
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA nanotweezers for stabilizing and dynamically lighting up a lipid raft on living cell membranes and the activation of T cells†
Lele
Sun‡
ab,
Yingying
Su‡
a,
Jun-Gang
Wang
a,
Fei
Xia
 a,
Ying
Xu
c and
Di
Li
*a
a,
Ying
Xu
c and
Di
Li
*a
aSchool of Chemistry and Molecular Engineering, East China Normal University, 200241, Shanghai, China. E-mail: dli@chem.ecnu.edu.cn
bInstitute of Functional Nano & Soft Materials (FUNSOM), Collaborative Innovation Center of Suzhou Nano Science and Technology, Soochow University, Suzhou, 215123, China
cDepartment of Pathophysiology, Key Laboratory of Cell Differentiation and Apoptosis of Ministry of Education, Shanghai Jiao-Tong University School of Medicine, Shanghai, 200025, China
First published on 7th January 2020
Abstract
Lipid rafts are generally considered as nanodomains on cell membranes and play important roles in signaling, viral infection, and membrane trafficking. However, the raft hypothesis is still debated with many inconsistencies because the nanoscale and transient heterogeneous raft structure creates difficulties in its location and functional analysis. In the present study, we report a DNA nanotweezer composed of a cholesterol-functionalized DNA duplex that stabilizes transient lipid rafts, which facilitate the further analysis of the raft component and its functions via other spectroscopy tools. The proposed DNA nanotweezer can induce clustering of raft-associated components (saturated lipids, membrane protein and possibly endogenous cholesterol), leading to the T cell proliferation through clustering of a T-cell antigen receptor (TCR). The flexibility of random sequence noncoding DNA provides versatile possibilities of manipulating lipid rafts and activating T cells, and thus opens new ways in a future T cell therapy.
Introduction
Lipid raft hypothesis suggests the existence of transient nanodomains with a high content of sphingolipids, sterols and specific proteins on the cell membrane.1–4 Lipid rafts are believed to have important roles in cell signaling through changes in the raft composition.5–8 However, not only the structure and composition of a lipid raft but also mechanisms that govern its genesis remain highly controversial.9,10 The transient and dynamic nanoscale structure of a lipid raft creates difficulties in the correlation between raft composition and its associated cellular physiological functions.2,11 For example, cholesterol is considered to play important roles in the formation of lipid rafts with preferential associations between cholesterol and saturated lipids.12–14 The role of cholesterol in lipid rafts has long been studied by removing cholesterol with depleting agents, such as methyl-β-cyclodextrin (MβCD),15 which often results in dramatic changes of the plasma membrane and functional outcomes associated with the dissolution of rafts.16 The ability to rationally manipulate the distribution of cholesterol on the plasma membrane may have great potentials in exploring the function of lipid rafts.The development of new technologies, including fluorescence microscopy,18–20 force spectroscopy,21,22 X-ray diffraction,23 mass spectrometry24,25 and solid state NMR,26 has advanced the understanding of mysterious lipid rafts.17 Among them, the major advantage of fluorescence microscopy is its high spatial resolution.2 However, owing to the putative small and dynamic nature of lipid rafts, classical fluorescence raft probes, including fluorophore-labeled lipids and proteins,27–31 suffer difficulties in exploring the detailed composition and functions of rafts in plasma membranes.32 As a result, the potent correlation between lipid rafts and cellular functions are still not fully understood. Therefore, specific labeling of lipid rafts on cell membranes remains one of the foremost challenges.2
Recently, DNA nanostructures with lipidic anchors were demonstrated to be promising membrane protein mimics that enable one to monitor molecular encounter events on living cell membranes.33–41 In the present study, we demonstrate that DNA nanotweezers could stabilize and dynamically light up lipid rafts on living cell membranes. The proposed DNA nanotweezers create an exogenous cholesterol-rich region on living cell membranes, which results in the recruitment of raft-associated lipids and proteins, and possibly also endogenous cholesterol. To further address the biological function of lipid rafts, we demonstrate that the DNA nanotweezers could activate the T cell proliferation in a nonspecific activation manner. The strategy proposed in this work provides new ways of regulating cell behaviors using random sequence noncoding DNAs.
Results and discussion
The principle of DNA nanotweezer induced formation of lipid rafts is depicted in Fig. 1A. The DNA nanotweezers are composed of a pair of cholesterol-(chol) functionalized DNA duplexes. In detail, a 5′chol- and 3′Cy3-functionalized single-stranded (ss)-DNA (termed as 5′chol- and 3′Cy3 anchor ss-DNA, the sequences of DNA used in this work are listed in Table S1†) was anchored on the membrane of suspension cells (Jurkat T cell) through the insertion of cholesterol into lipids. The inserted ss-DNA were evenly distributed on the cell membrane (Fig. 1B, upper). Of note, we designed a hydrophilic PEG linker between cholesterol and DNA to diminish the influence of DNA on the behavior of endogenous cholesterol inside the cell membrane.42 The insertion of cholesterol-decorated ss-DNA did not perturb the viability of Jurkat T cells (Fig. S1A†).43–45 In order to restore more anchor ss-DNAs on cell surfaces, chlorpromazine (CPZ) was added into the medium to inhibit endocytosis (the cytotoxicity of CPZ toward Jurkat cells was also tested as shown in Fig. S1B†). Then, its complementary DNA with a 5′chol functionalization (termed as 5′chol-cDNA) was introduced to hybridize with the membrane-anchored ss-DNA. Interestingly, DNA hybridization resulted in a redistribution of Cy3 fluorescence on the surface of the cell membrane, as shown in the cross-section fluorescence images of the treated cells in Fig. 1B. Upon hybridization, the homogeneously distributed Cy3 fluorescence (Fig. 1B, top) was gradually transformed into clusters with aggregates ranging from several hundred nanometers to micrometers (Fig. 1B, middle) in 10 min. The corresponding 3D confocal microscopy images further confirmed that the fluorescent aggregates are discontinuously distributed on the cell surface (Fig. S2†). The clustering of the fluorescence domain could be more easily visualized from the bottom section of confocal images (Fig. 1C, pictures were taken by focusing on the interface between cell and the glass substrate). The dynamic process of the DNA hybridization induced fluorescence aggregation was recorded (see Video S1 in the ESI†). Therefore, we speculated that there might be some correlations between the clustered distributions of DNA duplexes on cell membrane with lipid rafts.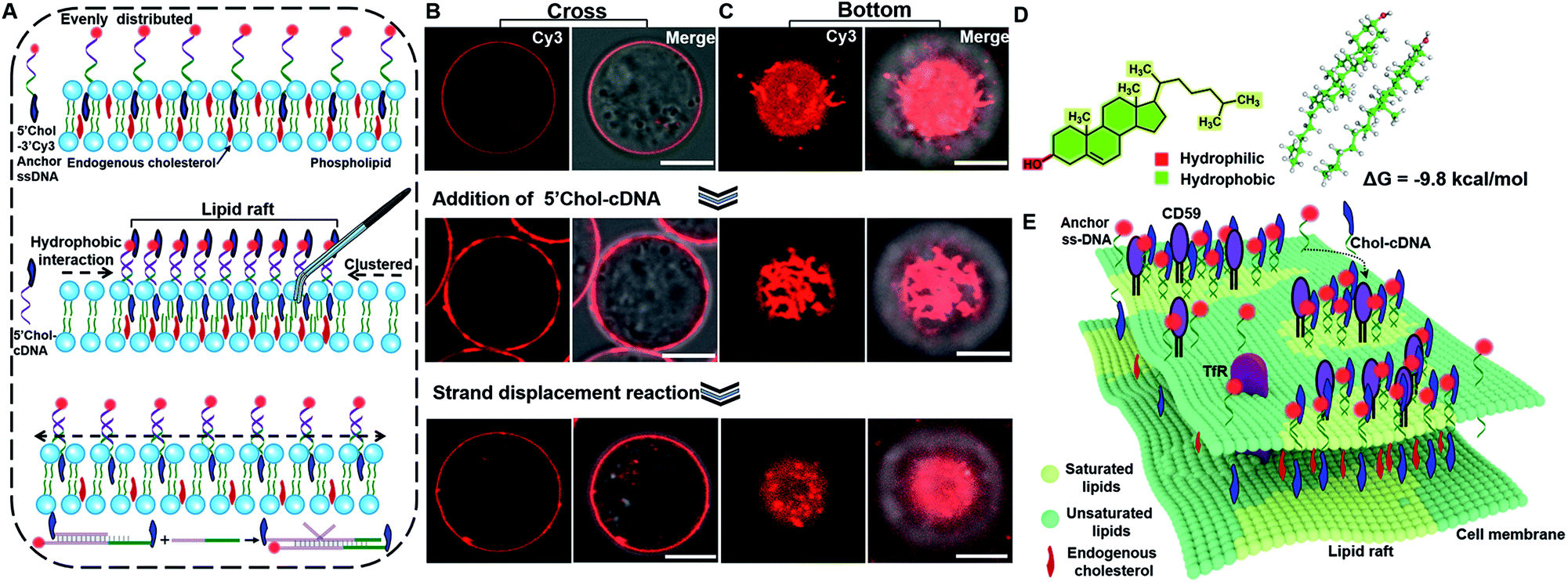 | ||
| Fig. 1 (A) Schematic illustrating the strategy of using DNA nanotweezers to manipulate cholesterol distribution on a living cell membrane, which stabilize and dynamically light up lipid rafts. (B and C) Confocal fluorescence microscopy images of the cells focused on cross (B) and bottom (C) section. The evenly distributed Cy3 fluorescence on cell membrane from 5′chol- and 3′Cy3 anchor ss-DNA (top), was clustered upon hybridization with 5′chol-cDNA (middle), which was then transferred back to an even distribution upon the introducing of a DNA strand displacement reaction that removes 5′chol-cDNA (bottom). Scale bar is 5 μm. (D) Optimized structure of the cholesterol dimer in solution phase. (E) Schematic illustrating the proposed strategy of using DNA nanotweezers to recruit raft-associated saturated lipids, membrane and possibility endogenous cholesterol. | ||
We then performed several control experiments to investigate this hypothesis. These included: (1) the fluorescence of a control anchor ss-DNA of the same sequence but with both 3′ and 5′chol functionalization and a Cy3 fluorophore modified in the middle was evenly distributed on the cell membrane (Fig. S3A†), suggesting either one or two cholesterol groups are inserted into the cell membrane. Moreover, upon adding a control cDNA with the same sequence but without 5′chol modification, the fluorescent domain also transformed into clusters (Fig. S3B†); (2) upon hybridizing with another control cDNA with same sequence but with a Cy5 modification near 5′chol, besides the aggregation of Cy3 fluorescence, a Cy3 to Cy5 FRET was also observed. The FRET further confirmed the hybridization of cDNA with anchor ss-DNA (Fig. S4†). Control experiments depicted in Fig. S3 and S4† indicated that DNA hybridization is pivotal for the cluster formation; (3) the fluorescent aggregates were absent if a control cDNA with the same sequence but without 5′chol modification was used to hybridize with the membrane-anchored ss-DNA (Fig. S5†); (4) upon hybridizing with another control cDNA of the same sequence but with 3′chol functionalization, the fluorescence of Cy3 was still evenly distributed on cell membranes (Fig. S6†), suggesting that the 5′chol functionalization of cDNA also plays a critical role in the formation of fluorescent aggregates; (5) the degree of fluorescence aggregation on the cell membrane was positively dependent on the amounts of 5′chol cDNA (Fig. S7†); (6) the DNA duplex-induced fluorescent aggregates were still preserved when we used MβCD to extract endogenous cholesterol from the cell membrane, suggesting that the aggregation of DNA duplexes on cell membrane was largely independent of endogenous cholesterol (Fig. S8†).
We thus concluded that DNA hybridization and the weak interactions between the pending cholesterol groups induce the gathering of DNA duplexes. We performed quantum mechanics calculations on cholesterol dimers using the semiempirical PM6 method.46 Upon the formation of a cholesterol dimer, the calculated solvation energy of dimer in a solution is −9.8 kcal mol−1, indicating that the aqueous solution could stabilize the cholesterol dimer via the hydrophobic effects (Fig. 1D). As a result, the inserted cholesterol groups are also gathered in cell membranes. According to the raft hypothesis, cholesterol is believed to be responsible for domain formation due to its high affinity toward saturated phospholipids. Thus, we speculated that the aggregated inserting cholesterol may also recruit raft-associated components (saturated lipids, membrane proteins and possibly endogenous cholesterol), which stabilize and enlarge the original raft domain. Therefore, we concluded that a synergistic effect of DNA hybridization and hydrophobic interactions of cholesterol groups result in a tweezer-like function, leading to seizing and enlarging an original cholesterol-rich raft domain and simultaneously lighting it up (Fig. 1E). We further investigated these DNA nanotweezers with other cell lines (L1210, HeLa) and found similar phenomena (Fig. S9 and S10†), suggesting the broad utility of the proposed strategy.
The programmable effect of DNA hybridization enables one to rationally control the distribution of DNA nanotweezers on the cell membrane. We introduced a strand displacement reaction to highlight the possibility of redistributing the raft domain.47 As shown in Fig. 1A (bottom), upon introducing a trigger DNA strand to initiate the strand displacement reaction, the DNA nanotweezers induced fluorescent aggregates were transferred back to an even distribution (Fig. 1B and C, bottom, and Video S2†). Therefore, DNA provides a unique capability of reversibly manipulating the distribution of raft-associated components on a cell membrane, which opens up new possibility of rationally controlling the on-off of the raft-associated reactions with external DNA-related stimuli.
To further confirm that the DNA nanotweezer-seized domain is not only associated with lipid rafts in spatial distribution but also in chemical components, we next investigated the chemical composition of the DNA lightened domain. We first explored the lipid components in the DNA nanotweezer-lightened region. Previous studies indicated that endogenous cholesterol favours interactions with saturated hydrocarbon chains, such as sphingolipids and gangliosides, and these lipids are considered as important components of lipid rafts.48,49 Ganglioside GM1, a receptor for cholera toxin on cell surfaces, is suggested to be transiently trapped within areas <20 nm in the plasma membrane in a cholesterol-dependent fashion and recognized as a raft-resident marker.47 We, thus, stained Jurkat T cells with Alexa 488-labeled cholera toxin B, and the even distribution of cholera toxin B suggests that original lipid rafts are extremely small and dynamic (Fig. S11†). Moreover, in another control experiment the cells were treated simultaneously with Alexa 488-labeled cholera toxin B and 5′chol- and 3′Cy3 anchor ss-DNA; we found both were evenly distributed on the cell surfaces (Fig. S12†). Upon treatment with DNA nanotweezers, the colocalization of Alexa 488 and Cy3 (Fig. 2A and B) indicated that the DNA nanotweezer-seized region coincided with GM1 on the cell membrane. We then extracted endogenous cholesterol with MβCD to reduce its availability. As a result, the colocalization of DNA nanotweezers and cholera toxin B was reduced (Fig. 2C and D). The quantitative analysis in Fig. 2E further confirms the strong colocalization of DNA nanotweezers with GM1.50 These results imply that (1) the region lightened by DNA nanotweezers possesses a similar higher content of GM1 as in lipid rafts; (2) the DNA nanotweezer-lightened region might also be rich in endogenous cholesterol.
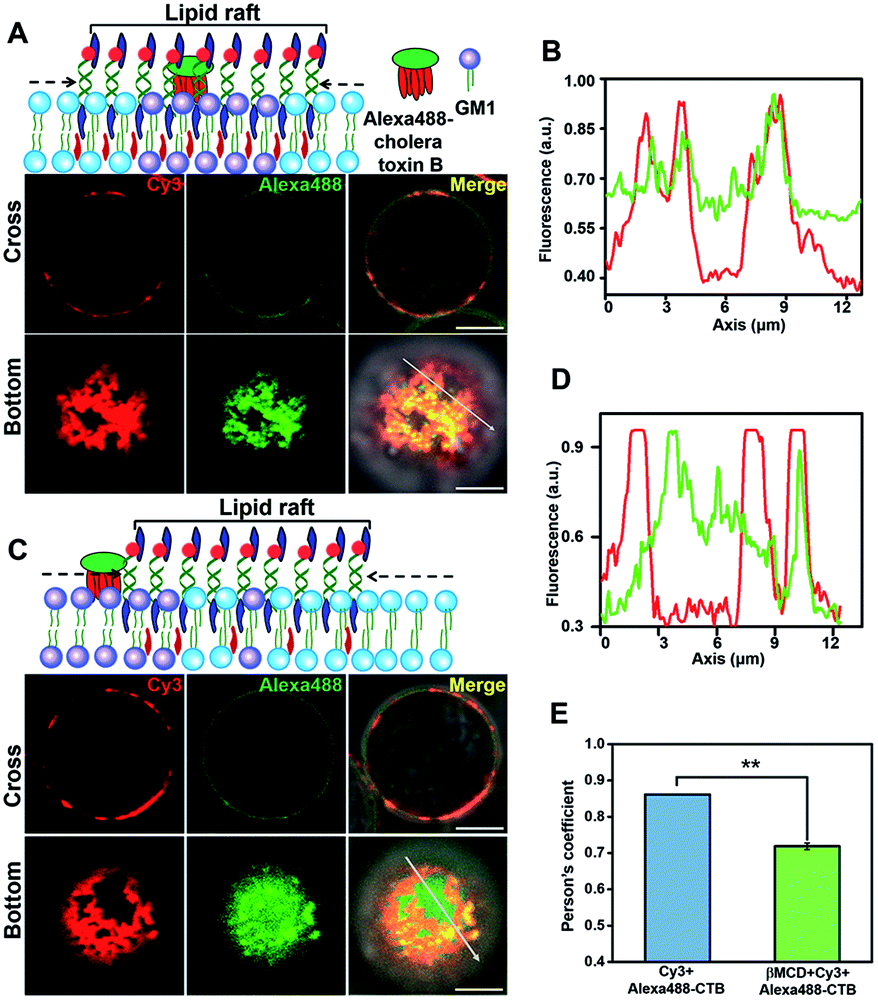 | ||
| Fig. 2 The DNA nanotweezer-lightened regions are similar with lipid rafts in lipid components. (A) Upper: scheme of the colocalization of cholera toxin B and DNA nanotweezers. Lower: confocal fluorescence microscopy images show the overlap of Cy3 and Alexa 488 in both cross and bottom fluorescence profiles. (B) Colocalization between cholera toxin B and DNA nanotweezers was analyzed by line profiling the fluorescence intensity of Cy3 (red) and Alexa 488 (green) along the line selected in the bottom fluorescence profile. (C) Upper: scheme of the reduced colocalization of cholera toxin B and DNA nanotweezers upon treatment with MβCD to reduce the availability of endogenous cholesterol. Lower: confocal fluorescence microscopy images show the reduced overlap of Cy3 (red) and Alexa 488 (green). Scale bar is 5 μm. (D) Reduced colocalization of cholera toxin B and DNA nanotweezers was analyzed by line profiling the fluorescence intensity of Cy3 (red) and Alexa 488 (green) along the line selected in the bottom fluorescence profile. (E) Classical colocalization analysis of Images in (A) and (C) using the coloc2-plugin of extended ImageJ version Fiji. The mean of Person's R values is 0.86 of (A) and 0.70 of (C), respectively (data represent mean of 20 images, asterisks indicate a significant difference with p < 0.001). | ||
We next investigated the protein component in the lightened region. Lipid rafts are platforms for membrane protein interactions,51 and among them glycosylphosphatidylinositol (GPI)-anchored membrane proteins are intensively studied.52 We selected CD59, a GPI-anchored protein, as a model protein to investigate its presence in the DNA-lightened region. We stained Jurkat T cells with FITC-labeled CD59 monoclonal antibodies and found that CD59 was homogeneously distributed on the cell surface (Fig. S13†), further confirming the dynamic nature of lipid rafts. Then, the CD-59 antibody-labeled cells were treated with 5′chol- and 3′Cy3-anchor ss-DNA, and FITC and Cy3 were both evenly distributed on the cell surfaces (Fig. S14†). Once 5′chol-cDNA was introduced to form the DNA nanotweezers, the Cy3 fluorescence region revealed a strong overlapping with that of FITC (Fig. 3A and B), indicating that the DNA nanotweezer-accumulated region could recruit CD59. We performed another control experiment to verify that protein components in the DNA nanotweezer-seized domain are associated with lipid rafts. Transferrin receptor (TfR) is an extensively studied non-lipid component on living cell membranes.53 Therefore, we stained Jurkat cells with APC-labeled TfR monoclonal antibodies. The fluorescence of APC could not be overlapped with Cy3 (Fig. 3C and D), suggesting that TfR was not recruited into the DNA nanotweezer-seized domain. In another control experiment with MβCD extraction to reduce the availability of endogenous cholesterol, we found a reduction in the colocalization of CD59 and DNA nanotweezers (Fig. 3E and F). These results confirmed that the DNA nanotweezer-lightened regions are largely identical to lipid rafts in both lipids and protein compositions. The quantitative analysis in Fig. 3G confirms the colocalization of DNA nanotweezers with CD59, while it does not support the overlapping of DNA nanotweezers with TfR.
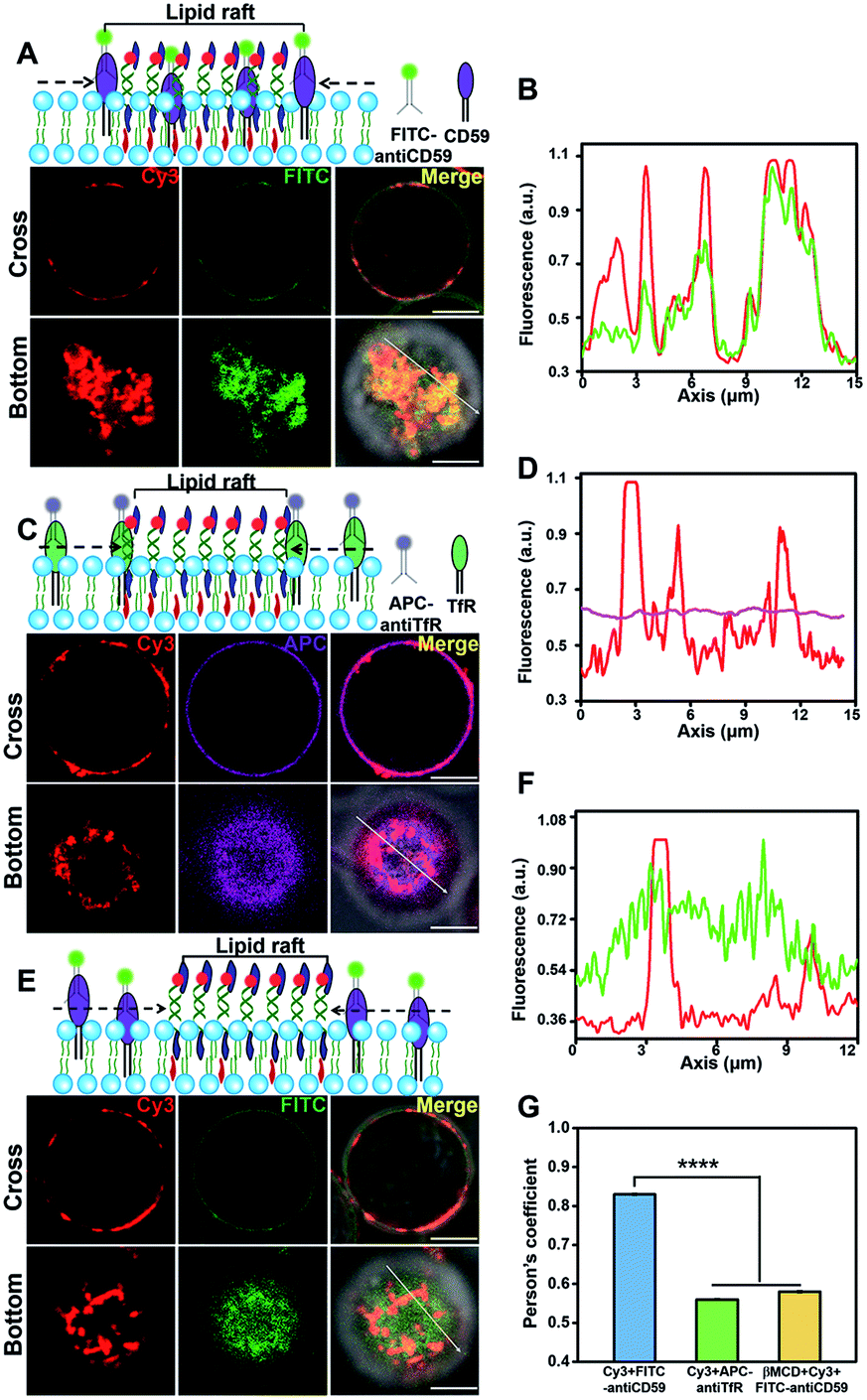 | ||
| Fig. 3 The DNA nanotweezers lightened region are similar with lipid raft in protein component. (A) Upper: scheme of the colocalization of CD59 and DNA nanotweezers. Lower: confocal fluorescence microscopy images show the distribution of Cy3 and FITC in both cross and bottom fluorescence profiles. (B) Colocalization between CD59 and DNA nanotweezers was analyzed by line profiling the fluorescence intensity of Cy3 (red) and FITC (green) along the line selected in the bottom fluorescence profile. (C) Upper: scheme of the distribution of TfR and DNA nanotweezers. Lower: confocal fluorescence microscopy images show the distribution of Cy3 and APC in both cross and bottom fluorescence profiles. (D) Colocalization between TfR and DNA nanotweezers was analyzed by line profiling the fluorescence intensity of Cy3 (red) and APC (purple) along the line selected in the bottom fluorescence profile. (E) Upper: scheme of the reduced colocalization of CD 59 and DNA nanotweezers upon treatment with MβCD to reduce the availability of endogenous cholesterol, lower: confocal fluorescence microscopy images show the distribution of Cy3 and FITC. (F) Reduced colocalization of CD59 and DNA nanotweezers was analyzed by line profiling the fluorescence intensity of Cy3 (red) and FITC (green) along the line selected in the bottom fluorescence profiles. Scale bar is 5 μm. (G) Classical colocalization analysis of images in (A), (C) and (E). The mean of Person's values is 0.83 of (A), 0.56 of (C) and 0.58 of (E), respectively (data represent mean of 20 images, asterisks indicate a significant difference with p < 0.0001). | ||
We then demonstrated the ability of DNA nanotweezers to manipulate cellular functions. An important role of lipid rafts is their function in receptor signal transduction and activation of lymphocytes.54 On the surface of T cells, a T-cell antigen receptor (TCR) is an important multi-subunit immune recognition receptor that engages lipid rafts during signaling, and transmembrane TCR-CD3 complexes are responsible for transmitting the stimulation of antigens to the intracellular effector proteins.11 It has been reported that cholesterol can directly bind to the transmembrane domain of the TCR-β chain to mediate TCR clustering on the T cell surface, and CD3 antibodies can produce the first signal of T cell activation by crosslinking TCR-CD3 complexes (Fig. 4A).55,56 Therefore, we speculated that the DNA nanotweezers could also behave as CD3 antibodies that are associated with initial signaling events in rafts. We thus stained the DNA nanotweezer-treated cells with APC-labeled CD3 antibodies and found that APC was also clustered with partial overlapping with Cy3 (Fig. 4B and S15†), indicating that the clustering of TCR-CD3 complexes is closely associated with DNA nanotweezers. We wondered if the DNA nanotweezer-induced assembly of the TCR-CD3 complexes also results in activation of the T cell receptor signaling pathway. LAT (Linker for Activation of T cells) is an important adaptor protein in T cell activation and could be phosphorylated by ZAP-70/Syk protein tyrosine kinases following the stimulation of TCR. Upon treatment with DNA nanotweezers, we found that the phosphorylation level of LAT was significantly increased (Fig. 4C), while the content of LAT remained constant, suggesting that the DNA nanotweezers indeed nonspecifically activate T cells.
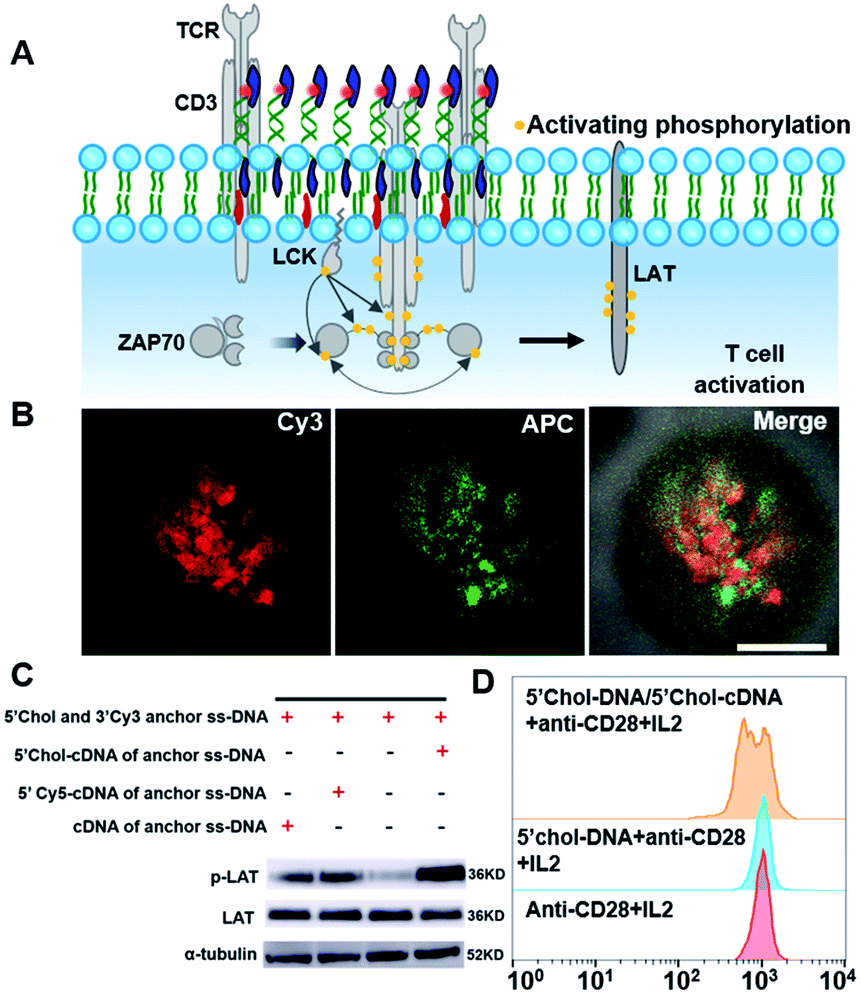 | ||
| Fig. 4 The DNA nanotweezers induce an activation of T cell. (A) Schematic of TCR signaling cascade after the engagement of the TCR-CD3 complexes. (B) Confocal fluorescence microscopy images show the distribution of Cy3 and APC in both cross and bottom fluorescence profiles. (C) The phosphorylation level of LAT from Jurkat cells was increased upon treatment with DNA nanotweezers, as determined by western blotting. (D) Flow cytometry data showing T cell proliferation after activation by DNA nanotweezers. Native T cells were stained by CFSE for monitoring proliferation. | ||
Therefore, we further explored if the DNA nanotweezer-induced T cell activation could lead to T cell proliferation. Since Jurkat cells are immortal cell lines with rapid cell division, it is not suitable for studying activation-induced T cell proliferation. We thus employed native T cells (CD8+ T cells from mouse spleen) to investigate the DNA nanotweezer-induced cell proliferation. CD28 antibodies and IL2 were used as co-stimulators to stimulate the DNA nanotweezer-treated cells,57 and the proliferation of native CD8+ T cell was studied using a carboxyfluorescein in succinimidyl ester (CFSE) cell division tracking kit using flow cell cytometry.58,59 This CFSE kit relies on the ability of CFSE to label long-lived intracellular molecules with a highly fluorescent dye, carboxyfluorescein. Following each cell division, the equal distribution of these fluorescent molecules to progeny cells results in the halving of the fluorescence of daughter cells.56 The flow cytometric analysis of CD8+ T cells was performed after 72 h of activation (Fig. 4D). Clearly, only DNA nanotweezer-treated CD8+ T cells led to profound cell proliferation. Therefore, we confirmed that the proposed DNA nanotweezers could induce aggregation of the TCR-CD3 complexes and subsequent T cell proliferation, indicating a strong correlation between lipid rafts and T cell activation.
Conclusions
In conclusion, we report a DNA tool to stabilize and dynamically light up lipid rafts on the plasma membrane, which enables further functional analysis of the lipid rafts via other spectroscopies. These DNA nanotweezers enable one to manipulate the distribution of raft-associated components on the cell membrane. In addition, the DNA nanotweezers could nonspecifically activate T cells through rationally controlling raft distribution. These results show great potentials of delicately manipulating cell fate through controlling the distribution of raft-associated components on living cell membranes. To the best of our knowledge, this is the first example of using dynamic DNA nanodevices to seize lipid rafts and to activate the associated T cell signaling pathway. The flexibility of noncoding DNA provides versatile possibilities of activating T cells; for example, stimuli-induced programmable activation with aptamers or light-induced activation with photo-responsive nucleotides thus opening up new ways in T cell therapy.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by NSFC (21675166, 21874046, 31671459, 21902050) and National Postdoctoral Program for Innovative Talents (BX20190118). The Shanghai Municipal Commission for Science and Technology (19JC1411800). The authors thank Prof. Wei Lv of Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences for helpful discussion.Notes and references
- D. Lingwood and K. Simons, Science, 2010, 327, 46–50 CrossRef CAS PubMed.
- E. Sezgin, I. Levental, S. Mayor and C. Eggeling, Nat. Rev. Mol. Cell Biol., 2017, 18, 361–374 Search PubMed.
- J. F. Hancock, Nat. Rev. Mol. Cell Biol., 2006, 7, 456–462 CrossRef CAS PubMed.
- I. W. Hamley and V. Castelletto, Angew. Chem., Int. Ed., 2007, 46, 4442–4455 CrossRef CAS PubMed.
- R. G. W. Anderson and K. Jacobson, Science, 2002, 296, 1821–1825 CrossRef CAS PubMed.
- A. D. Douglass and R. D. Vale, Cell, 2005, 121, 937–950 CrossRef CAS PubMed.
- M. Dykstra, A. Cherukuri, H. W. Sohn, S. J. Tzeng and S. K. Pierce, Annu. Rev. Immunol., 2003, 21, 457–481 CrossRef CAS PubMed.
- W. H. Binder, V. Barragan and F. M. Menger, Angew. Chem., Int. Ed., 2003, 42, 5802–5827 CrossRef CAS PubMed.
- I. Levental and S. L. Veatch, J. Mol. Biol., 2016, 428, 4749–4764 CrossRef CAS PubMed.
- E. Sevcsik and G. J. Schuetz, Bioessays, 2016, 38, 129–139 CrossRef CAS PubMed.
- K. Simons and D. Toomre, Nat. Rev. Mol. Cell Biol., 2000, 1, 31–39 CrossRef CAS PubMed.
- R. Ziblat, K. Kjaer, L. Leiserowitz and L. Addadi, Angew. Chem., Int. Ed., 2009, 48, 8958–8961 CrossRef CAS PubMed.
- S. R. Shaikh, V. Cherezov, M. Caffrey, W. Stillwell and S. R. Wassall, Biochemistry, 2003, 42, 12028–12037 CrossRef CAS PubMed.
- S. Mahammad, J. Dinic, J. Adler and I. Parmryd, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2010, 1801, 625–634 CrossRef CAS PubMed.
- E. P. C. Kilsdonk, P. G. Yancey, G. W. Stoudt, F. W. Bangerter, W. J. Johnson, M. C. Phillips and G. H. Rothblat, J. Biol. Chem., 1995, 270, 17250–17256 CrossRef CAS PubMed.
- F. M. Goni, Chem. Phys. Lipids, 2019, 218, 34–39 CrossRef CAS PubMed.
- K. Simons and M. J. Gerl, Nat. Rev. Mol. Cell Biol., 2010, 11, 688–699 CrossRef CAS PubMed.
- A. Honigmann, V. Mueller, H. Ta, A. Schoenle, E. Sezgin, S. W. Hell and C. Eggeling, Nat. Commun., 2014, 5, 5412 CrossRef CAS PubMed.
- D. M. Owen, D. J. Williamson, A. Magenau and K. Gaus, Nat. Commun., 2012, 3, 1256 CrossRef PubMed.
- Q. Y. Yan, Y. T. Lu, L. L. Zhou, J. L. Chen, H. J. Xu, M. J. Cai, Y. Shi, J. G. Jiang, W. Y. Xiong, J. Gao and H. D. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7033–7038 CrossRef CAS PubMed.
- Y. Shan and H. Wang, Chem. Soc. Rev., 2015, 44, 3617–3638 RSC.
- S. Chiantia, N. Kahya, J. Ries and P. Schwille, Biophys. J., 2006, 90, 4500–4508 CrossRef CAS PubMed.
- S. R. Shaikh, V. Cherezov, M. Caffrey, S. P. Soni, D. LoCascio, W. Stillwell and S. R. Wassall, J. Am. Chem. Soc., 2006, 128, 5375–5383 CrossRef CAS PubMed.
- M. M. Lozano, Z. Liu, E. Sunnick, A. Janshoff, K. Kumar and S. G. Boxer, J. Am. Chem. Soc., 2013, 135, 5620–5630 CrossRef CAS PubMed.
- M. M. Lozano, J. S. Hovis, F. R. Moss and S. G. Boxer, J. Am. Chem. Soc., 2016, 138, 9996–10001 CrossRef CAS PubMed.
- S. L. Veatch, O. Soubias, S. L. Keller and K. Gawrisch, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17650–17655 CrossRef CAS PubMed.
- H. B. Bhat, T. Kishimoto, M. Abe, A. Makino, T. Inaba, M. Murate, N. Dohmae, A. Kurahashi, K. Nishibori, F. Fujimori, P. Greimel, R. Ishitsuka and T. Kobayashi, J. Lipid Res., 2013, 54, 2933–2943 CrossRef CAS PubMed.
- M. Kinoshita, K. G. N. Suzuki, N. Matsumori, M. Takada, H. Ano, K. Morigaki, M. Abe, A. Makino, T. Kobayashi, K. M. Hirosawa, T. K. Fujiwara, A. Kusumi and M. Murata, J. Cell Biol., 2017, 216, 1183–1204 CrossRef CAS PubMed.
- A. S. Klymchenko and R. Kreder, Cell Biol., 2014, 21, 97–113 CAS.
- T. Harder, P. Scheiffele, P. Verkade and K. Simons, J. Cell Biol., 1998, 141, 929–942 CrossRef CAS PubMed.
- E. Sezgin, I. Levental, M. Grzybek, G. Schwarzmann, V. Mueller, A. Honigmann, V. N. Belov, C. Eggeling, U. Coskun, K. Simons and P. Schwille, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 1777–1784 CrossRef CAS PubMed.
- P. Sengupta, T. Jovanovic-Talisman, D. Skoko, M. Renz, S. L. Veatch and J. Lippincott-Schwartz, Nat. Methods, 2011, 8, 969–975 CrossRef CAS PubMed.
- M. X. You, Y. F. Lyu, D. Han, L. P. Qiu, Q. L. Liu, T. Chen, C. S. Wu, L. Peng, L. Q. Zhang, G. Bao and W. H. Tan, Nat. Nanotechnol., 2017, 12, 453–459 CrossRef CAS PubMed.
- L. L. Sun, Y. J. Gao, Y. Xu, J. Chao, H. J. Liu, L. H. Wang, D. Li and C. H. Fan, J. Am. Chem. Soc., 2017, 139, 17525–17532 CrossRef CAS PubMed.
- R. Z. Peng, H. J. Wang, Y. F. Lyu, L. J. Xu, H. Liu, H. L. Kuai, Q. Liu and W. H. Tan, J. Am. Chem. Soc., 2017, 139, 12410–12413 CrossRef CAS PubMed.
- P. Chidchob, D. Offenbartl-Stiegert, D. McCarthy, X. Luo, J. N. Li, S. Howorka and H. F. Sleiman, J. Am. Chem. Soc., 2019, 141, 1100–1108 CrossRef CAS PubMed.
- K. Zhang, H. Gao, R. Deng and J. Li, Angew. Chem., Int. Ed., 2018, 131, 4840–4849 CrossRef.
- H. Li, M. Wang, T. H. Shi, S. H. Yang, J. H. Zhang, H. H. Wang and Z. Nie, Angew. Chem., Int. Ed., 2018, 57, 10226–10230 CrossRef CAS PubMed.
- A. Kurz, A. Bunge, A. K. Windeck, M. Rost, W. Flasche, A. Arbuzova, D. Strohbach, S. Mueller, J. Liebscher, D. Huster and A. Herrmann, Angew. Chem., Int. Ed., 2006, 45, 4440–4444 CrossRef CAS PubMed.
- A. Lopez and J. Liu, Langmuir, 2018, 34, 15000–15013 CrossRef CAS PubMed.
- Y. L. Chen, H. P. Liu, Y. Y. Xiong and H. X. Ju, Angew. Chem., Int. Ed., 2018, 57, 785–789 CrossRef CAS PubMed.
- N. Momin, S. Lee, A. K. Gadok, D. J. Busch, G. D. Bachand, C. C. Hayden, J. C. Stachowiak and D. Y. Sasaki, Soft Matter, 2015, 11, 3241–3250 RSC.
- L. L. Sun, Y. J. Gao, Y. G. Wang, Q. Wei, J. Y. Shi, N. Chen, D. Li and C. H. Fan, Chem. Sci., 2018, 9, 5967–5975 RSC.
- R. Z. Peng, X. F. Zheng, Y. F. Lyu, L. J. Xu, X. B. Zhang, G. L. Ke, Q. L. Liu, C. J. You, S. Y. Huan and W. H. Tan, J. Am. Chem. Soc., 2018, 140, 9793–9796 CrossRef CAS PubMed.
- P. Shi, N. Zhao, J. P. Lai, J. Coyne, E. R. Gaddes and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 6800–6804 CrossRef CAS PubMed.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- N. Komura, K. G. N. Suzuki, H. Ando, M. Konishi, M. Koikeda, A. Imamura, R. Chadda, T. K. Fujiwara, H. Tsuboi, R. Sheng, W. Cho, K. Furukawa, K. Furukawa, Y. Yamauchi, H. Ishida, A. Kusumi and M. Kiso, Nat. Chem. Biol., 2016, 12, 402–410 CrossRef CAS PubMed.
- B. Ramstedt and J. P. Slotte, Biophys. J., 1999, 76, 908–915 CrossRef CAS PubMed.
- K. W. Dunn, M. M. Kamocka and J. H. McDonald, Am. J. Physiol.: Cell Physiol., 2011, 300, C723 CrossRef CAS PubMed.
- M. Yanez-Mo, O. Barreiro, M. Gordon-Alonso, M. Sala-Valdes and F. Sanchez-Madrid, Trends Cell Biol., 2009, 19, 434–446 CrossRef CAS PubMed.
- D. A. Brown and J. K. Rose, Cell, 1992, 68, 533–544 CrossRef CAS PubMed.
- T. Harder, P. Scheiffele, P. Verkade and K. Simons, J. Cell Biol., 1998, 141, 929–942 CrossRef CAS PubMed.
- X. Cheng and J. C. Smith, Chem. Rev., 2019, 119, 5849–5880 CrossRef CAS PubMed.
- C. D. Tsoukas, B. Landgraf, J. Bentin, M. Valentine, M. Lotz, J. H. Vaughan and D. A. Carson, J. Immunol., 1985, 135, 1719–1723 CAS.
- M. F. V. F. Neurath, B. L. Kelsall, E. Stuber and W. Strober, J. Exp. Med., 1995, 182, 1281–1290 CrossRef CAS PubMed.
- S. Zumerle, B. Molon and A. Viola, Front. Immunol., 2017, 8, 1467 CrossRef PubMed.
- B. J. C. Quah, H. S. Warren and C. R. Parish, Nat. Protoc., 2007, 2, 2049–2056 CrossRef CAS PubMed.
- A. B. Lyons, J. Immunol. Methods, 2000, 243, 147–154 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06203c |
| ‡ L. L. Sun, and Y. Y. Su equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2020 |