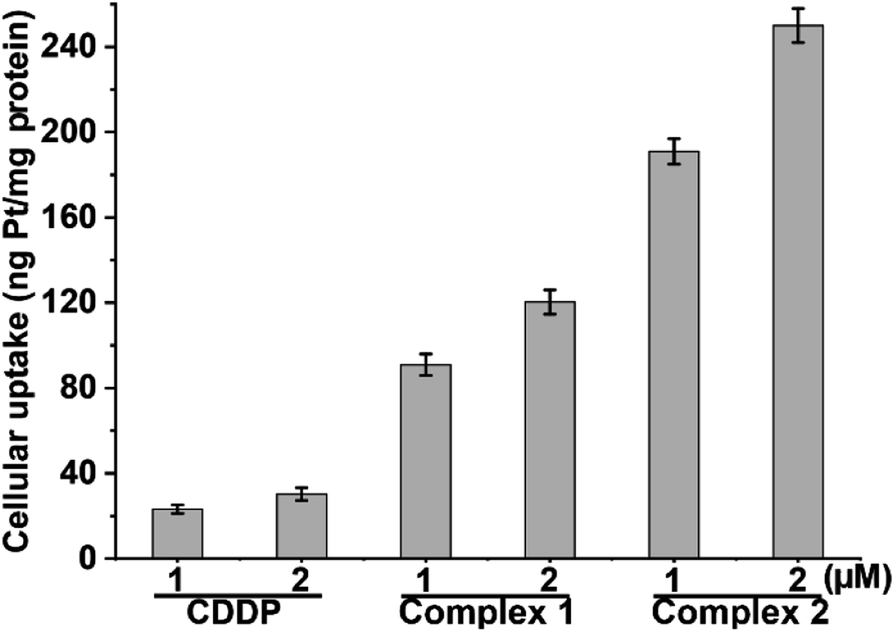
Owing to the potent cytotoxicity of 1 and 2 against CDDP-resistant cancer cells, the cellular Pt contents after incubation with A549/DDP cells for 12 h were measured by means of inductively coupled plasma mass spectrometry (ICP-MS). The results are shown in Fig. 2. The cellular Pt accumulation follows an order of 2 > 1 > CDDP, which is in line with their lipophilicity and cytotoxicity sequences. Apparently, the conjugation of L1 or L2 with the PtIV scaffold greatly improves the lipophilicity and cellular uptake of the complexes, which may contribute to the elevated cytotoxicity. Further, we measured the cellular Pt content in A549/DDP cells after incubation with different concentrations of CDDP, 1 and 2 for 36 h. The cellular uptake of Pt is positively correlated with the concentration of the complexes (see Table S3†). The Pt contents in A549/DDP cells after treatment with 12 μM CDDP, 1.4 μM 1, and 0.7 μM 2 are similar, suggesting that the cell-penetrating ability of 1 and 2 is greater than that of CDDP.
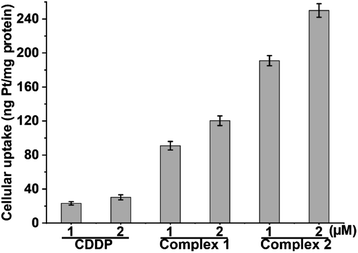 |
| | Fig. 2 Cellular uptake of Pt in A549/DDP cells after exposure to CDDP, 1 and 2 for 12 h. | |
DNA damage
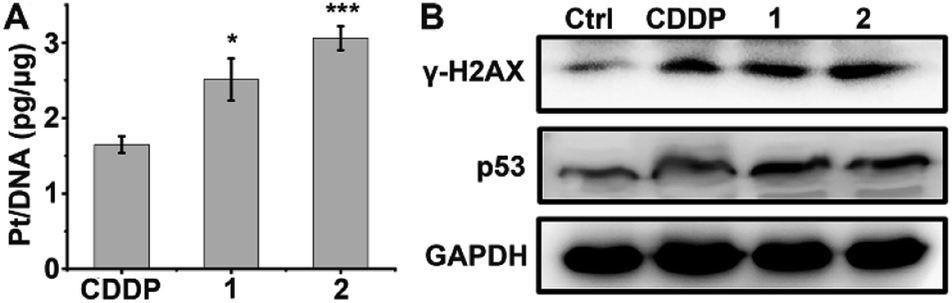
DNA is believed to be the major cellular target of platinum complexes, so the ability of 1 and 2 to platinize nuclear DNA was assessed. After drug treatment, the genomic DNA of A549/DDP cells was extracted by a DNA isolation reagent (DNAzol) and the quantity of bound Pt was measured by ICP-MS. As shown in Fig. 3A, the platination of DNA by 1 and 2 is greater than that by CDDP, which may be due to the higher cellular uptake of 1 and 2 in A549/DDP cells. γ-H2AX, the phosphorylated form of the histone protein H2AX, is a known biomarker of DNA DSBs caused by CDDP and can be detected by immunoblotting.46–48Fig. 3B shows that the expression of γ-H2AX in drug-treated A549/DDP cells increased significantly as compared with the control cells, indicating that the genomic DNA was severely damaged by the platinum complexes. The expression of p53, a downstream effector of DNA damage,49 also increased after treatment with complexes 1, 2 or CDDP (Fig. 3B), suggesting that the DNA damage could not be repaired and that p53 began to initiate the apoptosis process.
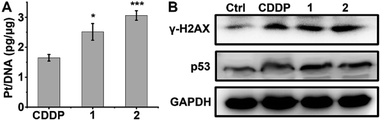 |
| | Fig. 3 Platination of cellular DNA (A), expression of γ-H2AX and p53 (B) in A549/DDP cells after incubation with 1 μM CDDP, 1 and 2 for 36 h. * p < 0.05, *** p < 0.001 (versus CDDP-treated group). | |
Cell cycle arrest
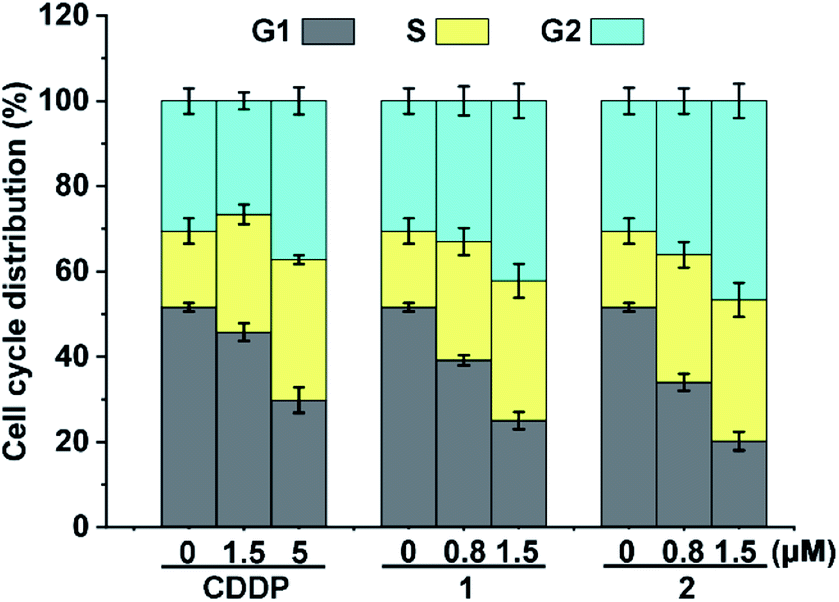
Cellular DNA damage could induce cell cycle arrest.43 We hence examined the cell cycle arrest of A549/DDP cells by flow cytometry after treatment with the complexes. As shown in Fig. 4, in comparison with the control, the cell distribution in the S and G2 phases increased moderately after the treatment with 1 and 2. CDDP also arrested the cell cycle mainly in the S and G2 phases, which is consistent with other reports.43 The similarity of the cell cycle arrest among these complexes suggests that the active form and DNA damage mode of 1 and 2 are similar to those of CDDP.
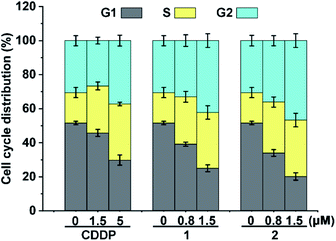 |
| | Fig. 4 Cell cycle arrest of A549/DDP cells after incubation with CDDP, 1 and 2 for 36 h. | |
The results associated with cellular DNA damage response and cycle arrest demonstrate that complexes 1 and 2 were efficiently reduced in A549/DDP cells to release PtII species for DNA binding, which constituted the basis for their remarkable cytotoxicity.
Expression of Mcl-1 and apoptotic proteins
In the absence of complexes, the expression of Mcl-1 in A549/DDP and A2780/DDP cells is significantly higher than that in A549 and A2780 cells (see Fig. S8†); hence, we examined the expression of Mcl-1 in A549/DDP cells in the presence of 1 or 2 using WB. As shown in Fig. 5A, the expression of Mcl-1 in A549/DDP cells is significantly inhibited, while it is slightly upregulated by CDDP as compared with the control cells. The downregulation of Mcl-1 could arise from the higher cellular uptake of 1 and 2 than that of CDDP (see Fig. 2). To examine this possibility, we studied the expression of Mcl-1 in A549/DDP cells after incubation with 1.4 μM 1, 0.7 μM 2, and 12 μM CDDP, which ensured similar Pt accumulation in the cells (vide supra). The results show that the expression of Mcl-1 decreased after incubation with 1 and 2, but increased after incubation with CDDP as compared with the control (see Fig. S11†), thus proving that the downregulation of cellular Mcl-1 by 1 and 2 is mainly due to the introduction of axial ligands rather than the higher cellular accumulation of Pt. Actually, ligand L1 or L2 alone could downregulate the cellular Mcl-1 at high concentrations (see Fig. S12†). The concentrations of 1 and 2 (ca. 1 μM) required to inhibit cellular Mcl-1 are much lower than those of L1 and L2 (ca. 200 μM), implying that it may be hard for the negatively charged ligands to enter the cells, and hence showing the advantages of 1 and 2 as integrated functional entities. In fact, the IC50 values of the mixtures of CDDP and L1 or L2 are similar to that of CDDP (see Table 1), showing no superiority in cytotoxicity.
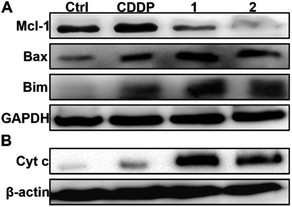 |
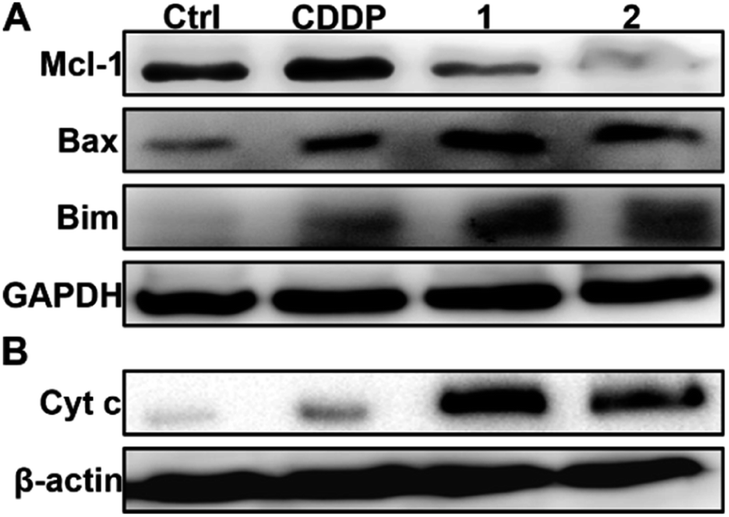
| | Fig. 5 Expression of Mcl-1, Bax and Bim (A), and Cyt c (B) in A549/DDP cells after incubation with complexes 1 and 2 (1 μM) for 36 h. | |
It is known that Mcl-1 prevents the activation of apoptosis effector proteins such as Bax, either through direct interaction or by inhibiting proapoptotic BH3-only proteins like Bim.50 Therefore, the downregulation or inhibition of Mcl-1 could result in the activation of several proapoptotic proteins and facilitate the release of cytochrome c (Cyt c) into the cytosol, thereby activating caspase and triggering an apoptotic response.50 The upregulation of Bim and Bax as well as the release of Cyt c in A549/DDP cells after treatment with 1 and 2 are shown in Fig. 5. The release of Cyt c can activate apoptotic protease and then cleave the caspase-3 zymogen, leading to apoptosis.51 Therefore, we further measured the level of cleaved caspase-3 in A549/DDP cells after incubation with CDDP, 1, and 2. As expected, cleaved caspase-3 was observed in cancer cells after the treatment with complexes 1 and 2 (see Fig. S13†). All these events are conducive to apoptosis or suppressing resistance to apoptosis.
Apoptosis
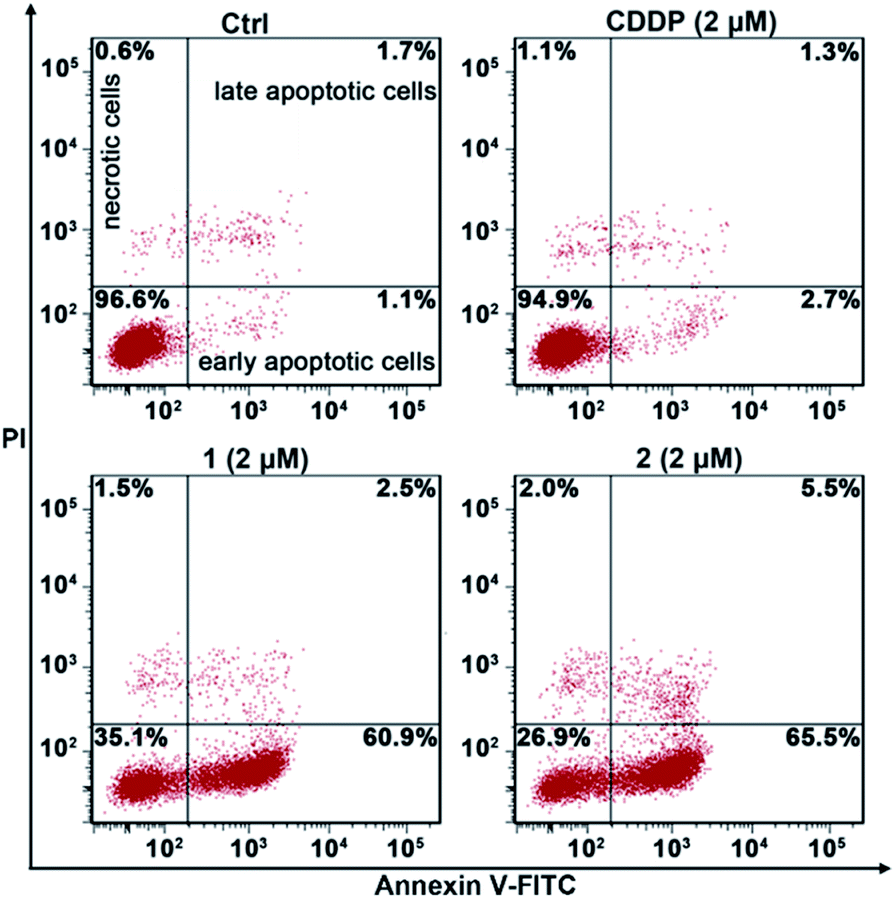
The apoptosis of A549/DDP cells after treatment with these complexes for 72 h and staining with annexin V-FITC and propidium iodide (PI) was investigated. The cellular density plots are shown in Fig. 6. Complexes 1 and 2 efficiently induced 60.9% and 65.5% of A549/DDP cells to go into early apoptosis, respectively. By contrast, CDDP only triggered 2.7% of the cells to undergo early apoptosis, which agrees well with the CDDP-resistant characteristics of A549/DDP cells. The sharp contrast of 1 and 2 to CDDP in promoting apoptosis reveals that these PtIV complexes possess great potential to conquer CDDP resistance.
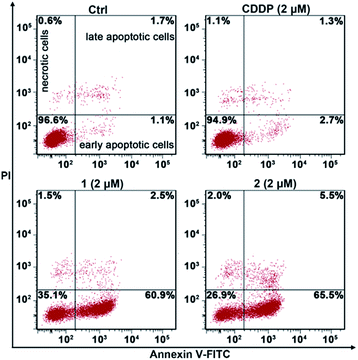 |
| | Fig. 6 Apoptotic analysis of A549/DDP cells by flow cytometry after incubation with the complexes for 72 h. | |
Expression of HR proteins RAD51 and BRCA2
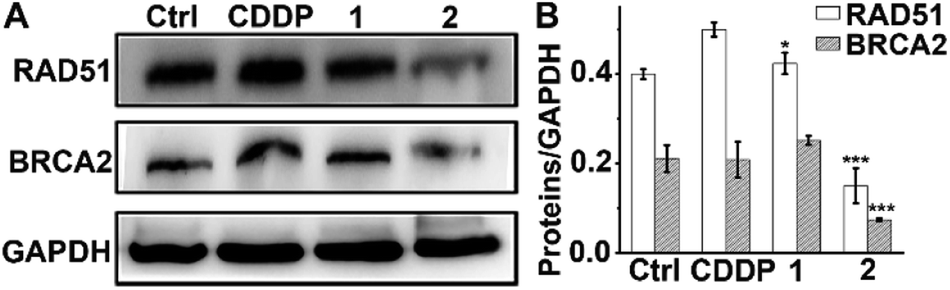
In addition to the antiapoptotic function, Mcl-1 also plays a key role in the DNA damage response.52,53 Cellular Mcl-1 depletion can impede the repair of DNA DSBs by impairing HR and decreasing the expression of related proteins.15 RAD51 and BRCA2 play essential roles in HR by promoting homology search and stimulating strand invasion into the sister chromatid. Specifically, following DNA-end resection, BRCA2 directs RAD51 filament nucleation onto single-stranded DNA (ssDNA), which catalyzes a strand exchange reaction to initiate HR repair.54,55 We hence measured the levels of RAD51 and BRCA2 in A549/DDP cells after exposure to complexes 1 and 2 or CDDP to evaluate their impact on the HR ability. As shown in Fig. 7, CDDP induced an increase in RAD51 expression, presumably due to the damage to genetic DNA, and barely influenced the expression of BRCA2. The levels of RAD51 and BRCA2 in 1-treated cells were slightly higher than those in the control cells, suggesting that 1 had almost no effect on the expression of RAD51 and BRCA2. Impressively, the expression of RAD51 and BRCA2 decreased markedly after treatment with 2 as compared with both the control and CDDP-treated cells. The downregulation of RAD51 implies that the repair of DSBs by HR is inactivated, and that DNA damage could remain to produce lethal effects to cancer cells. It has been reported that BRCA2-deficient cancer cells are hypersensitive to DNA-crosslinking agents such as CDDP due to the impaired HR, while BRCA2-proficient cancer cells are refractory to Pt-based drugs owing to the effective HR.56,57 Knockdown of BRCA2 can enhance the cytotoxicity of CDDP in ovarian cancer cells.58 Therefore, BRCA2 is a potential target for defeating CDDP resistance. The decreased expression of BRCA2 indicates that 2 can reduce the HR ability of A549/DDP cells. Oddly, complex 1 only differs slightly from complex 2 in terms of ligands, but it hardly influenced the expressions of RAD51 and BRCA2 in A549/DDP cells. Therefore, we suppose that the 6-methyl in L2 may play a key role in suppressing the expression of these proteins; however, the detailed action is unknown at the moment. The results exemplify that even a minute change in the structure of PtIV complexes could produce significant impact on their cytostatic mechanism and antitumor potency.
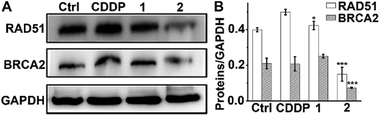 |
| | Fig. 7 Expression of RAD51 and BRCA2 in A549/DDP cells after incubation with each complex (1 μM) for 36 h (A), and the relative expression levels of RAD51 and BRCA2 to GAPDH (B). * p < 0.05, *** p < 0.001 (versus CDDP-treated group). | |
Formation of RAD51 foci
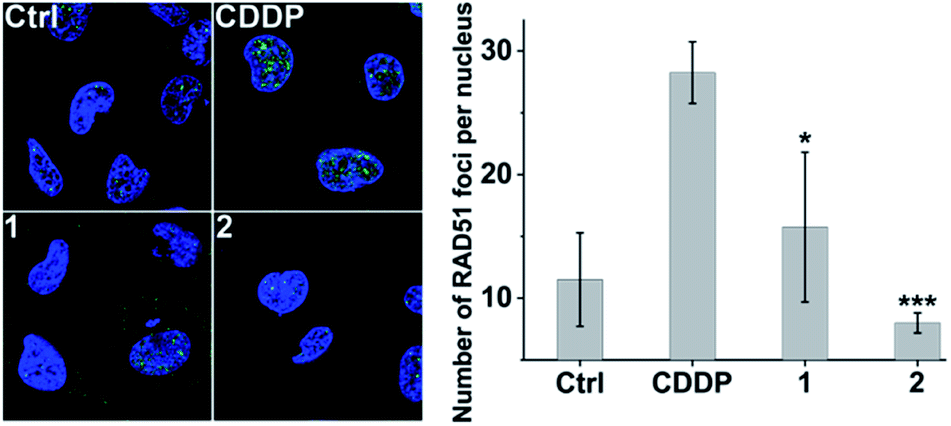
Furthermore, immunofluorescence staining was used to study the formation of RAD51 foci, which is regarded as a critical step and functional biomarker in HR.59,60 As shown in Fig. 8, CDDP triggered increased RAD51 foci (green) as compared with the control due to the DNA damage. However, the number of RAD51 foci in the nucleus induced by 2 is markedly less than that induced by CDDP even though the DNA damage caused by 2 is more severe than that caused by CDDP (see Fig. 3 and S9†). The results show that complex 2 can prevent the formation of RAD51 foci, which would hinder the HR repair of DSBs, and further sensitize cancer cells to DNA damage.
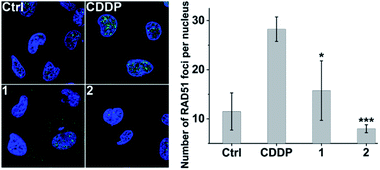 |
| | Fig. 8 Representative images of the formation of RAD51 foci (green) in the nucleus (blue) induced by different complexes (1 μM) after incubation with A549/DDP cells for 36 h (left), and the number of RAD51 foci per nucleus (right). * p < 0.05, *** p < 0.001 (versus CDDP-treated group). | |
Acute toxicity and in vivo antitumor activity
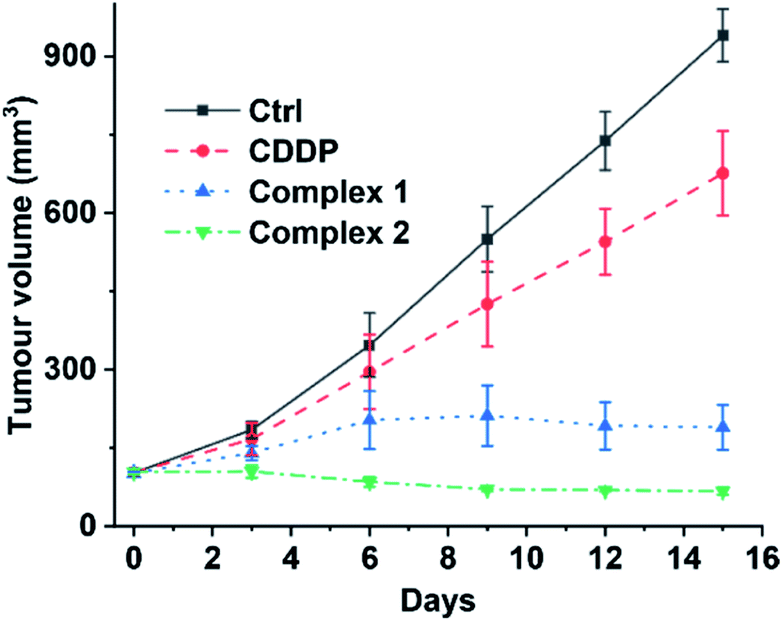
Finally, we evaluated the acute toxicity and in vivo antitumor efficacy of 1 and 2. The median lethal dose (LD50), body weight change and histological images of major organs after intravenous injection of each complex were investigated. The LD50 values of 1 and 2 are 16.24 ± 2.89 and 12.31 ± 1.31 mg kg−1, respectively (see Fig. S14†), much higher than that of CDDP (4.06 ± 1.02 mg kg−1). No obvious change in body weight or damage to major organs was observed in the 1- or 2-treated mice (see Fig. S15 and S16†). By contrast, the CDDP-treated mice showed significant weight loss as compared with the control (see Fig. S15†). These results indicate that complexes 1 and 2 are less toxic to the body or more biocompatible than CDDP. Furthermore, mice implanted with A549 cells were used as xenograft models to evaluate the in vivo antitumor efficacy of 1 and 2. As shown in Fig. 9 and S17,† strong inhibition of tumor growth was observed after the treatment. On day 15, the average tumor volume was 940.26 ± 50.55 and 676.25 ± 80.64 mm3 for the control and CDDP groups, respectively. Remarkably, complexes 1 and 2 effectively suppressed the tumor growth. The average tumor volume in the 1- and 2-treated groups after 15 d was 189.82 ± 43.17 and 67.29 ± 6.71 mm3, respectively, only reaching 20% and 7% of the control size. These results indicate that complexes 1 and 2 are more efficient inhibitors of tumor growth and less toxic to mice than CDDP, and hence are promising drug candidates for cancer therapy.
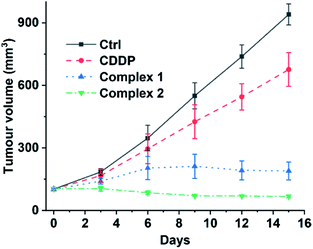 |
| | Fig. 9 Volume of A549 tumours in mice treated intravenously with CDDP (1.3 mgPt kg−1), 1, and 2 (1.1 mgPt kg−1) every three days for 15 d. | |
Conclusions
The mechanism of CDDP resistance is complicated and involves several stages.6,7 Intervening in one stage alone may not necessarily overcome resistance effectively because of the compensatory mechanism, and thus metal complexes with multiple targets are very attractive to researchers.61–63 Since CDDP primarily damages nuclear DNA to induce apoptosis,3 targeting proteins relevant to DNA repair or apoptosis seems to be an effective way to enhance cytotoxicity. Some examples of PtIV prodrugs targeting DNA damage repair or apoptosis-related proteins have been reported.31,35,39 However, efforts have rarely been dedicated to the development of Pt complexes interfering synchronously with these two processes. In this study, we designed two PtIV prodrugs that could target the myeloid cell leukemia-1 (Mcl-1) protein, hoping to circumvent cisplatin resistance. Both complexes showed enhanced cytotoxicity against cisplatin-resistant non-small-cell lung and ovarian cancer cells. More importantly, they exhibited remarkable anticancer potential toward lung cancer xenografts and low toxicity in mice. In particular, complex 2 not only inhibited the expression of Mcl-1, but also downregulated the homologous recombination proteins RAD51 and BRCA2, thereby significantly promoting apoptosis. Apoptosis evasion is an important hallmark of cancers64 and limits the efficacy of CDDP.6,7 Although Mcl-1 has received much attention due to its unique role in mediating apoptosis and chemoresistance,11,65,66 PtIV complexes targeting Mcl-1, and further targeting RAD51 and BRCA2 have not been reported so far. This study provides uncommon examples of the design of such complexes; also, it gives some new insight into the cytostatic mechanism of such complexes and broadens the way to designing Pt complexes for overcoming CDDP resistance. Regrettably, the molecular details of the interactions between the complexes and the proteins are unclear at the moment.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (Grants 21731004, 91953201, 21877059, 31570809, and 31700714), and the Excellent Research Program of Nanjing University (Grant ZYJH004).
Notes and references
- K. M. Deo, D. L. Ang, B. McGhie, A. Rajamanickam, A. Dhiman, A. Khoury, J. Holland, A. Bjelosevic, B. Pages, C. Gordon and J. R. Aldrich-Wright, Coord. Chem. Rev., 2018, 375, 148–163 CrossRef CAS.
- J. S. Butler and P. J. Sadler, Curr. Opin. Chem. Biol., 2013, 17, 175–188 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202–224 RSC.
- L. Galluzzi, I. Vitale, J. Michels, C. Brenner, G. Szabadkai, A. Harel-Bellan, M. Castedo and G. Kroemer, Cell Death Dis., 2014, 5, e1257 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- S. Maji, S. Panda, S. K. Samal, O. Shriwas, R. Rath, M. Pellecchia, L. Emdad, S. K. Das, P. B. Fisher and R. Dash, Adv. Cancer Res., 2018, 137, 37–75 CrossRef PubMed.
- A. Ray Chaudhuri, E. Callen, X. Ding, E. Gogola, A. A. Duarte, J. E. Lee, N. Wong, V. Lafarga, J. A. Calvo, N. J. Panzarino, S. John, A. Day, A. V. Crespo, B. Shen, L. M. Starnes, J. R. de Ruiter, J. A. Daniel, P. A. Konstantinopoulos, D. Cortez, S. B. Cantor, O. Fernandez-Capetillo, K. Ge, J. Jonkers, S. Rottenberg, S. K. Sharan and A. Nussenzweig, Nature, 2016, 535, 382–387 CrossRef PubMed.
- R. M. Perciavalle and J. T. Opferman, Trends Cell Biol., 2013, 23, 22–29 CrossRef CAS PubMed.
- A. De Blasio, R. Vento and R. Di Fiore, J. Cell. Physiol., 2018, 233, 8482–8498 CrossRef CAS PubMed.
- I. E. Wertz, S. Kusam, C. Lam, T. Okamoto, W. Sandoval, D. J. Anderson, E. Helgason, J. A. Ernst, M. Eby, J. Liu, L. D. Belmont, J. S. Kaminker, K. M. O’Rourke, K. Pujara, P. B. Kohli, A. R. Johnson, M. L. Chiu, J. R. Lill, P. K. Jackson, W. J. Fairbrother, S. Seshagiri, M. J. Ludlam, K. G. Leong, E. C. Dueber, H. Maecker, D. C. Huang and V. M. Dixit, Nature, 2011, 471, 110–114 CrossRef CAS PubMed.
- J. Ma, Z. Zhao, K. Wu, Z. Xu and K. Liu, Gene, 2016, 587, 147–154 CrossRef CAS PubMed.
- J. Michels, F. Obrist, I. Vitale, D. Lissa, P. Garcia, P. Behnam-Motlagh, K. Kohno, G. S. Wu, C. Brenner, M. Castedo and G. Kroemer, Biochem. Pharmacol., 2014, 92, 55–61 CrossRef CAS PubMed.
- A. R. Mattoo, R. K. Pandita, S. Chakraborty, V. Charaka, K. Mujoo, C. R. Hunt and T. K. Pandita, Mol. Cell. Biol., 2017, 37, e00535-16 CrossRef PubMed.
- J. Her and S. F. Bunting, J. Biol. Chem., 2018, 293, 10502–10511 CrossRef CAS PubMed.
- A. J. Deans and S. C. West, Nat. Rev. Cancer, 2011, 11, 467–480 CrossRef CAS PubMed.
- X. Wang, X. Wang, S. Jin, N. Muhammad and Z. Guo, Chem. Rev., 2019, 119, 1138–1192 CrossRef CAS PubMed.
- R. G. Kenny, S. W. Chuah, A. Crawford and C. J. Marmion, Eur. J. Inorg. Chem., 2017, 2017, 1596–1612 CrossRef CAS.
- Z. Wang, Z. Deng and G. Zhu, Dalton Trans., 2019, 48, 2536–2544 RSC.
- D. Gibson, J. Inorg. Biochem., 2019, 191, 77–84 CrossRef CAS PubMed.
- E. Petruzzella, R. Sirota, I. Solazzo, V. Gandin and D. Gibson, Chem. Sci., 2018, 9, 4299–4307 RSC.
- L. Ma, N. Wang, R. Ma, C. Li, Z. Xu, M. K. Tse and G. Zhu, Angew. Chem., Int. Ed., 2018, 57, 9098–9102 CrossRef CAS PubMed.
- D. Lo Re, D. Montagner, D. Tolan, C. Di Sanza, M. Iglesias, A. Calon and E. Giralt, Chem. Commun., 2018, 54, 8324–8327 RSC.
- S. Jin, Y. Hao, Z. Zhu, N. Muhammad, Z. Zhang, K. Wang, Y. Guo, Z. Guo and X. Wang, Inorg. Chem., 2018, 57, 11135–11145 CrossRef CAS PubMed.
- E. Gabano, M. Ravera, F. Trivero, S. Tinello, A. Gallina, I. Zanellato, M. B. Gariboldi, E. Monti and D. Osella, Dalton Trans., 2018, 47, 8268–8282 RSC.
- Z. Xu, J. Zhao, S. Gou and G. Xu, Chem. Commun., 2017, 53, 3749–3752 RSC.
- V. Reshetnikov, S. Daum and A. Mokhir, Chem. – Eur. J., 2017, 23, 5678–5681 CrossRef CAS PubMed.
- E. Petruzzella, J. P. Braude, J. R. Aldrich-Wright, V. Gandin and D. Gibson, Angew. Chem., Int. Ed., 2017, 56, 11539–11544 CrossRef CAS PubMed.
- S. H. Josef Mayr, B. Koblmüller, M. H. M. Klose, K. Holste, B. Fischer, K. Pelivan, W. Berger, P. Heffeter, C. R. Kowol and B. K. Keppler, J. Biol. Inorg Chem., 2017, 22, 591–603 CrossRef PubMed.
- Z. Wang, Z. Xu and G. Zhu, Angew. Chem., Int. Ed., 2016, 55, 15564–15568 CrossRef CAS PubMed.
- R. M. Gregory Thiabaud, G. He, J. F. Arambula, Z. H. Siddik and J. L. Sessler, Angew. Chem., Int. Ed., 2016, 55, 12626–12631 CrossRef PubMed.
- D. Y. Q. Wong, J. H. Lim and W. H. Ang, Chem. Sci., 2015, 6, 3051–3056 RSC.
- S. G. Awuah, Y. R. Zheng, P. M. Bruno, M. T. Hemann and S. J. Lippard, J. Am. Chem. Soc., 2015, 137, 14854–14857 CrossRef CAS PubMed.
- K. Suntharalingam, Y. Song and S. J. Lippard, Chem. Commun., 2014, 50, 2465–2468 RSC.
- R. K. Pathak, S. Marrache, J. H. Choi, T. B. Berding and S. Dhar, Angew. Chem., Int. Ed., 2014, 53, 1963–1967 CrossRef CAS PubMed.
- Q. Cheng, H. Shi, H. Wang, Y. Min, J. Wang and Y. Liu, Chem. Commun., 2014, 50, 7427–7430 RSC.
- J. Kasparkova, H. Kostrhunova, O. Novakova, R. Krikavova, J. Vanco, Z. Travnicek and V. Brabec, Angew. Chem., Int. Ed., 2015, 54, 14478–14482 CrossRef CAS PubMed.
- S. Zhang, H. Yuan, Y. Guo, K. Wang, X. Wang and Z. Guo, Chem. Commun., 2018, 54, 11717–11720 RSC.
- A. Friberg, D. Vigil, B. Zhao, R. N. Daniels, J. P. Burke, P. M. Garcia-Barrantes, D. Camper, B. A. Chauder, T. Lee, E. T. Olejniczak and S. W. Fesik, J. Med. Chem., 2013, 56, 15–30 CrossRef CAS PubMed.
- J. L. Yap, L. Chen, M. E. Lanning and S. Fletcher, J. Med. Chem., 2017, 60, 821–838 CrossRef CAS PubMed.
- J. Z. Zhang, P. Bonnitcha, E. Wexselblatt, A. V. Klein, Y. Najajreh, D. Gibson and T. W. Hambley, Chem. – Eur. J., 2013, 19, 1672–1676 CrossRef CAS PubMed.
- Y. R. Zheng, K. Suntharalingam, T. C. Johnstone, H. Yoo, W. Lin, J. G. Brooks and S. J. Lippard, J. Am. Chem. Soc., 2014, 136, 8790–8798 CrossRef CAS PubMed.
- N. Muhammad, N. Sadia, C. Zhu, C. Luo, Z. Guo and X. Wang, Chem. Commun., 2017, 53, 9971–9974 RSC.
- M. D. Hall, H. R. Mellor, R. Callaghan and T. W. Hambley, J. Med. Chem., 2007, 50, 3403–3411 CrossRef CAS PubMed.
- I. Revet, L. Feeney, S. Bruguera, W. Wilson, T. K. Dong, D. H. Oh, D. Dankort and J. E. Cleaver, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8663–8667 CrossRef CAS PubMed.
- W. M. Bonner, C. E. Redon, J. S. Dickey, A. J. Nakamura, O. A. Sedelnikova, S. Solier and Y. Pommier, Nat. Rev. Cancer, 2008, 8, 957–967 CrossRef CAS PubMed.
- S. Burma, B. P. Chen, M. Murphy, A. Kurimasa and D. J. Chen, J. Biol. Chem., 2001, 276, 42462–42467 CrossRef CAS PubMed.
- M. I. Sheau-Yann Shieh, Y. Taya and C. Prives, Cell, 1997, 91, 325–334 CrossRef.
- J. M. Adams and S. Cory, Cell Death Differ., 2018, 25, 27–36 CrossRef CAS PubMed.
- M. J. White, K. McArthur, D. Metcalf, R. M. Lane, J. C. Cambier, M. J. Herold, M. F. van Delft, S. Bedoui, G. Lessene, M. E. Ritchie, D. C. Huang and B. T. Kile, Cell, 2014, 159, 1549–1562 CrossRef CAS PubMed.
- S. Jamil, C. Stoica, T. L. Hackett and V. Duronio, Cell Cycle, 2010, 9, 2843–2855 CrossRef CAS PubMed.
- G. Chen, A. T. Magis, K. Xu, D. Park, D. S. Yu, T. K. Owonikoko, G. L. Sica, S. W. Satola, S. S. Ramalingam, W. J. Curran, P. W. Doetsch and X. Deng, J. Clin. Invest., 2018, 128, 500–516 CrossRef PubMed.
- L. Ranjha, S. M. Howard and P. Cejka, Chromosoma, 2018, 127, 187–214 CrossRef CAS PubMed.
- M. Roberti, F. Schipani, G. Bagnolini, D. Milano, E. Giacomini, F. Falchi, A. Balboni, M. Manerba, F. Farabegoli, F. De Franco, J. Robertson, S. Minucci, I. Pallavicini, G. Di Stefano, S. Girotto, R. Pellicciari and A. Cavalli, Eur. J. Med. Chem., 2019, 165, 80–92 CrossRef CAS PubMed.
- K. Gudmundsdottir and A. Ashworth, Oncogene, 2006, 25, 5864–5874 CrossRef CAS PubMed.
- J. Han, C. Ruan, M. S. Y. Huen, J. Wang, A. Xie, C. Fu, T. Liu and J. Huang, Nat. Commun., 2017, 8, 1470 CrossRef PubMed.
- B. Wan, L. Dai, L. Wang, Y. Zhang, H. Huang, G. Qian and T. Yu, Endocr.-Relat. Cancer, 2018, 25, 69–82 CAS.
- C. Cruz, M. Castroviejo-Bermejo, S. Gutierrez-Enriquez, A. Llop-Guevara, Y. H. Ibrahim, A. Gris-Oliver, S. Bonache, B. Morancho, A. Bruna, O. M. Rueda, Z. Lai, U. M. Polanska, G. N. Jones, P. Kristel, L. de Bustos, M. Guzman, O. Rodriguez, J. Grueso, G. Montalban, G. Caratu, F. Mancuso, R. Fasani, J. Jimenez, W. J. Howat, B. Dougherty, A. Vivancos, P. Nuciforo, X. Serres-Creixams, I. T. Rubio, A. Oaknin, E. Cadogan, J. C. Barrett, C. Caldas, J. Baselga, C. Saura, J. Cortes, J. Arribas, J. Jonkers, O. Diez, M. J. O’Connor, J. Balmana and V. Serra, Ann. Oncol., 2018, 29, 1203–1210 CrossRef CAS PubMed.
- M. Špírek, J. Mlčoušková, O. Beláň, M. Gyimesi, G. M. Harami, E. Molnár, J. Novacek, M. Kovács and L. Krejci, Nucleic Acids Res., 2018, 46, 3967–3980 CrossRef PubMed.
- R. G. Kenny and C. J. Marmion, Chem. Rev., 2019, 119, 1058–1137 CrossRef CAS PubMed.
- X. Wang, X. Wang and Z. Guo, Acc. Chem. Res., 2015, 48, 2622–2631 CrossRef CAS PubMed.
- N. Muhammad and Z. Guo, Curr. Opin. Chem. Biol., 2014, 19, 144–153 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- R. Yamaguchi, L. Lartigue and G. Perkins, Pharmacol. Ther., 2018, 195, 13–20 CrossRef PubMed.
- Y. Wan, N. Dai, Z. Tang and H. Fang, Eur. J. Med. Chem., 2018, 146, 471–482 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic routes, NMR, UV-vis, HPLC, cyclic voltammogram, protein expression, acute toxicity and images of tumours. See DOI: 10.1039/d0sc00197j |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Zijian
Guo
*b and
Zijian
Guo