
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-controllable bioorthogonal chemistry for spatiotemporal control of bio-targets in living systems
Jinbo
Li
 ,
Hao
Kong
,
Chenghong
Zhu
and
Yan
Zhang
*
,
Hao
Kong
,
Chenghong
Zhu
and
Yan
Zhang
*
State Key Laboratory of Analytical Chemistry for Life Sciences, Jiangsu Key Laboratory of Advanced Organic Materials, School of Chemistry and Chemical Engineering, Chemistry and Biomedicine Innovation Center (ChemBIC), Nanjing University, Nanjing 210023, China. E-mail: njuzy@nju.edu.cn
First published on 10th March 2020
Abstract
The establishment of bioorthogonal chemistry is one of the most significant advances in chemical biology using exogenous chemistry to perturb and study biological processes. Photo-modulation of biological systems has realized temporal and spatial control on biomacromolecules in living systems. The combination of photo-modulation and bioorthogonal chemistry is therefore emerging as a new direction to develop new chemical biological tools with spatiotemporal resolution. This minireview will focus on recent development of bioorthogonal chemistry subject to spatiotemporal control through photo-irradiation. Different strategies to realize photo-control on bioorthogonal bond-forming reactions and biological applications of photo-controllable bioorthogonal reactions will be summarized to give a perspective on how the innovations on photo-chemistry can contribute to the development of optochemical biology. Future trends to develop more optochemical tools based on novel photochemistry will also be discussed to envision the development of chemistry-oriented optochemical biology.
1. Introduction
Molecular events in living systems are usually dynamic and therefore difficult to decipher. Technologies with spatiotemporal resolution have found profound applications in exploring dynamic biological processes.1 Photo-irradiation as a conventional external stimulus can be tuned with excellent spatiotemporal resolution as well as biocompatibility.2 Optogenetics that uses light to modulate intracellular molecular events in a spatiotemporal manner has been considered as a revolutionary technique in neuroscience.3 However, limited types of genetically encoded proteins that are able to respond rapidly to light irradiation by changing conformation to alter cell behaviour restricted the broad application of optogenetics. In addition, optogenetic technology is not applicable to biomacromolecules that are not able to be genetically engineered. Alternatively, optochemical methods using photo-controllable chemical moieties integrated with various types of biomacromolecules provide promising spatiotemporal methods to explore dynamic biological systems.4Three types of photo-induced reactions have been applied to optochemical biology to develop molecular tools with spatiotemporal resolution. Photo-isomerization has been applied to the development of photo-switches to reversibly regulate a wide range of biological targets including transmembrane proteins, lipid membranes, nucleic acids, and so on.5 Photo-induced bond cleavage reactions have been applied to photo-uncaging for transient activation of specific bioactivity of “caged” biomolecules modified with photo-removable groups.6–8 Photo-induced bond-forming reactions as a major class of photoreactions of small molecules have rarely been used to develop molecular tools in optochemical biology.9,10 Efforts to revisit photo-induced bond-forming reactions under bio-mimetic conditions have demonstrated great potential in the development of biocompatible or bioorthogonal tools for spatiotemporal labelling of bio-targets.11
Bioorthogonal chemistry has presented an unprecedented approach for the in situ labelling and modulation of bio-targets in living systems without disturbing other “innocent” biomolecules.12–14 The development of bioorthogonal reactions to expand the bioorthogonal tool box has been of intense research interest.15–18 However, it remains a great challenge to establish new bioorthogonal reactions that are orthogonal to the most commonly used bioorthogonal reactions such as azide alkyne cycloaddition (AAC) catalysed by Cu (CuAAC) or promoted by strain (SPAAC).19,20 Inverse electron-demand Diels–Alder cycloaddition (IEDDAC) reactions using various strained alkenes have been established as new bioorthogonal tools that are orthogonal to current AAC chemistry. The importance of photo-controllable bioorthogonal chemistry has encouraged the development of UV or visible light-triggered cycloadditions as biocompatible or bioorthogonal reactions. In this review, we will focus on the recent progress of photo-triggered bioorthogonal chemistry, including different strategies to realize photo-control of bioorthogonal bond-forming reactions and the biological applications of photo-controllable bioorthogonal reactions.
2. Strategies to realize photo-control on bioorthogonal bond-forming reactions
Typical bioorthogonal bond-forming reactions that proceed via distinct mechanisms are listed in Fig. 1. Some of the catalyst-free bioorthogonal reactions, including SPAAC and IEDDAC, have been applied for the in situ labelling of bio-targets in living cells or even in mice models.21,22 However, the precise control of these bioorthogonal bond-forming reactions at single cell or subcellular levels is difficult to achieve, because biomacromolecules modified with bioorthogonal functionalities were hard to introduce into a population with single cellular or subcellular resolution. One potential approach to define where the bioorthogonal reaction will occur, such as at single-cellular or subcellular levels, is to use an external stimulus with spatial resolution to trigger the bioorthogonal reaction inside a specific area of living subjects. Light irradiation is a typical external stimulus with high spatiotemporal resolution and minimal damage to living subjects. Therefore, different strategies to trigger bioorthogonal reactions with light irradiation have been developed, which will be introduced in detail in the following sections.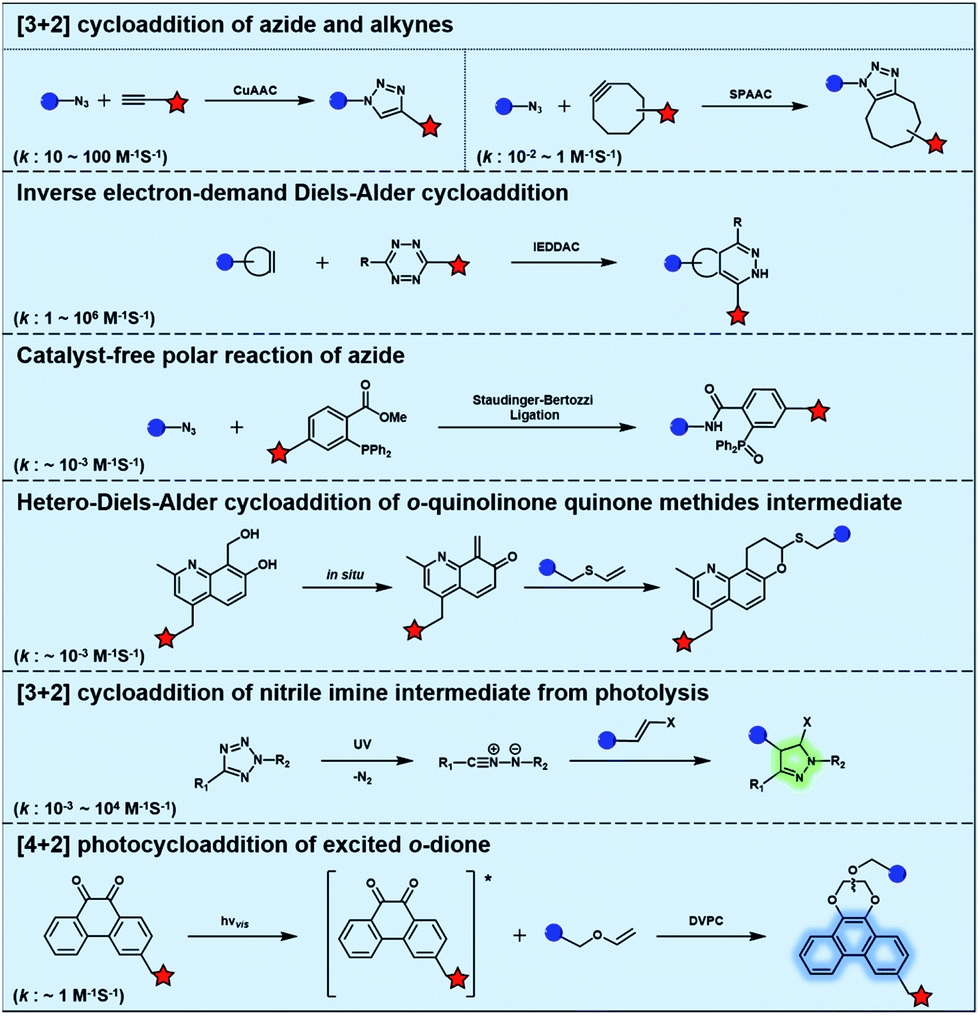 | ||
| Fig. 1 Typical bioorthogonal bond-forming reactions that proceed via distinct mechanisms. | ||
2.1 Bioorthogonal dione–vinyl ether photocycloaddition (DVPC) reaction triggered by visible light
To provide bond-forming bioorthogonal reactions under photo-control, the most straightforward strategy is to use photo-excitable bioorthogonal functionality that reacts with its partner via a specific photoreaction pathway leading to a single ligation product under biocompatible conditions. However, photoreactions that proceed via photo-induced electron transfer (PeT) from the high-energy excited state to form highly reactive radical species usually lead to a complex mixture of products. To develop new types of photo-controllable bioorthogonal reactions via PeT, the key challenge is to realize the control of both the PeT process of high-energy excited states and highly reactive radical species under complex biological environments.Revisiting classical photoreactions such as the Paterno–Büchi reaction involving alkenes or alkynes provides a feasible approach to develop a prototype of photo-triggered bioorthogonal reactions. Inspired by the photoreactions of excited o-diones with π-bonds,23 we recently established visible-light driven dione–vinyl ether photocycloaddition (DVPC), which is the first bioorthogonal reaction via the PeT pathway (Fig. 2A).24 The photo-controllable bioorthogonal functionality in DVPC is the o-dione moiety such as 9,10-phenanthrenequinone (PQ). PQ has an absorption peak between 400 and 500 nm and is able to be efficiently excited by focused visible light from light sources such as LED lamps. The two excited states of PQ, nπ* and ππ* excited states, are energetically close and have a similar conformational structure.25 In protic solvents such as water, the first excited state of PQ (PQ*) transits from nπ* to ππ*. The ππ* excited state of PQ* is inert to various reactive species abundant in biological systems. It is therefore possible to use water that is highly abundant in biological systems as a “solvent cage” to block side reactions that proceed via the excited states of o-diones, provided that there is any photocycloaddition substrate undergoing efficient PeT with the ππ* excited state of PQ*. With calculations on the Gibbs free energy on PeT of PQ* in ππ* with various substrates, we found that the highly electron-rich alkene vinyl ether (VE) has a strong tendency to proceed via PeT with PQ* in ππ* in aqueous solution. As shown in Fig. 2, the reaction of PQ* with VE proceeded via a rapid PeT process to form the first covalent bond with high specificity. The resulting biradical intermediate then proceeded exclusively via an intramolecular radical recombination to form the [4 + 2] photo-cycloadduct with two regioisomers. With this strategy, we addressed the challenge to control both the PeT process of excited states and the following radical-involved pathways under complex biological environments.
 | ||
| Fig. 2 (A) Visible-light driven bioorthogonal dione–vinyl ether photocycloaddition (DVPC) reaction via the PeT pathway; (B) orthogonal protein labelling achieved by SPAAC and DVPC that are orthogonal to each other. | ||
The successful establishment of the visible-light driven DVPC reaction added a new type of bioorthogonal bond-forming reaction to the current bioorthogonal tool-kit. While highly electron-rich alkenes such as VE reacted with the highest reaction rate with excited PQ* in aqueous solution, strained alkenes such as trans-cyclooctene also showed modest reactivity. However, neither terminal alkynes nor strained alkynes were able to undergo PeT with PQ*. DVPC and SPAAC were therefore able to be used together as orthogonal bioorthogonal reactions for orthogonal labelling of biomacromolecules such as proteins (Fig. 2B). Biomacromolecules on live cells can be labelled with high spatiotemporal resolution using the DVPC reaction driven by visible light with a tunable wavelength, intensity and irradiation area. Because light irradiation can be precisely confined at the single-cell or even the subcellular level, single-cellular or subcellular level labelling of targeted biomacromolecules is thus feasible using the bioorthogonal DVPC reaction.
2.2 Photo-release of bioorthogonal functionalities or nitrile imine for spatial control on the following reactions
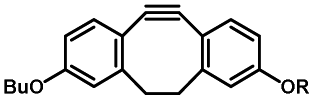
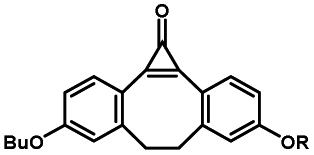
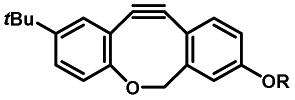
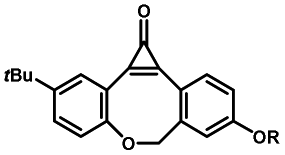
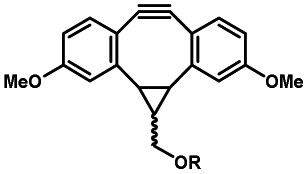
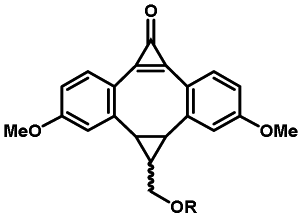
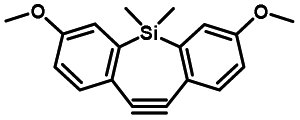
Photo-responsive precursors of established bioorthogonal functionalities have been designed and optimized to realize photo-control on several well-established bioorthogonal reactions such as SPAAC and Staudinger ligation. For example, photochemical generation of reactive dibenzocyclooctynes was developed by Popik et al. from the known photochemical decarbonylation of thermally stable diarylsubstituted cyclopropenones.26 Pioneered by this work, various photo-labile precursors of strained alkynes have been explored for photo-controllable generation of strained alkynes as the biorthogonal functionality.27–30 Visible-light triggered active alkyne generation from cyclopropenones was established with the aid of visible light-responsive photocatalysts.31 Caged phosphines have also been designed and prepared for UV or even visible light triggered generation of active phosphine for light-controllable Staudinger–Bertozzi ligation.32,33 Photo-caging of cyclopropenes has also been reported to provide different versions of cyclopropenes with different ligation kinetics with s-tetrazines.34,35Table 1 summarizes typical photo-labile precursors that have been reported to generate known bioorthogonal functionalities under different conditions for photolysis, with general comments on the advantages and limits for biological applications.| Bioorthogonal functionality obtained after photolysis | Photo-labile precursor | Typical conditions for photolysis | Advantages/limits for biological applications |
|---|---|---|---|
|
|
1–3 min' irradiation in a mini-Rayonet photoreactor equipped with multiple 350 nm fluorescent tubes26,27 | Photo-SPAAC enabled photo-control on labeling azido-bearing biomolecules in live cells, with concern on diffusion of the in situ formed strained alkyne due to fast kinetics in photolysis but a relatively slow rate of SPAAC |
|
|
||
|
|
5 min' irradiation at 365 nm from a UV lamp VL-215.L (ref. 28) | Photo-IEDDAC reactions with fast kinetics enabled rapid labeling of tetrazine-bearing proteins within minutes at low μM concentrations. Future applications on mammalian cells need more biocompatible light than UV |
|
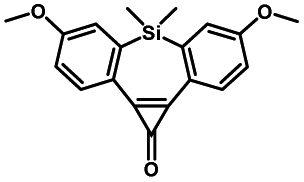
|
Pulsed laser at 375 nm (ref. 29) | Ultra-fast and efficient photo-release of dibenzosilacyclohept-4-yne achieved well-balanced combination of reactivity and stability in aqueous solutions. Photo-stability of the released alkyne exposed to a laser at the irradiation wavelength was a concern |
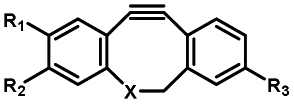
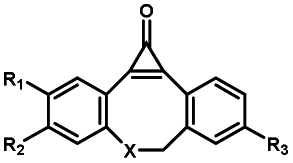
|
|
Femtosecond laser for two-photon (690 nm) or thee-photon (1050 nm) excitation30 | Multiphoton activation of strained alkynes allowed MP-SPAAC with low phototoxicity and deep penetration in biological systems, with a limit in the region of interest irradiated with a two-photon or three-photon laser |
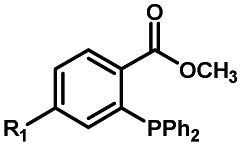
|
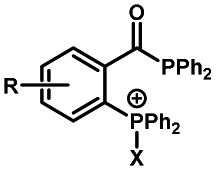
|
30–180 min' UV light irradiation with a 350 W medium pressure Hg arc lamp32 | Simple uncaging approach helped to realize photo-control on the highly specific Staudinger–Bertozzi ligation, with concern on the biocompatibility of the light irradiation conditions with live cells |
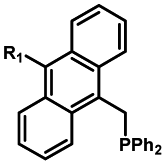
|
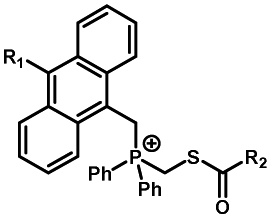
|
20–40 min' irradiation with broadband visible light (>420 nm) from a 500 W Hg lamp33 | Photo-uncaging quantum efficiency of Staudinger ligation triggered by visible light is lower than UV uncaging |
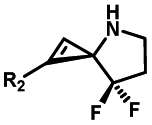
|
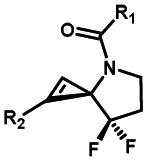
|
Hours of irradiation under UV34 | Module photo-cage of cyclopropane needs to be further optimized for real biological applications |
Photolysis or uncaging of the listed precursors together with the subsequent bioorthogonal reactions such as SPAAC or IEDDAC has been termed photo-SPAAC, photo-IEDDAC, etc. Photo-triggered release of the bioorthogonal functionality helps to control when and where the bioorthogonal partners react with each other. If the photolysis proceeds fast enough to generate the reactive bioorthogonal group, diffusion of molecules bearing the photo-released bioorthogonal functionalities would not jeopardize the spatial resolution of the following bioorthogonal reaction in living systems. The photophysical and photochemical properties of these precursors have been investigated extensively to optimize the photolysis step.26,29,30,32,33 The absorption wavelength and quantum yield are two important parameters that have been considered for the purpose of photo-release of bioorthogonal functionalities, because light with a longer wavelength and lower intensity is more compatible with living systems. Fast rates of photolysis and the following bioorthogonal ligation process are important for the spatiotemporal resolution of a photo-controlled reaction.28,29,34,35 It should be noted that this type of photo-controllable bioorthogonal reaction is only a modified version of known bioorthogonal reactions. Cross-talk of the original version and photo-controllable version of the same bioorthogonal reactions is therefore not avoidable.
Nitrile imines were reported by Lin et al. to react rapidly with alkenes via 1,3-dipolar cycloaddition in aqueous solution.36 Tetrazole–ene photo-click chemistry (TEPC) was therein developed using mild UV light to trigger the fast generation of nitrile imines from 2,5-biaryltetrazoles. TEPC has been tested in living cells with unique fluorescence turn-on accompanied by the formation of the pyrazoline product.11 To make the reaction more bio-compatible, tetrazoles substituted with thiophene37 or naphthalene38 had been used to realize visible light triggered TEPC. Other than tetrazoles, diarylsydnones were also reported to generate nitrile imines upon light irradiation.39,40 To address the issue of competitive reactions of nitrile imine intermediates with carboxylic acid and hydroxyl groups in living systems,41,42 efforts have been made to design and prepare tetrazole with bulky substitution groups to minimize side reactions in competition with TEPC (Fig. 3).43 Lin et al. designed and prepared di(o-2′-N-Boc-pyrrole)-substituted tetrazole and found that it upon UV irradiation formed nitrile imines with an extraordinarily long half-life (up to 120 s). The sterically shielded nitrile imine showed significantly improved selectivity between 1,3-dipolar addition with alkenes and nucleophilic addition (Fig. 3). The TEPC reaction of this bulky tetrazole was successfully applied to rapid (∼1 min) bioorthogonal labeling of the glucagon receptor in mammalian cells. Other than nitrile imine, nitrile ylide generated by azirine through UV irradiation has also been reported to undergo 1,3-dipolar cycloaddition with alkenes,44 which has been applied to protein bioconjugation.
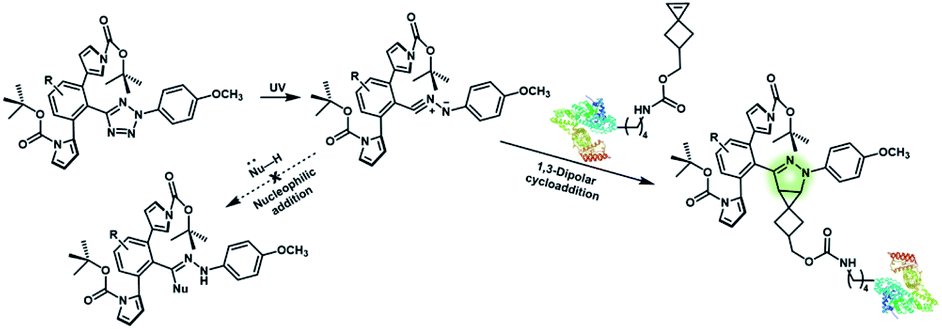 | ||
| Fig. 3 Strategy to improve the bioorthogonal TEPC reaction to block the side reaction of nucleophilic addition to nitrile imine by using a sterically shielded tetrazole substrate. | ||
2.3 Photocatalytic activation of biocompatible or bioorthogonal reactions
Light with longer wavelengths is more compatible with biological systems, and it is therefore attractive to use visible, red or near infrared (IR) light to control bioorthogonal reactions.38,45 Photocatalysts sensitive to red light have been utilized to trigger the formation of bioorthogonal substrates such as tetrazine for the subsequent bioorthogonal IEDDAC reactions (Fig. 4).46 Visible-light catalysis has attracted great research interest in synthetic chemistry in recent years,47 which resulted in a large pool of visible light-catalysed bond-forming reactions. Because visible light photo-redox catalysis based on ruthenium complexes has been applied to various chemical biological studies,48 the adaptability of this large family of organic reactions to bioorthogonal reactions is worth exploring.49–51 Fluorescent dyes such as fluorescein and rhodamine derivatives have been reported to catalyse visible light driven reactions in mammalian cells including neurons.52 They may also be applied to photocatalytic activation of bioorthogonal reactions.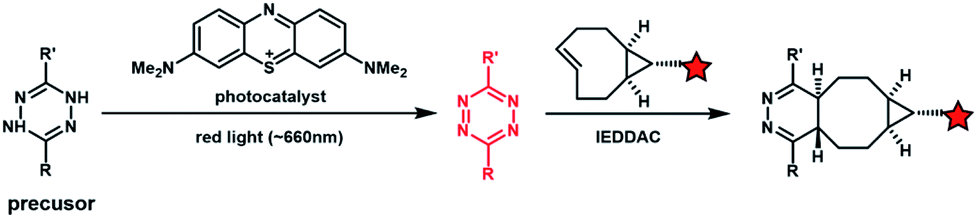 | ||
| Fig. 4 Photocatalytic activation of the IEDDAC reaction. | ||
3 Biological applications of photo-controllable bioorthogonal reactions
Most of the naturally occurring photo-switches in biological systems are based on photo-isomerization, which has been reviewed elsewhere.53 Except for photo-switches based on photo-isomerizable double bonds, bioorthogonal molecular tools based on bioorthogonal bond-forming as well as bond-cleavage reactions have also been utilized to explore biological systems.4 Small bioorthogonal handles have been successfully inserted into endogenous bio-targets using chemical biological techniques, such as genetic engineering with unnatural amino acid54 and metabolic incorporation.13 Spatiotemporal labelling or modulation of these bio-targets has been achieved by using photo-controllable bioorthogonal reactions.3.1 Photo-controlled labelling of specific bio-targets in live cells
In situ labelling of specific bio-targets in live cells or in mice has been achieved by using bioorthogonal reactions such as SPAAC and IEDDAC.55,56 With the photo-controllable version of these bioorthogonal reactions, in situ labelling of bio-targets in live cells has been achieved with spatiotemporal resolution. By genetically introducing a tetrazine group into the outer-membrane OmpC protein, Lang et al. used a photo-IEDDAC of a cyclopropenone-caged dibenzoannulated bicyclo-[6.1.0]nonyne probe to realize UV-controllable labelling of tetrazine-bearing OmpC in live E. coli.57 Carrico et al. applied a photo-Staudinger–Bertozzi reaction for spatial labelling of cell-surface azidoglycoproteins in fixed cells and zebrafish.32 We utilized the visible light-triggered DVPC reaction to label the epidermal growth factor receptor (EGFR) protein on a live cell membrane bound with VE modified Cetuximab (Fig. 5).24 The labelling process was spatiotemporally performed on living A549 cells by changing the irradiation time and the area of live cells exposed to a focused visible light beam from a hand-held LED lamp.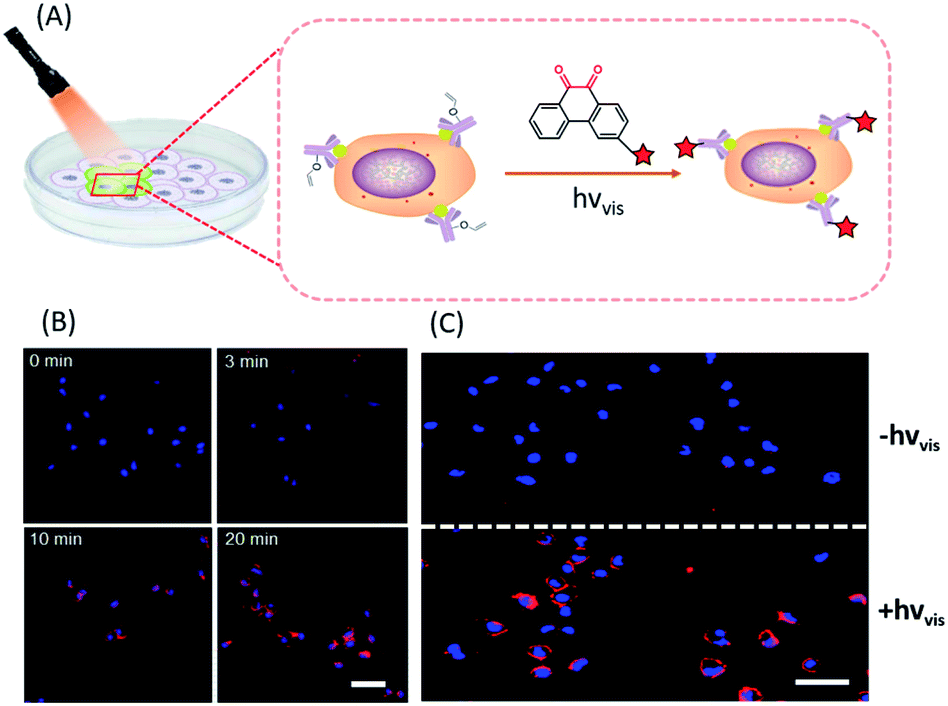 | ||
| Fig. 5 Spatiotemporal labelling of live cells realized by the visible-light driven DVPC reaction (scale bar: 50 μm). | ||
Nucleic acids inside live cells have also been spatiotemporally labelled by various photo-controllable ligation reactions. Azide-modified nucleosides were metabolically incorporated into nascent RNAs in subpopulations of live cells. Selective UV irradiation of cyclopropenone-caged oxadibenzocyclooctynes induced photo-SPAAC reactions with azide-modified RNAs.27 Tetrazole-modified microRNAs after being transfected into live cells were found to be functional to suppress target gene expression.58 We therefore applied intramolecular TEPC reactions for spatiotemporal labelling of microRNAs inside cells by controlling the UV irradiated area and time on live cells.59 Our attempts to trigger intermolecular TEPC reactions of tetrazole-modified microRNAs with monomethyl fumarate in live cells did not result in intracellular fluorescence turn-on, which indicated that the efficiency of the intermolecular TEPC reaction in live cells was much lower than that of intramolecular TEPC.
3.2 Spatiotemporal regulation of intracellular biological functions
Manipulating the biological function of intracellular biomolecules with spatiotemporal resolution is pivotal to deciphering dynamic molecular events occurring within a specific region of live cells. Various biological activities, such as gene expression, protein modification and cell signalling, have been regulated with spatiotemporal resolution using optochemical tools based on bioorthogonal photo-uncaging or photo-isomerization reactions. Because Deiters and co-workers have summarized the significant advances of optochemical control of various biological processes in cells and animals reported before 2018,60 we will briefly introduce the recent advances achieved thereafter in this section.Site-specific incorporation of photo-removable blocking groups into active residues of a target protein has been achieved via unnatural amino acid insertion. This approach, combined with bioorthogonal photo-uncaging, has become a canonical strategy to spatiotemporal regulation of protein functions. While the key residues of most proteins are not amenable to genetic expanding strategies, Chen et al. recently reported a computationally aided and genetically encoded proximal decaging (CAGE-prox) strategy to block residues close to active sites for subsequent photo-activation.61 Time-resolved activation of a broad range of proteins was achieved by this strategy using UV irradiation to remove blocking groups in proximity to the functional residue of specific proteins. Spatiotemporal control on the activity of Cas9 protein for gene editing has been demonstrated by Choudhary et al. They fused Cas9 with the destabilized domains of dihydrofolate reductase (DHFR) and used a photo-caged ligand for DHFR, trimethoprim, to realize precise control on the activity of Cas9 through light irradiation.62 Small molecule ligands that can degrade target proteins via the proteolysis targeting chimera (PROTAC) strategy have also been used together with photo-decaging or photo-isomerization strategies for light-induced or reversible regulation of protein expression.63,64
3.3 Spatiotemporal modulation of microenvironments to trigger cellular responses
Cells are known to respond to dynamic changes of microenvironments. Photo-modulation of cellular microenvironments engineered with photo-controllable units may trigger cellular responses.65 To mimic native cellular microenvironments, cells have been encapsulated in three dimensional microenvironments that are supramolecular or polymer hydrogels engineered with photo-controllable bioorthogonal functionalities.66,67 A UV-triggered intramolecular TEPC reaction has been adopted to modulate the mechanical properties of supramolecular hydrogel encapsulating stem cells.66 Photo-induced bioorthogonal bond-cleavage reactions has been used together with bioorthogonal ligation reactions such as SPAAC to construct a 3D cellular microenvironment.67 Photo-reversible protein-patterning of the 3D cellular microenvironment to guide the stem cell fate has been demonstrated.68 Recently, photo-reversible immobilization for the 4D patterning of cellular microenvironments has been realized by using site-specifically modified proteins bearing bioorthogonal functionalities.65 Photo-release of soluble proteins in engineered cellular microenvironments promoted canonical EGFR activation and associated membrane endocytosis of the encapsulated cells.4. Future outlook
Nature has developed precise mechanisms for living systems to sense and utilize sunlight. Through learning from mother nature how to use biologically occurring photo-switches, optogenetics has made significant contributions to decipher molecular events in complex and dynamic biological systems. With the development of versatile optochemical tools, a new era of optochemical biology is emerging. Based on diversified photochemistry of small molecules in combination with chemical biological methods, the development of photo-controllable bioorthogonal chemistry is of great importance in optochemical biology. With the establishment of bioorthogonal photocycloadditions via controllable excited states induced by visible light, a variety of optochemical molecular tools can be developed to explore biological processes at a single cellular or even at a subcellular level. Meanwhile, how to use light with a long wavelength to control bioorthogonal reactions is worth exploring because of potential applications in deep tissues of living animals.5. Conclusions
The recent development of bioorthogonal chemistry subject to photo-control to realize spatiotemporal resolution was reviewed. Different strategies to develop photo-controllable bioorthogonal bond-forming reactions were summarized. A visible-light driven DVPC reaction that proceeds via the excited state of o-dione with the electron-rich alkene vinyl ether was introduced in detail. The successful establishment of a bioorthogonal reaction via PeT of o-dione at ππ* excited states to its partner bioorthogonal functionality represented a new direction to develop light-driven bioorthogonal reactions. Alternative ways to provide bioorthogonal bond-forming reactions under photo-control, such as using photo-uncaging or photo-activation for in situ formation of known bioorthogonal functionalities, were also introduced such as photo-SPAAC, photo-IEDDAC, etc. Recent examples of optochemical tools based on photo-controllable bioorthogonal chemistry were briefly mentioned with respect to their applications in the spatiotemporal labelling or regulation of bio-targets in living systems. A future outlook on the development of new photo-controllable biorthogonal chemistry as well as new optochemical tools envisioned the capacity of chemistry-oriented optochemical biology to create powerful spatiotemporal molecular tools for precise modulation of biological systems. Moreover, significant challenges including shifting the activation wavelength of photo-controllable biorthogonal chemistry from the UV region to the visible light region and extending their applications into intracellular microenvironments in living cells are yet to be addressed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge financial support from the National Natural Science Foundation of China (91753116, 21977043, and 21877058).References
- S. Aubert, M. Bezagu, A. C. Spivey and S. Arseniyadis, Nat. Rev. Chem., 2019, 3, 706–722 CrossRef CAS.
- K. Glusac, Nat. Chem., 2016, 8, 734–735 CrossRef CAS PubMed.
- K. Deisseroth, Nat. Methods, 2011, 8, 26–29 CrossRef CAS PubMed.
- N. Ankenbruck, T. Courtney, Y. Naro and A. Deiters, Angew. Chem., Int. Ed., 2018, 57, 2768–2798 CrossRef CAS PubMed.
- K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710–10747 CrossRef PubMed.
- H. Yu, J. Li, D. Wu, Z. Qiu and Y. Zhang, Chem. Soc. Rev., 2010, 39, 464–473 RSC.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- M. J. Hansen, W. A. Velema, M. M. Lerch, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2015, 44, 3358–3377 RSC.
- G. W. Prestonab and A. J. Wilson, Chem. Soc. Rev., 2013, 42, 3289–3301 RSC.
- H. Wu and J. Kohler, Curr. Opin. Chem. Biol., 2019, 53, 173–182 CrossRef CAS PubMed.
- C. P. Ramil and Q. Lin, Curr. Opin. Chem. Biol., 2014, 21, 89–95 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- B. Cheng, R. Xie, L. Dong and X. Chen, ChemBioChem, 2016, 17, 11–27 CrossRef CAS PubMed.
- H. W. Shih, D. N. Kamber and J. A. Prescher, Curr. Opin. Chem. Biol., 2014, 21, 103–111 CrossRef CAS PubMed.
- M. F. Debets, S. S. van Berkel, J. Dommerholt, A. J. Dirks, F. P. J. T. Rutjes and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS PubMed.
- F. Liu, Y. Liang and K. N. Houk, Acc. Chem. Res., 2017, 50, 2297–2308 CrossRef CAS PubMed.
- E. Kim and H. Koo, Chem. Sci., 2019, 10, 7835–7851 RSC.
- D. N. Kamber, S. S. Nguyen, F. Liu, J. S. Briggs, H. W. Shih, R. D. P. J. M. Johnson, A. Halpin, T. Morizumi, V. I. Prokhorenko, O. P. Ernst and R. J. Dwayne Miller, Nat. Chem., 2015, 7, 980–986 CrossRef PubMed.
- D. M. Patterson and J. A. Prescher, Curr. Opin. Chem. Biol., 2015, 28, 141–149 CrossRef CAS PubMed.
- R. D. Row and J. A. Prescher, Acc. Chem. Res., 2018, 51, 1073–1081 CrossRef CAS PubMed.
- H. J. Jeonga, R. J. Yoob, J. K. Kima, M. H. Kimb, S. H. Parka, H. Kima, J. W. Lima, S. H. Doc, K. C. Leeb, Y. J. Leeb and D. W. Kim, Biomaterials, 2019, 199, 32–39 CrossRef PubMed.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 CrossRef CAS PubMed.
- L. Wang, Y. Huang, Y. Liu, H. Fun, Y. Zhang and J. Xu, J. Org. Chem., 2010, 75, 7757–7768 CrossRef CAS PubMed.
- J. Li, H. Kong, L. Huang, B. Cheng, K. Qin, M. Zheng, Z. Yan and Y. Zhang, J. Am. Chem. Soc., 2018, 140, 14542–14546 CrossRef CAS PubMed.
- V. R. Kumar, N. Rajkumar, F. Ariese and S. Umapathy, J. Phys. Chem. A, 2015, 119, 10147–10157 CrossRef CAS PubMed.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS PubMed.
- S. Nainar, M. Kubota, C. McNitt, C. Tran, V. V. Popik and R. C. Spitale, J. Am. Chem. Soc., 2017, 139, 8090–8093 CrossRef CAS PubMed.
- S. V. Mayer, A. Murnauer, M. K. von Wrisberg, M. L. Jokisch and K. Lang, Angew. Chem., Int. Ed., 2019, 58, 15876–15882 CrossRef CAS PubMed.
- M. Martínek, L. Filipová, J. Galeta, L. Ludvíková and P. Klán, Org. Lett., 2016, 18, 4892–4895 CrossRef PubMed.
- C. D. McNitt, H. Cheng, S. Ullrich, V. V. Popik and M. Bjerknes, J. Am. Chem. Soc., 2017, 139, 14029–14032 CrossRef CAS PubMed.
- K. Mishiro, T. Kimura, T. Furuyama and M. Kunishima, Org. Lett., 2019, 21, 4101–4105 CrossRef CAS PubMed.
- L. Shah, S. T. Laughlin and I. S. Carrico, J. Am. Chem. Soc., 2016, 138, 5186–5189 CrossRef CAS PubMed.
- P. Hu, K. Berning, Y. Lam, I. H. Ng, C. Yeung and M. H. Lam, J. Org. Chem., 2018, 83, 12998–13010 CrossRef CAS PubMed.
- P. Kumar, O. Zainul, F. M. Camarda, T. Jiang, J. A. Mannone, W. Huang and S. T. Laughlin, Org. Lett., 2019, 21, 3721–3725 CrossRef CAS PubMed.
- P. Kumar, T. Jiang, S. Li, O. Zainul and S. T. Laughlin, Org. Biomol. Chem., 2018, 16, 4081–4085 RSC.
- A. Herner and Q. Lin, Top. Curr. Chem., 2016, 374, 1–31 CrossRef CAS PubMed.
- P. An, Z. Yu and Q. Lin, Chem. Commun., 2013, 49, 9920–9922 RSC.
- Z. Yu, T. Y. Ohulchanskyy, P. An, P. N. Prasad and Q. Lin, J. Am. Chem. Soc., 2013, 135, 16766–16769 CrossRef CAS PubMed.
- L. Zhang, X. Zhang, Z. Yao, S. Jiang, J. Deng, B. Li and Z. Yu, J. Am. Chem. Soc., 2018, 140, 7390–7394 CrossRef CAS PubMed.
- X. Zhang, X. Mu, S. Jiang, J. Gao, Z. Yao, J. Deng, L. Zhang and Z. Yu, Chem. Commun., 2019, 55, 7187–7190 RSC.
- Z. Li, L. Qian, L. Li, J. C. Bernhammer, H. V. Huynh, J. S. Lee and S. Yao, Angew. Chem., Int. Ed., 2016, 55, 2002–2006 CrossRef CAS PubMed.
- Y. Tian, M. P. Jacinto, Y. Zeng, Z. Yu, J. Qu, W. R. Liu and Q. Lin, J. Am. Chem. Soc., 2017, 139, 6078–6081 CrossRef CAS PubMed.
- P. An, T. M. Lewandowski, T. G. Erbay, P. Liu and Q. Lin, J. Am. Chem. Soc., 2018, 140, 4860–4868 CrossRef CAS PubMed.
- R. K. V. Lim and Q. Lin, Chem. Commun., 2010, 46, 7993–7995 RSC.
- R. J. Mart and R. K. Allemann, Chem. Commun., 2016, 52, 12262–12277 RSC.
- H. Zhang, W. S. Trout, S. Liu, G. A. Andrade, D. A. Hudson, S. L. Scinto, K. T. Dicker, Y. Li, N. Lazouski, J. Rosenthal, C. Thorpe, X. Jia and J. M. Fox, J. Am. Chem. Soc., 2016, 138, 5978–5983 CrossRef CAS PubMed.
- Q. Liu and L. Wu, Natl. Sci. Rev., 2017, 4, 359–380 CrossRef CAS.
- S. Angerani and N. Winssinger, Chem.–Eur. J., 2019, 25, 6661–6672 CrossRef CAS PubMed.
- H. Huang, G. Zhang, L. Gong, S. Zhang and Y. Chen, J. Am. Chem. Soc., 2014, 136, 2280–2283 CrossRef CAS PubMed.
- C. Hu and Y. Chen, Tetrahedron Lett., 2015, 56, 884–888 CrossRef CAS.
- J. Yang, J. Zhang, L. Qi, C. Hua and Y. Chen, Chem. Commun., 2015, 51, 5275–5278 RSC.
- H. Wang, W. Li, K. Zeng, Y. Wu, Y. Zhang, T. Xu and Y. Chen, Angew. Chem., Int. Ed., 2019, 58, 561–565 CrossRef CAS PubMed.
- P. J. M. Johnson, A. Halpin, T. Morizumi, V. I. Prokhorenko, O. P. Ernst and R. J. Dwayne Miller, Nat. Chem., 2015, 7, 980–986 CrossRef CAS PubMed.
- Z. Hao, S. Hong, X. Chen and P. R. Chen, Acc. Chem. Res., 2011, 44, 742–751 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16–20 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- S. V. Mayer, A. Murnauer, M. K. von Wrisberg, M. L. Jokisch and K. Lang, Angew. Chem., 2019, 131, 16023–16029 CrossRef.
- J. Li, L. Huang, X. Xiao, Y. Chen, X. Wang, Z. Zhou, C. Zhang and Y. Zhang, J. Am. Chem. Soc., 2016, 138, 15943–15949 CrossRef CAS PubMed.
- L. Huang, Y. Chen, L. Chen, X. Xiao, X. Wang, J. Li and Y. Zhang, Chem. Commun., 2017, 53, 6452–6455 RSC.
- A. Bardhan and A. Deiters, Curr. Opin. Struct. Biol., 2019, 57, 164–175 CrossRef CAS PubMed.
- J. Wang, Y. Liu, Y. Liu, S. Zheng, X. Wang, J. Zhao, F. Yang, G. Zhang, C. Wang and P. R. Chen, Nature, 2019, 569, 509–513 CrossRef CAS PubMed.
- D. Manna, B. Maji, S. A. Gangopadhyay, K. J. Cox, Q. Zhou, B. K. Law, R. Mazitschek and A. Choudhary, Angew. Chem., Int. Ed., 2019, 58, 6285–6289 CrossRef CAS PubMed.
- G. Xue, K. Wang, D. Zhou, H. Zhong and Z. Pan, J. Am. Chem. Soc., 2019, 141, 18370–18374 CrossRef CAS PubMed.
- P. Pfaff, K. T. G. Samarasinghe, C. M. Crews and E. M. Carreira, ACS Cent. Sci., 2019, 5, 1682–1690 CrossRef CAS PubMed.
- J. A. Shadish, G. M. Benuska and C. A. DeForest, Nat. Mater., 2019, 18, 1005–1014 CrossRef CAS PubMed.
- M. He, J. Li, S. Tan, R. Wang and Y. Zhang, J. Am. Chem. Soc., 2013, 135, 18718–18721 CrossRef CAS PubMed.
- C. A. DeForest and K. S. Anseth, Nat. Chem., 2011, 3, 925–931 CrossRef CAS PubMed.
- C. A. DeForest and D. A. Tirrell, Nat. Mater., 2015, 14, 523–531 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |