
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the electronic and mechanistic roles of the μ4-sulfur atom in a synthetic CuZ model system†
Suresh C.
Rathnayaka
a,
Shahidul M.
Islam
a,
Ida M.
DiMucci
b,
Samantha N.
MacMillan
 b,
Kyle M.
Lancaster
*b and
Neal P.
Mankad
*a
b,
Kyle M.
Lancaster
*b and
Neal P.
Mankad
*a
aDepartment of Chemistry, University of Illinois at Chicago, 845 W. Taylor St., Chicago, IL 60607, USA. E-mail: npm@uic.edu
bDepartment of Chemistry & Chemical Biology, Cornell University, Baker Laboratory, Ithaca, NY 14853, USA. E-mail: kml236@cornell.edu
First published on 17th February 2020
Abstract
Nitrous oxide (N2O) contributes significantly to ozone layer depletion and is a potent greenhouse agent, motivating interest in the chemical details of biological N2O fixation by nitrous oxide reductase (N2OR) during bacterial denitrification. In this study, we report a combined experimental/computational study of a synthetic [4Cu:1S] cluster supported by N-donor ligands that can be considered the closest structural and functional mimic of the CuZ catalytic site in N2OR reported to date. Quantitative N2 measurements during synthetic N2O reduction were used to determine reaction stoichiometry, which in turn was used as the basis for density functional theory (DFT) modeling of hypothetical reaction intermediates. The mechanism for N2O reduction emerging from this computational modeling involves cooperative activation of N2O across a Cu/S cluster edge. Direct interaction of the μ4-S ligand with the N2O substrate during coordination and N–O bond cleavage represents an unconventional mechanistic paradigm to be considered for the chemistry of CuZ and related metal–sulfur clusters. Consistent with hypothetical participation of the μ4-S unit in two-electron reduction of N2O, Cu K-edge and S K-edge X-ray absorption spectroscopy (XAS) reveal a high degree of participation by the μ4-S in redox changes, with approximately 21% S 3p contribution to the redox-active molecular orbital in the highly covalent [4Cu:1S] core, compared to approximately 14% Cu 3d contribution per copper. The XAS data included in this study represent the first spectroscopic interrogation of multiple redox levels of a [4Cu:1S] cluster and show high fidelity to the biological CuZ site.
Introduction
Metal–sulfide clusters represent a common motif in bioinorganic chemistry. The most studied examples are iron–sulfur clusters (e.g. [2Fe:2S], [4Fe:4S], etc.) that serve as ubiquitous electron transfer sites in a wide range of metalloproteins.1 Other scenarios such as the [NiFe] and [MoCu] catalytic sites of carbon monoxide dehydrogenases (CODHs),2,3 the H-cluster found in [FeFe] hydrogenases, and the [FeMo]-cofactor of nitrogenases4 involve multinuclear metal–sulfide clusters facilitating multielectron/multiproton catalytic transformations. Typically, the bridging sulfido (S2−) ligands in these clusters are thought to be crucial for electronically coupling the transition metal sites, thereby facilitating electron delocalization and lowering barriers towards electron transfer either to/from a catalytic site or along an electron transport chain. However, only in rare cases are the sulfur centers proposed to play a direct rather than spectator role with regard to bond activation and/or bond formation. In the case of the [FeMo]-cofactor of nitrogenase, various hypotheses have been put forward in which sulfide ligands might act as redox-active proton relays5 or even that sulfide(s) may serve in a gating mechanism to nitrogenase activity.6 In the case of the [MoCu] catalytic site of aerobic CODH, one mechanistic hypothesis based on crystallographic studies with substrate analogues involves the μ2-sulfide ligand actively participating in CO activation via transient S–C bond formation.2During bacterial denitrification, nitrous oxide (N2O) is converted to N2 + H2O in a 2e−/2H+ reaction catalyzed by the metalloenzyme, nitrous oxide reductase (N2OR).7 The catalytic site of N2OR is a tetranuclear copper–sulfur cluster, CuZ, which has been structurally characterized in both [4Cu:1S] and [4Cu:2S] forms.8,9 Both forms show N2O reductase activity to some extent, and both require physiological reduction to their most reduced redox states to activate N2O: the 4CuI (“fully reduced”) state for the [4Cu:1S] cluster and the 3CuI:1CuII (“1-hole”) state for the [4Cu:2S] cluster.10 For the [4Cu:1S] cluster, Solomon has proposed N2O binding across a dicopper(I) cluster edge, with the N2O molecule occupying a μ-1,3 binding mode, based on computational modeling (Fig. 1a).11 For the [4Cu:2S] form, Einsle has reported crystallographic data on N2O-pressurized crystals of N2OR showing a N2O molecule within van der Waals contact of CuZ, but the N2O molecule was not found within coordination distance of CuZ and had not undergone significant activation (Fig. 1b).9 In neither case has experimental data emerged to probe the nature of N2O activation by the copper–sulfur clusters.
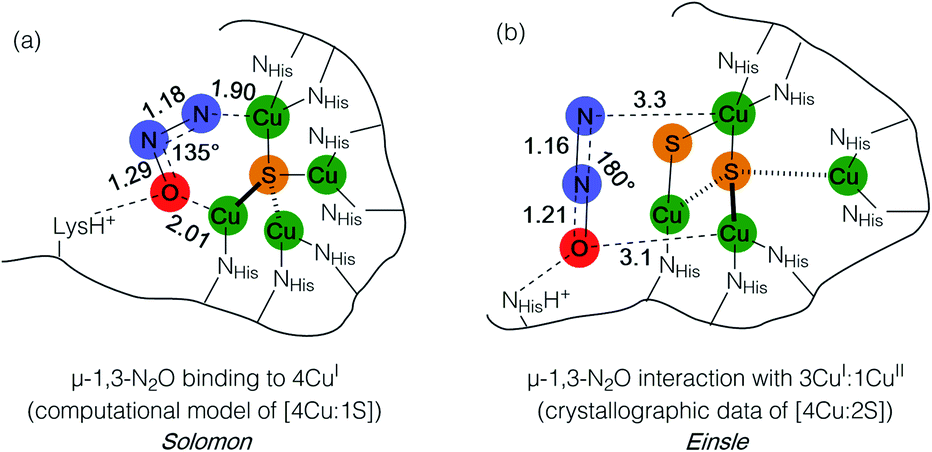 | ||
| Fig. 1 N2O interactions with the CuZ site of nitrous oxide reductase proposed by (a) Solomon and (b) Einsle. Distances are given in Å. | ||
Studying synthetic model systems can aid understanding of how these unusual inorganic copper–sulfur functional groups behave,12 which is particularly crucial knowledge in the context of N2O's significant impact as a greenhouse gas and an ozone layer depleting agent.13,14 Among the synthetic copper compounds and materials known to activate N2O,15–18 one of our groups has reported the only examples of N2O activation by copper sulfide clusters. In one case, a dicuprous [Cu2S] cluster with an unsupported μ2-sulfide bridge19 was found to reduce multiple N2O equivalents to N2, resulting in exhaustive oxidation of the sulfur center to a μ2-sulfate ligand (Scheme 1a).20 Here, the copper centers remained redox inactive while the μ2-sulfide ligand was not only the redox-active center but also acted as an oxygen atom acceptor. In another case, a phosphine-supported tetranuclear [Cu4S] cluster in its 4CuI state showed reactivity towards N2O reduction,21 but the cluster lost structural integrity during the reaction, losing the sulfur atom to unknown products in the reaction medium and thus limiting insight that can be gained about its role. Finally, a formamidinate-supported [Cu4S] cluster in its formally 3CuI:1CuII ([4Cu:1S]1−) state was found to reduce 15N2O to 15N2 (Scheme 1b).22,23 Here the μ4-sulfide bridge remained intact during a formal oxidation to the 2CuI:2CuII ([4Cu:1S]0) redox state of the cluster, allowing us to establish a closed cycle for N2O reduction. Based on these results, the potential role (or lack thereof) of the bridging sulfide ligand in copper–sulfur clusters merits further investigation.
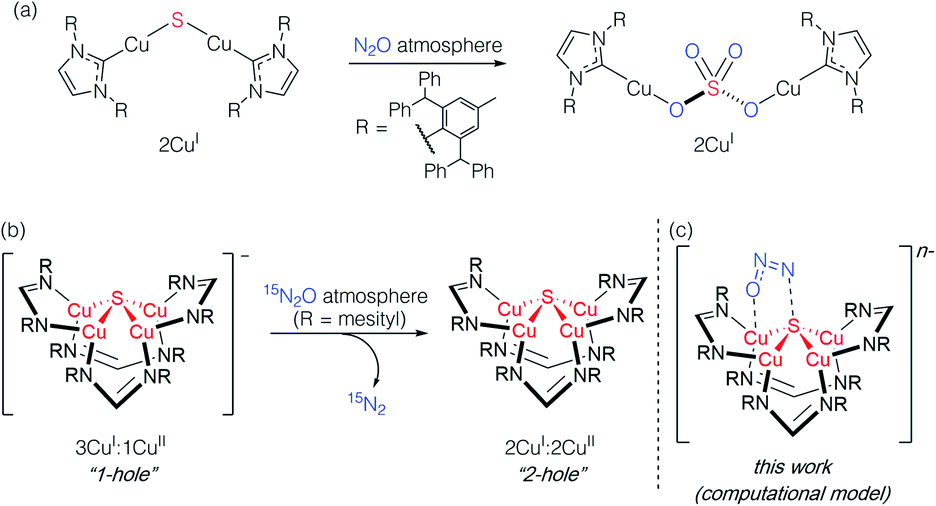 | ||
| Scheme 1 Active participation of bridging sulfide ligands in N2O activation by (a) binuclear and (b) and (c) tetranuclear synthetic copper sulfide models. | ||
In this report, we disclose a combined experimental/computational study of the latter system that collectively implicates the μ4-sulfide ligand as participating in redox changes and directly interacting with N2O during its activation (Scheme 1c). Our data includes the first spectroscopic interrogation of multiple [4Cu:1S] redox levels, which has proven challenging in the metalloenzyme system,7,24 and highlights the fidelity of our synthetic model to the biological CuZ site. Additionally, the direct interaction of N2O with the bridging sulfur atom(s) in CuZ has not been proposed before. Such reaction pathways should be considered for the chemistry of CuZ and related metal–sulfide clusters in light of the synthetic model studies reported herein.
Results and discussion
In our previous report of N2O reduction by the anionic complex [Cu4(μ4-S)(μ2-NCN)4]1− (the 1-hole cluster, referred to here as [4Cu:1S]1−) as its [K(18-crown-6)]+ salt (NCN = [MesNC(H)NMes]1−),22 we were able to use NMR spectroscopy, isotopic labeling experiments, and post situ electrophilic trapping to establish the presence of three products: neutral [Cu4(μ4-S)(μ2-NCN)4] (the 2-hole cluster, referred to here as [4Cu:1S]0), N2, and O2−. However, we were unable to definitively establish the reaction stoichiometry at that time. Since then, we have undertaken quantitative GC-MS analysis of the reaction headspace to determine the yield of N2. According to this analysis (see ESI†), 0.53 ± 0.06 mol of N2 are produced per mol of the [4Cu:1S]1− complex. When combining this result with our previous observations, we can now confidently propose the balanced reaction shown in Scheme 2 as the dominant pathway. Based on this reaction stoichiometry, we proceeded with the working hypothesis that one equivalent of [4Cu:1S]1− is responsible for N2O activation while a second equivalent is acting as a sacrificial reductant, thus accounting for the overall two-electron redox reaction.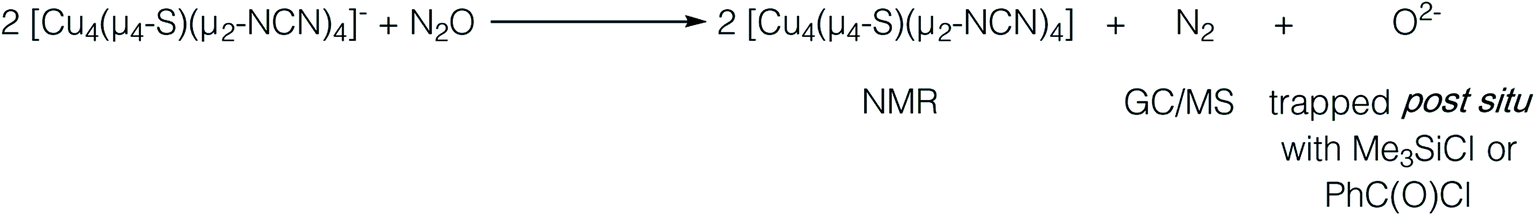 | ||
| Scheme 2 Overall balanced reaction being studied. | ||
Next, because we have been unable to detect any intermediates experimentally, we sought to examine the binding mode of N2O using DFT modeling at the B3LYP/6-31G(d) level in the gas phase. To save computational time, the mesityl groups on the supporting NCN ligands were replaced with methyl groups. After attempting to simulate several types of adducts between the [4Cu:1S]1− model complex and N2O, we were able to locate minima associated with N2O coordination to both the [4Cu:1S]1− model (intermediate [A]1−) and to its closed-shell, fully reduced [4Cu:1S]2− analogue (intermediate [A]2−). In both cases N2O occupied a μ-1,3 binding mode, but to our surprise the N2O molecule was found to bridge one of the Cu centers and the S atom (Fig. 2a). In each case, one of the other Cu centers has moved away from the S atom to facilitate its direct interaction with N2O. An alternative, μ3-1,2 binding mode in which the N2O molecule bridges two Cu centers as well as the S atom also was located but was determined to be significantly higher in energy by +11.8 kcal mol−1 on the Gibbs free energy surface (see ESI†). The preferred binding mode for this model system is distinct from the μ-1,3 bridging between two Cu centers that is proposed for CuZ (see Fig. 1a), where the μ4-S2− ligand is not proposed to interact directly with N2O. It should be noted that a mononuclear intermediate in which N2O bridges across a terminal nickel–sulfide bond has been isolated and crystallographically characterized by Hayton and coworkers.25,26 The accord between the metrical parameters of the activated N2O in our computational model with Hayton's experimental data (Fig. 2b) lends further support to the intermediacy of [A]1−. Binding of N2O to the 1-hole [4Cu:1S]1− model to form [A]1− was calculated to be endothermic by +18.5 kcal mol−1, consistent with our inability to observe an N2O-bound intermediate experimentally.
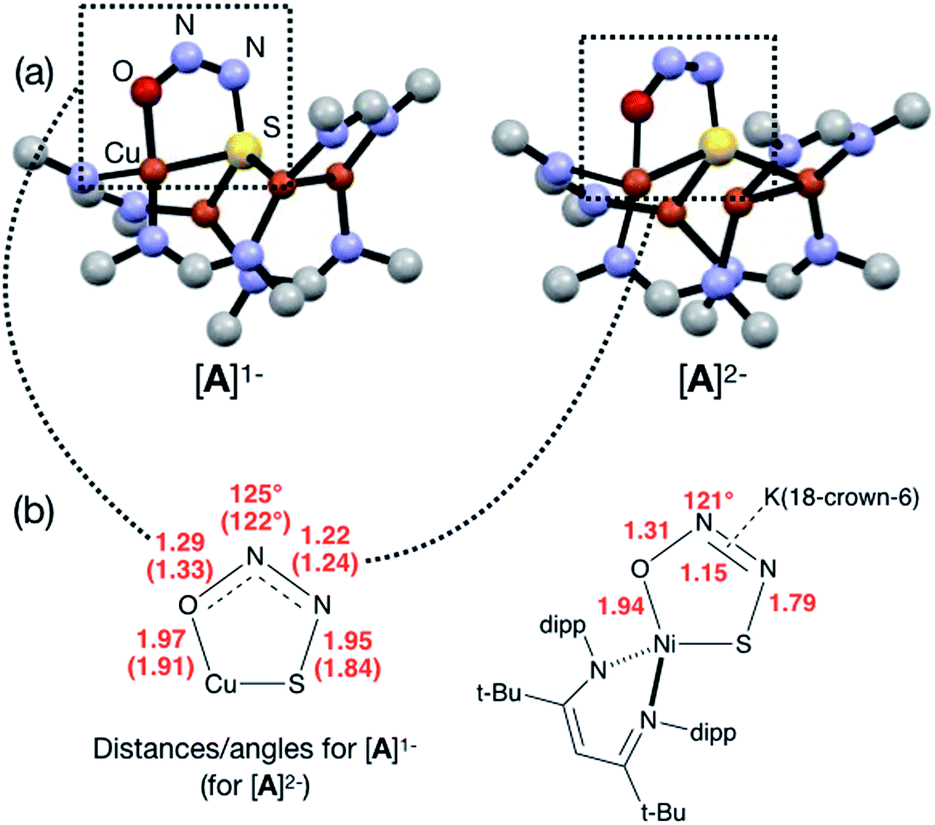 | ||
| Fig. 2 (a) Optimized structures of N2O-activated intermediates [A]1− and [A]2− (color code: Cu, orange; S, yellow; O, red; N, blue; C, gray). (b) Comparison of the cyclic core structures of [A]n− and a related mononuclear Ni complex characterized by Hayton;25 distances are given in Å. | ||
Assuming that small equilibrium concentrations of an N2O-bound intermediate akin to [A]1− form under N2O atmosphere, we next considered the potential reaction pathway to N2 + O2− (Scheme 3). We expect that N2O binding to the [4Cu:1S]1− complex would raise its reduction potential, due to the π-accepting nature of N2O.27 Thus, there would be a driving force for [A]1− to undergo reduction by a sacrificial 1-hole complex to provide [A]2−. As in the [4Cu:1S] form of CuZ,11 [A]2− is in the fully-reduced 4CuI state and thus is expected to π-backdonate sufficient electron density into the N2O π* manifold to induce N–O bond cleavage. Conversion to the resulting intermediate [B]2− from N2 loss was calculated to be exothermic relative to [A]2−. Further energy lowering was found by shifting the terminal O2− ligand in [B]2− to a μ2-bridging position in [C]2−. In the case of CuZ, Solomon has reported that the on-cycle intermediate CuZ° formed after N2 loss features a terminal oxygen ligand stabilized by hydrogen bonding with a nearby lysine residue, and has found that disruption of hydrogen bonding produces the off-cycle intermediate CuZ* in which the oxygen ligand occupies its thermodynamically preferred bridging position.11 Because we propose O2− to be a stoichiometric product of our aprotic model reaction, we assume that O2− dissociates from either [B]2− or [C]2−.
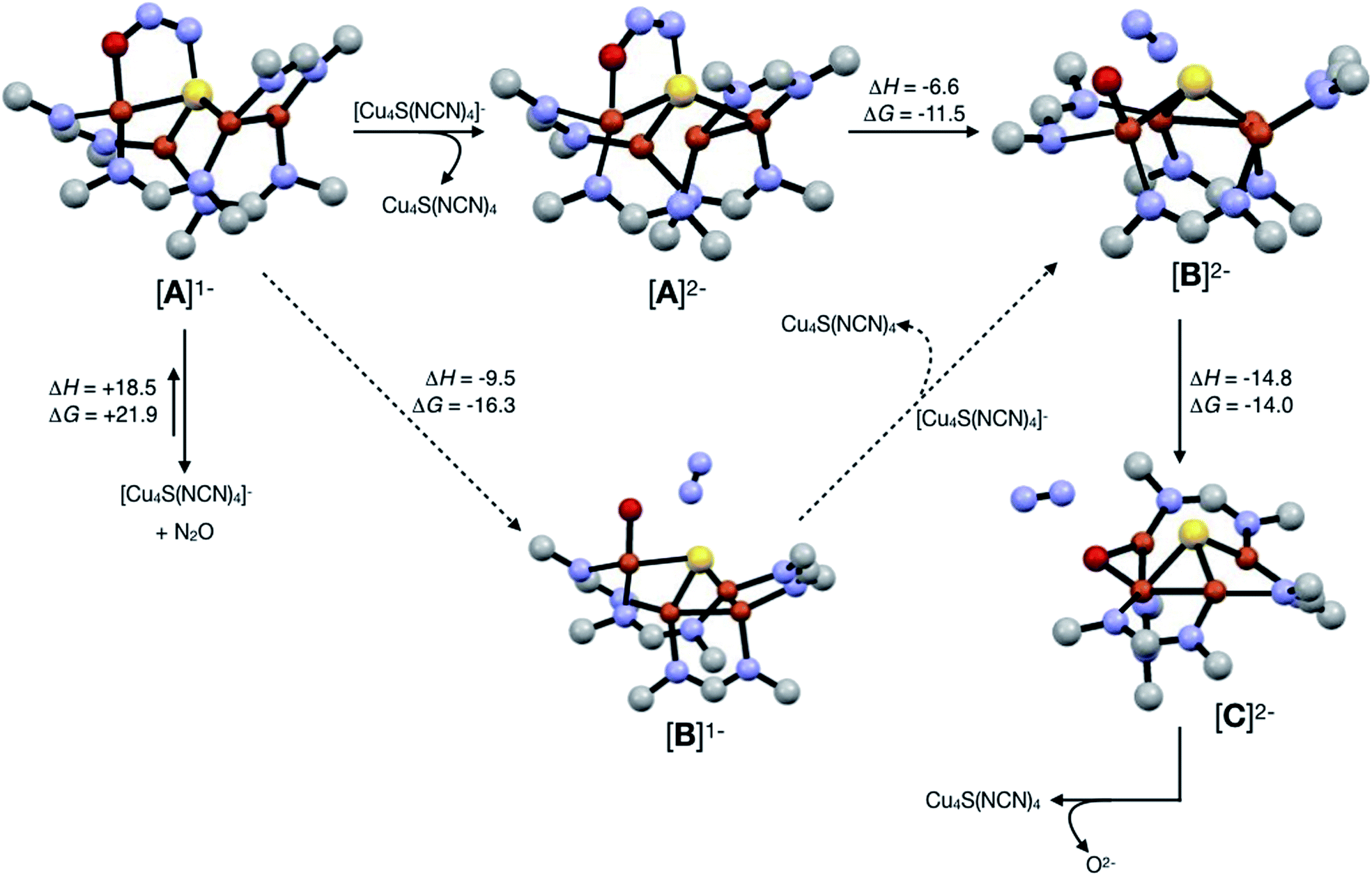 | ||
| Scheme 3 Reaction pathways modeled by DFT (B3LYP/6-31G(d)). Energies at 298 K are shown in kcal mol−1. The favored pathway is shown with solid arrows, and the disfavored pathway with dotted arrows. | ||
An alternative pathway (Scheme 3, dotted arrows) would involve N2 loss directly from 1-hole [A]1− prior to reduction, producing intermediate [B]1−. Reduction of [B]1− by a sacrificial [4Cu:1S]1− complex would then produce intermediate [B]2− that is common to both pathways. However, because O2− is expected to lower the reduction potential of the tetracopper cluster due to its π-donor character, it should be unfavorable for [B]1− to undergo reduction by the sacrificial 1-hole species. Indeed, [A]1− was calculated to be more oxidizing than [B]1− by 0.21 V. Thus, we consider this alternative pathway to be unlikely, but we cannot rule it out definitively.
Because the μ4-sulfide ligand seems to play a crucial and direct role in N2O activation according to our DFT modeling, we wondered whether the frontier orbitals of these synthetic [4Cu:1S] complexes have notable sulfur character. In order to validate our mechanistic model, we thus undertook multi-edge X-ray absorption spectroscopy (XAS) combined with higher-level computational modeling to interrogate the electronic structural changes underpinning the [4Cu:1S]0/1− redox process.
Cu K-edge XAS data obtained for [4Cu:1S]1− and [4Cu:1S]0 are shown in Fig. 3a. Spectral subtraction was carried out to remove a minor contribution of [4Cu:1S]0 in the spectrum of the monoanion (vide infra). Neither spectrum presents a resolved pre-edge (1s → 3d) feature, although both spectra feature a shoulder that gives a peak in the second derivative spectrum at 8979.8 eV, consistent with the presence of Cu 3d vacancies (Fig. 3b). The rising edges of the two spectra have qualitatively similar fine structure including maxima at ca. 8983 eV suggesting the presence of CuI centers,28 although the spectrum of the [4Cu:1S]1− cluster is shifted, with inflection points occurring at 0.8 to 1.1 eV lower energy relative to [4Cu:1S]0. Given the effectively identical coordination environments between the two species, the shift in rising edge position largely reflects some Cu participation in the redox process. Moreover, the lack of dramatic intensity changes for the rising edge features suggests a delocalized redox process, i.e. that a localized [2CuI:2CuII]/[3CuI:1CuII] description is not appropriate.
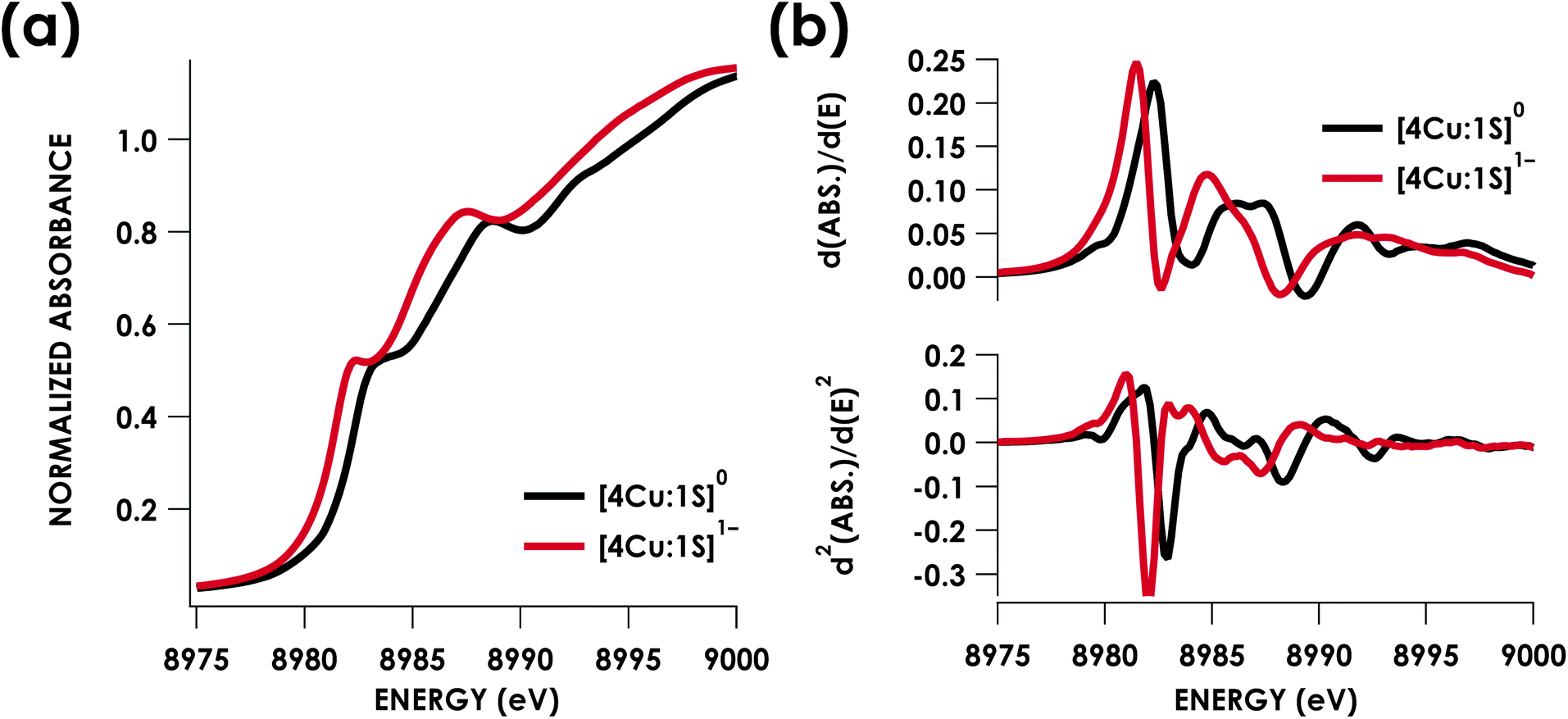 | ||
| Fig. 3 (a) Cu K-edge XAS spectra obtained for the [4Cu:1S]0/1− redox couple. (b) First (top) and second (bottom) derivative Cu K-edge XAS spectra. Isoenergetic pre-edge (1s → 3d) excitations are evident in the second-derivative spectra at 8979.8 eV. Rising edge inflection points occur at 8982.3 and 8985.9 eV for [4Cu:1S]0 and 8981.5 and 8984.8 eV for [4Cu:1S]1−. | ||
Quantitative estimates of S participation in the redox-active molecular orbital (RAMO) can be gleaned through analysis of S K-edge XAS data29 obtained for the two clusters, which are presented in Fig. 4. Well-resolved pre-edge peaks are apparent in both spectra, occurring at 2470.2 eV for [4Cu:1S]0 and 2469.5 eV in the spectrum of [4Cu:1S]1−. A ca. 18% [4Cu:1S]0 impurity was evident in the spectrum of [4Cu:1S]1− which was removed by subtraction and re-normalization as carried out by Solomon and co-workers to remove S K-edge XAS contributions from CuA in N2OR30,31 (Fig. S15†). Notably, the 2469.5 eV [4Cu:1S]1− pre-edge peak energy value closely matches pre-edge peak energies reported by Solomon and co-workers for the CuZ sites of resting Achromobacter cycloclastes31 and Paracoccus denitrificans30 N2OR at 2469.2 and 2469.0 eV, respectively. On the basis of Cu K-edge XAS analysis, Solomon and co-workers assigned resting CuZ as a 3CuI:1CuII cluster,30 consistent with the formal oxidation state distribution expected for the [4Cu:1S]1− cluster.
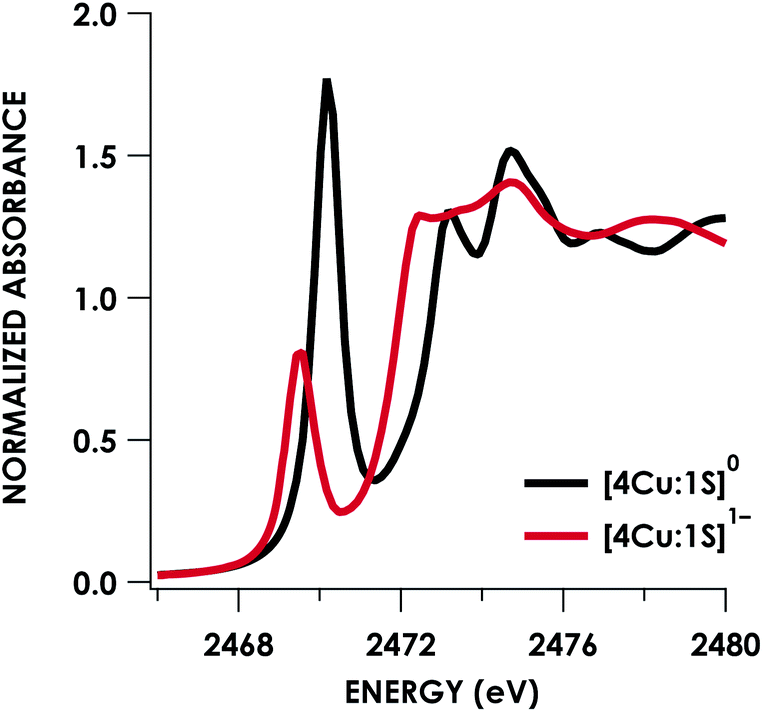 | ||
| Fig. 4 S K-edge XAS data obtained for the [4Cu:1S]0/1− redox couple. Pre-edge peaks corresponding to S 1s → ψ* excitations are located at 2470.2 eV for [4Cu:1S]0 and 2469.5 eV for [4Cu:1S]1−. | ||
Pre-edge peaks in the S K-edge XAS spectra of metal complexes and clusters bearing S-donor ligands reflect excitations from S 1s → ψ*, where ψ*, the acceptor MO, is an anti-bonding ligand field MO born of metal–sulfur mixing:
| ψ* ≈ α2S 3p − (1 − α)2M 3d | (1) |
Pre-edge peak intensities (D0) are then given by the relationship:
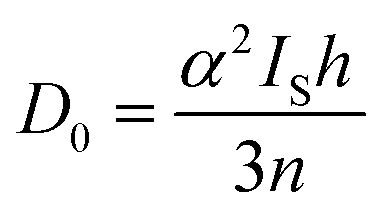 | (2) |
Fitting pseudo-Voigt peaks to the pre-edge peaks in the S K-edge data give integrated peak areas D0 for the two clusters of 2.03 ± 0.01 for [4Cu:1S]0 and 0.91 ± 0.02 for [4Cu:1S]1−. The ca. twofold decrease in D0 upon reduction confirms S 3p contribution to the RAMO shared by the redox couple. Application of eqn (2) then gives 20.5 ± 0.1% S 3p in the RAMO of [4Cu:1S]0 and 21.1 ± 0.5% S 3p in the RAMO of [4Cu:1S]1−. The latter values are comparable to the estimate given by Solomon and co-workers for the RAMO of the CuZ site in resting Paracoccus denitrificans N2OR at 15–22%.30
DFT calculations were carried out to further interrogate the nature of the RAMO in the [4Cu:1S]0/1− redox couple. Calculations were carried out on truncated models as described above and employed the B3LYP hybrid density functional with the CP(PPP) basis set34,35 on Cu and the scalar relativistically recontracted ZORA-def2-TZVP(-f)36 basis on all other atoms. The LUMO of [4Cu:1S]0 and SOMO of [4Cu:1S]1− are depicted in Fig. 5. These are qualitatively similar, indicating that the RAMO is a highly delocalized orbital featuring effectively equal participation of Cu 3d from all 4 metal centers along with a significant contribution from S 3p. Equal participation of all three Cu centers in the SOMO was previously indicated by simulation of experimental EPR parameters.22 The equal contributions from Cu are also in accord with observation that the Cu K-edge XANES shift in energy but do not exhibit differences in fine structure. Calculated S 3p contributions are 20.6% for [4Cu:1S]0 and 21.1% for [4Cu:1S]1−, in splendid agreement with experiment as well as with previous EPR analysis of the [4Cu:1S]1− species that indicated anomalously small Cu hyperfine coupling.22 Moreover, TDDFT calculations37 of the S K-edge XAS for both species initiated from the aforementioned single-point DFT calculations give spectra that nicely reproduce the energy and intensity differences encountered in the experimental data (Fig. 6).
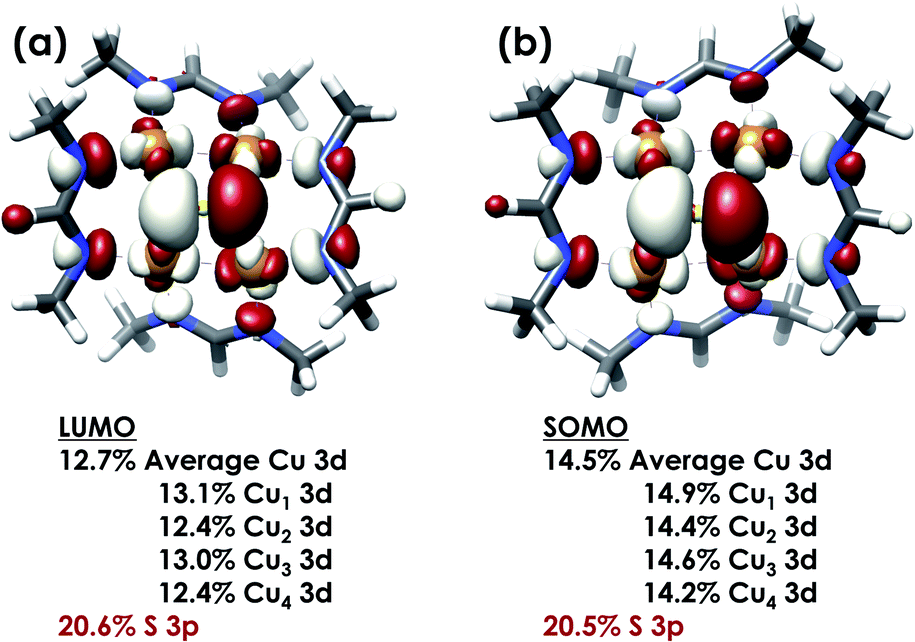 | ||
| Fig. 5 (a) Restricted Kohn–Sham LUMO for [4Cu:1S]0 and (b) quasi-restricted (QRO)33 SOMO for [4Cu:1S]1−. Both MOs were calculated for truncated models using the B3LYP hybrid density functional with the CP(PPP) basis set on Cu and the scalar-relativistically recontracted ZORA-def2-TZVP(-f) basis set on all other atoms. Orbitals are plotted at an isovalue of 0.03 au. | ||
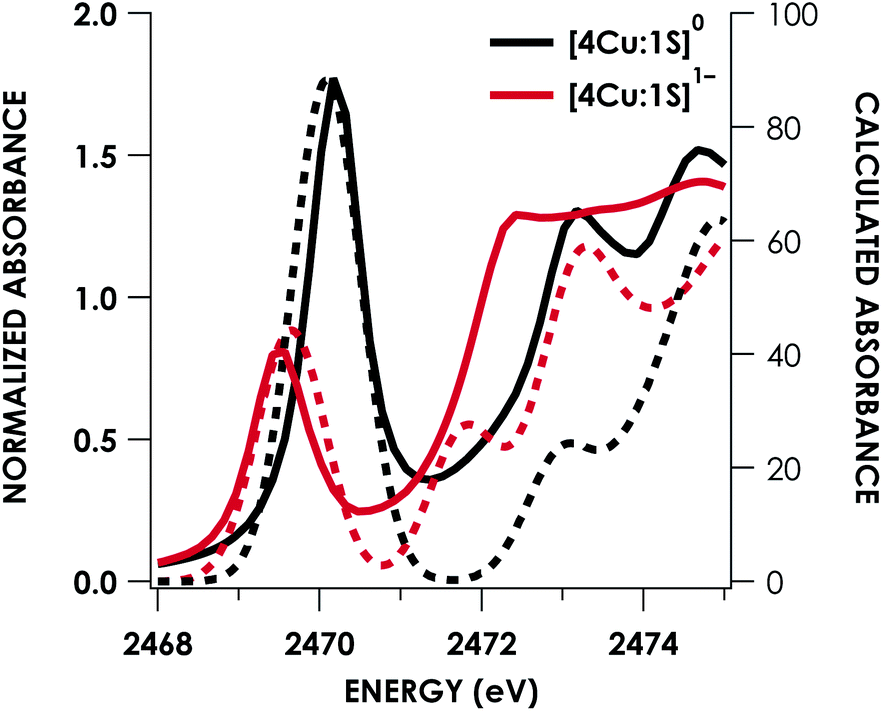 | ||
| Fig. 6 Overlay of TDDFT-calculated (dashed) S K-edge XAS with experimental (solid) spectra obtained for the [4Cu:1S]0/1− redox series. TDDFT calculations were initiated from B3LYP single-point calculations with the CP(PPP)34 basis set on Cu and the ZORA-def2-TZVP(-f)38 basis set on all other atoms. Calculated spectra are shifted by +40.4 eV to correct for inaccurate core potential modeling endemic to standard hybrid DFT calculations. | ||
Conclusions
We previously reported that the 1-hole [4Cu:1S]1− model cluster is oxidized to its 2-hole state by N2O with N2 evolution.22 Here, we have measured the reaction stoichiometry, allowing us to conclude that the overall 2-electron reduction of N2O requires two equivalents of the [4Cu:1S]1− cluster molecule, with each equivalent mediating a 1-electron redox process individually. Under the assumption that one equivalent activates N2O while the other acts as a sacrificial reductant, a computational model of the reaction intermediates indicated cooperative Cu/S coordination of N2O.This cooperative binding mode implies direct participation of the bridging S-atom in N2O activation and N–O cleavage, in contrast to the passive role of bridging S-atoms in typical metal–sulfur active sites. Consistent with this proposal, XAS analysis of the 1-hole and 2-hole clusters indicated that the μ4-S center contributes appreciably to the redox-active molecular orbital. Crucially, the S K-edge energies and estimated S-atom participation in redox chemistry closely match previous characterization of the biological CuZ site, making this synthetic system a faithful model in terms of electronic structure as well as atomic connectivity and chemical reactivity. Moreover, to our knowledge this data represents the first spectroscopic interrogation of multiple redox levels of a conserved [4Cu:1S] cluster.
Key to the model cluster's reactivity, and in particular to the μ4-S center's active participation in N2O activation and reduction, is the high degree of covalency within the [4Cu:1S] core. This Cu/S covalency allows the S-atom to exhibit characteristics typically associated with transition metals, such as the ability to simultaneously accept and donate electron density to/from the substrate and to vary its oxidation level during a chemical process, that are necessary for a catalytic active site mediating a multielectron redox process. Thus, it is important to consider both metal/metal and metal/ligand cooperation when interrogating highly covalent multinuclear catalysts such as CuZ and related systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding was provided by NIH/NIGMS (R01 GM116820 to N. P. M. and R35 GM124908 to K. M. L.). K. M. L. is an A. P. Sloan Research Fellow. Dr Chia-Wei Hsu assisted with preparation of XAS samples. This research was supported in part through computational resources provided by the Advanced Cyberinfrastructure for Education and Research (ACER) at UIC. XAS data were obtained at SSRL, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract no. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the Department of Energy's Office of Biological and Environmental Research, and by NIH/NIGMS (including P41GM103393).Notes and references
- H. Beinert, R. H. Holm and E. Mu, Science, 2009, 277, 653–659 CrossRef PubMed.
- H. Dobbek, L. Gremer, R. Kiefersauer, R. Huber and O. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15971–15976 CrossRef CAS PubMed.
- J. H. Jeoung and H. Dobbek, Science, 2007, 318, 1461–1464 CrossRef CAS PubMed.
- J. W. Peters and J. B. Broderick, Annu. Rev. Biochem., 2012, 81, 429–450 CrossRef CAS PubMed.
- R. Y. Igarashi, M. Laryukhin, P. C. Dos Santos, H. I. Lee, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, J. Am. Chem. Soc., 2005, 127, 6231–6241 CrossRef CAS PubMed.
- T. Spatzal, K. A. Perez, O. Einsle, J. B. Howard and D. C. Rees, Science, 2014, 345, 1620–1623 CrossRef CAS PubMed.
- S. R. Pauleta, S. Dell'Acqua and I. Moura, Coord. Chem. Rev., 2013, 257, 332–349 CrossRef CAS.
- K. Brown, M. Tegoni, M. Prudêncio, A. S. Pereira, S. Besson, J. J. Moura, I. Moura and C. Cambillau, Nat. Struct. Biol., 2000, 7, 191–195 CrossRef CAS PubMed.
- A. Pomowski, W. G. Zumft, P. M. H. Kroneck and O. Einsle, Nature, 2011, 477, 234–237 CrossRef CAS PubMed.
- E. M. Johnston, S. Dell'Acqua, S. Ramos, S. R. Pauleta, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2014, 136, 614–617 CrossRef CAS PubMed.
- E. M. Johnston, C. Carreira, S. Dell'Acqua, S. G. Dey, S. R. Pauleta, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2017, 139, 4462–4476 CrossRef CAS PubMed.
- R. H. Holm and E. I. Solomon, Chem. Rev., 2004, 104, 347–348 CrossRef CAS PubMed.
- A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Science, 2009, 326, 123–125 CrossRef CAS PubMed.
- A. J. Thomson, G. Giannopoulos, J. Pretty, E. M. Baggs and D. J. Richardson, Philos. Trans. R. Soc., B, 2012, 367, 1157–1168 CrossRef CAS PubMed.
- I. Bar-Nahum, A. K. Gupta, S. M. Huber, M. Z. Ertem, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2009, 131, 2812–2814 CrossRef CAS PubMed.
- C. Esmieu, M. Orio, S. Torelli, L. Le Pape, J. Pécaut, C. Lebrun and S. Ménage, Chem. Sci., 2014, 5, 4774–4784 RSC.
- M.-L. Tsai, R. G. Hadt, P. Vanelderen, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, J. Am. Chem. Soc., 2014, 136, 3522–3529 CrossRef CAS PubMed.
- V. Zhuravlev and P. J. Malinowski, Angew. Chem., Int. Ed., 2018, 57, 11697–11700 CrossRef CAS PubMed.
- J. Zhai, A. S. Filatov, G. L. Hillhouse and M. D. Hopkins, Chem. Sci., 2016, 7, 589–595 RSC.
- S. Bagherzadeh and N. P. Mankad, Chem. Commun., 2018, 54, 1097–1100 RSC.
- C.-W. Hsu, S. C. Rathnayaka, S. M. Islam, S. N. MacMillan and N. P. Mankad, Angew. Chem., Int. Ed., 2020, 59, 627–631 CrossRef CAS PubMed.
- B. J. Johnson, W. E. Antholine, S. V. Lindeman, M. J. Graham and N. P. Mankad, J. Am. Chem. Soc., 2016, 138, 13107–13110 CrossRef CAS PubMed.
- B. J. Johnson, W. E. Antholine, S. V. Lindeman and N. P. Mankad, Chem. Commun., 2015, 51, 11860–11863 RSC.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- N. J. Hartmann, G. Wu and T. W. Hayton, Angew. Chem., Int. Ed., 2015, 54, 1–5 CrossRef.
- N. J. Hartmann, G. Wu and T. W. Hayton, Chem. Sci., 2018, 326, 123–129 Search PubMed.
- W. C. Trogler, Coord. Chem. Rev., 1999, 187, 303–327 CrossRef CAS.
- L. S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2001, 109, 6433–6442 CrossRef.
- T. Glaser, B. Hedman, K. O. Hodgson and E. I. Solomon, Acc. Chem. Res., 2000, 33, 859–868 CrossRef CAS PubMed.
- P. Chen, S. I. Gorelsky, S. Ghosh and E. I. Solomon, Angew. Chem., Int. Ed., 2004, 43, 4132–4140 CrossRef CAS PubMed.
- S. Ghosh, S. I. Gorelsky, S. D. George, J. M. Chan, I. Cabrito, D. M. Dooley, J. J. G. Moura, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 3955–3965 CrossRef CAS PubMed.
- R. Sarangi, S. DeBeer George, D. Jackson Rudd, R. K. Szilagyi, X. Ribas, C. Rovira, M. Almeida, K. O. Hodgson, B. Hedman and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 2316–2326 CrossRef CAS PubMed.
- F. Neese, Inorg. Chem., 2005, 44, 2245–2254 CrossRef PubMed.
- F. Neese, Inorg. Chim. Acta, 2002, 337, 181–192 CrossRef.
- S. Sinnecker, L. D. Slep, E. Bill and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef PubMed.
- D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS PubMed.
- S. DeBeer George and F. Neese, Inorg. Chem., 2010, 49, 1849–1853 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral, analytical, and computational data. See DOI: 10.1039/c9sc06251c |
| This journal is © The Royal Society of Chemistry 2020 |