
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition-metal-free C(sp3)–H/C(sp3)–H dehydrogenative coupling of saturated heterocycles with N-benzyl imines†
Zhengfen
Liu‡
a,
Minyan
Li‡
*b,
Guogang
Deng
a,
Wanshi
Wei
a,
Ping
Feng
a,
Quanxing
Zi
a,
Tiantian
Li
ac,
Hongbin
Zhang
 *a,
Xiaodong
Yang
*a and
Patrick J.
Walsh
*b
*a,
Xiaodong
Yang
*a and
Patrick J.
Walsh
*b
aKey Laboratory of Medicinal Chemistry for Natural Resources, Ministry of Education and Yunnan Province, State Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan, School of Chemical Science and Technology, Yunnan University, Kunming, 650091, P. R. China. E-mail: xdyang@ynu.edu.cn; zhanghb@ynu.edu.cn
bRoy and Diana Vagelos Laboratories Penn/Merck Laboratory for High-Throughput Experimentation Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, PA, USA. E-mail: pwalsh@sas.upenn.edu; liminyan@sas.upenn.edu
cDepartment of Soil and Water Science, University of Florida, 2181 McCarty Hall A, Gainesville, FL 32611-0290, USA
First published on 31st March 2020
Abstract
A unique C(sp3)–H/C(sp3)–H dehydrocoupling of N-benzylimines with saturated heterocycles is described. Using super electron donor (SED) 2-azaallyl anions and aryl iodides as electron acceptors, single-electron-transfer (SET) generates an aryl radical. Hydrogen atom transfer (HAT) from saturated heterocycles or toluenes to the aryl radical generates alkyl radicals or benzylic radicals, respectively. The newly formed alkyl radicals and benzylic radicals couple with the 2-azaallyl radicals with formation of new C–C bonds. Experimental evidence supports the key hydrogen-abstraction by the aryl radical, which determines the chemoselectivity of the radical–radical coupling reaction. It is noteworthy that this procedure avoids the use of traditional strong oxidants and transition metals.
Introduction
Cyclic ethers and amines are fundamental structural motifs in nature and are found in a vast number of bioactive compounds, small-molecule drugs1 and functional materials. As such, tremendous effort has been devoted to their synthesis.2 Despite significant progress in the synthesis of saturated heterocycles, approaches to their C–H activation and subsequent C–C bond formation remain in great demand. The high C–H bond dissociation energies (∼90–100 kcal mol−1)3 and low acidity (pKa's usually >45) of saturated heterocycles have significantly hindered the development of methods to functionalize these robust C(sp3)–H bonds.Recently, a variety of tactics have been introduced to tackle the functionalization of saturated heterocycles. Directing group facilitated C(sp3)–H activation has evolved into an enabling strategy to functionalize heterocycles. Recently, Yu4 and Sanford5 developed the arylation of nitrogen-containing heterocycles with the aid of thioamide and perfluorinated amide directing groups (Scheme 1a). In these examples, the C–H bonds of the substrates were cleaved by Pd catalysts, presumably through a concerted metalation-deprotonation (CMD) mechanism.6 These reactions are quite challenging, and even with highly optimized directing groups, require very high reaction temperatures (150 °C). Since 2011, visible-light-induced C–H bond functionalization has emerged as a powerful strategy for the activation of heterocycles,7 as shown in the representative example8 in Scheme 1b. Mechanistically, photoredox-driven single-electron transfer and α-C–H deprotonation enabled the activation of C–H bonds adjacent to nitrogen. Other strategies to activate the C(sp3)–H bonds of heterocycles include application of hydrogen atom transfer (HAT).9 For example, Porta et al. developed a radical coupling where HAT of aryl radical from THF was the key step for radical generation. More recently, Doyle and co-workers found that photolysis of a Ni(III)–Cl intermediate generated chlorine radicals that underwent HAT with THF10 to achieve a unique THF arylation (Scheme 1c). In the same year, MacMillan and co-workers11 used photoredox catalysis to oxidize 3-acetoxyquinuclidine to the radical cation, which promoted HAT with cyclic amines (Scheme 1c). Wu's team12 developed a Eosin Y photocatalytic HAT activation of tetrahydrofurans.
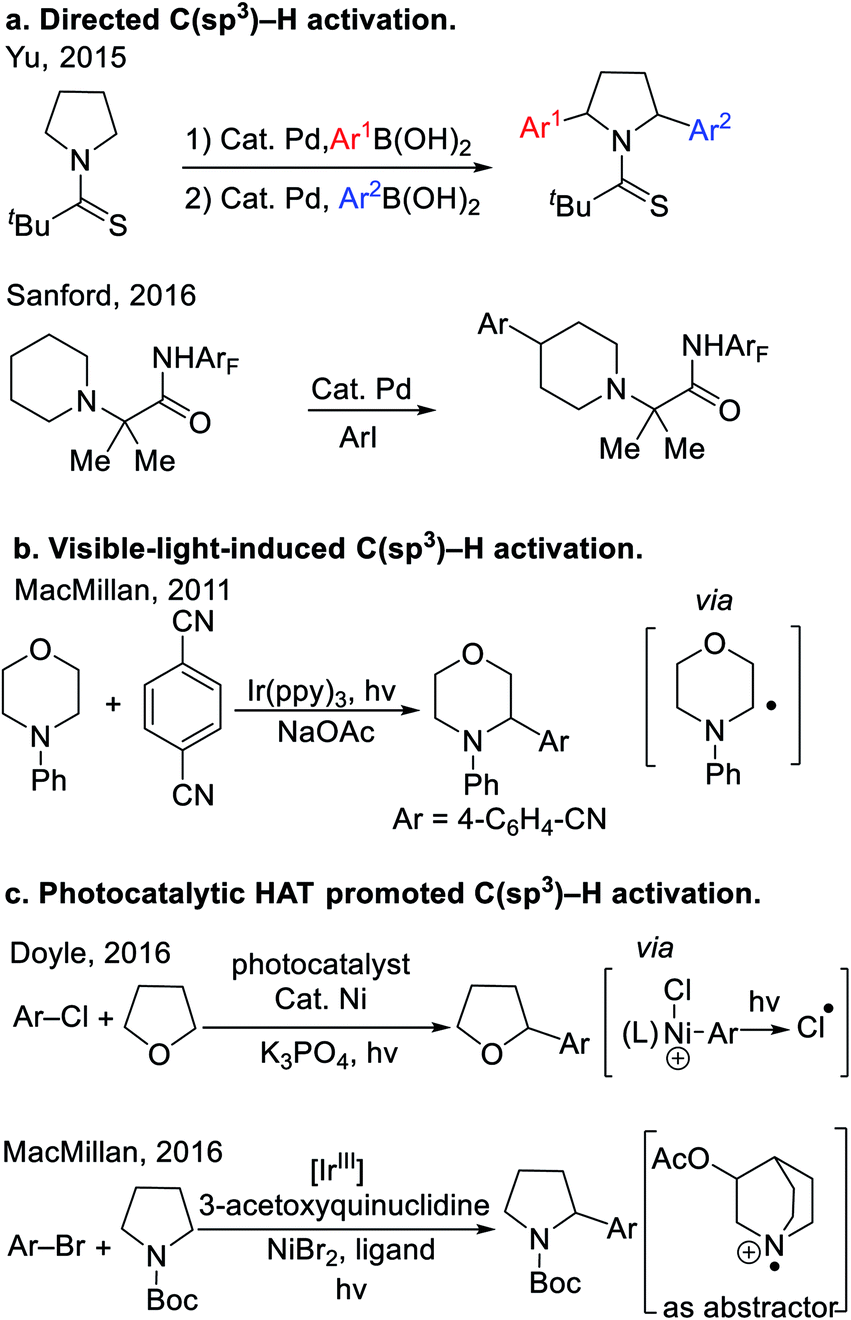 | ||
| Scheme 1 General strategies in C(sp3)–H activation of heterocyclic alkanes. (a) Directed C(sp3)–H activation. (b) Visible-light-induced C(sp3)–H activation. (c) Photocatalytic HAT promoted C(sp3)–H activation. | ||
These impressive and impactful advances inspired us to ponder the prospect of exploiting so called super electron donors (SED)13 to functionalize C(sp3)–H bonds in the absence of transition metals or photoredox catalysts. Our team has been pursuing the fascinating chemistry of 2-azaallyl anions,14 which are easily generated by deprotonation of readily accessible N-benzyl benzophenone imines.15 We identified the inherent reducing nature of the highly colored 2-azaallyl anions, which enabled the introduction of a series of transition metal free coupling reactions (Scheme 2a). In its simplest form, the 2-azaallyl anion reduces aryl iodides or tertiary alkyl iodides and bromides to generate aryl or alkyl radicals and the 2-azaallyl radical.14f These two radicals then undergo selective coupling with each other to generate a new C–C bond, providing diarylmethyl amines and benzylic amines after hydrolysis. More sophisticated radical cyclization/coupling reactions to afford benzofurylethylamines (Scheme 2b) and their isochromene analogues have also been accomplished.14a,b Given the high reactivity of aryl radicals, we hypothesized that if the aryl radical16 coupling with the 2-azaallyl radical in Scheme 2a could be sufficiently slowed, perhaps the aryl radical would preferentially undergo HAT with other organic compounds possessing weaker C(sp3)–H bonds. Such a HAT would generate new radicals that we envisioned would couple with the 2-azaallyl radical (Scheme 2c). The net result would be a cross-dehydrogenative coupling reaction17 between two C(sp3)–H bonds to form a C(sp3)–C(sp3) bond under mild and transition metal-free conditions. Although much of this study is dedicated to mapping the reactivity of the HAT and coupling steps, the synthetic utility of this process is also explored. The products of these reactions are heterocyclic methylamine derivatives, which are of importance in the pharmaceutical industry,18 but would be otherwise difficult to access under mild conditions.
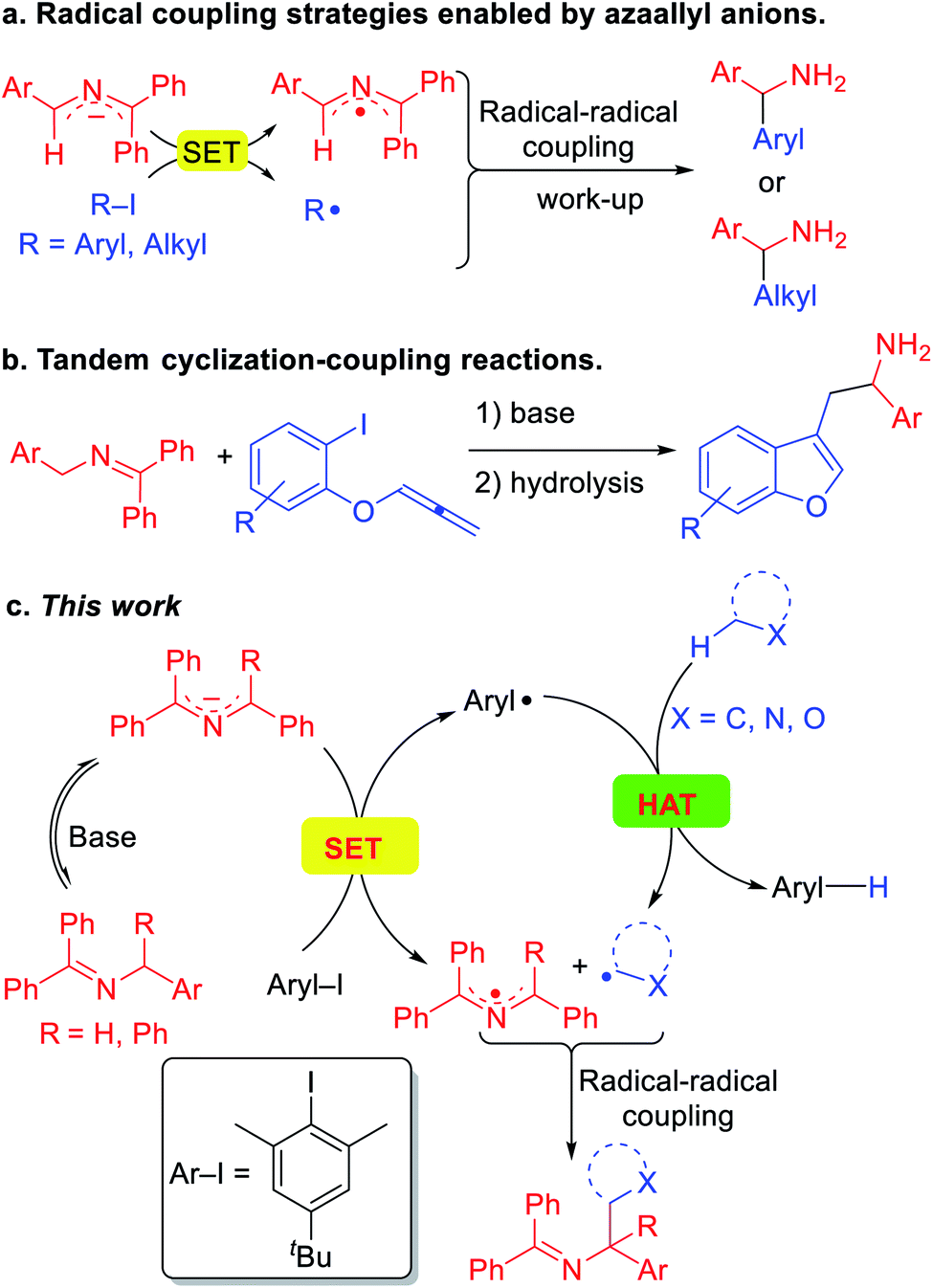 | ||
| Scheme 2 Application of 2-azaallyl anions in radical coupling reactions. (a) Radical coupling strategies enabled by 2-azaallyl anions as super-electron-donors. (b) Tandem reactions for the synthesis of heterocycles. (c) Radical relay design for the synthesis of heterocyclic amine derivatives. | ||
Herein, we report a unique activation strategy of relatively inert heterocyclic C(sp3)–H bonds. The strategy is founded on the reducing character of 2-azaallyl anions and the HAT abstraction feature of aryl radicals (Scheme 2c). To inhibit the aryl radical from coupling with the 2-azaallyl radical, as in Scheme 2a, a sterically protected aryl radical precursor was employed and found to undergo single-electron-transfer with diverse 2-azaallyl anions. The resulting hindered aryl radical effectively abstracts hydrogen from the heterocycles' C(sp3)–H bond, generating alkyl radicals in a reaction akin to an intermolecular radical translocation process.19 The coupling of alkyl radicals and 2-azaallyl radicals furnishes valuable heterocyclic benzylic amines. This HAT protocol employs a sacrificial aryl radical to overcome the limitation of redox potentials and acidities. Finally, we disclose that toluene derivatives can also be activated and coupled with 2-azaallyl intermediates in this transformation.
Results and discussion
With the mechanistic hypothesis in Scheme 2c in mind, we first investigated the aryl iodides as precursors to hydrogen abstractors. The benzophenone imine 1a and tetrahydrofuran solvent (2a) were chosen as the model substrates for the coupling studies at 80 °C (Table 1). When iodobenzene 3a was used, the target coupling product was obtained in an overall 35% assay yield (AY, determined by 1H NMR integration against an internal standard). We next examined substituent effects on the aryl motif. The electron donating alkyl of 1-(tert-butyl)-4-iodobenzene (3b) resulted in an increase in the overall assay yield to 56%. The more electron donating 4-OMe (3c) or halogenated 4-Cl (3d) exhibited decreased AY of 46 and 35%, respectively. Although it is known that the phenyl radical will react with THF very quickly (k = 4.8 × 106 M−1 s−1),20 coupling products derived from reaction of the aryl radical with the 2-azaallyl radical were still observed (10–20% AY). To discourage the direct coupling event, sterically hindered substrates possessing two ortho-substituents were examined. While 2-iodo-1,3-dimethyl benzene (3e) afforded little improvement (combined AY of 43%), addition of a tert-Bu group, which proved beneficial in the reaction of 3bvs.3a, again resulted in an increase in the AY. Thus, 5-(tert-butyl)-2-iodo-1,3-dimethylbenzene (3f) reacted to generate the coupling products with a combined AY of 59%. Further increasing the size of the flanking substituents on the aryl radical to isopropyl (3g) led to a combined AY of 48%. As might be expected, neopentyl iodide (3h) as HAT precursor did not participate in the C–H activation pathway, implying that sacrificial HAT abstractors derived from C(sp3)–X bonds are not viable in our approach.| a Reactions conducted on a 0.1 mmol scale. Assay yield determined by 1H NMR spectroscopy of the crude reaction mixtures using C2H2Cl4 as an internal standard. |
|---|
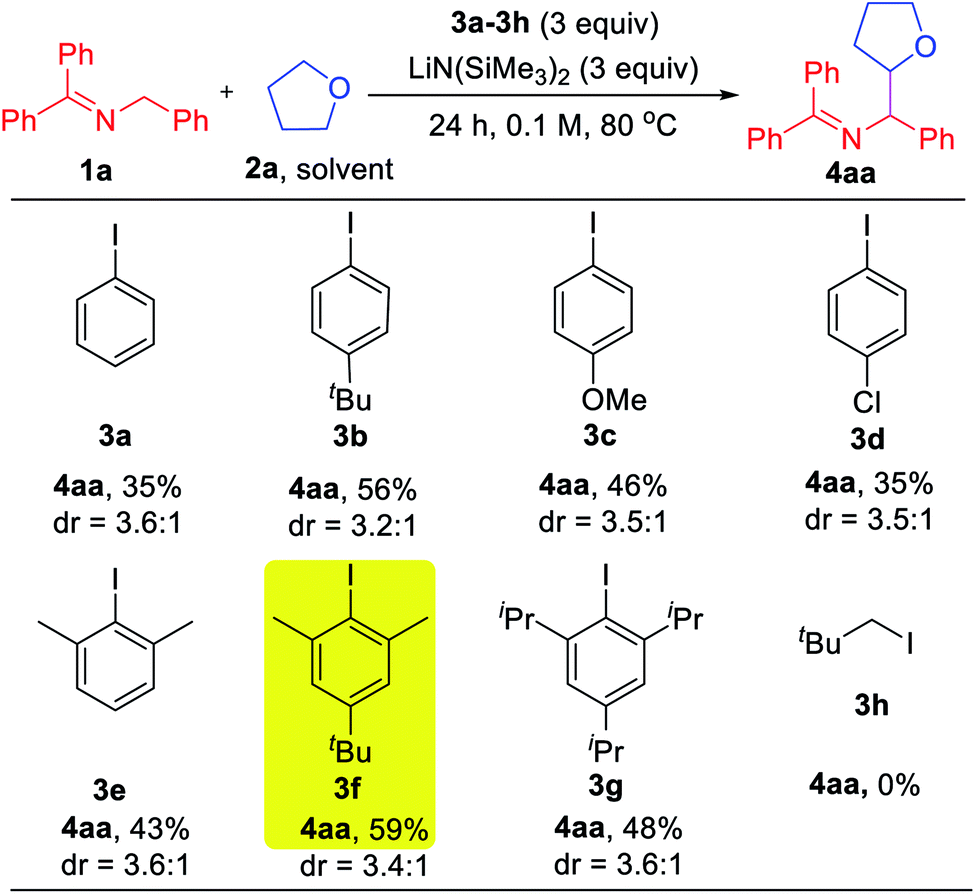
|
Based on these initial studies, we continued the optimization with aryl iodide 3f as the hydrogen abstractor precursor. We next set out to determine the optimal reaction time and found that decreasing from 24 h to 1 h led to an increase in the AY from 49 to 73% (Table 2, entry 1–5). Replacing LiN(SiMe3)2 with NaN(SiMe3)2 led to an overall 81% AY (entry 6). For reasons that are not clear, using KN(SiMe3)2 resulted in formation of a complex mixture with no expected product detected (entry 7).
|
|
||||
|---|---|---|---|---|
| Entry | M = | Time/h | T/°C | Yield of 4aa/% |
| a Reactions conducted on a 0.1 mmol scale. Assay yields determined by 1H NMR spectroscopy of the crude reaction mixture using C2H2Cl4 as an internal standard. b Diastereomeric ratio (dr) of alpha coupling product between 1a and 2a determined by HPLC. The beta coupling product was observed in trace amounts by HPLC but the dr could not be determined. c 0.2 M. d 3f (2 equiv.). e Isolated yield and diastereomeric ratio after chromatographic purification. | ||||
| 1 | Li | 24 | 80 | 49 (dr = 3.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
| 2 | Li | 12 | 80 | 58 (dr = 2.8:1) |
| 3 | Li | 6 | 80 | 64 (dr = 2.7:1) |
| 4 | Li | 3 | 80 | 68 (dr = 2.8:1) |
| 5 | Li | 1 | 80 | 73 (dr = 2.8:1) |
| 6 | Na | 1 | 80 | 81 (dr = 1.6:1) |
| 7 | K | 1 | 80 | Complex mixture |
| 8 | Na | 1 | 100 | 52 (dr = 1.5:1) |
| 9 | Na | 1 | 60 | 69 (dr = 1.7:1) |
| 10c | Na | 1 | 80 | 81 (dr = 1.7:1) |
| 11c,d | Na | 1 | 80 | 84 (80)e (dr = 1.7:1) |
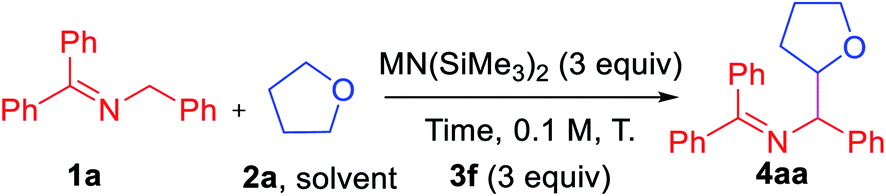
Under the same conditions with NaN(SiMe3)2 but increasing or decreasing the temperature resulted in diminished AY (entries 8–9). Finally, tuning the concentration to 0.2 M and decreasing the aryl iodide 3f to 2 equiv. afforded product 4aa in overall 84% assay yield and 80% isolated yield (dr = 1.7:1, entry 11).
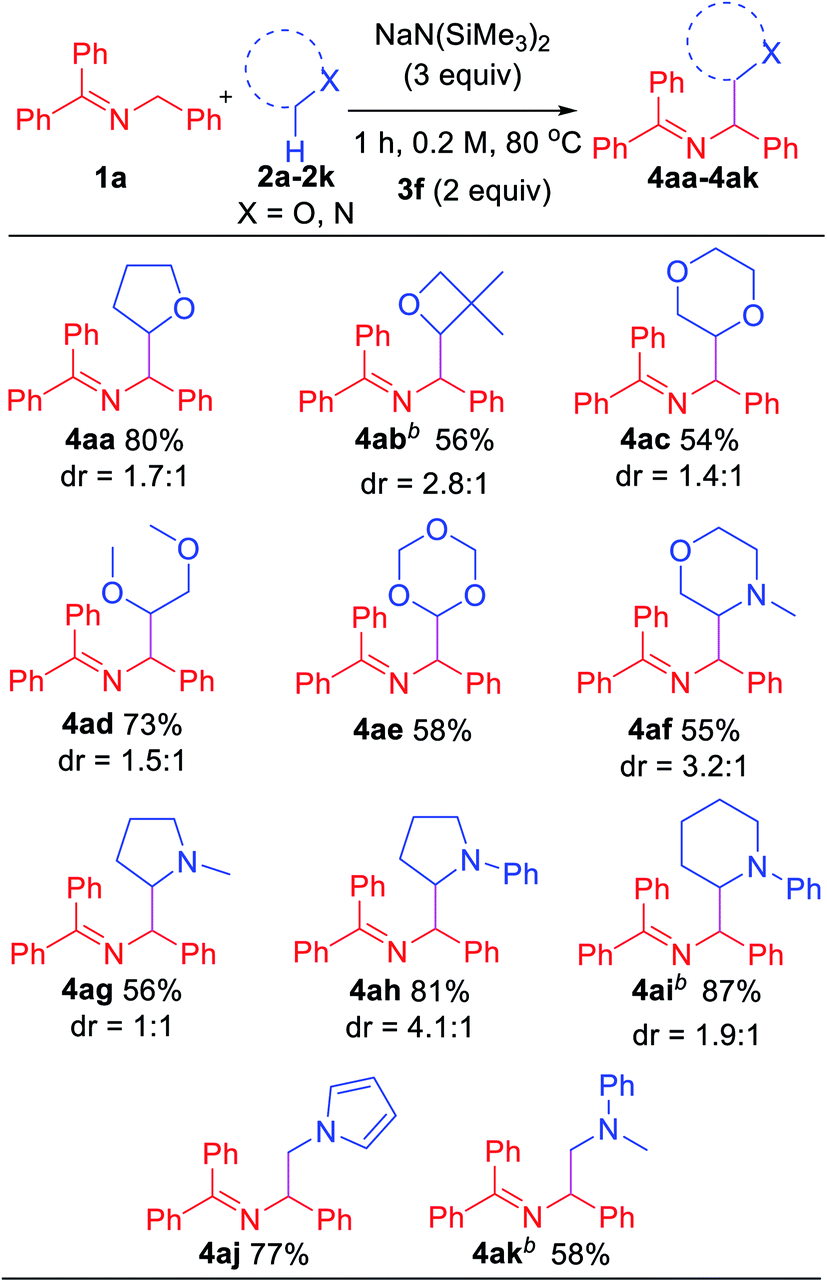
In an effort to explore the scope of this transformation, we first examined saturated heterocycles. In general, we found that a wide variety of substrates with ring sizes ranging from 4 to 6 underwent the C–H functionalization/coupling process (54–87% isolated yields). The scope of the reaction is outlined in Table 3. With more expensive substrates, we explored the coupling reactions in benzene solvent. Thus, 5 equiv. of 3,3-dimethyloxetane (2b) coupled with ketimine 1a in 56% yield (2.8:1 dr). As for the dioxolanes, 1,4-dioxane (2c) furnished 54% yield (1.4:1 dr) of the product isomers. The acyclic analogue 1,2-dimethoxyethane (DME, 2d) was found to couple with ketimine 1a in 73% yield exclusively at the methylene. The symmetrical 1,3,5-trioxane 2e participated in the coupling reaction to furnish the protected α-amino aldehyde 4ae in 58% yield. We were also interested in exploring selectivity between C–H's positioned α to oxygen vs. α to nitrogen. We were pleased to find that coupling of N-methylmorpholine 2f provided the coupling product 4af in 55% yield, with coupling observed only adjacent to the amino group (3.2:1 dr). Likewise, N-methyl pyrrolidine 2g, N-phenyl pyrrolidine 2h and N-phenyl piperidine 2i coupled in 56, 81 and 87% yields (1:1, 4.1:1 and 1.9:1 dr), respectively. Moreover, N-methyl pyrrole 2j afforded product 4aj in 77% yield. Finally, 5 equiv. of N,N-dimethylaniline (2k) in benzene coupled with ketimine 1a in 58% yield. These latter two examples highlight the potential utility of this chemistry for the synthesis of diamines. Coupling products with the following H-atom donor either failed or gave trace products: DMF, DMA, N,N-dimethylbenzamide and 1,3-dimethylimidazolidin-2-one afforded only tract product. Sulfur-containing substrates, such as tetrahydrothiophene and DMSO did not lead to coupling products under the standard conditions.
Encouraged by the results with saturated heterocycles, we selected tetrahydrofuran 2a as coupling partner and investigated the imine scope. As shown in Table 4, the reaction proceeded with good overall yields (up to 88%). N-Benzyl imines bearing substituents on the arene, such as 4-tBu (1b), generated the product 4ba in 81% yield. For electron donating substituents (1c, 4-OMe and 1d, dioxol), good yields of the coupled products were obtained (4ca, 84% yield and 4da, 75% yield). The halide substituted ketimines (1e, 4-F, 1f, 4-Cl and 1g, 4-Br) underwent coupling with 2a in lower yield (26–58%). Although the reason for this is not obvious, it could be attributed to the weaker reducing power of azaallyl anions or destabilization of the 2-azaallyl radical by the halide. Reaction with biphenyl substrate (1h) proceeded in 67% yield. Unfortunately, ketimines derived from heterocyclic benzylic amines (2-,3-,4-pyridyl, 2-furanyl and 2-thiophene) did not afford coupling products under our standard conditions.
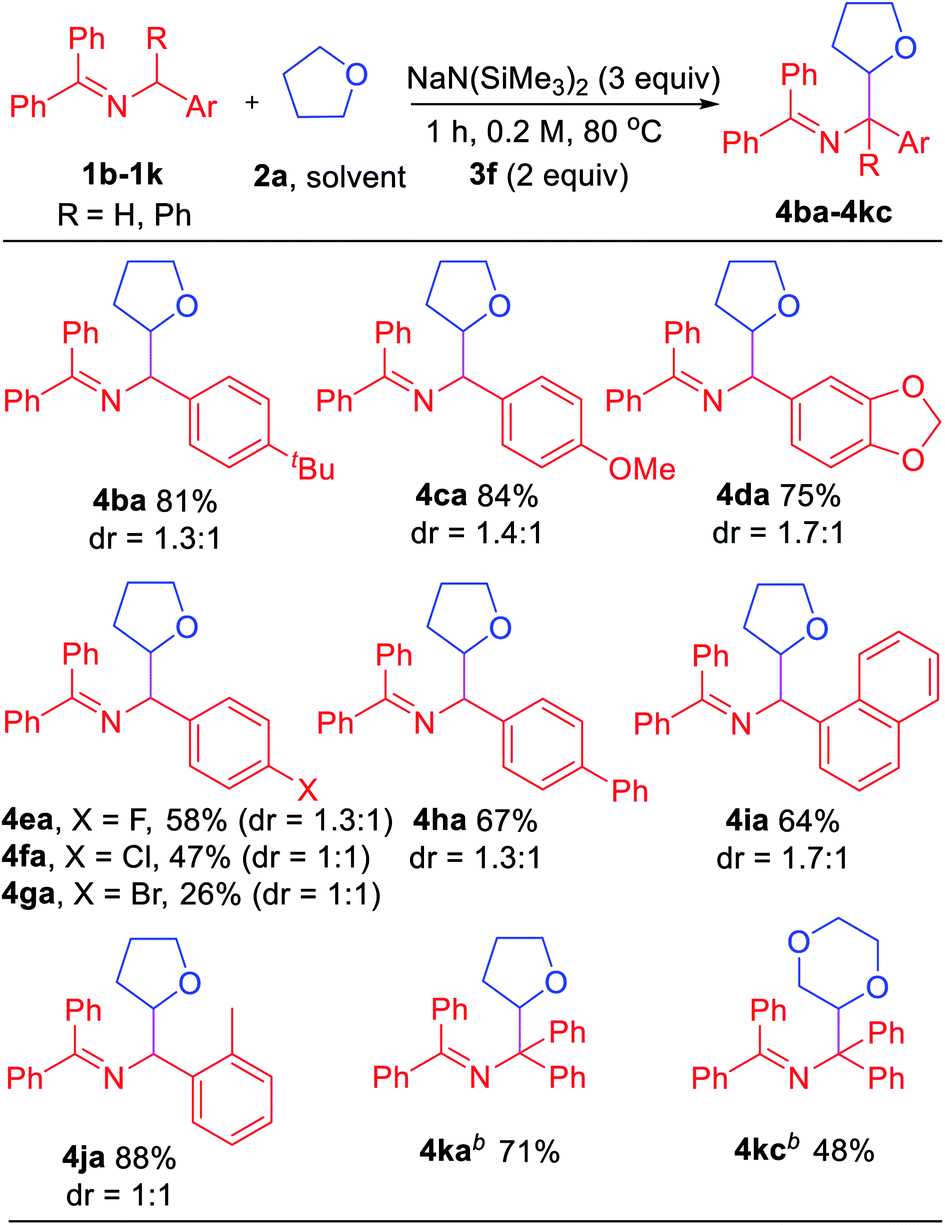
The impact of more sterically hindered N-benzylimines was next probed. Interestingly, 1-naphthyl (1i) and 2-Me (1j) ketimines coupled with tetrahydrofuran in high yields (64% and 88%, respectively). Interestingly, tetraphenyl ketimine 1k, which has proven unreactive in our previous radical coupling studies, successfully coupled with 2a in 71% yield. This result demonstrates that the tetraphenyl 2-azaallyl anion is also strongly reducing. We also tested the coupling of tetraphenyl imine 1k with 1,4-dioxane (2c) and isolated 48% yield of the coupled product. The final two entries in Table 4 would be challenging to efficiently prepare by other methods.
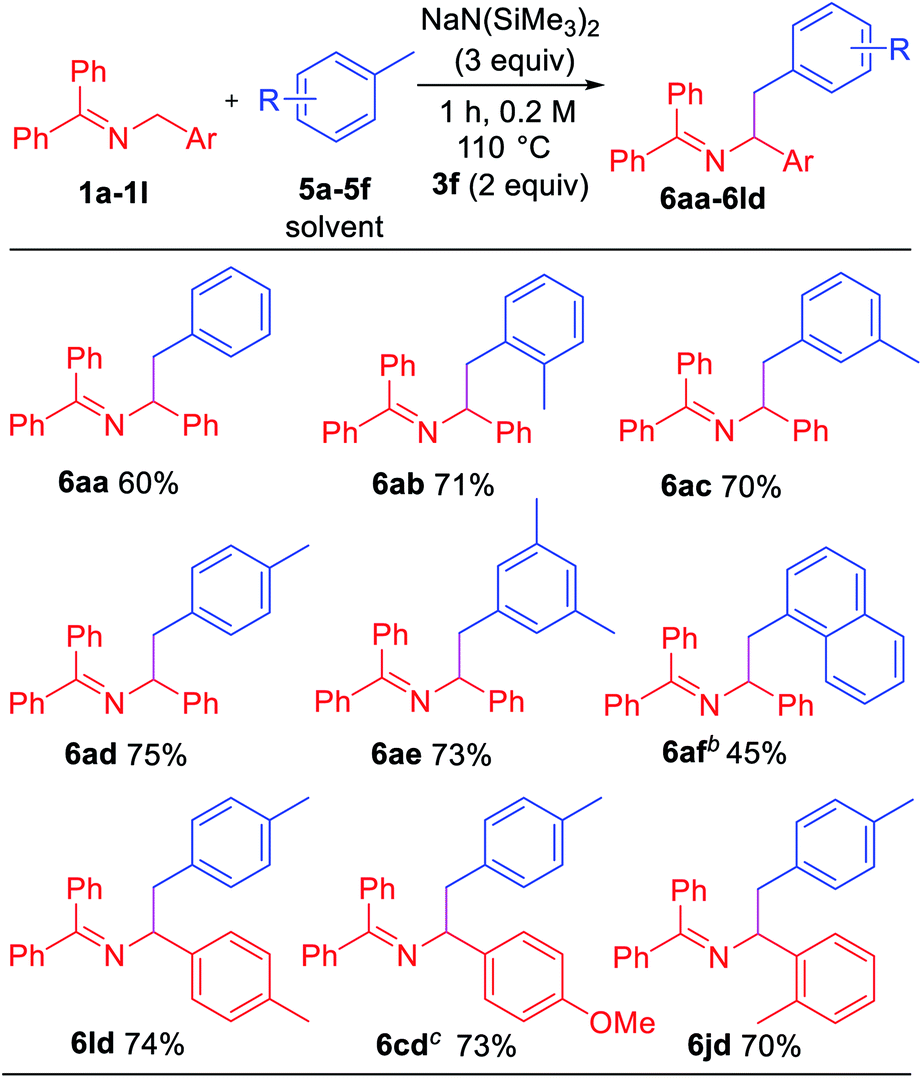
Toluene derivatives are abundant and inexpensive components of petroleum distillates, and we have been interested in their functionalization via deprotonation under relatively mild conditions.21 Here, however, we hypothesized that the aryl radical intermediate would selectively abstract a benzylic hydrogen to generate a benzylic radical. Subsequent radical–radical coupling between the benzyl radical and 2-azaallyl radical would give rise to benzylated imines. As shown in Table 5, under slightly modified conditions (110 °C), net dehydrocoupling of ketimine 1a and toluene 5a gave 60% yield of the tetrasubstituted imine. Ortho-xylene 5b reacted with 1a to form the desired product 6ab in 71% yield. Similarly, meta-xylene 5c and para-xylene 5d afforded products 6ac and 6ad in 70 and 75% yields, respectively. Mesitylene 5e formed the coupling product 6ae in 73% yield. Sterically hindered 1-methylnaphthalene 5f underwent coupling with 1a to give 6af in 45% yield. To demonstrate the versatility, ketimine derivatives with 4-Me (1l), 4-OMe (1c) and 2-Me (1j) groups were tested and gave yields of 70–74%.
Scalability test and product hydrolysis were next conducted. We first formed the ketimine 1a in gram scale (Scheme 3a). Next, treatment of unpurified 1a with tetrahydrofuran 2a and 3 equiv. NaN(SiMe3)2 following the standard procedure afforded 1.17 g of 4aa (86% over 2 steps). It's worth noting that the β-coupling product was isolated together with α-coupling product at gram scale (8:1 regioselectivity). Using a single diastereomer of α-4aa as example, hydrolysis of the product afforded α-7aa in 96% yield (Scheme 3b).
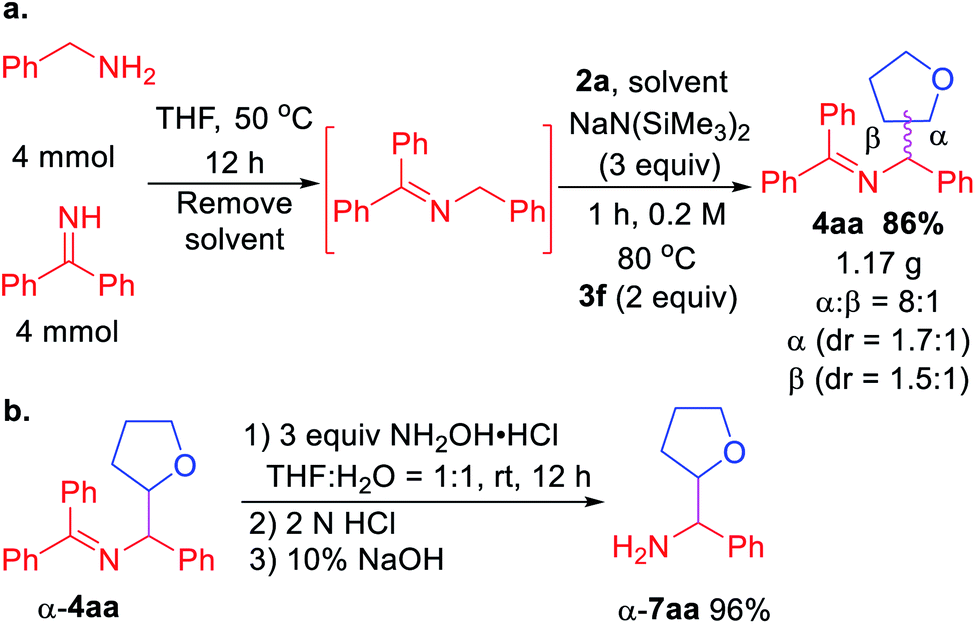 | ||
| Scheme 3 (a) Gram scale telescoped imine formation and coupling. (b) Product hydrolysis. | ||
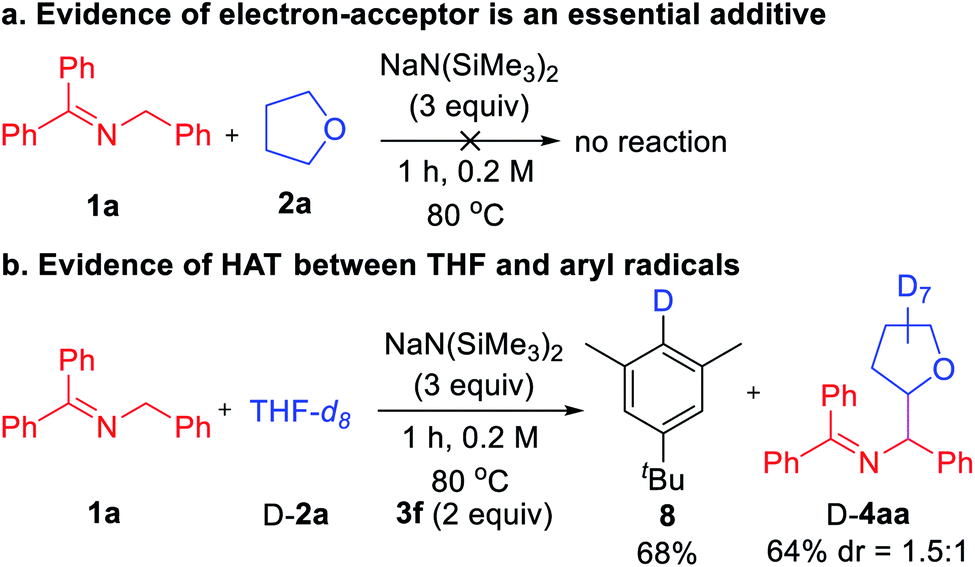
Finally, we turned our attention to mechanistic studies to confirm the role of aryl iodide as a hydrogen abstractor. A control experiment was conducted in the absence of aryl iodide 3f that led to no coupled product, confirming the essential role of aryl iodide (Scheme 4a). Next, subjecting THF-d8 (D-2a) and ketimine 1a to the optimized conditions afforded deuterated arene 8 in 68% yield (Scheme 4b). Thus, we favor the mechanism outlined in Scheme 2c with key steps being SET from the 2-azaallyl anion to the aryl iodide leading to generation of the sterically protected aryl radical and the persistent 2-azaallyl radical.22 The aryl radical then reacts with the ether, amine, or toluene derivatives via HAT to generate the alkyl radical, which couples with the 2-azaallyl radical to furnish the products.
 | ||
| Scheme 4 Mechanistic experiments. | ||
Conclusions
In summary, a new cross-dehydrogenative coupling reaction between C(sp3)–H bonds of saturated heterocycles and the benzylic C–H of benzophenone imines has been described. Unlike many past successful advances, which are based on photoredox catalysts and their specialized reactors or on use of peroxides as radical initiators, this approach employs a novel reactivity from a readily accessible organic super electron donor, 2-azaallyl anions. The key advance in this system is the use of a sterically protected sacrificial hydrogen atom abstractor that enables redirection of the reactivity from coupling with the 2-azaallyl radical (Scheme 2b)14f to H-abstraction from a donor (ether, amine, or toluene derivative, Scheme 2c). This reaction is highly efficient without traditional strong oxidants and tolerates a range of functional groups. We anticipate that the conceptual advances outlined herein can serve as the bases for introduction of new, transition metal-free strategies to cross-dehydrogenative coupling processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the NSFC (21662043, 21572197 and U1702286), Program for Changjiang Scholars and Innovative Research Teams in Universities (IRT17R94) and IRTSTYN, the NSF of Yunnan Province (2019FY003010), the DongLu Scholar & Excellent Young Talents of Yunnan University, and the YunLing Scholar of Yunnan Province. P. J. W. thanks the US National Science Foundation (CHE-1902509) for financial support.Notes and references
-
(a) R. D. Taylor, M. MacCoss and A. D. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed
; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed
-
(a) K. W. Armbrust, M. G. Beaver and T. F. Jamison, J. Am. Chem. Soc., 2015, 137, 6941–6946 CrossRef CAS PubMed
-
Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, Boca Raton, FL, 2007 Search PubMed
- J. E. Spangler, Y. Kobayashi, P. Verma, D. H. Wang and J. Q. Yu, J. Am. Chem. Soc., 2015, 137, 11876–11879 CrossRef CAS PubMed
- J. J. Topczewski, P. J. Cabrera, N. I. Saper and M. S. Sanford, Nature, 2016, 531, 220–224 CrossRef CAS PubMed
-
(a) D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749–823 CrossRef CAS PubMed
-
(a) C. Raviola and D. Ravelli, Synlett, 2019, 30, 803–808 CrossRef CAS
- A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed
-
(a) S. Yang, X. Wu, S. Wu and C. Zhu, Org. Lett., 2019, 21, 4837–4841 CrossRef CAS PubMed
- B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed
- M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Science, 2016, 352, 1304–1308 CrossRef CAS PubMed
- X. Z. Fan, J. W. Rong, H. L. Wu, Q. Zhou, H. P. Deng, J. D. Tan, C. W. Xue, L. Z. Wu, H. R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS PubMed
-
(a) J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, L. E. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402–7410 CrossRef CAS PubMed
-
(a) K. Yu, M. Li, G. Deng, C. Liu, J. Wang, Z. Liu, H. Zhang, X. Yang and P. J. Walsh, Adv. Synth. Catal., 2019, 361, 4354–4359 CrossRef CAS
- S. Tang, X. Zhang, J. Sun, D. Niu and J. J. Chruma, Chem. Rev., 2018, 118, 10393–10457 CrossRef CAS PubMed
-
(a) H. Tian, H. Yang, C. Zhu and H. Fu, Sci. Rep., 2016, 6, 19931 CrossRef PubMed
-
(a) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed
- M. D. Delost, D. T. Smith, B. J. Anderson and J. T. Njardarson, J. Med. Chem., 2018, 61, 10996–11020 CrossRef CAS PubMed
-
(a) D. P. Curran, D. Kim, H. T. Liu and W. Shen, J. Am. Chem. Soc., 1988, 110, 5900–5902 CrossRef CAS
- J. C. Scaiano and L. C. Stewart, J. Am. Chem. Soc., 1983, 105, 3609–3614 CrossRef CAS
-
(a) H. Jiang, S. C. Sha, S. A. Jeong, B. C. Manor and P. J. Walsh, Org. Lett., 2019, 21, 1735–1739 CrossRef CAS PubMed
-
(a) A. Studer, Chem.–Eur. J., 2001, 7, 1159–1164 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1943984 and 1944046. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00031k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |