
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioinspired chemistry at MOF secondary building units
James R.
Bour†
 ,
Ashley M.
Wright†
,
Xin
He
and
Mircea
Dincă
*
,
Ashley M.
Wright†
,
Xin
He
and
Mircea
Dincă
*
Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. E-mail: mdinca@mit.edu
First published on 23rd January 2020
Abstract
The secondary building units (SBUs) in metal–organic frameworks (MOFs) support metal ions in well-defined and site-isolated coordination environments with ligand fields similar to those found in metalloenzymes. This burgeoning class of materials has accordingly been recognized as an attractive platform for metalloenzyme active site mimicry and biomimetic catalysis. Early progress in this area was slowed by challenges such as a limited range of hydrolytic stability and a relatively poor diversity of redox-active metals that could be incorporated into SBUs. However, recent progress with water-stable MOFs and the development of more sophisticated synthetic routes such as postsynthetic cation exchange have largely addressed these challenges. MOF SBUs are being leveraged to interrogate traditionally unstable intermediates and catalytic processes involving small gaseous molecules. This perspective describes recent advances in the use of metal centers within SBUs for biomimetic chemistry and discusses key future developments in this area.
Biomimetic chemistries in crystalline porous materials
Chemists have long sought to understand and emulate the remarkable chemical reactions catalyzed by metalloenzymes. Synthetic biomimetic inorganic chemistry aims to understand enzymatic function through the creation of structural and functional models of active sites.1 Through replication of the active site in synthetic systems (molecular & solid-state), scientists aim to achieve metalloenzyme function without the limitations of natural enzymes. To date, most biomimetic inorganic chemistry has been studied with solution phase model complexes. The power of this strategy is rooted in the highly tunable ligand fields enabled through organic synthesis and interrogated through solution phase spectroscopies (e.g. nuclear magnetic resonance). More recently, however, porous crystalline solids such as zeolites and mesoporous silicas have emerged as complementary platforms for the study of biomimetic inorganic chemistry.2Several important parallels exist between metalloenzymes and metal sites in permanently porous crystalline materials. For instance, the restricted translation or complete immobilization of metal centers in these materials is expected to limit or completely inhibit interactions between metals. This property mimics the shielding effect of the large peptide support in metalloenzymes. Indeed, the enormous bulk of the protein often provides local steric and extended spatial protection of the active site thereby reducing its interactions with other metals. The extended three-dimensional structure of the protein protects the active site and modulates active site accessibility through steric or polarity gating of substrates, thereby enforcing product selectivity. With careful material choice and design, the solid-state structures of porous solids can simulate the reactivity and selectivity resulting from the greater steric and polar environment of the metalloenzyme.3
Although differences between metalloenzymes and porous solid supported metals can present notable challenges and limitations, such disparities can also engender new analytical and synthetic approaches.2 For example, porous materials can support highly coordinatively unsaturated metals that are difficult to produce in solution phase. Furthermore, spectroscopic studies of reactive intermediates and biologically relevant gaseous small molecules on solid-supported metals can be performed without the interference of solvent. Permanent porosity also enables the use of reaction media in which traditional model complexes may exhibit limited solubility, stability, or other unfavorable interactions. Finally, solid-supported metals are often more stable than the metalloproteins they emulate and may thus be compatible with harsher or more convenient reagents. In other words, unnatural reactants can be used to generate the key biologically inspired active species (e.g. accessing [Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O] with N2O), which lends these materials to potentially robust catalytic applications.
O] with N2O), which lends these materials to potentially robust catalytic applications.
Despite the numerous opportunities for porous solids in bioinspired chemistries, this promising area of research has arguably lagged behind advances made in other applications of crystalline porous solids such as gas separations, gas storage, and catalysis. Precise knowledge of ligand sphere and tunable ligand properties has proven important in the realization and rationalization of bio-relevant reactivity of molecular models. Corresponding characterization and controlled manipulation of the ligand sphere of solid-supported metals (e.g. zeolites, mesoporous silicas, etc.) is comparatively difficult.2,4 Moreover, even modest tuning of secondary sphere interactions remains a considerable challenge in most porous solids.
Metal–organic frameworks (MOFs) stand out among microporous solids because they are atomically and periodically precise, much like zeolites in this sense. Unlike zeolites or other porous solids, MOFs span a broad compositional space that covers essentially the entire periodic table and provides unparalleled chemical and structural tunability. As such, they hold substantial potential to address the long-standing challenges of biomimetic chemistries in the solid state.5,6
A variety of different strategies are amenable for incorporating biomimetic metal active sites into MOFs. These approaches include metallolinkers,7 non-covalent encapsulation of molecular compounds and enzymes,8,9 templated metal/ligand assemblies,10 and the use of the metal ions or clusters comprising the secondary building units (SBUs). This perspective describes recent developments and future directions in bioinorganic chemistry and biomimetic catalysis centered at metal–organic framework SBUs. Our contribution is not intended to be comprehensive. Rather, we focus on recent advances that highlight the unique properties of SBU-based metals in their reactions with biologically important gaseous molecules (e.g. NO, CO2, etc.). Accordingly, we do not discuss bioinspired chemistry centered primarily at the organic linker, MOF-enzyme composites, or molecular species encapsulated in MOF pores. These are conceptually distinct approaches that have been extensively reviewed elsewhere.9,11–15
SBU-based biomimetic chemistry
Metal ions comprising the SBUs share many structural and electronic characteristics with the active sites of metalloenzymes (Fig. 1). They are site-isolated and are typically coordinated to the organic linker by carboxylate, azolate, imidazolate, phenolate, or thiolate donors. These functionalities are similar, or in some cases, identical to the carboxylate, histidine, thiolate, or phenolate moieties that often form the coordination sphere of active sites in metalloenzymes. These weak field ligands commonly result in high-spin electronic configurations, again similar to those found in metalloenzymes. Although the similarity between MOF SBUs and enzyme active sites has long been recognized, the use of SBUs for inner-sphere small molecule chemistry is a relatively recent development. Practical challenges associated with hydrolytic stability and incorporation of redox active ions at the SBU has historically impeded progress in this area.16 Many of these obstacles have now been overcome through better understanding of the kinetic stabilization of MOF structures.76 Furthermore, a wide variety of effective synthetic methods for control of the SBU primary structure and secondary structure properties now enable robust bioinspired chemistries in MOFs.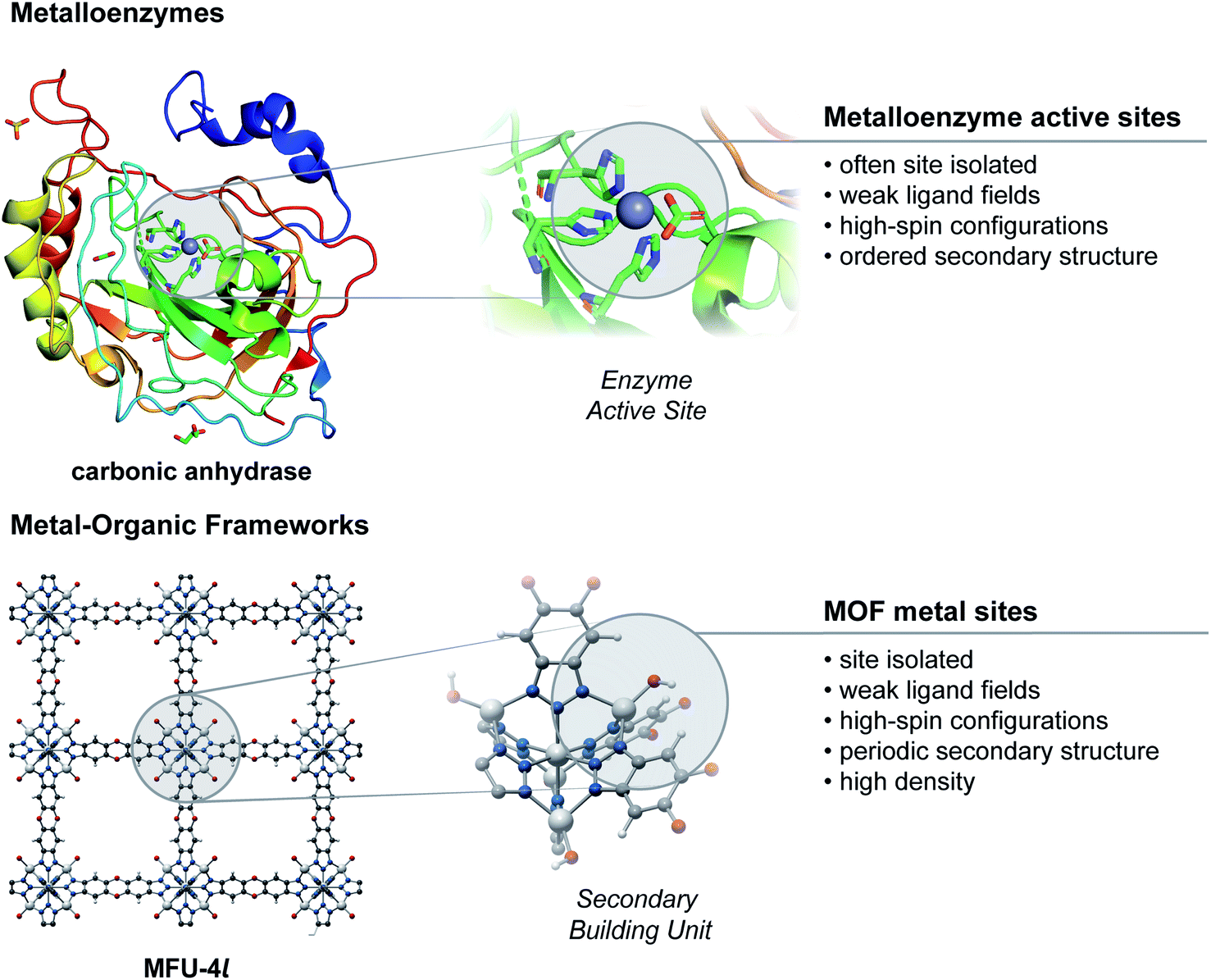 | ||
| Fig. 1 Comparison of properties between metalloenzyme actives sites and metal sites in metal–organic frameworks. | ||
Synthetic approaches to control primary structure
Incorporation of biologically relevant transition metal ions into MOF SBU is generally achieved through one of three strategies: direct synthesis, post-synthetic cation exchange, and post-synthetic deposition/grafting on the SBU (Fig. 2). Direct syntheses are a synthetic regime in which the metal ion is present during the synthesis of the material, usually through solvothermal reaction between a common metal salt and free organic linker. This strategy is step efficient and generally leads to high metal content of the desired ion. However, this approach is fundamentally limited by the deterministic reaction and crystallization profiles for a given metal/linker combination. Finely balanced kinetic criteria must often be met for crystallization of the material and synthetic conditions to satisfy these criteria are difficult to determine a priori. Direct synthetic methods generally require extensive condition screening. Most limiting is the fact that direct synthesis gives rise to SBUs preferred by the metal/ligand combination. These characteristic SBUs rarely possess the desired metal or oxidation state for a targeted application in small molecule reactivity. One strategy that circumvents the reliance on labor-intensive and structurally inflexible direct synthesis methods is the post-synthetic exchange of metal ions into pre-formed SBUs. Termed cation exchange, this strategy enables the integration of metals into SBUs that cannot be formed by the same metals via direct synthesis. Practically, a parent material with the desired SBU is self-assembled from a “sacrificial” parent metal ion, which is subsequently exchanged by the chemically functional metal ion either partially or completely, with retention of the parent SBU and MOF structure.17 First discovered serendipitously in a systematic study of MOFs used for hydrogen storage,18 this cation exchange strategy proved to be applicable across a wide variety of MOFs, including notoriously dynamic materials such as MOF-5. Cation exchange generally only refers to instances where a parent metal ion that is part of an SBU is swapped post-synthetically. Other strategies involving exchange of ionically paired cations or grafting of cations on the SBU have also been pursued.19,20 Although post-synthetic grafting of heterometals on the SBU is operationally simple and widely compatible with numerous materials, it can result in less well-defined coordination environments and therefore does not benefit from the structural fidelity enabled by SBU cation exchange. Nevertheless, taken together, these strategies enable the site isolation of a wide variety of metal ions within or on MOF SBUs.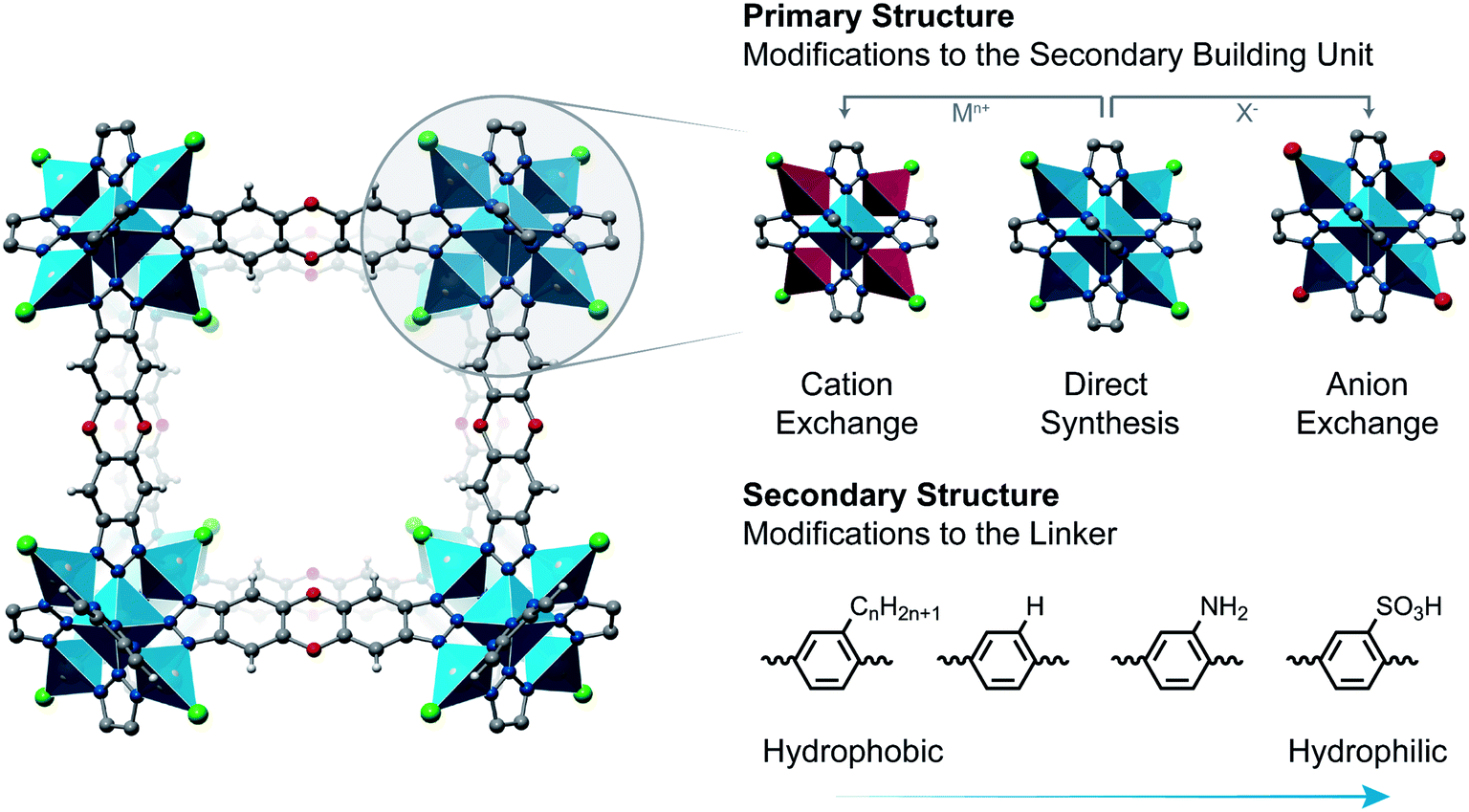 | ||
| Fig. 2 Modification of the MOF primary structure and secondary structure. | ||
An additional key component of the SBU primary structure is the non-structural ligands of the material, typically simple hard anions (OH−, Cl−, OAc−) or solvent molecules (DMF, water, etc.). These ligands are also incorporated directly during the material synthesis but can sometimes be replaced via post-synthetic exchange. Post-synthetic ligand exchanges of non-structural anions or solvent ligands represent a modular way to tune the electronic structure and reactivity of a metal site of interest.21 Modulation of framework properties through variation of non-structural ligands has already been used to finely tune the adsorption properties of MOFs and has recently been used to enable new bioinspired reactivities at MOF SBUs.22,23
Synthetic approaches to control secondary structure
Although it is currently not widely used in the context of biomimetic chemistry, changing the secondary structural properties of the MOF through substitution on the organic linker can directly impact the reaction outcome and selectivity.24 One potential benefit of using MOFs to functionally mimic metalloenzymes is the control over environmental components such as hydrophilicity. Modulation of the secondary structural properties through variation of the organic linker is, in principle, only limited by the organic chemistry needed to make the desired linker and the linker's compatibility with crystallization. However, systematic structure–function relationship studies on linker functionality are highly labor intensive; minor linker substitutions can prevent or enhance MOF crystallization.25 Post-synthetic modifications have been developed as an alternative strategy to direct syntheses. For sufficiently labile linkers, treatment of the MOF with a new linker can result in linker exchange with preserved structural integrity.26 In a separate strategy, reactive handles on a linker (such as NH2) and the metal node can be systematically functionalized after MOF synthesis.27 This approach can be highly effective for late-stage derivatization but is itself limited by the nature of the reactive handle and its compatibility with MOF synthesis.Chemistry of bioactive small molecules at MOF SBUs
Here, we highlight examples of biomimetic small molecule chemistry at the SBU.Carbon dioxide
A bioinspired strategy for the capture of CO2 is being explored as an alternative approach to conventional amine-based absorption materials such as liquid amines or alcohols.28 Carbonic anhydrases (CA) are a family of metalloenzymes that catalyze the hydration and dehydration of carbon dioxide (H2O + CO2 ⇌ HCO3− + H+).29 The critical elementary step in this transformation is the reaction of a terminal nucleophilic metal hydroxide, typically a zinc hydroxide in a N(His)3ZnOH coordination environment, with CO2. The reversibility of insertion of CO2 into the metal–hydroxide bond makes Zn–OH moieties an attractive target for energy efficient capture of CO2 and subsequent release.MOFs have long been studied as adsorbent materials for CO2 capture.30,31 Conventional strategies include physisorption, reaction with appended amines, and encapsulation of molecular catalysts.32 Using bioinspired chemistry at the SBU, Zhang and co-workers installed terminal hydroxide groups by reacting hydrogen peroxide with redox active metals in MII2Cl2(bbta) (M = Mn, Co; H2bbta = 1H,5H-benzo(1,2-d:4,5-d′)bistriazole).33 Inclusion of the terminal hydroxide in the partial oxidation product, MIIMIII(OH)Cl2(bbta), augments CO2 sorption capacities by a factor of 4 or 5 over the parent MII2Cl2(bbta) materials at low pressures (0.15 bar). Infrared (IR) spectroscopic analysis of the CO2-loaded material revealed formation of a bicarbonate moiety similar to the first step in CA chemistry. Notably, the material adsorbs CO2 even in high relative humidity (RH = 82%) conditions, an improvement over physisorption materials. Although MIIMIII(OH)Cl2(bbta) has similar features that mimic CA, it is not structurally analogous.
In contrast, the triazolate MOFs, Zn5(OH)4(bibta)3 (CFA-1-(OH), H2bibta = 5,5′-bibenzotriazole)34 and Zn5(OH)4(btdd) (MFU-4l-(OH), H2btdd = bis(1,2,3-triazolo[4,5-b],[4′,5′-i])dibenzo[1,4]dioxin)35 feature SBUs that are close structural homologues of the CA active site (Fig. 3). In both materials, SBUs are Kuratowski clusters, which feature a central octahedral zinc(II) with six bridging azolate ligands and four peripheral tetrahedral zinc(II) sites with an exchangeable X-ligand.36 The peripheral zinc(II) site is structurally and electronically similar to the active site in CA; it features an N3ZnX coordination environment comprised of three triazolate nitrogen donors, which are electronically similar to histidines. In addition to their structural mimicry, these materials also engage in reactivity that functionally mimics CA. The parent CFA-1 material, Zn5(OAc)4(bibta), hydrolyzes para-nitrophenyl acetate and CO2 under basic conditions.37 In a separate study, CFA-1-(OH) was directly prepared by anion exchange of the acetate ligand in CFA-1 with bicarbonate followed by heating to 100 °C to induce decarboxylation.22 Sorption studies on CFA-1 and CFA-1-(OH) revealed significant improvement in CO2 uptake in the hydroxide material. Interestingly, because CFA-1 is a chiral material, the Zn–X sites in the SBU are not all equivalent, and periodic calculations showed that once formed, a bicarbonate may interact with a second Zn–OH group or Zn–CO3H group at an adjacent SBU representing an unusual example of secondary coordination sphere effects in MOFs. The ensuing hydrogen bonding interaction enhances the initial CO2 sorption profile.22
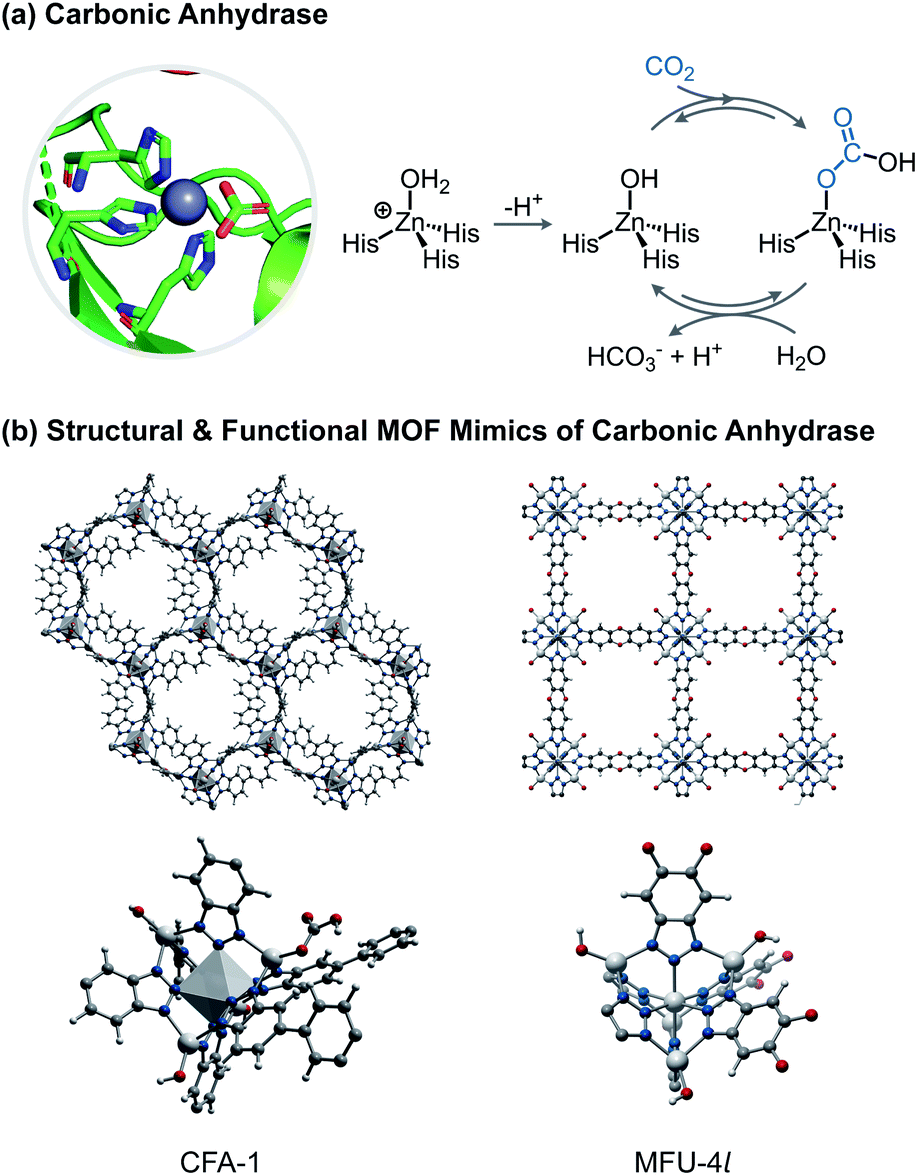 | ||
| Fig. 3 (a) Depiction of the carbonic anhydrase active site and schematic of the hydration mechanism. (b) DFT optimized structures of CFA-1 (left) and MFU-4l-(OH) (right) along with an expanded view of the secondary building unit/metal node. | ||
Exhibiting the same SBUs as CFA-1, MFU-4l differs from the former in being a cubic MOF with equally spaced SBUs and N3ZnX sites (Fig. 3). As with CFA-1, terminal hydroxides can be installed in MFU-4l (Zn5Cl4(btdd)3) by anion exchange with [tBu4N][OH].23 MFU-4l-(OH) exhibits enhanced CO2 sorption over its parent chloride material. Further studies demonstrated that MFU-4l-(OH) acts as functional mimic of CA: it catalyzes the isotopic exchange between H218O and CO2 as well as the hydrolysis of para-nitrophenyl acetate. Detection of the bicarbonate ligand after reaction with CO2 by IR spectroscopy suggests that the isotopic exchange mechanism is similar to that in CA, where CO2 reversibly inserts into the Zn–OH bond.
These CA models highlight important opportunities and limitations of SBU-based model systems. Some of the primary advantages of these systems are the high density and site isolation of the zinc centers, which allow convenient spectroscopic characterization of the CO2 insertion process without complications arising from bimolecular processes. Solution phase CA models can undergo bimolecular decomposition through the formation of dizinc carbonate products.38 This undesired reaction is not observed with the native enzyme. Although a high density of metal active sites can facilitate characterization, it also creates challenges. Both MFU-4l-(OH) and CFA-1-(OH) possess four accessible Zn–OH motifs per SBU. CO2 sorption measurements suggest that each Zn–OH unit is not fully electronically isolated from the other Zn centers in the SBU. The first two equivalents of CO2 insert with a much higher affinity than the second two equivalents. This effect was attributed to an overall reduction in electron density at the remaining Zn–OH sites in the SBU following initial CO2 insertions. This example serves as a reminder of potential interplay between multiple metal centers at the SBU and that such interactions should be always considered. Nonetheless, CFA-1-(OH) and MFU-4l-(OH) provide important insight into the nature of CO2 insertion into Zn–OH bonds and thus serve as functional mimics of carbonic anhydrase.
Methane & ethane
The functionalization of methane is a critical component in realigning the chemical feedstock landscape from long aliphatic chain feedstocks to gaseous ones.39 Selective and incomplete oxidation of the strong C–H bonds in methane and ethane is an outstanding objective for the valorization of these new feedstock landscapes. Biology serves as a major inspiration in the development of selective lower alkane oxidation reactions. For instance, particulate (pMMO) and soluble (sMMO) methane monooxygenases catalyze the oxidation of methane to methanol. The wealth of research aimed at understanding and mimicking MMO reactivity has been reviewed elsewhere.40 Leading structural proposals for the active sites invoke mono or dinuclear copper centers in pMMO, and dinuclear iron sites in sMMO.41–43 These motifs are found in a variety of Cu/Fe MOF SBUs and have accordingly been investigated for methane/ethane oxidation activity. Other examples of alkane oxidation by abiological metals and/or oxidizing metal species (e.g. cobalt superoxos) are not discussed here.Yaghi and co-workers employed a strategy involving post-synthetic modification of the SBU to position biologically relevant ligands within the pore of MOF-808, a zirconium MOF.10 The adamantane shaped pore structure of MOF-808 features an SBU with hydroxide, formate and aquo ligands; post-synthetic installation of carboxylate ligands with pendant imidazoles followed by loading with Cu(I) ions under air resulted in bridging dimeric copper(II) species located within the pore.10 Oxo-bridged copper centers have been proposed (but recently called into question, vide infra) as intermediates in the oxidation of methane to methanol by pMMO.42 Sequential isothermal treatments of 3% N2O in He followed by flow of CH4 for one hour at 150 °C resulted in the formation of methanol with productivities ranging from ∼1.5–2.5 mmol MeOH per mol Cu dependent on the imidazole ligand (Table 1), with no loss of MOF crystallinity. A Cu2O2 active site proposed from density functional theory (DFT) was supported by X-ray absorption spectroscopy. Although precise mechanistic and structural features of this reaction are still unclear, this example represents an important development in the use of the MOF pore structure to template the formation of inorganic assemblies that mimic proposed enzyme active sites.
| MOF | Oxidant | Methanol productivity (μmol MeOH per g MOF) | Methanol productivity (mmol MeOH per mol Cu) | Ref. |
|---|---|---|---|---|
| NU-1000 | O2 | 0.3 | 45 | |
| Cu-2.9-NU-1000 | O2 | 4.1 | 9.7 | 45 |
| Cu-1.9-NU-1000 | O2 | 2.0 | 6.2 | 45 |
| Cu-0.6-NU-1000 | O2 | 0.15 | 1.4 | 45 |
| MOF-808-His-Cu | N2O | 31.7 | ∼13 | 10 |
| MOF-808-Iza-Cu | N2O | 61.8 | ∼22 | 10 |
| MOF-808-Bzz-Cu | N2O | 71.8 | ∼23 | 10 |
| MIL-53(Al, Fe) | H2O2 | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
46 |
In an alternative strategy, Lercher and co-workers deposited Cu(II) ions onto the zirconium SBU of NU-1000.44,45 The modified MOFs outperform the unmodified ones in methane oxidation (Table 1). Activation occurs by exposure to 1 atm of O2 followed by 1–40 bar methane at 150–200 °C for 0.5 to 3 hours. Attempts to determine the coordination environment of the copper ions using X-ray absorption spectroscopy and DFT revealed multiple trimeric, dimeric, and monomeric copper sites, with the distribution dependent on copper loading: at low Cu loadings mononuclear sites dominate whereas higher Cu loadings give a preponderance of higher nuclearity species. In situ structural characterization revealed oxidation state and nuclearity of copper sites remain unchanged during the O2 treatment. The authors concluded that dinuclear oxyl centers are the active site for methane oxidation, which is supported by stoichiometric activity, in situ structural characterization, and DFT.
In copper-grafted MOF-808 and NU-1000 materials, the preferential designation of the active site is a dimeric Cu(II). Although the pMMO active site was also long thought to feature two or more copper ions,47 recent studies suggest that it is most likely a mononuclear site.42 Future studies of monocopper sites at SBUs may provide insight into the nature of such species and should be studied in due course. Overall, these studies complement additional investigations of copper-mediated and catalyzed methane oxidation chemistry within other porous materials.48–51
Unlike pMMO, where the nuclearity of the active site is still debated, the hydroxylase subunit of soluble methane monooxygenase (sMMO) is generally agreed to feature a dimeric Fe active site.40 Suitably spaced diiron motifs have been reported in MOFs and evaluated for oxidase-like activity. Gascon and co-workers recently reported that the controlled incorporation of Fe into MIL-53(Al) results in a catalyst with high selectivity for methane oxidation in the presence of H2O2.46 Preparation of MIL-53(Al, Fe) was most easily controlled using an electrochemical synthetic approach. Fe loadings of 0.3–5.5 wt% with homogeneous distributions were reliably achieved without generation of extraframework Fe species such as Fe2O3. Using DFT, the same authors identified key dimeric Fe sites which were best for activating H2O2 and oxidizing methane. Critically, MIL-53(Al, Fe) reproduces the key features of sMMO and, unlike molecular models, the rigid MOF structure enforces the dimeric state, thereby limiting decomposition to inactive monomeric species. Furthermore, in contrast to the sensitivity of sMMO enzymes towards H2O2, MIL-53(Al, Fe) is stable in the reaction media which facilitates the design of a robust MOF-based catalyst for methane activation.
The compatibility of MIL-53(Al, Fe) with H2O2 underscores two important distinctions between biomimetic MOFs and the enzymes they emulate. First, the key active species can be accessed with reagents and conditions that are too harsh for most enzymatic catalysis. Secondly, the high thermal and chemical stability of many MOFs can be exploited to augment the reactivity of biologically inspired intermediates beyond that of the native enzymes. In 2014 Long and coworkers similarly leveraged both of these properties to effect the oxidation of ethane using Fe0.1Mg1.9(dobdc), a bimetallic MOF-74 analog. Treatment of this material with N2O (an oxygen atom transfer reagent) at elevated temperatures resulted in the formation of a reactive intermediate capable of oxidizing ethane.52,53 DFT calculations implicate a high-spin S = 2, Fe(IV)O species as the active oxidant, which is similar to those proposed for many mononuclear non-heme Fe oxidases.54,55 In contrast to the diiron active site of sMMO, mononuclear non-heme oxidases are not generally thought to be competent in the oxidation of strong C–H bonds of ethane or methane. The observed oxidation of ethane by the putative Fe(IV)O species is likely enabled, at least in part, by the high thermal and chemical stability of Fe0.1Mg1.9(dobdc).53
Oxygen
Dioxygen is a thermodynamically potent oxidant that drives nearly all metabolic processes in aerobic organisms. As a resonance stabilized ground-state triplet, it is kinetically sluggish in the oxidation of singlet substrates.56 Consequently, most synthetically important transformations involving O2 activation require enzymes bearing open-shell transition metals such as Fe or Cu.57,58 Key questions still remain over how these enzymes convert kinetically stable molecular O2 into strongly oxidizing metal–oxygen species (e.g. M–oxo/oxyls, M–hydroperoxos, etc.) or how subtle changes in ligand field perturb elementary aspects of O2 activation. SBU-based Fe and Cu centers provide opportunities to probe detailed features of O2 activation that underlie some of the most remarkable transformations catalyzed by O2-dependent oxidases. The permanent porosity and crystallinity of MOFs enable investigation of O2 binding through gas sorption studies and crystal-to-crystal transformations. This section highlights select examples of O2 binding and activation at metal sites bearing ligand fields similar to those found in important O2-activating metalloenzymes.Some of the earliest steps in studying detailed aspects of O2 activation with MOFs were reported in 2011 by Long and coworkers, who characterized the temperature-dependent O2 bonding to the open metal sites in Fe2(dobdc) (Fe-MOF-74, dobdc4− = 2,5-dioxido-1,4-benzenedicarboxylate).59 At 298 K, approximately half of the Fe(II) sites bind O2 irreversibly. Lowering the temperature to 211 K results in reversible O2 binding and full occupancy of the available Fe(II) sites. The difference in bonding mode at variable temperature was further evidenced by neutron diffraction studies under an O2 atmosphere at 94 K and 220 K (Fig. 4). At 94 K, partial electron transfer from the FeII site to O2 generates Fe2(O2)2(dobdc). Warming this material to 220 K results in formal electron transfer from Fe to O2 to produce a high-spin side-on FeIII–peroxide, as supported by Mössbauer and IR spectroscopy, as well as Rietveld refinement of neutron diffraction data. Although this study was originally described in the context of gas separations, these observations are relevant to the chemistry of non-heme diiron oxygenases. The SBU of Fe-MOF-74 enforces a comparable Fe–Fe distance and an all-oxygen ligand field similar to that of oxygenases.42 The exact nature of O2 activation in these enzymes is not completely understood.60 Related studies of O2 activation by other diiron motifs in MOFs may shed new light on the mechanisms through which diiron oxidases function.
 | ||
| Fig. 4 Depiction of O2 binding to Fe2(dobdc) at 90 and 220 K as determined from Rietveld analysis of neutron diffraction data.59 Orange, grey, and red spheres are Fe, C, and O atoms, respectively. | ||
The binding of O2 to an unsaturated CuI center in Cu-exchanged MFU-4l has also been probed through sorption isotherm studies.61 O2 binds to the unsaturated CuI metal site in MFU-4l with a heat of adsorption of −53 kJ mol−1. Density functional theory calculations corroborate this value and suggest that O2 bonds to copper in an end-on rather than side-on (η2) fashion. Both the energies and geometry of O2 binding to the Cu(I) sites of MFU-4l are in good agreement with thermochemical and structural parameters of solution phase models of monocopper oxidases.61,62 We also note that determination of bonding enthalpies and coordination modes of O2 in traditional solution phase model complexes is non-trivial. Competitive formation of dicopper peroxos (Cu–O–O–Cu) or oxidative degradation of the supporting ligand often precludes precise characterization of this elementary step.63 Such obstacles are not observed in the formation of Cu superoxos in Cu-exchanged MFU-4l and are not expected in other site-isolated metal centers of MOFs; additional studies of these systems may yield new fundamental insight into the activation of O2 by copper-based metalloenzymes.
Nitrogen monoxide
Nitrogen monoxide (NO), also known as nitric oxide, is a highly regulated bio-signaling agent that is crucial for physiological functions such as vasodilation and immune response, and a potent environmental pollutant. Many transformations involving NO require metalloenzymes whose mechanism is not yet completely elucidated.64 Owing to their inherent site isolation, MOF SBUs are an attractive platform to gain insight into the reactivity of NO by targeting the isolation of reactive intermediates that are difficult to study in the natural systems or in solution. The majority of NO studies with MOFs have focused on NO sorption and on creating NO-releasing materials for therapeutic applications.65 A variety of MOFs featuring open metal sites, such as, HKUST-1, the MOF-74 (CPO-27) family, and MIL-88(Fe) derivatives, have been explored for the purpose of NO storage and release, but these do not enable further NO reactivity.66–73Of relevance here are reports where NO engages in reactivity mimicking that of metalloenzymes. For instance, cation exchange of Fe(II) into MOF-5 yields Fe-MOF-5 featuring a FeZn3O cluster in the SBU.74,75 The Fe(II) ion occupies a pseudo-tetrahedral site and reacts with NO to affect stoichiometric NO disproportionation as evidenced by the formation of nitrous oxide, N2O, and a Fe(III)–NO2 group. By monitoring the stoichiometric addition of NO by diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), Dincă and co-workers were able to identify two intermediates in the reaction mechanism, an iron nitrosyl as well as a radical hyponitrite species (N2O2˙−) (Fig. 5). The latter had not been previously observed spectroscopically and highlights the potential of MOFs as a platform for gaining new insights into complex redox reactivity of small molecules with transition metals. Uniquely to MOFs and their well-defined metal coordination environments, the detection of the intermediate is likely facilitated by site isolation of the Fe(II) site within the MOF structure, which prevents bimetallic reaction pathways.
 | ||
| Fig. 5 NO disproportionation at Fe-MOF-5. | ||
Importantly, NO disproportionation reactions at Fe centers in MOFs do not occur when the metal coordination sphere is saturated after NO binding, suggesting that at least two open coordination sites are necessary for this reactivity. For instance, Fe-MOF-74, which features a square-pyramidal Fe center with a single open site adsorbs NO strongly to form an iron nitrosyl, as evidenced by the high uptake capacity (6.21 mmol NO per g MOF) at low pressure (0.04 mbar), but does not undergo further reactivity with NO even at a pressure of 7 bar.70 Fe-MIL-88, also exhibiting a single open coordination site at iron, adsorbs NO to form an iron nitrosyl, but no additional reaction with NO has been reported.69 The availability of more than one coordinatively unsaturated metal site is thus critical for NO disproportionation and is a feature that is rare in other solids. Taking advantage of multiply unsaturated metal sites in SBUs is a promising avenue for future studies in small molecule reactivity with MOFs.
Concluding remarks & outlook
In this perspective, we have described developments in the study of bioinspired reactivity at MOF SBUs. When treated as electronically and spatially isolated molecular entities, these become powerful platforms for interrogating small molecule reactivity in an atomically precise, well-defined porous matrix. These studies are the equivalent of matrix-isolation chemistry that is accessible at a wide range of temperatures and without specialized equipment. The ligand fields enabled by the typical MOF ligands are electronically – and sometimes structurally – analogous to those encountered in natural metalloenzymes. Thus, even though enzymes and MOFs are clearly very different classes of chemicals, they share more commonalities: site isolation, ligand fields, electronic structure, open coordination sites, than one would suspect. These unique properties of MOFs are now being leveraged to detect traditionally unstable intermediates and realize functionally biomimetic catalysts. The relatively recent advances made in this area were largely made possible through progress in synthetic methodologies in MOFs over the past 10 years.We expect future developments in this field to be similarly contingent upon continued advances in the synthesis of readily tunable materials and new MOFs with exchangeable primary structural features and/or multiple open coordination sites. More specifically, the opportunities to affix secondary reaction partners (cofactors) near the SBU is currently underdeveloped in the context of bioinspired chemistries. Current examples of SBU-based biomimetic chemistries are limited to relatively simple systems that are not critically reliant on cooperativity with other motifs. Additional highly tunable materials are needed to realize higher order mimicry of metalloenzymes. Materials featuring functional handles that react in a predictable way with minimal byproducts will enable more advanced mimicry to be realized.
Secondly, discovery of new MOFs with exchangeable primary features (metal cation and non-structural ligands) and multiple unsaturated metal sites will drastically increase the scope of reactivity that can be probed and the number of enzymes that can be functionally mimicked in a stable MOF environment. A large proportion of MOFs feature either inaccessible metals or unexchangeable primary features. MOFs with SBUs featuring unsaturated metal sites, in diverse geometries, and with other pore architectures will significantly improve the breadth of achievable chemistry. These are strong arguments for continued research in fundamentally new MOF topologies and especially new SBU geometries/nuclearities.
With additional suitable materials, improved correlations between the nature of small molecule activation with reactivity can determined. To date, bioinspired chemistries at MOF SBUs have largely focused on either the detailed aspects of small molecule bonding at the SBU or the catalytic reactivity of a material. Fundamental insights that connect elementary bonding with substrate functionalization, catalysis, and mechanism are still rare. We believe that the full potential of this approach will be realized when the unique analytical techniques amenable to MOFs are merged with the catalytic applications of these materials. Such studies will complement existing model platforms to shed new light on the remarkable transformations catalyzed by metalloenzymes and ultimately address outstanding synthetic challenges.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for support from the National Science Foundation (DMR-1452612) for various fundamental aspects of cation exchange and small molecule chemistry in MOFs. JRB thanks the Arnold and Mabel Beckman Foundation for a Postdoctoral Fellowship.Notes and references
- R. H. Holm and E. I. Solomon, Chem. Rev., 2004, 104, 347–348 CrossRef CAS PubMed.
- B. E. R. Snyder, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Chem. Rev., 2018, 118, 2718–2768 CrossRef CAS PubMed.
- D. J. Xuereb and R. Raja, Catal. Sci. Technol., 2011, 1, 517–534 RSC.
- C. Y. Chen and S. I. Zones, Zeolites Catal., 2010, 1, 155–170 CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- C. H. Hendon, A. J. Rieth, M. D. Korzyński and M. Dincǎ, ACS Cent. Sci., 2017, 3, 554–563 CrossRef CAS PubMed.
- S. M. Cohen, Z. Zhang and J. A. Boissonnault, Inorg. Chem., 2016, 55, 7281–7290 CrossRef CAS PubMed.
- H. An, M. Li, J. Gao, Z. Zhang, S. Ma and Y. Chen, Coord. Chem. Rev., 2019, 384, 90–106 CrossRef CAS.
- R. J. Drout, L. Robison and O. K. Farha, Coord. Chem. Rev., 2019, 381, 151–160 CrossRef CAS.
- J. Baek, B. Rungtaweevoranit, X. Pei, M. Park, S. C. Fakra, Y. S. Liu, R. Matheu, S. A. Alshmimri, S. Alshehri, C. A. Trickett, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 18208–18216 CrossRef CAS PubMed.
- Z.-Y. Gu, J. Park, A. Raiff, Z. Wei and H.-C. Zhou, ChemCatChem, 2014, 6, 67–75 CrossRef CAS.
- M. Zhao, S. Ou and C.-D. Wu, Acc. Chem. Res., 2014, 47, 1199–1207 CrossRef CAS PubMed.
- Y. Chen and S. Ma, Dalton Trans., 2016, 45, 9744–9753 RSC.
- I. Nath, J. Chakraborty and F. Verpoort, Chem. Soc. Rev., 2016, 45, 4127–4170 RSC.
- K. Chen and C.-D. Wu, Coord. Chem. Rev., 2019, 378, 445–465 CrossRef CAS.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H. C. Zhou, Adv. Mater., 2018, 1704303, 1–35 Search PubMed.
- C. K. Brozek and M. Dincǎ, Chem. Soc. Rev., 2014, 43, 5456–5467 RSC.
- M. Dincǎ and J. R. Long, J. Am. Chem. Soc., 2007, 129, 11172–11176 CrossRef PubMed.
- D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586–10589 CrossRef CAS PubMed.
- M. J. Hardie, Encycl. Inorg. Bioinorg. Chem., 2014, 1–19 CAS.
- A. J. Rieth, A. M. Wright, G. Skorupskii, J. L. Mancuso, C. H. Hendon and M. Dincă, J. Am. Chem. Soc., 2019, 141, 13858–13866 CrossRef CAS PubMed.
- C. E. Bien, K. K. Chen, S.-C. Chien, B. R. Reiner, L.-C. Lin, C. R. Wade and W. S. W. Ho, J. Am. Chem. Soc., 2018, 140, 12662–12666 CrossRef CAS PubMed.
- A. M. Wright, Z. Wu, G. Zhang, J. L. Mancuso, R. J. Comito, R. W. Day, C. H. Hendon, J. T. Miller and M. Dincă, Chem, 2018, 4, 2894–2901 CAS.
- D. J. Xiao, J. Oktawiec, P. J. Milner and J. R. Long, J. Am. Chem. Soc., 2016, 138, 14371–14379 CrossRef CAS PubMed.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- O. Karagiaridi, W. Bury, J. E. Mondloch, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2014, 53, 4530–4540 CrossRef CAS PubMed.
- Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2007, 129, 12368–12369 CrossRef CAS PubMed.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- V. M. Krishnamurthy, G. K. Kaufman, A. R. Urbach, I. Gitlin, K. L. Gudiksen, D. B. Weibel and G. M. Whitesides, Chem. Rev., 2008, 108, 946–1051 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- P. C. Sahoo, Y. N. Jang and S. W. Lee, J. Cryst. Growth, 2013, 373, 96–101 CrossRef CAS.
- P.-Q. Liao, H. Chen, D.-D. Zhou, S.-Y. Liu, C.-T. He, Z. Rui, H. Ji, J.-P. Zhang and X.-M. Chen, Energy Environ. Sci., 2015, 8, 1011–1016 RSC.
- P. Schmieder, D. Denysenko, M. Grzywa, B. Baumgärtner, I. Senkovska, S. Kaskel, G. Sastre, L. van Wüllen and D. Volkmer, Dalton Trans., 2013, 42, 10786 RSC.
- D. Denysenko, M. Grzywa, M. Tonigold, B. Streppel, I. Krkljus, M. Hirscher, E. Mugnaioli, U. Kolb, J. Hanss and D. Volkmer, Chem.–Eur. J., 2011, 17, 1837–1848 CrossRef CAS PubMed.
- S. Biswas, M. Tonigold, M. Speldrich, P. Kögerler, M. Weil and D. Volkmer, Inorg. Chem., 2010, 49, 7424–7434 CrossRef CAS PubMed.
- C. Jin, S. Zhang, Z. Zhang and Y. Chen, Inorg. Chem., 2018, 57, 2169–2174 CrossRef CAS PubMed.
- A. Looney, R. Han, K. McNeill and G. Parkin, J. Am. Chem. Soc., 1993, 115, 4690–4697 CrossRef CAS.
- H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 10096–10115 CrossRef CAS PubMed.
- V. C. C. Wang, S. Maji, P. P. Y. Chen, H. K. Lee, S. S. F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574–8621 CrossRef CAS PubMed.
- R. L. Lieberman and A. C. Rosenzweig, Nature, 2005, 434, 177–182 CrossRef CAS PubMed.
- M. O. Ross, F. MacMillan, J. Wang, A. Nisthal, T. J. Lawton, B. D. Olafson, S. L. Mayo, A. C. Rosenzweig and B. M. Hoffman, Science, 2019, 364, 566–570 CrossRef CAS PubMed.
- A. C. Rosenzweig, C. A. Frederick, S. J. Lippard and P. Nordlund, Nature, 1993, 366, 537–543 CrossRef CAS PubMed.
- T. Ikuno, J. Zheng, A. Vjunov, M. Sanchez-Sanchez, M. A. Ortuño, D. R. Pahls, J. L. Fulton, D. M. Camaioni, Z. Li, D. Ray, B. L. Mehdi, N. D. Browning, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and J. A. Lercher, J. Am. Chem. Soc., 2017, 139, 10294–10301 CrossRef CAS PubMed.
- J. Zheng, J. Ye, M. A. Ortuño, J. L. Fulton, O. Y. Gutiérrez, D. M. Camaioni, R. K. Motkuri, Z. Li, T. E. Webber, B. L. Mehdi, N. D. Browning, R. L. Penn, O. K. Farha, J. T. Hupp, D. G. Truhlar, C. J. Cramer and J. A. Lercher, J. Am. Chem. Soc., 2019, 141, 9292–9304 CrossRef PubMed.
- D. Y. Osadchii, A. I. Olivos-Suarez, Á. Szécsényi, G. Li, M. A. Nasalevich, I. A. Dugulan, P. S. Crespo, E. J. M. Hensen, S. L. Veber, M. V. Fedin, G. Sankar, E. A. Pidko and J. Gascon, ACS Catal., 2018, 8, 5542–5548 CrossRef CAS.
- R. Balasubramanian, S. M. Smith, S. Rawat, L. A. Yatsunyk, T. L. Stemmler and A. C. Rosenzweig, Nature, 2010, 465, 115–119 CrossRef CAS PubMed.
- K. T. Dinh, M. M. Sullivan, P. Serna, R. J. Meyer, M. Dincǎ and Y. Román-Leshkov, ACS Catal., 2018, 8, 8306–8313 CrossRef CAS.
- K. T. Dinh, M. M. Sullivan, K. Narsimhan, P. Serna, R. J. Meyer, M. Dincă and Y. Román-Leshkov, J. Am. Chem. Soc., 2019, 141, 11641–11650 CrossRef CAS PubMed.
- B. E. R. Snyder, L. H. Böttger, M. L. Bols, J. J. Yan, H. M. Rhoda, A. B. Jacobs, M. Y. Hu, J. Zhao, E. Ercan Alp, B. Hedman, K. O. Hodgson, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4565–4570 CrossRef CAS PubMed.
- M. B. Park, E. D. Park and W.-S. Ahn, Front. Chem., 2019, 7, 1–7 CrossRef PubMed.
- D. J. Xiao, E. D. Bloch, J. A. Mason, W. L. Queen, M. R. Hudson, N. Planas, J. Borycz, A. L. Dzubak, P. Verma, K. Lee, F. Bonino, V. Crocellà, J. Yano, S. Bordiga, D. G. Truhlar, L. Gagliardi, C. M. Brown and J. R. Long, Nat. Chem., 2014, 6, 590–595 CrossRef CAS PubMed.
- P. Verma, K. D. Vogiatzis, N. Planas, J. Borycz, D. J. Xiao, J. R. Long, L. Gagliardi and D. G. Truhlar, J. Am. Chem. Soc., 2015, 137, 5770–5781 CrossRef CAS PubMed.
- E. I. Solomon, K. M. Light, L. V. Liu, M. Srnec and S. D. Wong, Acc. Chem. Res., 2013, 46, 2725–2739 CrossRef CAS PubMed.
- E. I. Solomon, A. Decker and N. Lehnert, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3589–3594 CrossRef CAS PubMed.
- W. T. Borden, R. Hoffmann, T. Stuyver and B. Chen, J. Am. Chem. Soc., 2017, 139, 9010–9018 CrossRef CAS PubMed.
- N. Kitajima and Y. Moro-oka, Chem. Rev., 1994, 94, 737–757 CrossRef CAS.
- A. J. Jasniewski and L. Que, Chem. Rev., 2018, 118, 2554–2592 CrossRef CAS PubMed.
- E. D. Bloch, L. J. Murray, W. L. Queen, S. Chavan, S. N. Maximoff, J. P. Bigi, R. Krishna, V. K. Peterson, F. Grandjean, G. J. Long, B. Smit, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14814–14822 CrossRef CAS PubMed.
- J. B. Vincent, G. L. Olivier-Lilley and B. A. Averill, Chem. Rev., 1990, 90, 1447–1467 CrossRef CAS.
- D. Denysenko, M. Grzywa, J. Jelic, K. Reuter and D. Volkmer, Angew. Chem., Int. Ed., 2014, 53, 5832–5836 CrossRef CAS PubMed.
- M. P. Lanci, V. V. Smirnov, C. J. Cramer, E. V. Gauchenova, J. Sundermeyer and J. P. Roth, J. Am. Chem. Soc., 2007, 129, 14697–14709 CrossRef CAS PubMed.
- C. E. Elwell, N. L. Gagnon, B. D. Neisen, D. Dhar, A. D. Spaeth, G. M. Yee and W. B. Tolman, Chem. Rev., 2017, 117, 2059–2107 CrossRef CAS PubMed.
- I. M. Wasser, S. de Vries, P. Moënne-Loccoz, I. Schröder and K. D. Karlin, Chem. Rev., 2002, 102, 1201–1234 CrossRef CAS PubMed.
- L. K. Keefer, Nat. Mater., 2003, 2, 357–358 CrossRef CAS.
- B. Xiao, P. S. Wheatley, X. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, I. L. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203–1209 CrossRef CAS PubMed.
- A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson, R. E. Morris, A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2008, 130, 10440–10444 CrossRef CAS PubMed.
- F. Bonino, S. Chavan, J. G. Vitillo, E. Groppo, G. Agostini, C. Lamberti, P. D. C. Dietzel, C. Prestipino and S. Bordiga, Chem. Mater., 2008, 20, 4957–4968 CrossRef CAS.
- A. C. McKinlay, J. F. Eubank, S. Wuttke, B. Xiao, P. S. Wheatley, P. Bazin, J.-C. Lavalley, M. Daturi, A. Vimont, G. De Weireld, P. Horcajada, C. Serre and R. E. Morris, Chem. Mater., 2013, 25, 1592–1599 CrossRef CAS.
- E. D. Bloch, W. L. Queen, S. Chavan, P. S. Wheatley, J. M. Zadrozny, R. Morris, C. M. Brown, C. Lamberti, S. Bordiga and J. R. Long, J. Am. Chem. Soc., 2015, 137, 3466–3469 CrossRef CAS PubMed.
- K. Peikert, L. J. McCormick, D. Cattaneo, M. J. Duncan, F. Hoffmann, A. H. Khan, M. Bertmer, R. E. Morris and M. Fröba, Microporous Mesoporous Mater., 2015, 216, 118–126 CrossRef CAS.
- D. Cattaneo, S. J. Warrender, M. J. Duncan, C. J. Kelsall, M. K. Doherty, P. D. Whitfield, I. L. Megson and R. E. Morris, RSC Adv., 2016, 6, 14059–14067 RSC.
- R. R. Haikal, C. Hua, J. J. Perry, D. O'Nolan, I. Syed, A. Kumar, A. H. Chester, M. J. Zaworotko, M. H. Yacoub and M. H. Alkordi, ACS Appl. Mater. Interfaces, 2017, 9, 43520–43528 CrossRef CAS PubMed.
- C. K. Brozek, J. T. Miller, S. A. Stoian and M. Dincă, J. Am. Chem. Soc., 2015, 137, 7495–7501 CrossRef CAS PubMed.
- J. Jover, C. K. Brozek, M. Dincǎ and N. López, Chem. Mater., 2019, 31, 8875–8885 CrossRef CAS.
- A. J. Rieth, A. M. Wright and M. Dincă, Nat. Rev. Mater., 2019, 4, 708–725 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |