
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSize and charge dual-transformable mesoporous nanoassemblies for enhanced drug delivery and tumor penetration†
Liang
Chen
 a,
Tiancong
Zhao
a,
Mengyao
Zhao
a,
Wenxing
Wang
a,
Caixia
Sun
a,
Lu
Liu
a,
Qin
Li
b,
Fan
Zhang
a,
Dongyuan
Zhao
a and
Xiaomin
Li
*ab
a,
Tiancong
Zhao
a,
Mengyao
Zhao
a,
Wenxing
Wang
a,
Caixia
Sun
a,
Lu
Liu
a,
Qin
Li
b,
Fan
Zhang
a,
Dongyuan
Zhao
a and
Xiaomin
Li
*ab
aDepartment of Chemistry, Laboratory of Advanced Materials, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, State Key Laboratory of Molecular Engineering of Polymers, Collaborative Innovation Center of Chemistry for Energy Materials (2011-iChEM), Fudan University, Shanghai 200433, P. R. China. E-mail: lixm@fudan.edu.cn
bQueensland Micro- and Nanotechnology Centre, School of Engineering & Built Environment, Griffith University, Nathan, QLD 4111, Australia
First published on 3rd February 2020
Abstract
A series of biological barriers in a nanoparticle-formulated drug delivery process inevitably result in the current low delivery efficiency, limited tumor penetration and insufficient cellular internalization of drugs. These multiple biological barriers are intimately related to the physicochemical properties of nanoparticles, especially the contradictory demand on size and surface charge for long blood circulation (larger and negative) and deep tumor penetration (smaller) as well as efficient cellular internalization (positive). Herein, we report tumor microenvironment triggered size and charge dual-transformable nanoassemblies. The nanoassembly is realized by immobilizing positive up/downconverting luminescent nanoparticles (U/DCNPs) onto large mesoporous silica nanoparticles (MSNs) via acid-labile bonds to form core@satellite structured MSN@U/DCNPs nanoassemblies, and subsequent capping of charge reversible polymers. At physiological pH, the integrated nanoassemblies with a larger size (∼180 nm) and negative charge can effectively achieve a prolonged blood circulation and high tumor accumulation. While under an acidic tumor microenvironment, the charge reversal of outer polymers and cleavage of linkers between MSNs and U/DCNPs can induce disintegration of the nanoassemblies into isolated MSNs and smaller U/DCNPs, both with a positively charged surface, which thereby potentiate the tumor penetration and cell uptake of dissociated nanoparticles. Combined with the independent near-infrared (NIR)-to-visible and NIR-to-NIR luminescence of U/DCNPs and high surface area of MSNs, the nanoassemblies can implement NIR bioimaging guided chemo- and photodynamic combined therapy with remarkable antitumor efficiency because of the high accumulation and deep tumor penetration induced by the dual transformability of the nanoassemblies.
Introduction
The last two decades have witnessed remarkable progress of sophisticated nanoparticles for the targeted delivery of theranostic agents (drugs, imaging agents, etc.) to solid tumors.1 In comparison to traditional drugs, nanomedicine holds great promise for improving treatment efficacy and minimizing systemic toxicity due to the well-known enhanced permeability and retention (EPR) effect.2,3 Nanoparticle-formulated drugs typically go through several stages before reaching the tumor site, including blood circulation, tumor accumulation and penetration, cell internalization and intracellular drug release.4 To achieve effective tumor treatment, theranostic agents must accumulate and penetrate into the deep tumor tissue efficiently to access as many cancer cells as possible.5 However, a series of biological barriers in the delivery process inevitably prevent the nanocarriers from arriving at the targets,6 resulting in the low delivery efficiency, limited tumor penetration and insufficient cellular internalization of nanoagents.7The physiochemical properties of nanoparticles, including size, shape, rigidity, charge, surface chemistry etc., have profound effects on their pharmacokinetics and delivery efficiency.8 Even though substantial efforts have been devoted to incorporating multiple functionalities and moieties into various nanocarriers, many of these strategies fail to adequately address biological barriers in the delivery system.9 The main reason is that there are multiple irreconcilable requirements on the physiochemical properties of the theranostic nanoparticles at different transport stages.10 For example, a relatively large size (100–200 nm) enables prolonged blood circulation and tumor accumulation of nanoparticles, whereas the large size hinders their deep penetration into the tumor parenchyma.11 Contrastingly, small nanoparticles (<20 nm) can penetrate deeply and homogeneously into the tumor, but they are generally subjected to fast clearance and inferior tumor accumulation.12 Similarly, the surface charge of nanocarriers also needs to be balanced to meet variable requirements in transportation: the negatively charged surface of nanoparticles can mitigate the immune clearance effect as compared to positively charged nanoparticles, but the negative charge impairs cell internalization due to the weakened interaction with the cell membrane.13 To address this dilemma, much effort has been made to develop size-,14–18 charge-,19,20 or surface modification-transformable21 delivery systems in response to a given stimulus, such as pH,22 enzyme,23,24 NIR light,25,26etc. For instance, Ge and co-workers developed an endogenous stimuli-sensitive multistage polymeric micelleplex drug delivery system by encapsulating cisplatin conjugated dendrimers into pH-responsive micelles. The micelles can maintain a large size (∼100 nm) under physiological conditions for better blood circulation and tumor accumulation, while unloading small-sized dendrimer prodrugs under acidic conditions for deep tumor penetration.27 In another study, they reported light-triggered decomposable micelles that can release chlorin e6/cypate conjugated dendrimers for tumor penetration and phototherapy.28 Zhao et al. also used a lipid–dendrimer nanoassembly to overcome the conflicting requirements in the delivery process.29 However, most of these studies are focused on dealing with single contradictory requirements.30 It is urgently desired for smart nanomedicine with multiple transformable capability to adapt to the multiple biological barriers in the delivery system, which may further improve the drug delivery efficiency.31–33
Herein, we report a core@satellite structured nanoassembly of mesoporous silica nanoparticles (MSN) and up/downconverting nanoparticles (U/DCNPs) (denoted as MSN@U/DCNPs), which can undergo remarkable dual-transformation in size (from 180 to 20 nm) and surface charge (from negative to positive) in an acidic tumor microenvironment, for prolonged blood circulation, high tumor accumulation, and enhanced tumor penetration and cell uptake. As illustrated in Scheme 1, the core@satellite structured nanoassembly is constructed by the integration of four segments: a large amino-functionalized MSN core, small U/DCNPs satellites with a positively charged polyethyleneimine (PEI) surface, acid-labile linkers between the MSN and U/DCNPs, and an outermost pH-responsive charge reversible polymer layer of poly(allylamine)–dimethylmaleic anhydride–polyethylene glycol (PAH–DMMA–PEG). In blood circulation, the obtained MSN@U/DCNPs nanoassemblies with an optimized large size and negatively charged surface are very stable, which is conducive for prolonged blood circulation and enhanced tumor accumulation, while in the acidic tumor microenvironment in the tumor site, the charge reversal of the outermost PAH–DMMA–PEG from negative to positive can trigger the rapid detachment of the polymer from the surface of the nanoassemblies. At the same time, the benzoic-imine bonds between MSN and U/DCNPs are also cleaved which induces the disintegration of the nanoassemblies. The internalization efficiency of the dissociated MSNs and U/DCNPs by tumor cells is enhanced because of the positively charged surface properties. Furthermore, the detached U/DCNPs with a much smaller size can efficiently penetrate into the deep tumor tissue. The transportation and accumulation of the nanoassemblies can be carefully monitored based on the downconversion fluorescence of the U/DCNPs in the second NIR window (NIR-II, 1000–1700 nm), which has attracted widespread research interest in the past few years because of its remarkable merits over the conventional optical imaging in the first NIR window (NIR-I, 650–900 nm).34 On the other hand, when the MSNs and U/DCNPs segments were loaded with chemotherapeutic drugs and photosensitizers respectively, the nanoassemblies can implement NIR bio-imaging guided combined chemo- and photodynamic therapy (PDT). Taking advantage of NIR laser activated therapy35 and the dual transformability of the prepared nanoassemblies, the drug loaded MSN@U/DCNPs are expected to realize remarkable antitumor efficiency.
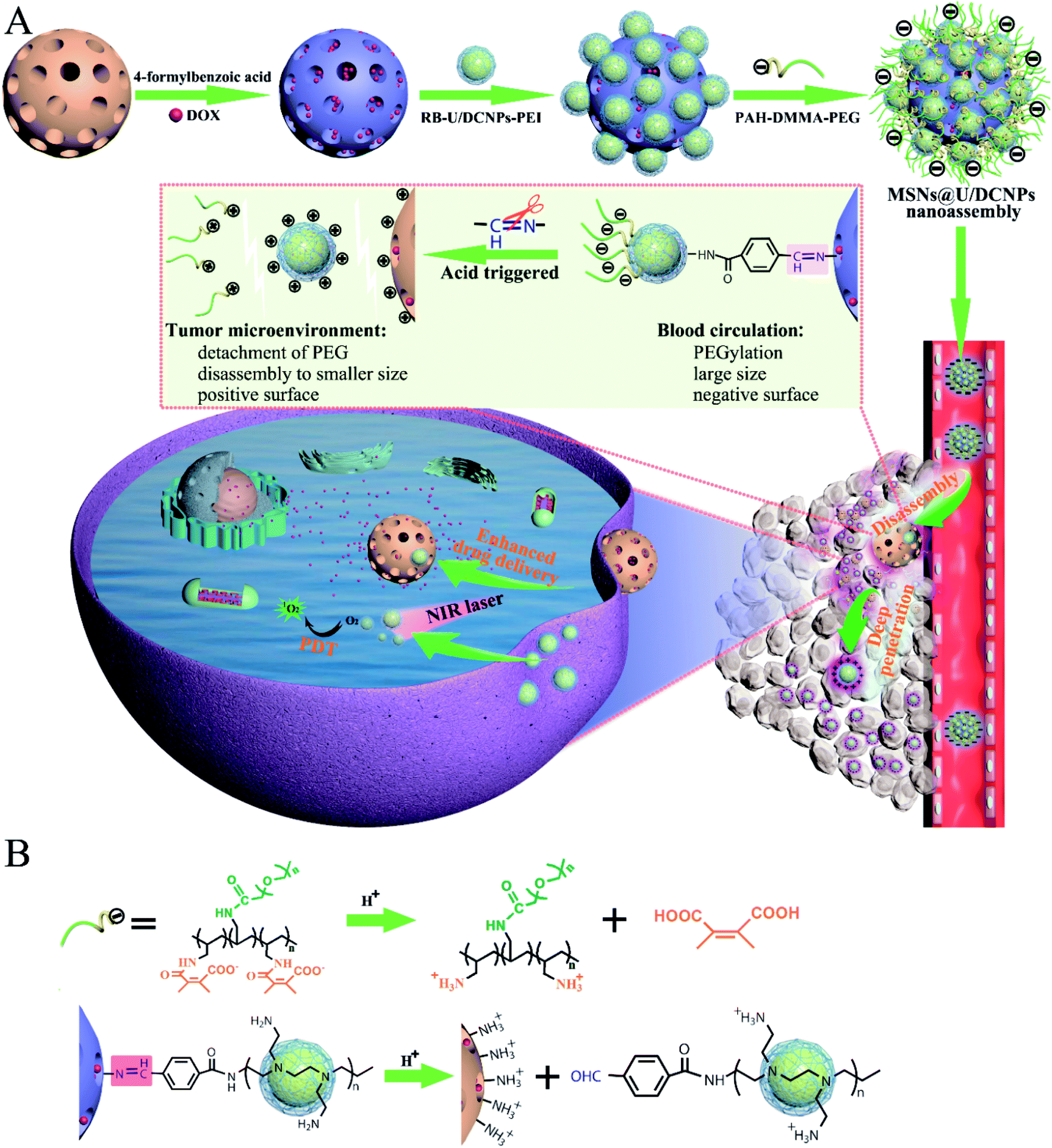 | ||
| Scheme 1 (A) Schematic illustration of the preparation of core@satellite nanoassemblies and the acidic tumor microenvironment triggered size/charge dual-transformability for combined chemo- and photodynamic therapy (PDT). The nanoassemblies maintain a negatively charged surface and PEGylation for prolonged blood circulation and tumor accumulation. They are disassembled in an acidic tumor microenvironment, which further results in size shifting from larger assemblies to smaller segments, and charge reversal from negative to positive for deep tumor penetration and improved cell uptake. (B) The working mechanism for the charge reversal of PAH–DMMA–PEG and breakage of benzoic-imine bonds between PEI modified U/DCNPs and amino-modified MSNs. | ||
Results and discussion
Fabrication and characterization of the nanoassemblies
The MSNs of the core@satellite structured nanoassemblies were synthesized by a bi-phase method36 and exhibit a uniform spherical morphology with a diameter of ∼150 nm (Fig. 1A and S1†). The core–multishell structured NaGdF4:Yb,Er@NaYF4@NaGdF4:Nd@NaYF4 U/DCNPs with a diameter of ∼20 nm (Fig. 1B) were fabricated and employed as satellites around large sized MSNs. In the core–multishell nanostructure of the as-prepared U/DCNPs (Fig. S2 and S3†), Er3+ doped nanoparticles were constructed as a core for the emission of visible upconversion luminescence under 980 nm excitation. The NaGdF4:Nd layer can emit NIR-II downconversion fluorescence under 800 nm excitation.37 To construct the core@satellite structured nanoassemblies, the amino functionalized MSNs were first modified with 4-formylbenzoic acid to form an acid-labile benzoic-imine group. Then cationic PEI modified U/DCNPs (Fig. S4 and S5†) were conjugated onto MSNs using amide groups (Fig. S6†). Finally, the charge reversible polymer PAH–DMMA–PEG (Fig. S7 and S8†) was capped onto the positively charged surface by electrostatic interaction.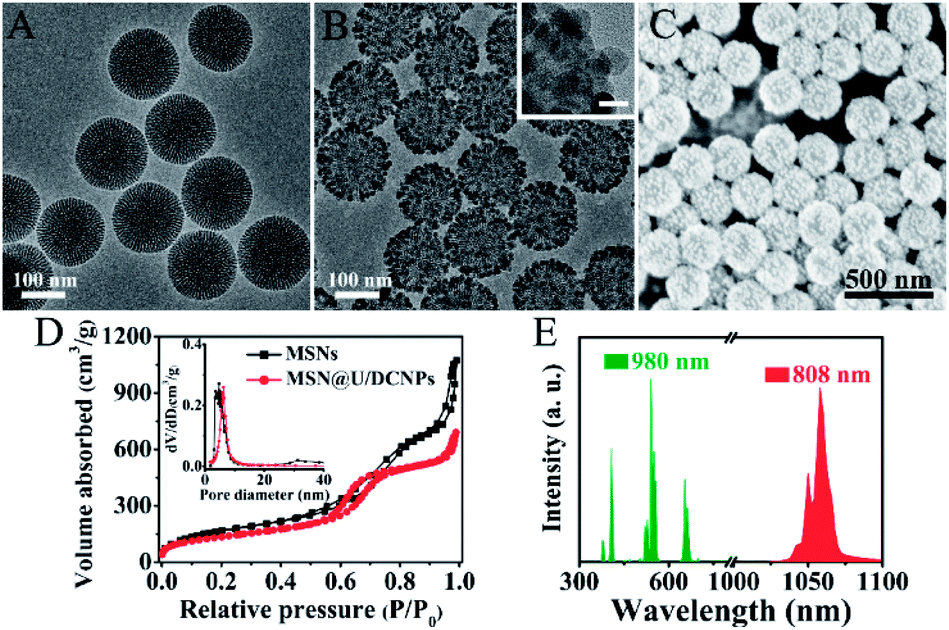 | ||
| Fig. 1 Transmission electron microscopy (TEM) images of (A) bare MSNs and (B) the MSN@U/DCNPs nanoassemblies (the inset shows the high-resolution TEM image of the nanoassemblies, scale bar: 20 nm). (C) Scanning electron microscopy (SEM) image of the prepared MSN@U/DCNPs nanoassemblies. (D) The nitrogen absorption–desorption isotherm of MSNs and MSN@U/DCNPs. (E) The up (green) and down (red) conversion emission spectra of MSN@U/DCNPs nanoassemblies under excitation of 980 and 808 nm lasers. | ||
The resultant MSN@U/DCNPs nanoassemblies display a clear core@satellite structure with a granular surface fully covered by U/DCNPs (Fig. 1B and C). Moreover, the nanoassemblies can also be prepared on smaller MSNs with a diameter of 100 nm (Fig. S9†). The zeta potential of the nanoassemblies also shows a negatively charged surface after capping with PAH–DMMA–PEG (Fig. S10†). N2 adsorption/desorption measurements demonstrate that both pure MSNs and MSN@U/DCNPs nanoassemblies exhibit typical type-IV isotherms with a rapid increase in the adsorption branch at a relative pressure of 0.5–0.8, clearly indicating the presence of uniform mesopores. The Brunauer–Emmett–Teller (BET) surface area of the MSN@U/DCNPs nanoassemblies is calculated to be ∼490 m2 g−1, which is much lower than that of the bare MSNs (730 m2 g−1) (Fig. 1D), confirming the attachment of U/DCNPs and charge reversible PAH–DMMA–PEG on the surface (Fig. S11 and S12†). Taking advantage of the independent dual-mode up and down conversion luminescence of the elaborately designed U/DCNPs, the nanoassemblies can exhibit both visible upconversion emission at 550/665 nm and NIR downconversion luminescence at 1060 nm under 980 and 808 nm excitations (Fig. 1E), respectively, which is crucial for NIR bio-imaging38 and NIR triggered PDT.39
Size and charge transformability of the nanoassemblies
The pH-responsive size and charge dual transformability of the MSN@U/DCNPs nanoassemblies were carefully studied. The core–satellite structure of nanoassemblies was very stable under neutral (pH 7.4) physiological conditions (Fig. 2A, S13 and S14†), while the small U/DCNPs rapidly detached from the MSNs after incubation at pH 6.5 (Fig. 2B). Dynamic light scattering (DLS) measurements show that the hydrodynamic diameter of the nanoassemblies shifts from single distribution at about ∼250 nm at pH 7.4 to dual distribution at ∼305 and ∼65 nm at pH 6.5 (Fig. 2C and D), indicating the disassembly of MSN@U/DCNPs in an acidic microenvironment. Not only the particle size was shifted by the dissociation of the nanoassemblies, the surface charge of the dissociated components can also be reversed. As shown in Fig. 2E, the negative charge of the initial nanoassemblies is maintained very well at pH 7.4, and it gradually shifts to positive after being incubated for several hours at pH 6.5. The shifting of the size and surface charge can be attributed to the hydrolysis of β-carboxylic amide in the outermost coverage of PAH–DMMA–PEG and the fracture of the benzoic-imine linkage between MSN and U/DCNPs under acidic conditions,40 which induce the re-exposure of the abundant positively charged amino groups on the U/DCNP and MSN surface, further resulting in the complete separation of those segments.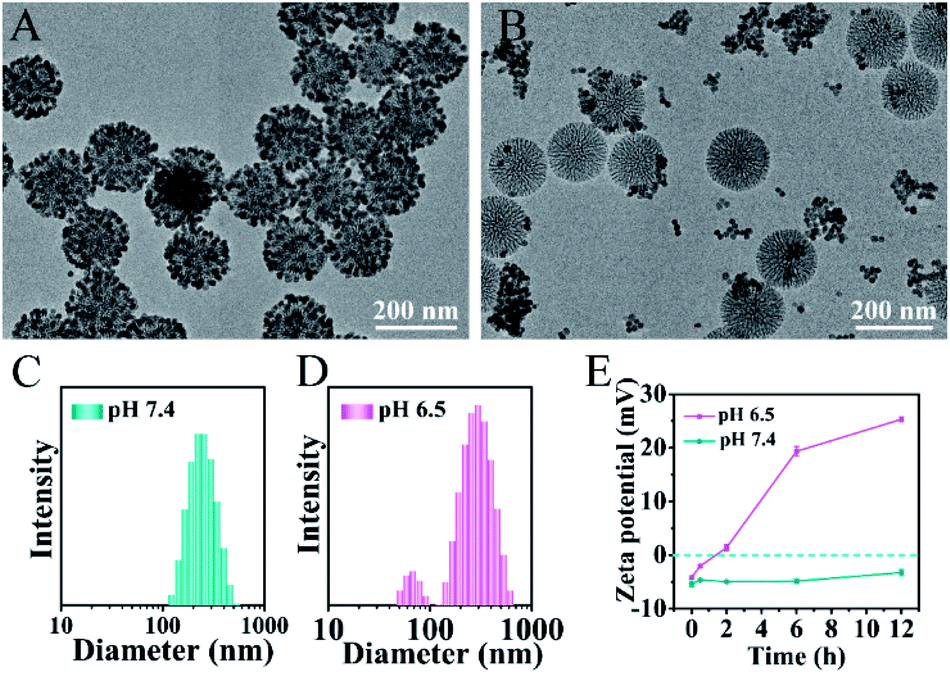 | ||
| Fig. 2 TEM images of the MSN@U/DCNPs nanoassemblies after being incubated at (A) pH 7.4 and (B) 6.5 for 6 h. Size distribution of the MSN@U/DCNPs nanoassemblies at (C) pH 7.4 and (D) 6.5 determined by DLS. (E) Zeta potential variation of the MSN@U/DCNPs nanoassemblies under different pH conditions. | ||
Cellular uptake and tumor penetration of the nanoassemblies
It has been demonstrated that the surface charge of nanoparticles has profound effects on the cell internalization efficiency. To inspect the intracellular uptake of MSN@U/DCNPs at different pH values, murine breast tumor cells (4T1) were incubated with the nanoassemblies at pH 7.4 and 6.5 for 4 h. The green upconversion emission of U/DCNPs can be used to evaluate the cellular uptake efficiency. After 4 h of incubation, the confocal laser scanning microscopy (CLSM) images show that the nanoassemblies can enter the cell under both pH conditions, but the uptake efficiency is distinctly different. At pH 6.5, the bright green fluorescence inside 4T1 cells is much stronger than that in the cells incubated at physiological pH 7.4 (Fig. 3A). Furthermore, quantification analysis was performed by measuring the intracellular Gd concentration with inductively coupled plasma atomic emission spectrometry (ICP-AES). When the incubation concentration of the nanoassemblies is 200 μg mL−1, the intracellular Gd amount is ∼0.58 μg mg−1 after 4 h of incubation at pH 6.5, and can reach as high as ∼1.17 μg mg−1 when the concentration increases to 400 μg mL−1, which is about two times higher than that at pH 7.4 (Fig. 3B). The results clearly demonstrate that the MSN@U/DCNPs exhibit more efficient cell internalization at pH 6.5 compared with that at pH 7.4, which can be attributed to the strong interaction between the negatively charged cell membrane and positively charged U/DCNPs segments after dissociation of the nanoassemblies under acidic conditions. Along with acid triggered charge reversal for improved cellular internalization, the size of the nanoassemblies also decreases dramatically from ∼180 nm of the intact nanoassemblies to ∼20 nm of the U/DCNPs segments, which is crucial for tumor penetration. Multicellular spheroids (MCSs) are versatile three-dimensional models for studying tumor biology due to their similarity in morphology and biological microenvironment to solid tumors. In our study, 4T1 cell derived MCSs were employed as an in vitro model to evaluate the tumor penetration of MSN@U/DCNPs at pH 7.4 and 6.5. After incubation with the nanoassemblies at pH 7.4 for 6 h, the green fluorescence of U/DCNPs appears only at the superficial layer of MCSs (Fig. 3C). In comparison, the green fluorescence is evenly distributed throughout the MCSs when incubated at pH 6.5, implying the homogeneous distribution of U/DCNPs in the whole MCSs under acidic tumor conditions. The Z-stack scanning CLSM results show that the green fluorescence inside the MCSs is clearly observed even at a scanning depth of 150 μm (Fig. 3D), while at the same depth under pH 7.4, it is very weak (Fig. 3E). The quantitative intensity of line scanning further confirms that the green fluorescence in the deep region under acidic conditions is as strong as that at the superficial layer of 4T1 MCSs (Fig. 3F). The results demonstrate that the deep penetration and uniform distribution of U/DCNPs in the deep area of MCSs under acidic conditions are because of the dissociation of the nanoassemblies.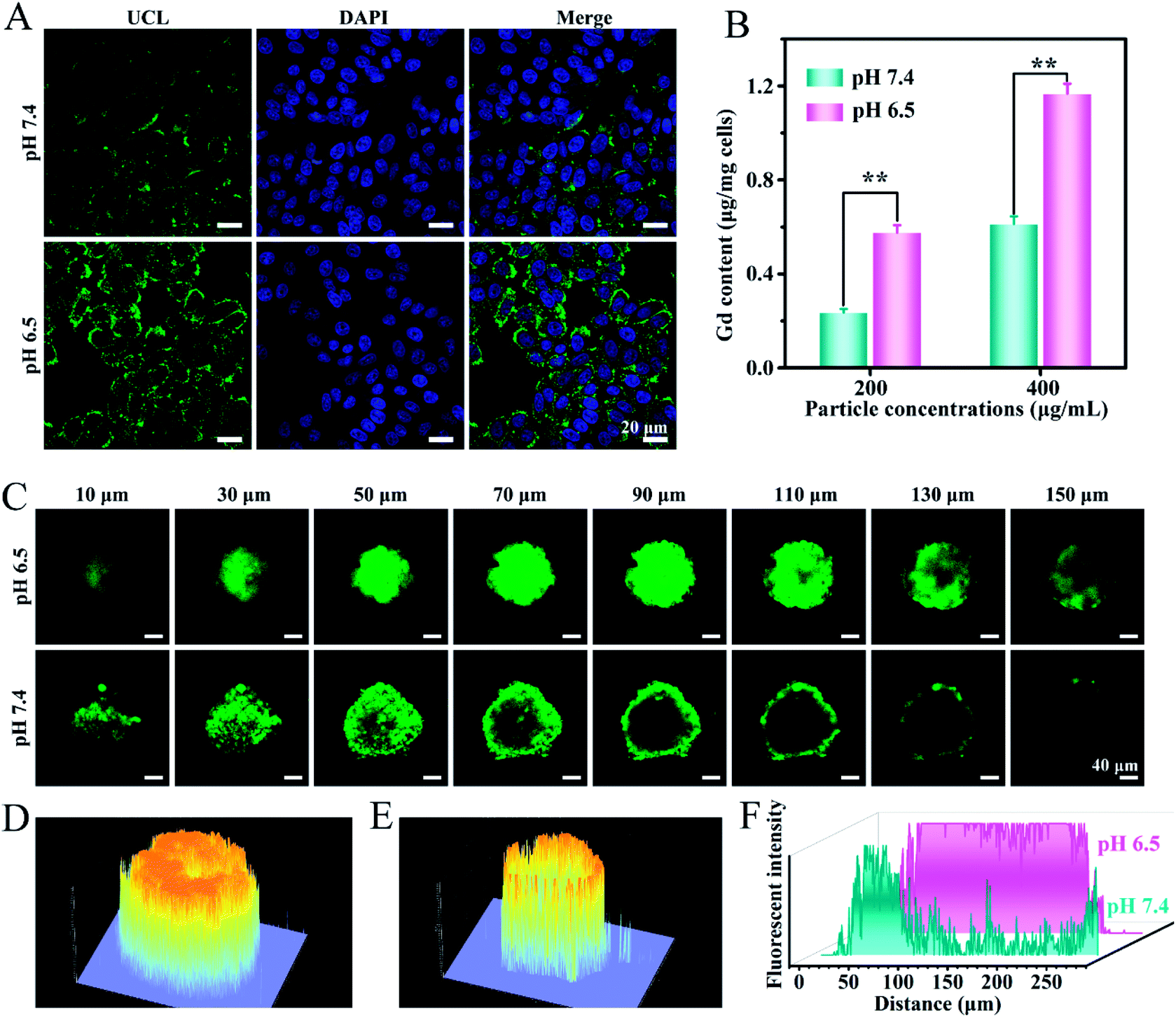 | ||
| Fig. 3 (A) CLSM images of 4T1 cells treated with MSN@U/DCNPs under different pH conditions. (B) The intracellular Gd amount of 4T1 cells exposed to different concentrations of MSN@U/DCNPs at pH 6.5 and 7.4. (C) Tumor penetration of MSN@U/DCNPs in 4T1 MCSs at pH 6.5 and 7.4 observed by Z-stack CLSM. (D, E) 2.5D fluorescence and (F) line intensity analysis of the CLSM images at a 90 μm depth. | ||
The robust deep tumor penetration and efficient cancer cell internalization of nanoparticles in an acidic tumor microenvironment endow the nanoassemblies with a much higher opportunity to access more cancer cells and increase cell-killing efficacy. Therefore, combined with the advantages of the high surface area of the MSN core and unique NIR to visible upconversion luminescence of U/DCNPs satellites, the chemotherapeutic drug (doxorubicin hydrochloride, DOX) and photosensitizer (rose bengal, RB) were respectively introduced into the MSN and U/DCNPs parts of the nanoassemblies (denoted as DOX–MSN@U/DCNPs and MSN@U/DCNPs–RB). The in vitro DOX release from MSN@U/DCNPs nanoassemblies was investigated first. Because of the attachment of U/DCNPs and the charge-reversible polymer on the MSN surface, the mesoporous channels of MSNs are partially blocked,41 which further results in an as low as 7.9% cumulative DOX release ratio within the first 4 h under physiological conditions. In stark contrast, the drug release at acidic pH is much faster than that at physiological pH (Fig. S15†). At pH 5.0 and 6.5, the cumulative DOX release ratio reached as high as 40.87% and 26.72% within the first 4 h, respectively. The dissociation of MSN@U/DCNP nanoassemblies under acidic conditions is also expected to influence the intracellular DOX delivery efficiency. 4T1 cells were incubated with DOX–MSN@U/DCNPs for 4 h at different pH values. Obviously, the red fluorescence of DOX in the cells at pH 6.5 is much brighter than that at pH 7.4 (Fig. S16†), implying that more drug molecules are transported into 4T1 cells under acidic conditions. The improved DOX concentration in cancer cells is mainly resulted from the positive surface charge of MSNs after disintegration of nanoassemblies, which could significantly enhance the interaction between the MSNs and negatively charged cell membrane.42
In vitro therapeutic effect of the nanoassemblies
The absorption band of RB is largely consistent with the upconversion emission of U/DCNPs (Fig. S17†), ensuring the NIR-activated photodynamic effect through Förster resonance energy transfer (FRET) or LRET.43 Since the acid-triggered disassembly of MSN@U/DCNPs can also improve the internalization of detached positively charged small U/DCNPs as mentioned above, the intracellular generation of reactive oxygen species (ROS) is evaluated. 1,3-Diphenylisobenzofuran (DPBF) was used as a probe to detect the generation of ROS from the MSN@U/DCNPs–RB nanoassemblies under NIR light irradiation. The maximum absorbance (∼420 nm) of DPBF is clearly decreased upon exposure to 980 nm irradiation, and over 60% of DPBF is degraded by the generated ROS after irradiation for 0.5 h (Fig. S18†). The intracellular ROS are also detected by the 2,7-dichlorodihydrofluorescein (DCFH-DA) probe at different pH values. Stronger green fluorescence is observed for the cells treated with NIR-activated PDT at pH 6.5 than that at pH 7.4 (Fig. 4A), which is consistent with the quantitative analysis results obtained by flow cytometry (Fig. 4B, C and S19†). Meanwhile, both the control group and the laser irradiation treated group display negligible green fluorescence, demonstrating that the ROS in cancer cells is generated by the NIR irradiation-activated photosensitizers. Thus, we can conclude that the disintegration of MSN@U/DCNPs–RB also improves the intracellular ROS generation ability under NIR irradiation.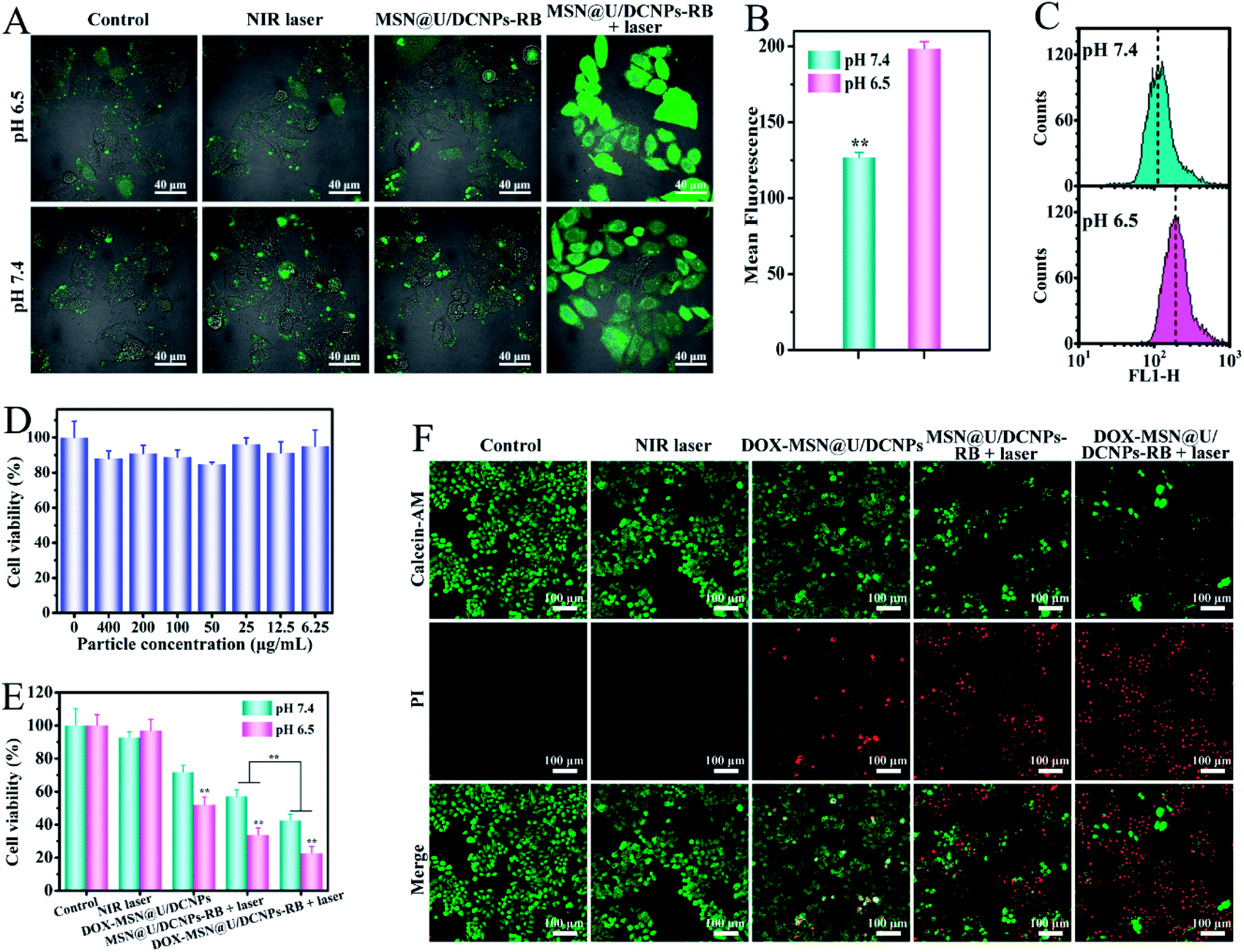 | ||
| Fig. 4 (A) CLSM images of DCFH-DA stained 4T1 cells after different treatments with a NIR laser, MSN@U/DCNPs–RB, and MSN@U/DCNPs–RB + NIR laser at different pH values. (B) Mean fluorescence intensity of 4T1 cells treated with MSN@U/DCNPs–RB + NIR laser at different pH values. (C) Quantitative analysis of the intensity of the green fluorescence of the cells by flow cytometry. (D) Viability of 4T1 cells incubated with bare MSN@U/DCNPs at different concentrations for 24 h. (E) Viability of 4T1 cells subjected to different treatments at different pH values. (F) The corresponding fluorescence images of live–dead stained 4T1 cells after different treatments at pH 6.5. | ||
Prior to studying therapeutic efficiency of drug loaded MSN@U/DCNPs, the cytotoxicity of pure nanoassemblies was first studied by using 4T1 cells and mouse macrophage cells (RAW 264.7). The results manifest that the nanoassemblies exhibit no obvious cytotoxicity, and over 80% of the cells were alive even when exposed to a high concentration of 400 μg mL−1 (Fig. 4D and S20†). Additionally, the MSN@U/DCNPs cause no damage to the cell skeleton of 4T1 and RAW 264.7 cells (Fig. S21†), implying the excellent biocompatibility of the as-prepared nanoassemblies. Then, as a proof of concept, 4T1 cells were selected to evaluate the cancer cell killing efficiency of DOX and RB co-loaded nanoassemblies (denoted as DOX–MSN@U/DCNPs–RB). In a normal physiological (pH 7.4) and acidic tumor (pH 6.5) microenvironment, 4T1 cells were subjected to different treatments with a NIR laser, DOX–MSN@U/DCNPs, MSN@U/DCNPs–RB + laser and DOX–MSN@U/DCNPs–RB + laser. The results show that the combined therapy of PDT and chemotherapy at pH 6.5 exhibits the highest cancer cell killing efficiency (∼78%) among all groups (Fig. 4E). In comparison, the mortality rate of mere chemotherapy or PDT is only ∼49% and ∼64%, respectively. Live–dead staining was also carried out to visualize the therapeutic efficiency of each group. The red spots that represent dead cells in the group of combined therapy are apparently more than those in mere chemotherapy or PDT groups (Fig. 4F). In addition, it is noticeable that the cancer cell killing efficiency at pH 6.5 is significantly higher than that at pH 7.4 in all groups (Fig. 4E), which can be attributed to the enhanced intracellular uptake and delivery efficiency of the drug loaded nanoassemblies due to the acid triggered size and charge dual transformability.
In vivo pharmacokinetics and therapy of the nanoassemblies
For in vivo assessment, a 4T1 xenografted tumor model was established on BALB/c mice to investigate the in vivo bio-distribution and tumor accumulation of the nanoassemblies based on the Nd3+ dominated down-conversion NIR-II fluorescence of the U/DCNPs at 1050 nm under 808 nm excitation. The prepared MSN@U/DCNPs display good colloid stability in various biological media (Fig. S23†). After 2 h of intravenously injecting ∼180 nm nanoassemblies with a negatively charged surface, the NIR II fluorescence signals at the tumor site can be easily detected and are much brighter than that of the free U/DCNPs injection group (Fig. 5A). It is noted that in order to avoid the interference from the signal of the non-interested area, a screener with only the tumor area exposed was used. The strongest NIR-II fluorescence signal at the tumor site is detected at 6 h post-injection, about twice that of the free U/DCNPs injection group at the same time point (Fig. 5B). The strong NIR-II signal for the nanoassembly group can last over 24 h, suggesting the effective tumor retention of the nanoassemblies. The biological distribution of the nanoassemblies and free U/DCNPs in different tissues of tumor-bearing mice was determined by ICP-AES. The results show that most of the nanoparticles, in both the nanoassemblies and free U/DCNPs groups, are accumulated in the liver and spleen. However, the accumulation amount of nanoassemblies in the liver and spleen is obviously lower than that of the free U/DCNPs group (Fig. 5C). On the other hand, the retention rate of nanoassemblies at the tumor site is about 6.94 ± 1.26% ID per g, which is about 1.8 times higher than that of free U/DCNPs (3.72 ± 0.63%). We consider that the effective tumor retention of the nanoassemblies mainly results from several aspects: (i) the negatively charged surface and PEGylation of the nanoassemblies can ensure prolonged blood circulation (Fig. 5D);44 (ii) the larger initial size of the nanoassemblies enhance their EPR effect; (iii) the small size of the dissociated U/DCNPs can promote penetration into deep regions of the tumor triggered by the acidic microenvironment;45 (iv) the positively charged surface of the dissociated U/DCNPs induces efficient tumor cell internalization.46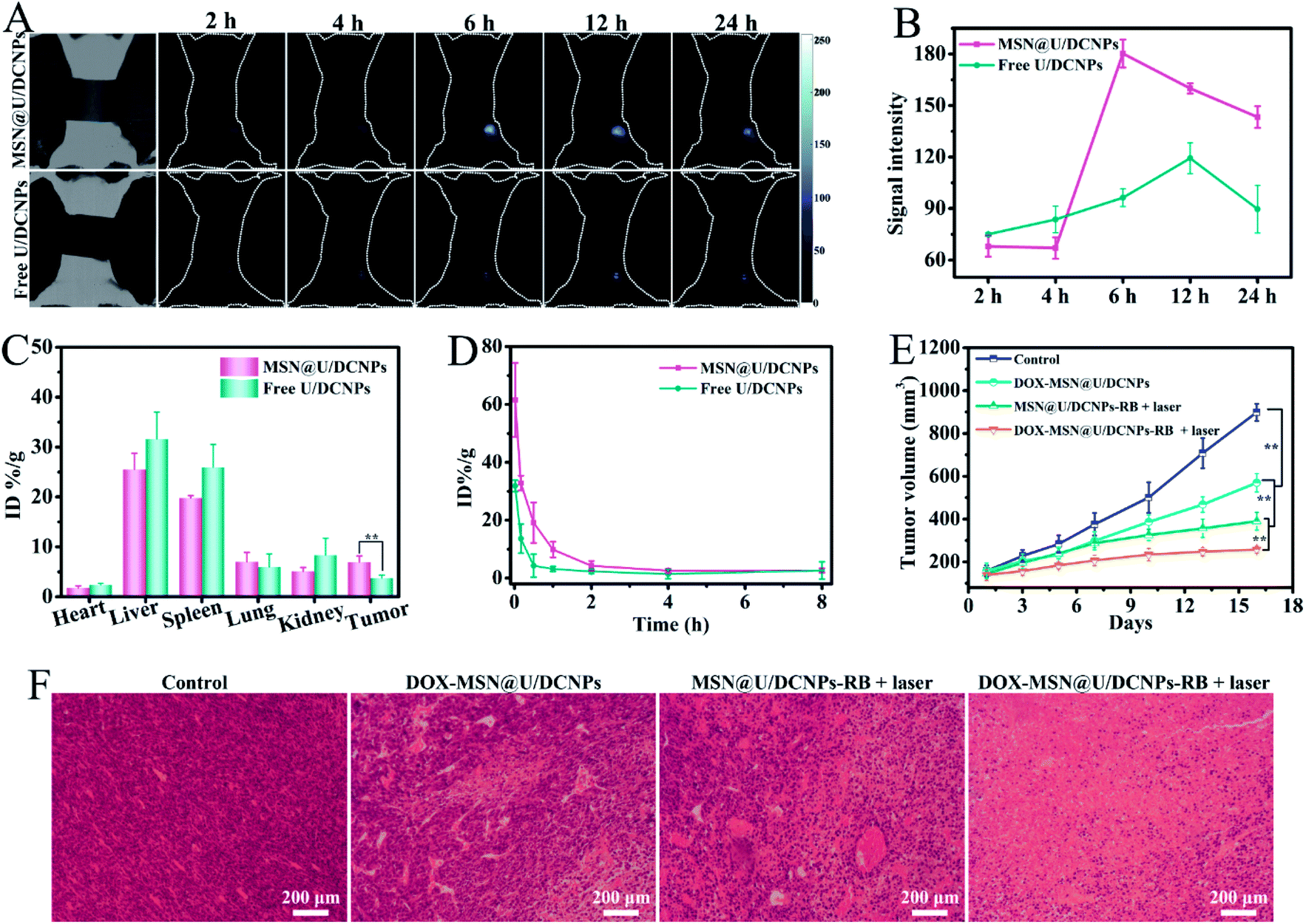 | ||
| Fig. 5 (A) The NIR fluorescence images of subcutaneous 4T1 tumor-bearing mice after intravenous injection of MSN@U/DCNPs and free U/DCNPs at predetermined time points. (B) The quantitative analysis of the fluorescence signal of the images in (A). (C) The bio-distribution of Gd in the main organs and tumor at 24 h post-injection of MSN@U/DCNPs and free U/DCNPs. (D) Time-dependent relative contents of the Gd element in blood after the injection of MSN@U/DCNPs and free U/DCNPs. (E) Tumor volume change in the experimented mice under different treatments and (F) H&E staining sections of the extracted tumor in different groups after treatments. | ||
The in vivo antitumor efficiency was also evaluated by using 4T1 tumor-bearing mice. The tumor growth was effectively suppressed upon treatment with DOX and RB co-loaded nanoassemblies under NIR laser irradiation. Owing to the negative charge and larger size induced high tumor accumulation efficiency, small size can promote deep tumor penetration, positive charge induces efficient cancer cell internalization as well as combined chemotherapy and PDT, the therapeutic effect is significantly improved compared with other groups (Fig. 5E and S25†). These results are consistent with that of the hematoxylin & eosin (H&E) staining analysis (Fig. 5F). No remarkable tissue damage or any other side effect is observed on the heart, liver, spleen, lung, kidney, and weight fluctuations (Fig. S26 and S27†), indicating excellent biocompatibility and negligible toxicity of these smart tumor microenvironment triggered size and charge dual transformable nanoassemblies.
Conclusions
In summary, we report core@satellite structured MSN@U/DCNPs nanoassemblies with a negatively charged surface, which can responsively disintegrate into positively charged MSNs and small-sized U/DCNPs in an acidic tumor microenvironment. Due to their optimized relatively large size (∼180 nm) and negatively charged surface, the nanoassemblies can effectively provide a high tumor accumulation efficiency of 6.94% (∼1.8 times higher than that of the control group). After size and charge dual transformation under acidic tumor microenvironment, the tumor penetration and intracellular uptake can be greatly enhanced because of the small size and positively charged surface of the dissociated segments. Combined with the unique NIR-to-NIR and NIR-to-UV/vis down/upconversion luminescence of U/DCNPs satellites and the high surface area of the MSN core, the nanoassemblies can realize NIR-II imaging guided chemo- and photo-dynamic combined therapy and exhibit remarkable antitumor efficiency. The smart delivery and NIR bioimaging guided combined therapy of these tumor microenvironment triggered size & charge dual-transformable nanoassemblies exhibit great potential toward a novel platform for next generation nano-theranostics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21875043, 21701027, 21733003 and 51961145403), Natural Science Foundation of Shanghai (18ZR1404600), National Key R&D Program of China (2018YFA0209401 and 2017YFA0207303), Key Basic Research Program of Science and Technology Commission of Shanghai Municipality (17JC1400100), China Postdoctoral Science Foundation (Grant No. 2018M641911), and Australia Research Council (ARC) (DP160104089). All the animal experiments were approved by the Shanghai Science and Technology Committee (SYXK2014-0029, Shanghai, China) and performed in agreement with the guidelines of the Department of Laboratory Animal Science, Fudan University.References
- J. J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- H. Maeda, Adv. Drug Delivery Rev., 2015, 91, 3–6 CrossRef CAS PubMed.
- P. P. Yang, S. L. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679–3698 RSC.
- Q. H. Sun, Z. X. Zhou, N. S. Qiu and Y. Q. Shen, Adv. Mater., 2017, 29, 1606628 CrossRef.
- Q. Zhou, S. Q. Shao, J. Q. Wang, C. H. Xu, J. J. Xiang, Y. Piao, Z. X. Zhou, Q. S. Yu, J. B. Tang, X. R. Liu, Z. H. Gan, R. Mo, Z. Gu and Y. Q. Shen, Nat. Nanotechnol., 2019, 14, 799 CrossRef CAS PubMed.
- E. Tasciotti, X. Liu, R. Bhavane, K. Plant, A. D. Leonard, B. K. Price, M. M.-C. Cheng, P. Decuzzi, J. M. Tour and F. Robertson, Nat. Nanotechnol., 2008, 3, 151–157 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- H. Meng, W. Leong, K. W. Leong, C. Y. Chen and Y. L. Zhao, Biomaterials, 2018, 174, 41–53 CrossRef CAS PubMed.
- L. Zhu, T. Wang, F. Perche, A. Taigind and V. P. Torchilin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17047–17052 CrossRef CAS.
- O. Veiseh, B. C. Tang, K. A. Whitehead, D. G. Anderson and R. Langer, Nat. Rev. Drug Discovery, 2015, 14, 45–57 CrossRef CAS PubMed.
- J. Q. Wang, W. W. Mao, L. L. Lock, J. B. Tang, M. H. Sui, W. L. Sun, H. G. Cui, D. Xu and Y. Q. Shen, ACS Nano, 2015, 9, 7195–7206 CrossRef CAS PubMed.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS PubMed.
- H. X. Wang, Z. Q. Zuo, J. Z. Du, Y. C. Wang, R. Sun, Z. T. Cao, X. D. Ye, J. L. Wang, K. W. Leong and J. Wang, Nano Today, 2016, 11, 133–144 CrossRef CAS.
- J. Tan, H. Li, X. X. Hu, R. Abdullah, S. T. Xie, L. L. Zhang, M. M. Zhao, Q. Luo, Y. Z. Li, Z. J. Sun, Q. Yuan and W. H. Tan, Chem, 2019, 5, 1775–1792 CAS.
- Y. Li, H. Y. Yang, T. Thambi, J. H. Park and D. S. Lee, Biomaterials, 2019, 217, 119299 CrossRef CAS PubMed.
- Z. Y. Wang, Y. M. Ju, Z. Ali, H. Yin, F. G. Sheng, J. Lin, B. D. Wang and Y. L. Hou, Nat. Commun., 2019, 10, 4418 CrossRef PubMed.
- P. F. Zhang, J. Q. Wang, H. Chen, L. Zhao, B. B. Chen, C. C. Chu, H. Liu, Z. N. Qin, J. Y. Liu, Y. Z. Tan, X. Y. Chen and G. Liu, J. Am. Chem. Soc., 2018, 140, 14980–14989 CrossRef PubMed.
- C. D. Ji, Q. Gao, X. H. Dong, W. Y. Yin, Z. J. Gu, Z. H. Gan, Y. L. Zhao and M. Z. Yin, Angew. Chem., Int. Ed., 2018, 57, 11384–11388 CrossRef CAS PubMed.
- H. J. Li, J. Z. Du, X. J. Du, C. F. Xu, C. Y. Sun, H. X. Wang, Z. T. Cao, X. Z. Yang, Y. H. Zhu, S. M. Nie and J. Wang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4164–4169 CrossRef CAS PubMed.
- T. L. Li, J. Zhou, C. L. Zhang, X. Zhi, J. Q. Niu, H. L. Fu, J. Song and D. X. Cui, NPG Asia Mater., 2018, 10, 1046–1060 CrossRef CAS.
- Q. Jin, Y. Y. Deng, X. H. Chen and J. Ji, ACS Nano, 2019, 13, 954–977 CAS.
- T. T. Wang, D. G. Wang, J. P. Liu, B. Feng, F. Y. Zhou, H. W. Zhang, L. Zhou, Q. Yin, Z. W. Zhang, Z. L. Cao, H. J. Yu and Y. P. Li, Nano Lett., 2017, 17, 5429–5436 CrossRef CAS PubMed.
- N. S. Qiu, X. R. Liu, Y. Zhong, Z. X. Zhou, Y. Piao, L. Miao, Q. Z. Zhang, J. B. Tang, L. Huang and Y. Q. Shen, Adv. Mater., 2016, 28, 10613 CrossRef CAS PubMed.
- C. Hu, X. L. Cun, S. B. Ruan, R. Liu, W. Xiao, X. T. Yang, Y. Y. Yang, C. Y. Yang and H. L. Gao, Biomaterials, 2018, 168, 64–75 CrossRef CAS PubMed.
- D. D. Li, Y. C. Ma, J. Z. Du, W. Tao, X. J. Du, X. Z. Yang and J. Wang, Nano Lett., 2017, 17, 2871–2878 CrossRef CAS PubMed.
- J. X. Wang, S. Shen, D. D. Li, C. Y. Zhan, Y. Y. Yuan and X. Z. Yang, Adv. Funct. Mater., 2018, 28, 1704806 CrossRef.
- J. J. Li, W. D. Ke, H. Li, Z. S. Zha, Y. Han and Z. S. Ge, Adv. Healthcare Mater., 2015, 4, 2206–2219 CrossRef CAS PubMed.
- J. J. Li, K. Wei, S. Zuo, Y. X. Xu, Z. S. Zha, W. D. Ke, H. B. Chen and Z. S. Ge, Adv. Funct. Mater., 2017, 27, 1702108 CrossRef.
- Q. H. Sun, X. R. Sun, X. P. Ma, Z. X. Zhou, E. L. Jin, B. Zhang, Y. Q. Shen, E. A. Van Kirk, W. J. Murdoch, J. R. Lott, T. P. Lodge, M. Radosz and Y. L. Zhao, Adv. Mater., 2014, 26, 7615–7621 CrossRef CAS PubMed.
- G. B. Yang, S. Z. F. Phua, W. Q. Lim, R. Zhang, L. Z. Feng, G. F. Liu, H. W. Wu, A. K. Bindra, D. Jana, Z. Liu and Y. L. Zhao, Adv. Mater., 2019, 31, 1901513 CrossRef PubMed.
- J. J. Chen, J. X. Ding, Y. C. Wang, J. J. Cheng, S. X. Ji, X. L. Zhuang and X. S. Chen, Adv. Mater., 2017, 29, 1701170 CrossRef PubMed.
- L. L. Dai, X. Li, X. L. Duan, M. H. Li, P. Y. Niu, H. Y. Xu, K. Y. Cai and H. Yang, Adv. Sci., 2019, 6, 1801807 CrossRef PubMed.
- X. D. Xue, Y. Huang, R. N. Bo, B. Jia, H. Wu, Y. Yuan, Z. L. Wang, Z. Ma, D. Jing, X. B. Xu, W. M. Yu, T. Y. Lin and Y. P. Li, Nat. Commun., 2018, 9, 3653 CrossRef PubMed.
- J. T. Xu, A. Gulzar, P. P. Yang, H. T. Bi, D. Yang, S. L. Gai, F. He, J. Lin, B. G. Xing and D. Y. Jin, Coord. Chem. Rev., 2019, 381, 104–134 CrossRef CAS.
- J. T. Xu, W. Han, P. P. Yang, T. Jia, S. M. Dong, H. T. Bi, A. Gulzar, D. Yang, S. L. Gai, F. He, J. Lin and C. X. Li, Adv. Funct. Mater., 2018, 28, 1803804 CrossRef.
- D. K. Shen, J. P. Yang, X. M. Li, L. Zhou, R. Y. Zhang, W. Li, L. Chen, R. Wang, F. Zhang and D. Y. Zhao, Nano Lett., 2014, 14, 923–932 CrossRef CAS PubMed.
- T. C. Zhao, P. Y. Wang, Q. Li, A. A. Al-Khalaf, W. N. Hozzein, F. Zhang, X. M. Li and D. Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 2611–2615 CrossRef CAS PubMed.
- M. Y. Zhao, B. H. Li, P. Y. Wang, L. F. Lu, Z. C. Zhang, L. Liu, S. F. Wang, D. D. Li, R. Wang and F. Zhang, Adv. Mater., 2018, 30, 1804982 CrossRef PubMed.
- F. Y. Li, Y. Du, J. A. Liu, H. Sun, J. Wang, R. Q. Li, D. Kim, T. Hyeon and D. S. Ling, Adv. Mater., 2018, 30, 1802808 CrossRef PubMed.
- X. Guo, X. Wei, Y. T. Jing and S. B. Zhou, Adv. Mater., 2015, 27, 6450–6456 CrossRef CAS PubMed.
- Q. Lei, S. B. Wang, J. J. Hu, Y. X. Lin, C. H. Zhu, L. Rong and X. Z. Zhang, ACS Nano, 2017, 11, 7201–7214 CrossRef CAS PubMed.
- T. Feng, X. Z. Ai, G. H. An, P. P. Yang and Y. L. Zhao, ACS Nano, 2016, 10, 4410–4420 CrossRef CAS PubMed.
- B. B. Ding, S. Shao, C. Yu, B. Teng, M. F. Wang, Z. Y. Cheng, K. L. Wong, P. A. Ma and J. Lin, Adv. Mater., 2018, 30, 1802479 CrossRef PubMed.
- X. X. Han, Y. Y. Li, Y. Xu, X. Zhao, Y. L. Zhang, X. Yang, Y. W. Wang, R. F. Zhao, G. J. Anderson, Y. L. Zhao and G. J. Nie, Nat. Commun., 2018, 9, 3390 CrossRef PubMed.
- L. L. Dai, K. Li, M. H. Li, X. J. Zhao, Z. Luo, L. Lu, Y. F. Luo and K. Y. Cai, Adv. Funct. Mater., 2018, 28, 1707249 CrossRef.
- S. Wang, G. C. Yu, Z. T. Wang, O. Jacobson, R. Tian, L. S. Lin, F. W. Zhang, J. Wang and X. Y. Chen, Adv. Mater., 2018, 30, 1803926 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06260b |
| This journal is © The Royal Society of Chemistry 2020 |