DOI:
10.1039/C9SC06328E
(Edge Article)
Chem. Sci., 2020,
11, 2193-2199
A well-directional three-dimensional DNA walking nanomachine that runs in an orderly manner†
Received
14th December 2019
, Accepted 8th January 2020
First published on 10th January 2020
Abstract
Herein, we report a three-dimensional (3D) DNA walking nanomachine innovatively constructed from a functionalized 3D DNA track, which runs in an orderly manner with favorable directionality to allow for programming certain pathways of information transduction for some target tasks. The nanomachine was constructed using a departure station of walker (UB + W) and a functionalized 3D track, which was made up of a rolling circle amplification (RCA)-generated backbone chain and numerous triangular rung units with stators (UA + S) assembled into a repeating array along the backbone. A specific domain (SD) was designed at the 5′-end of the backbone to capture the UB + W, and stators with specific RNA substrates were immobilized at the three UA corners for the DNA walker to travel on. Powered by 10–23 DNAzyme, the DNA walker started moving from the SD end to the other end of the track by the autonomous cleavage of RNA substrates. Significantly, the homogeneous distribution of stators in the longitudinal and horizontal extensions paved a specific path for each walker to move along the 3D track. This resulted in random and inactive self-avoiding walking; thus, the nanomachine exhibited good executive ability. These properties allowed the DNA walking nanomachine to program the certain pathways of information transduction for the stepwise and programmed execution of some target tasks, such as the synthesis of specific polyorganics and cargo delivery. We believe that such a 3D DNA walking nanomachine could enrich the concept in the field of dynamic DNA nanotechnology, and may improve the development of novel DNA nanomachines in cargo delivery and composite product synthesis.
Introduction
The DNA walking nanomachine, as one of the DNA nanomachines, has attracted intense attention with its remarkable performance of automaticity and controllability through converting chemical energy to walking locomotion.1–4 In terms of the track of the DNA walking machine, the majority of the emerging DNA walking machines utilized the track of a one-dimensional (1D) DNA footpath with the highest level of controllability and directionality.5–8 These travel distances of each DNA walker, however, were well confined at only a few walking steps,9 so that the 1D DNA walkers were of very limited processivity,10 which restricted the executive ability of such DNA walking nanomachines. By bringing in a second plane, two-dimensional (2D) DNA origami was designed to serve as the track,11–14 which increased the number of walking steps and degrees of freedom, but the travel space was still confined,15 as well as the processivity of the walker. To address these issues, researchers recently exploited particle-based three-dimensional (3D) DNA walking nanomachines, which enabled a broad moving space with abundant steps for each DNA walker,15–19 thus performing a high processivity compared to the 1D and 2D ones. Despite the prominent increase in the amount of DNA stators for such 3D DNA nanomachines, the attachment of the stators to the particles via coordination bond was stochastic; thus, the stators were at a heterogeneous and locally disordered surface.20,21 The disordered surface environments disturbed the movement of each walker. When the walker traveled to the sparse locations of the stators, the locomotion was terminated once the distance between two stators was too far for the walker to move forward.22 In this way, the other stators of the whole machine would be wasted to restrict the good execution extent and performance ability of each walker, eventually limiting the executive ability of this machine. Further, the disordered stators would guide these walkers to chaotically walk on a spherical surface without a certain direction,15–19 which was not beneficial to programming certain pathways for information transduction. In consequence, although the 3D DNA walking nanomachine has overcame the low processivity from 1D and introduced the third plane from 2D, it lost the high level of directionality and controllability from 1D and did not maximumly utilize the executive ability of each walker. There are many challenges to overcome in order to integrate the superiorities of the 1D and 3D walking nanomachines and develop a novel DNA nanomachine, which has not only a high executive ability, but also a relatively good controllability and directionality.
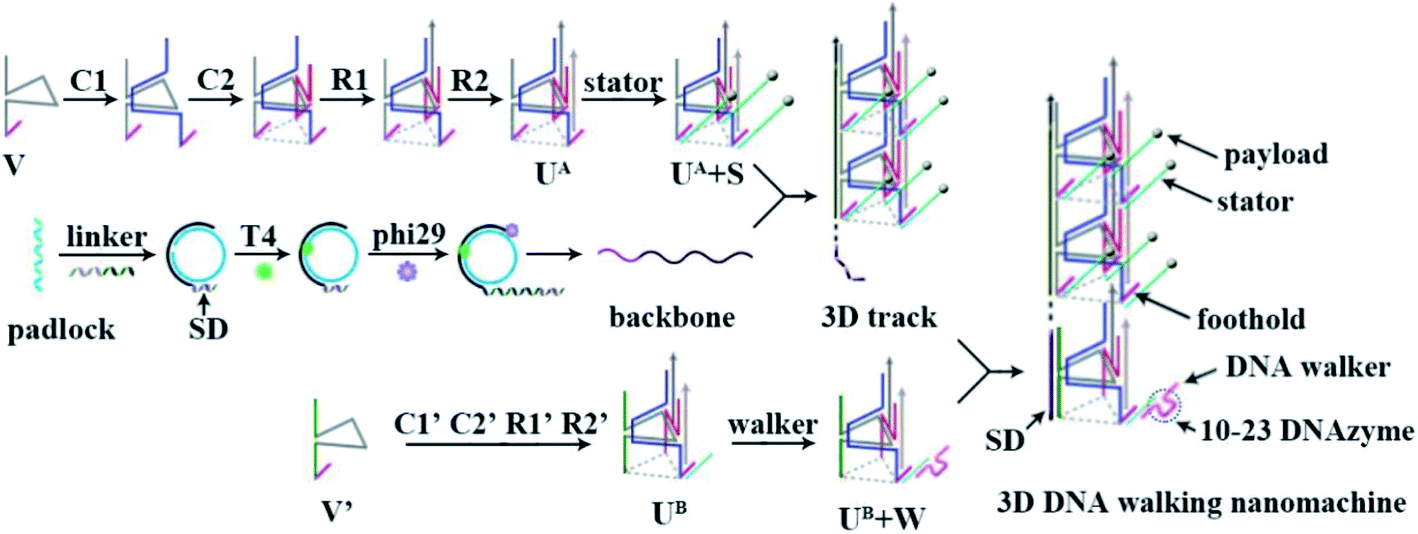
Herein, by directly using Watson–Crick base pairs, a 3D DNA walking nanomachine that can run in an orderly manner was constructed by designing a specific departure site and homogeneous steps for the DNA walker, which could pave a certain pathway for information transduction. The principle of this design is shown in Scheme 1. The 3D track of the walking machine was formed by numerous triangular rung units with payload-labeled stators (UA + S) assembled into a repeating array along an extremely long rolling circle amplification (RCA)-generated backbone with a specific domain (SD) at the 5′-end. Each triangular rung units (UA) had a foothold at every corner to immobilize three stators with specific RNA substrates for the DNA walker to move on, and the SD was especially designed to capture a departure station of the walker (UB + W). Fueled by the 10–23 DNAzyme,23–25 one DNA walker was able to move from the SD end to the other end of the track by cleaving the RNA substrates of stators at every step with the release of the payload. Since every side of UA + S had 20 invariable nt sequences and every layer between each UA + S contained constant sequences (40 nt), the DNA walker was programmed to follow a certain pathway. It would first move along the triangle, and then go to the next UA + S with a random and inactive self-avoiding walking. In this design, the departure position of each DNA walker was well-designed. The programmed 3D track provided a definite direction and invariable distance between each step for each DNA walker to travel with controllability. In principle, our design is beneficial to the stepwise and programmed execution of target tasks (by performing a series of multistep reactions), such as specific composite production synthesis and cargo delivery.
 |
| | Scheme 1 Schematic illustration of the principle of the 3D DNA walking nanomachine. | |
Results and discussion
Characterization of the designed nanomachine
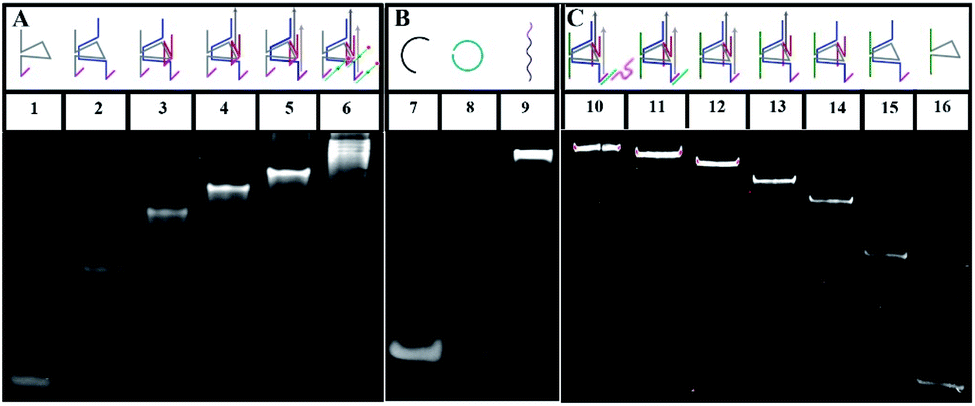
To verify the construction of the 3D DNA nanomachine, we evaluated the assembly products by polyacrylamide gel electrophoresis (PAGE, 8%). As exhibited in Fig. 1A, the number of chains hybridized together was increased stepwise and ascribed to lines 1 to 5, so that those products showed a stepwise slow mobility with stepwise higher molecular weight. In addition, line 6 demonstrated that UA (line 5) hybridized with the stator chains, indicating the formation of the proposed DNA construction of UA + S. Meanwhile, lines 7–9 (shown in Fig. 1B) showed the process of the rolling circle amplification (RCA). The RCA products, with extremely low mobility, exhibited a bright band in line 9, indicating that we obtained a long proposed DNA chain. Because of the low concentration of polyacrylamide and relatively long electrophoresis experimental time, the linker DNA in line 7 and padlock probe DNA in line 8 with low molecular weights both moved out from the bottom of the polyacrylamide gel. The result of the PAGE experiment of UB + W is exhibited in Fig. 1C. Lines 10 through 16 showed a stepwise slow mobility in the same manner as the PAGE images of UA + S, revealing the successful formation of the proposed UB + W construction. Sequence information of all DNA oligonucleotides used in our work are exhibited in Table S1 of the ESI.† Moreover, to investigate the morphology of our designed 3D nanomachine, atomic force microscopy (AFM) was performed in air. Single DNA nanomachines were observed and are shown in Fig. S6.† The AFM image indicated that the DNA nanomachines successfully formed with an approximate length of 0.30 μm.
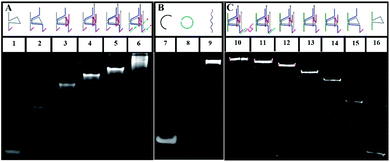 |
| | Fig. 1 (A) Native PAGE (8%) images of UA + S: lane 1, V (0.3 μM); lane 2, V + C1 (0.3 μM); lane 3, V + C1 + R1 (0.3 μM); lane 4, V + C1 + C2 + R1 (0.3 μM); lane 5, V + C1 + C2 + R1 + R2 (0.3 μM); lane 6, V + C1 + C2 + R1 + R2 + Cy5/3-S (0.3 μM); (B) native PAGE (8%) images of DNA backbone: lane 7, linker (1 μM), lane 8, padlock probe (1 μM), lane 9, DNA backbone. (C) Native PAGE (8%) images of UB + W: lane 10, V′ + C1′ + C2′ + R1′ + R2′ + h-S + walker (0.3 μM); lane 11, V′ + C1′ + C2′ + R1′ + R2′ + h-S; lane 12, V′ + C1′ + C2′ + R1′ + R2′ (0.3 μM); lane 13, V′ + C1′ + C2′ + R1′ (0.3 μM); lane 14, V′ + C1′ + C2′ (0.3 μM); lane 15, V′ + C1′ (0.3 μM); lane 16, V′ (0.3 μM). | |
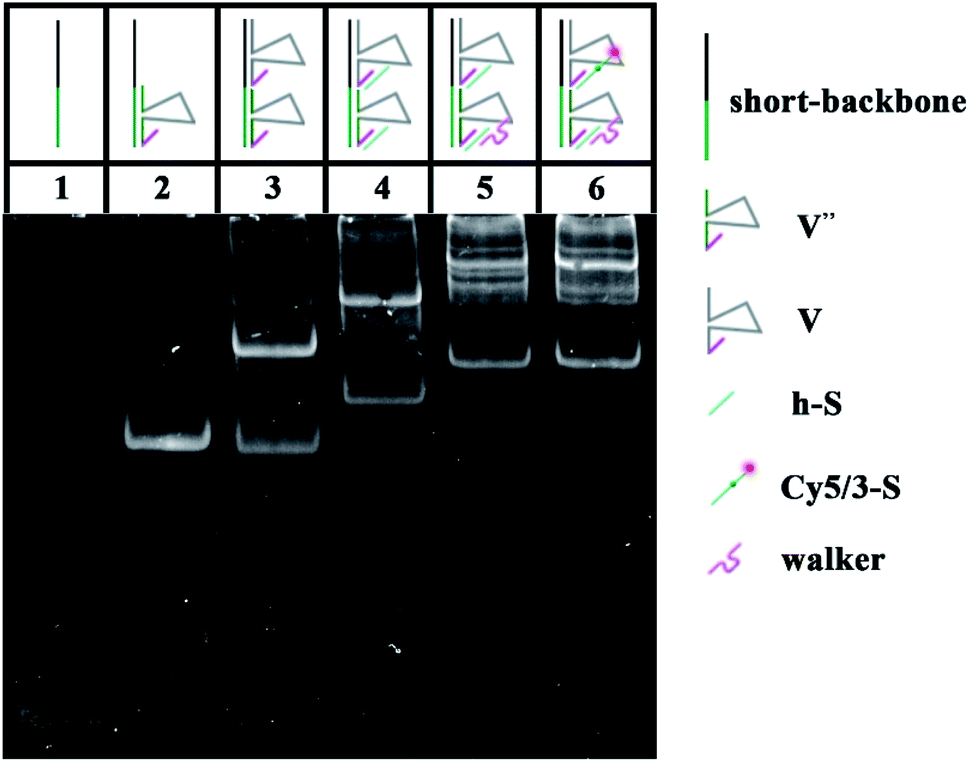
To verify the feasibility of the movement of the walker and the sequence specificity of the 10–23 DNAzyme, we employed PAGE to analyze whether the walker could successfully move from UB + W to the next layer (UA + S). The backbone is a long chain produced by RCA with a high molecular weight (approximately thousands of bases), which makes it difficult to observe and analyze such related products. Thus, to replace the long backbone chain, we introduced a short-backbone that only had one complementary region for UB + W and another one for UA + S. Although this structure was simplified to two layers by the introduction of the short-backbone, the triangle unit still had approximately 1200 nt from assembly by the short-backbone, UB + W and UA + S. Thus it required further simplification. Herein, we still assembled two layers to implement the experiment, but this was a simple two-layer combination with relatively low molecular weight. The two-layer combination was composed of a short-backbone, V′′ and V, which would not affect the proposed analysis due to no changes in the distance between the two new layers. As shown in Fig. 2, the lines from 1–5 exhibited the PAGE results of the successful assembly process. More importantly, the status of line 6 showed the reaction result of the walker walking, which is the same as that of line 5. This indicated the successful cleavage and release of the DNA substrate, and eventually the successful movement of the walker between the two layers.
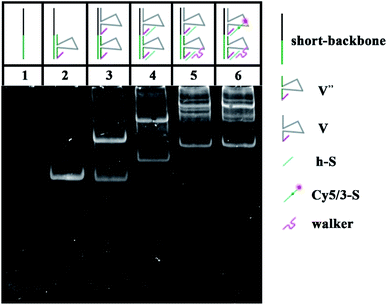 |
| | Fig. 2 Native PAGE (8%) images: lane 1, short-backbone (0.3 μM); lane 2, short-backbone + V′′ (0.3 μM); lane 3, short-backbone + V′′ + V (0.3 μM); lane 4, short-backbone + V′′ + V + h-S (0.3 μM); lane 5, short-backbone + V′′ + V + h-S + walker (0.3 μM); lane 6, short-backbone + V′′ + V + Cy5/3-S + walker (0.3 μM). | |
Construction of a fluorometric 3D DNA walking nanomachine
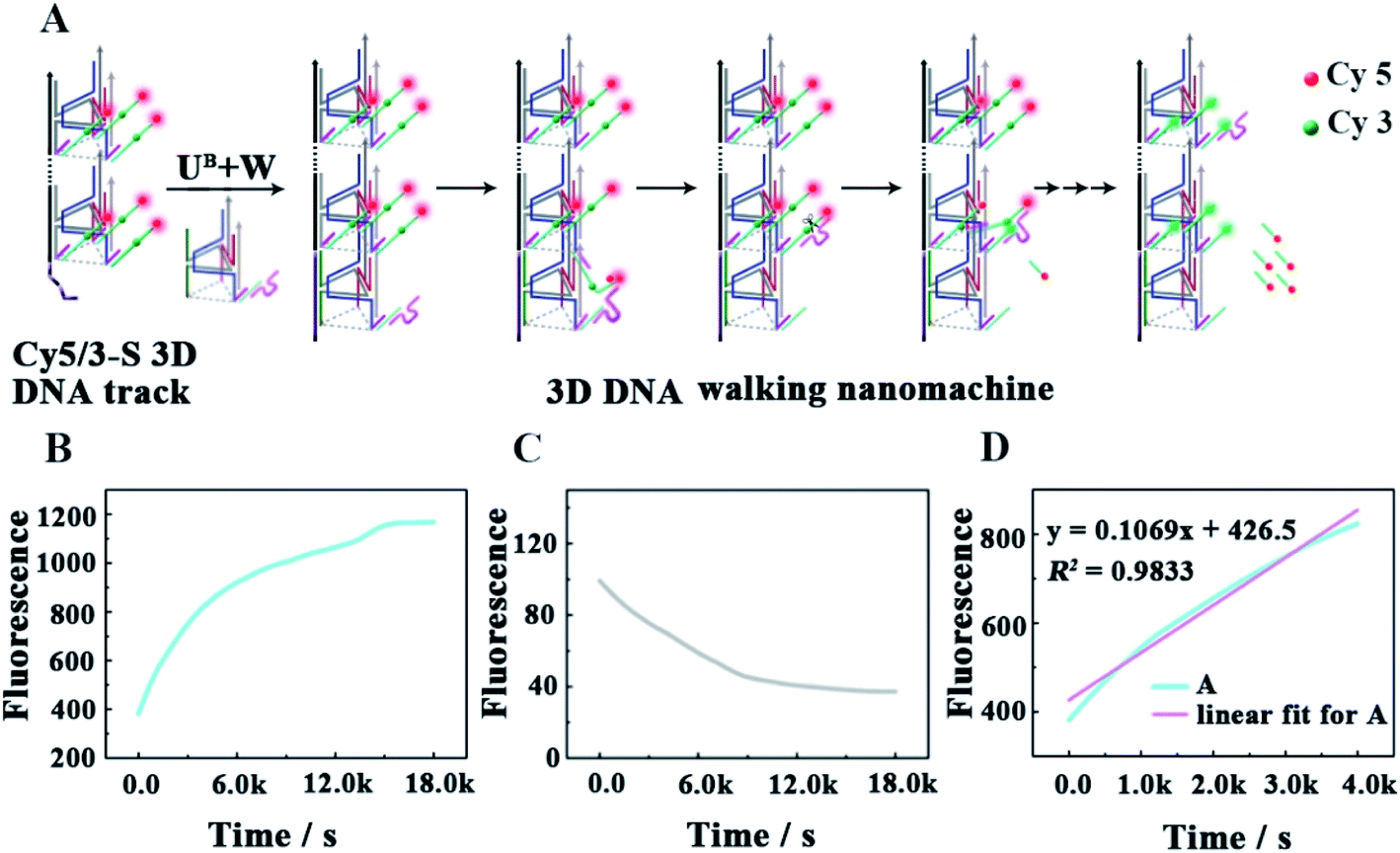
As shown in Fig. 3A, as a proof of principle, we first designed a 3D DNA walking nanomachine in which the fluorescence dye Cy5 labeled at the 5′-end of a stator served as the payload. Another fluorescence dye Cy3 was labeled at a position 17 nt away from Cy5, and the stator was denoted as Cy5/3-S. Initially, the distance between Cy3 and Cy5 was roughly 5 nm. Thus, a red fluorescence from Cy5 was observed due to the generation of fluorescence resonance energy transfer (FRET)26–28 between Cy3 (donor) and Cy5 (acceptor) with the excitation wavelength at 550 nm. Once this machine worked, each walker driven by 10–23 DNAzyme would achieve autonomous motion along the 3D DNA track with the release of the Cy5 payload, causing an apparent quenching of the Cy5 fluorescence and increasing of the Cy3 fluorescence owing to the far distance between the two fluorescent dyes, which is not beneficial for FRET.
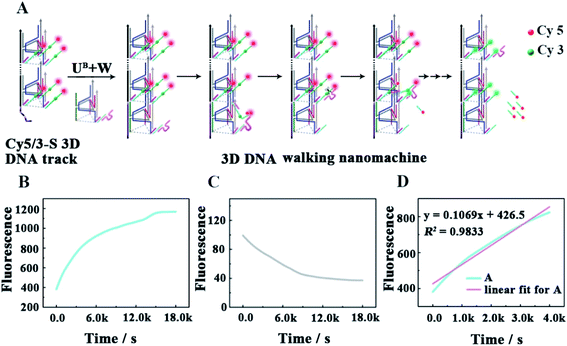 |
| | Fig. 3 (A) Schematic illustrating the locomotion mechanism of the elaborate 3D DNA walking machine. (B and C) The fluorescence increases of Cy3 and the fluorescence quenching of Cy5 as a function of time from the fluorescent 3D DNA walking machine (0.12 μM). (D) Monitoring the initial moving rate of each walker by linear fit for continuous fluorescence increases in the first 4000 s. | |
To quantitatively characterize the performance of this machine, the fluorescence increases of Cy3 and quenching of Cy5 in real time were studied (Fig. 3B and C, respectively). After the 0.24 μM UB + W was added to the 0.12 μM Cy5/3-S 3D DNA track, an obvious increase in the fluorescence intensity of Cy3 occurred due to the disappearance of FRET for the release of Cy5, which approached a saturation state after about 4 h. The initial locomotion rate of this DNA walking nanomachine was roughly estimated to be 1.80 × 10−11 M s−1. This rate was determined by the fluorescence changes for the first 4000 s (Fig. 3D). Moreover, we also examined the fluorescence emission of the Cy5/Cy3 system before and after walking to further check the operation of this machine. The images represented two typical peaks for Cy3 and Cy5 located at around 564 nm and 668 nm, respectively, in which the fluorescence intensity of Cy3 was increased and that of Cy5 was quenched after walking compared to the initial state (before walking) (Fig. S1†). Simultaneously, we employed a confocal microscope to synchronously monitor the results of the DNA walking at three channels in green, red and colocalization (Fig. 4). The images (A–C) were the results of fluorescence before walking and the images (D–F) were the results after walking. Comparing the results in images (A and B) with those of (D and E), the fluorescence intensity of the green spots in image (D) were more enhanced than that in (A), and the fluorescence intensity of the red spots in image (E) were apparently more quenched than that in (B). Meanwhile, the corresponding colocalized spots were shown in images (C and F). Moreover, the respective confocal fluorescence intensities of Cy3 and Cy5 before and after walking were plotted as histograms (Fig. S2†). Collectively, these results indicate the successful operation of the designed 3D DNA walking machine.
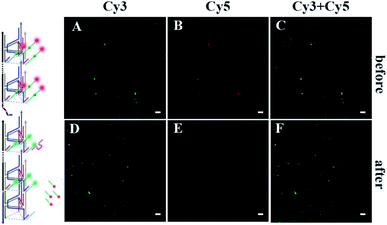 |
| | Fig. 4 Fluorescence comparison diagrams. (A–C) Confocal images of the DNA nanomachine before walker moving. (D–F) Confocal images of the DNA nanomachine after walker moving. The scale bar is 10 μm. | |
Feasibility investigation of the nanomachine
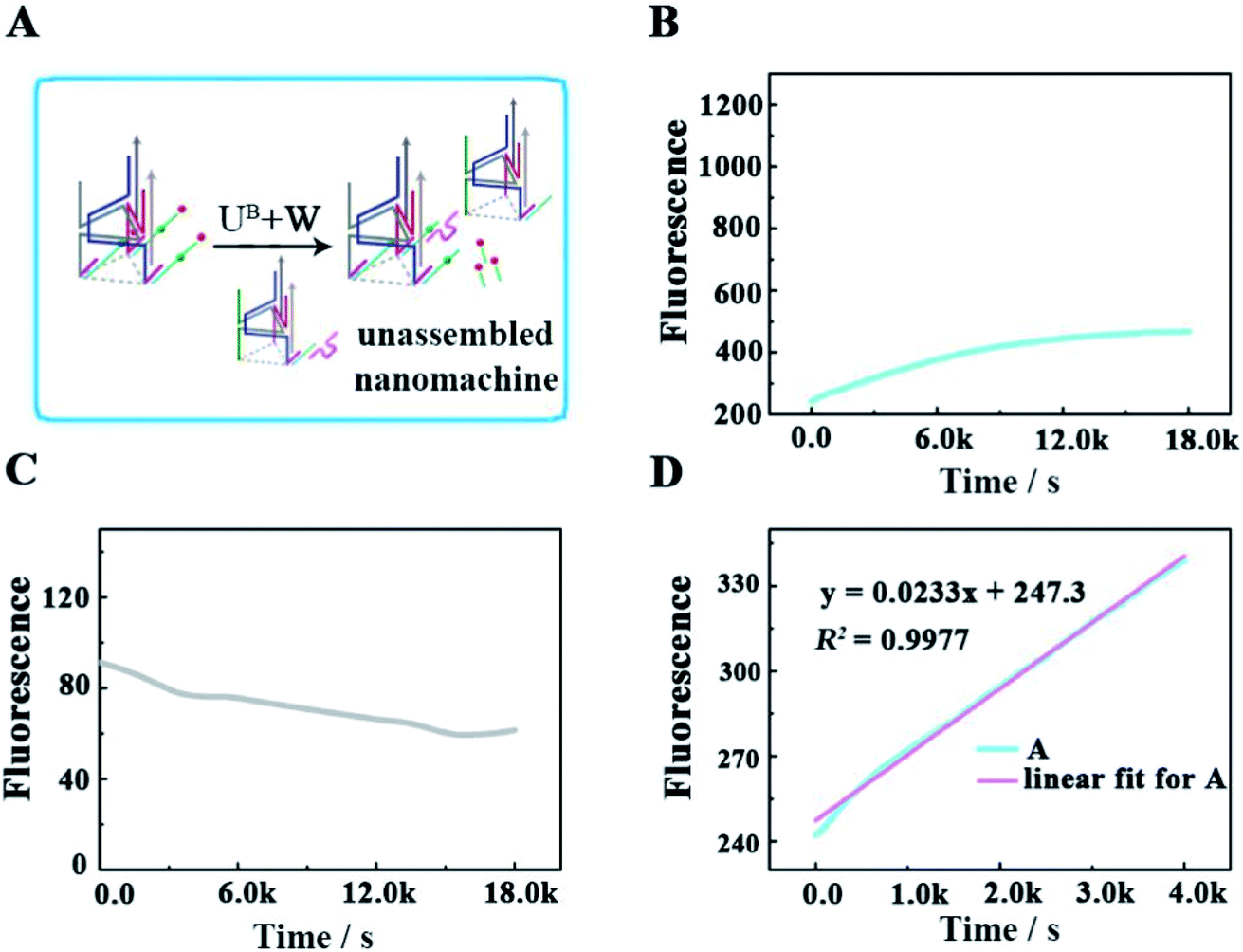
To verify that each DNA walker traveled along the 3D track rather than moving between the scattered UA + S, we designed a control experiment by hybridizing UA with three times the amount of Cy5/3-S, but without the backbone (Fig. 5A), to construct an unassembled nanomachine. For contrasting with the correct nanomachine in Fig. 3A, we also monitored the fluorescence changes of Cy3 and Cy5 in real time under the same analysis condition (Fig. 5B and C), and linear fitting was also performed for the first 4000 s. As shown in Fig. 5B and D, the total efficiency of this reaction was relatively slow. The saturation point did not occur after a reaction time of 5 h with an initial rate of 3.68 × 10−12 M s−1. We think the collision theory could be employed to explain this phenomenon. The reactants have to collide to make contact with each other, and then they may react. The collision frequency is proportional to the concentration of the walker and UA in this work.29,30 In the scattered situation, the local concentrations of the walker and UA + S were lower than that of being assembled together based on the backbone,31 resulting in a low collision efficiency and further giving rise to a depressed reaction efficiency between them. This result indicates that the construction of the 3D DNA track is a key component for improving the executive ability of this machine. The relevant fluorescence changes could be read out from the fluorescence spectra in Fig. S3.†
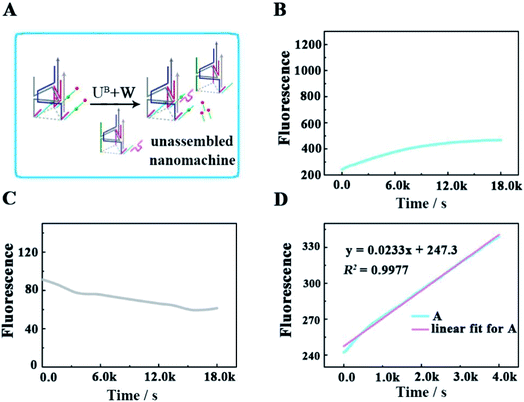 |
| | Fig. 5 (A) Schematic illustrating the locomotion mechanism of the unassembled 3D DNA walking nanomachine. (B and C) The respective fluorescence changes of Cy3 and Cy5 as a function of time. (D) Linear fit for the continuous increase in fluorescence during the first 4000 s. | |
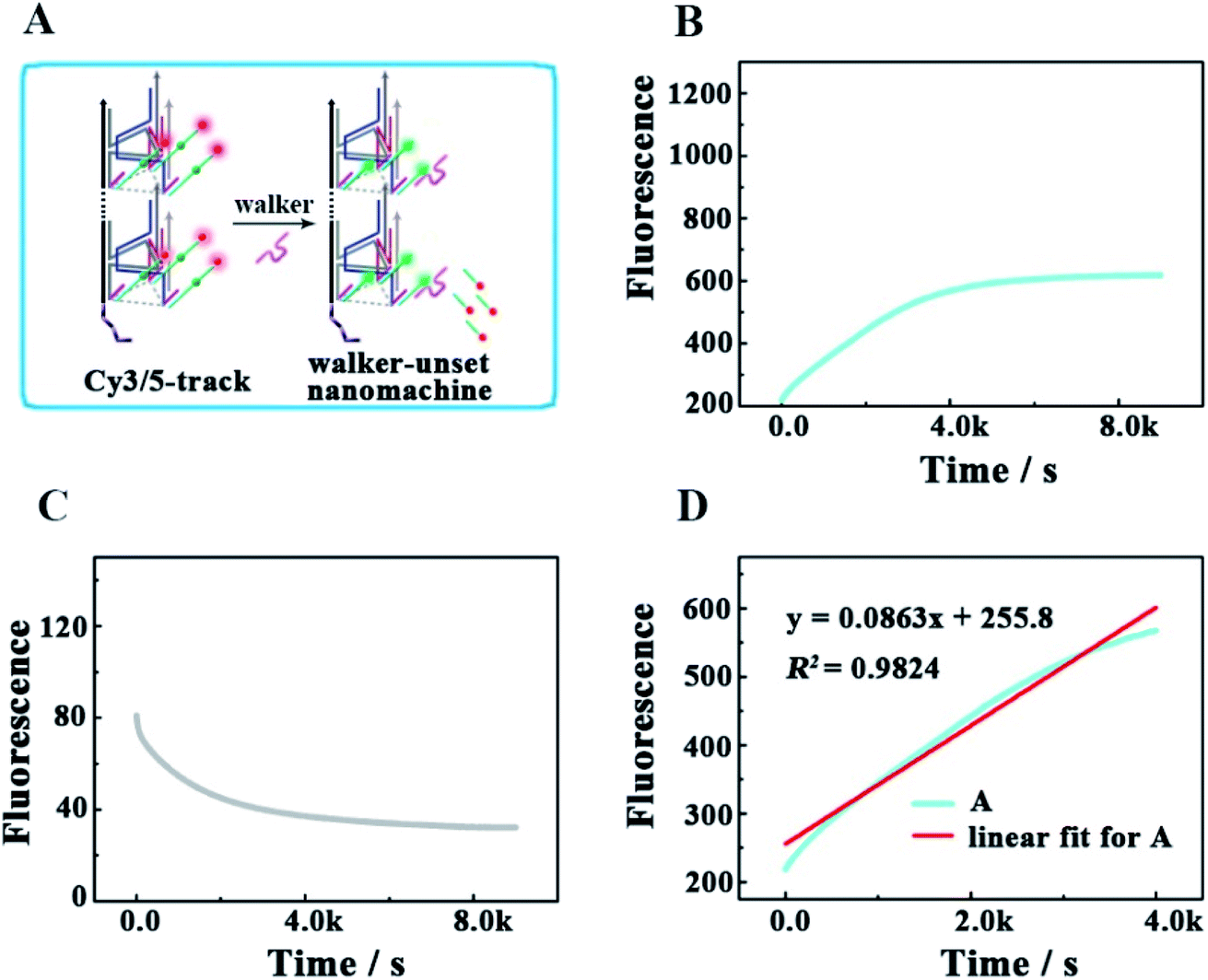
To check the influence of UB + W at one end of the track for realizing the directional movement of the walker, we also explored a second control experiment by adding only the walker to Cy3/5-track (Fig. 6). This enabled the construction of a walker-unset nanomachine, in which each walker was not involved in assembling UB + W, but could randomly hybridize with any Cy5/3-S. In this way, both the initial moving position for each walker and the number of walkers hybridized on one Cy3/5-track were uncertain; thus, an unquantifiable walker could start moving from anywhere on one Cy3/5-track to an uncertain direction. As shown in Fig. 6B and C, the completion time for the walker-unset nanomachine was about 1.1 h (roughly v = 1.33 × 10−11 M s−1), which was 3.6 times shorter than that of the correct nanomachine with a lower saturated fluorescence intensity. We think that the chaotic hybridization of the walkers and stators would make the walker start moving from the uncertain position towards one end of the track, while the Cy5 on the other end would not be released due to a lack of the nearest neighbors for the walker to move back to this end. In addition, some walker-unset nanomachines might be left that were not hybridized with any walker, so these machines would not work. Owing to the mentioned speculations above, a saturation state was approached earlier with a lower fluorescence intensity than that of the correct 3D DNA walking nanomachine. Therefore, such a departure station is a key point for the DNA walker to move along the track with a certain direction; thus, each walker could move in an orderly manner with controllability to exhibit the good execution extent and performance ability. Similarly, the relevant fluorescence spectra are shown in Fig. S4.†
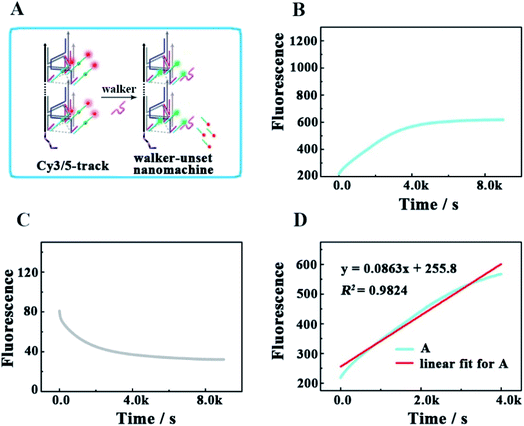 |
| | Fig. 6 (A) Schematic illustrating the locomotion mechanism of the walker-unset 3D DNA walking nanomachine. (B and C) The respective fluorescence changes of Cy3 and Cy5 as a function of time from the walker-unset 3D DNA walking nanomachine (0.12 μM). (D) Linear fit for the continuous increase in fluorescence during the first 4000 s. | |
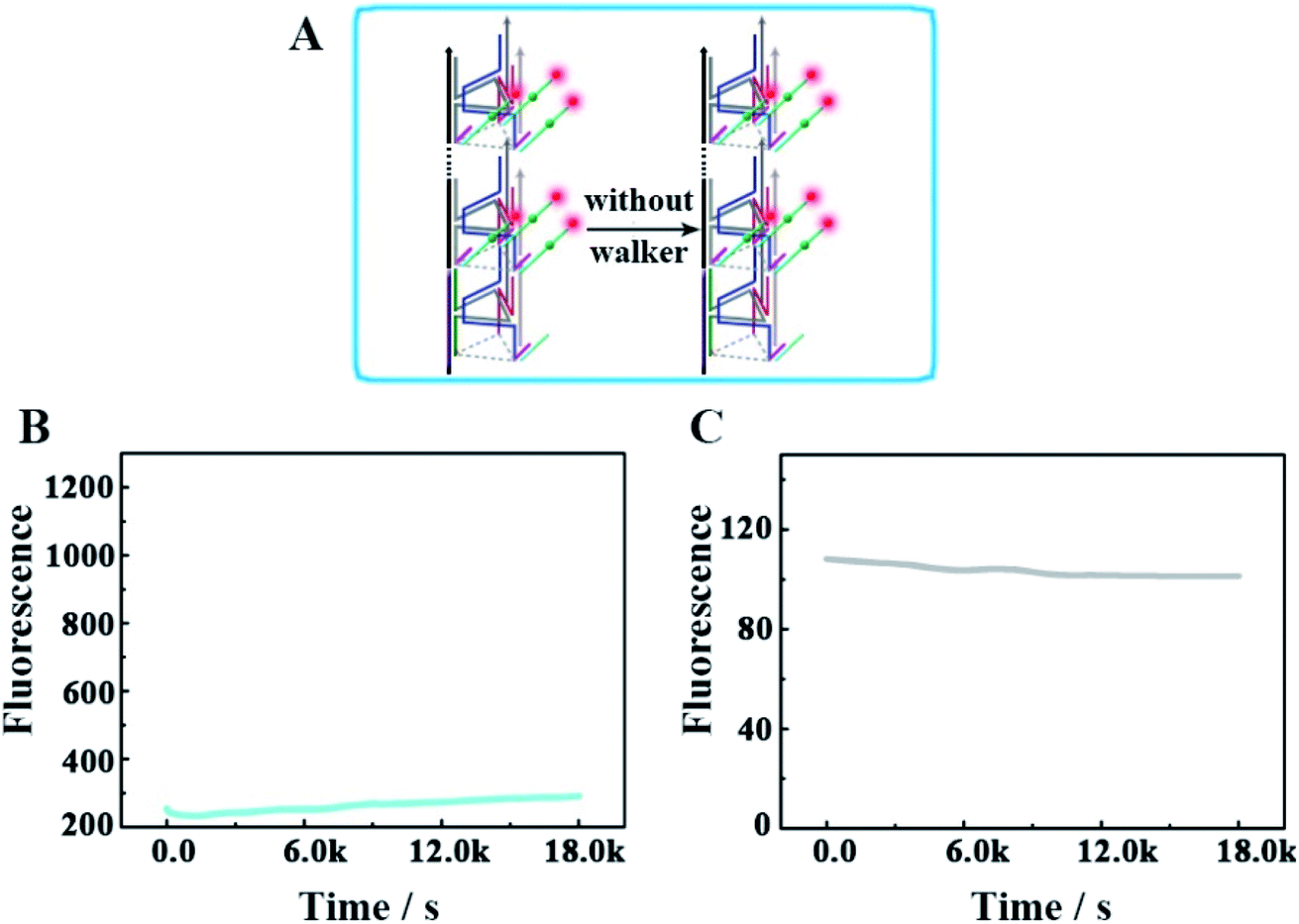
Furthermore, we designed another control experiment, in which the walker did not appear to move along the track, to check whether the changes of the fluorescence intensity occur because of the operation of this designed DNA nanomachine (Fig. 7). As shown in Fig. 7B and C, in the no-walker nanomachine system, the fluorescence intensities of both Cy3 and Cy5 showed almost no change during a continuous reaction time of 5 h compared with that of the correct walking nanomachine in Fig. 3B and C. These results confirm that the walker is the necessary factor to the successful release of the payload. The relevant fluorescence spectra in Fig. S5† also exhibited the same results.
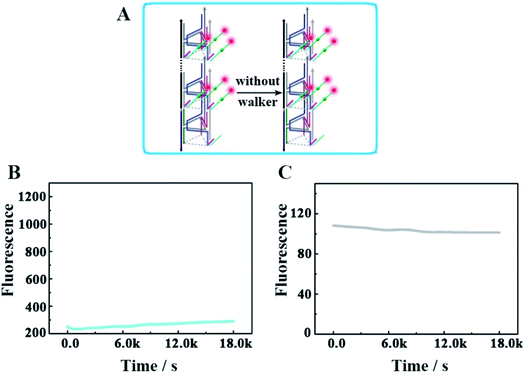 |
| | Fig. 7 (A) Schematic illustrating the mechanism of the no-walker 3D DNA walking nanomachine. (B and C) The respective fluorescence changes of Cy3 and Cy5 as a function of time from the no-walker 3D DNA walking nanomachine (0.12 μM). | |
Conclusions
Unlike the existing 3D DNA walking nanomachine whose direction of each moving step is random, our 3D DNA track-based nanomachine has a relatively high level of controllability and directionality. The track is well designed to have a departure station for the walker at one end, and the stators are orderly and homogeneously arranged along the track, which ensure a certain pathway for DNA walking. The new concept of integrating the directionality from the 1D DNA walking nanomachine and processivity from the 3D nanomachine is beneficial to extending the functionality of the DNA walking machine, such as specific polyorganic synthesis and cargo delivery. This protocol exhibited the promising application of the DNA nanostructure on improving the locomotion performance of the DNA walking machine, which may pave a significant avenue to the development of artificial DNA nanotechnology.
Reagents and materials
Phi29 DNA polymerase, 10× phi29 DNA polymerase buffer, and deoxyribonucleoside triphosphate (dNTPs) solution were obtained from Sangon Inc. (Shanghai, China). T4 DNA ligase and T4 DNA ligase buffer were purchased from TaKaRa (Dalian, China). DNA oligonucleotides (as shown in Table S1†) utilized in this work were synthesized by Sangon Inc. (Shanghai, China).
TAE/Mg2+ buffer system: 40 mM Tris, 10.0 mM Mg(OAc)2 and 2 mM EDTA with the pH value adjusted to 8.0 using glacial acetic acid.
Instrumentation
The polyacrylamide gel electrophoresis (PAGE) analysis was carried out with a BG-verMIDI standard vertical electrophoresis apparatus (Baygene, Beijing, China). The gel imaging was implemented using a Bio-Rad Gel Doc XR+ System (Bio-Rad, Hercules, CA). The morphologies of the designed DNA nanotube were characterized by atomic force microscopy (AFM, Bruker, Germany) with a Dimension ICON system. In addition, the fluorescence responses were acquired from an FL-7000 fluorescence spectrophotometer (Hitachi, Tokyo, Japan). The nanomachines were imaged using an LSM780 confocal microscope (Carl Zeiss), which was equipped with ZEN 2010 software.
Preparation of the DNA backbone based on rolling circle amplification (RCA)
The long DNA backbone was prepared by a modified method.32 The details are as follows: a volume of 2 μL of 10× T4 DNA ligase buffer, 9 μL of linker (4 μM) and 9 μL of the padlock probe (2 μM) were mixed together, and then subsequently annealed at 95 °C for 5 min and cooled down to 20 °C over 75 min. After the successful hybridization of linker and the padlock probe, 5 U of T4 DNA ligase was added to combine its 5′-end with the 3′-end of the padlock probe, and then the mixture was incubated at 16 °C for 12 h and thermally treated at 65 °C for 15 min. After that, 1 μL 10× phi29 DNA polymerase buffer, 0.5 U phi29 DNA polymerase and 2.5 μL dNTPs (2.5 mM) were reacted with every 6 μL of the prepared mixture at 37 °C for 2 h to generate the long DNA backbone. Then, the phi29 DNA polymerase was inactivated by thermal treatment at 65 °C for 15 min to stop the reaction.
The assembling of the designed 3D DNA walking nanomachine
The proposed DNA nanomachine consisted of the DNA backbone, a departure station of the walker (UB + W), and an abundant triangular rung unit with stators (UA + S). In brief, UB + W and UA + S were separately generated by the annealed product of V1′, C1′, C2′, R1′, R2′, h-S and the walker with an equal concentration and V1, C1, C2, R1, R2 with the stator at 3 times the concentration in TAE/Mg2+ buffer. Then, 10 μL of the prepared UA + S with a final concentration of 1.77 μM was combined with 5 μL DNA backbone by annealing the mixture from 44 °C to 20 °C over 75 min. This nanomachine was activated by adding 10 μL UB + W solution (final concentration of 0.24 μM) to the above solution mixture.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This paper was financially supported by the National Natural Science Foundation of China (21974108, 21775124, 21675129, 81370836 and 81873606).
References
- J. Valero, N. Pal, S. Dhakal, N. G. Walter and M. Famulok, Nat. Nanotechnol., 2018, 13, 496–503 CrossRef CAS PubMed.
- T. Omabegho, R. Sha and N. C. Seeman, Science, 2009, 324, 67–71 CrossRef CAS PubMed.
- T.-G. Cha, J. Pan, H. R. Chen, J. Salgado, X. Li, C. D. Mao and J. H. Choi, Nat. Nanotechnol., 2014, 9, 39–43 CrossRef CAS PubMed.
- H. Z. Gu, J. Chao, S. Xiao and N. C. Seeman, Nature, 2010, 465, 202–206 CrossRef CAS PubMed.
- M. You, Y. Chen, X. Zhang, H. Liu, R. Wang, K. Wang, K. R. Williams and W. Tan, Angew. Chem., 2012, 124, 2507–2510 CrossRef.
- Y. He and D. R. Liu, Nat. Nanotechnol., 2010, 5, 778–782 CrossRef CAS PubMed.
- L. Wang, R. J. Deng and J. H. Li, Chem. Sci., 2015, 6, 6777–6782 RSC.
- M. S. Xiao, X. E. Wang, L. Li and H. Pei, Anal. Chem., 2019, 91, 11253–11258 CrossRef CAS PubMed.
- Y. Yang, M. A. Goetzfried, K. Hidaka, M. You, W. Tan, H. Sugiyama and M. Endo, Nano Lett., 2015, 15, 6672–6676 CrossRef CAS.
- X. M. Qu, D. Zhu, G. B. Yao, S. Su, J. Chao, H. J. Liu, X. L. Zuo, L. H. Wang, J. Y. Shi, L. H. Wang, W. Huang, H. Pei and C. H. Fan, Angew. Chem., Int. Ed., 2017, 56, 1855–1858 CrossRef CAS PubMed.
- F. J. W. Shelley, B. Jonathan, K. Yousuke, E. Masayuki, H. Kumi, S. Hiroshi and J. T. Andrew, Nat. Nanotechnol., 2012, 7, 169–173 CrossRef PubMed.
- E. T. Toma, T. Roman, G. Yair, B. Yaron, L. Miran, A. Dorit, G. Doron and N. Eyal, ACS Nano, 2017, 11(4), 4002–4008 CrossRef PubMed.
- A. J. Thubagere, W. Li, R. F. Johnson, Z. B. Chen, S. y. Doroudi, Y. L. Lee, G. Izatt, S. Wittman, N. Srinivas, D. Woods, E. Winfree and L. L. Qian, Science, 2017, 357, eaan6558 CrossRef.
- K. Lund, A. J. Manzo, N. Dabby, N. Michelotti, A. JohnsonBuck, J. Nangreave, S. Taylor, R. Pei, M. N. Stojanovic, N. G. Walter, E. Winfree and H. Yan, Nature, 2010, 465, 206–209 CrossRef CAS PubMed.
- C. Jung, P. B. Allen and A. D. Ellington, Nat. Nanotechnol., 2016, 11, 157–163 CrossRef CAS PubMed.
- X. L. Yang, Y. A. Tang, S. D. Mason, J. B. Chen and F. Li, ACS Nano, 2016, 10, 2324–2330 CrossRef CAS PubMed.
- C. Jung, P. B. Allen and A. D. Ellington, ACS Nano, 2017, 11, 8047–8054 CrossRef CAS PubMed.
- F. Chen, J. Xue, M. Bai, J. Qin and Y. X. Zhao, Chem. Sci., 2019, 10, 3103–3109 RSC.
- M. S. Xiao, K. Zou, L. Li, Li. H. Wang, Y. Tian, C. H. Fan and H. Pei, Angew. Chem., 2019, 131, 15594–15600 CrossRef.
- P. Zhang, J. Jiang, R. Yuan, Y. Zhuo and Y. Q. Chai, J. Am. Chem. Soc., 2018, 140, 9361–9364 CrossRef CAS PubMed.
- H. Pei, N. Lu, Y. L. Wen, S. P. Song, Y. Liu, H. Yan and C. H. Fan, Adv. Mater., 2010, 22, 4754–4758 CrossRef CAS PubMed.
- D. F. Wang, C. Vietz, T. Schröder, G. Acuna, B. Lalkens and P. Tinnefeld, Nano Lett., 2017, 17, 5368–5374 CrossRef CAS.
- S. W. Santoro and G. F. Joyce, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4262 CrossRef CAS PubMed.
- Y. Tian, Y. He, Y. Chen, P. Yin and C. D. Mao, Angew. Chem., Int. Ed., 2005, 44, 4355–4358 CrossRef CAS.
- J. Nowakowski, P. J. Shim, G. S. Prasad, C. D. Stout and G. F. Joyce, Nat. Struct. Biol., 1999, 6, 151–156 CrossRef CAS.
- G. D. Hamblin, K. M. M. Carneiro, J. F. Fakhoury, K. E. Bujold and H. F. Sleiman, J. Am. Chem. Soc., 2012, 134, 2888–2891 CrossRef CAS PubMed.
- L. He, D. Q. Lu, H. Liang, S. T. Xie, C. Luo, M. M. Hu, L. J. Xu, X. B. Zhang and W. H. Tan, ACS Nano, 2017, 11(4), 4060–4066 CrossRef CAS PubMed.
- Y. F. He, M. L. Lu, J. Cao and H. P. Lu, ACS Nano, 2012, 6(2), 1221–1229 CrossRef CAS PubMed.
- C. Bettegowda, M. Sausen, R. J. Leary, I. Kinde, Y. Wang, N. Agrawal, B. R. Bartlett, H. Wang, B. Luber and R. M. Alani, Sci. Transl. Med., 2014, 6, 224ra224 Search PubMed.
- L. P. Xu, Y. X. Chen, G. Yang, W. X. Shi, B. Dai, G. N. Li, Y. H. Cao, Y. Q. Wen, X. J. Zhang and S. T. Wang, Adv. Mater., 2015, 27, 6878–6884 CrossRef CAS PubMed.
- K. W. Ren, Y. F. Xu, Y. Liu, M. Yang and H. X. Ju, ACS Nano, 2018, 12, 263–271 CrossRef CAS PubMed.
- J. L. Tang, Y. R. Yu, H. Shi, X. X. He, Y. L. Lei, J. F. Shangguan, X. Yang, Z. Z. Qiao and K. M. Wang, Anal. Chem., 2017, 89, 6637–6644 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06328e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Yin
Zhuo
*b,
Yin
Zhuo