
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLipshutz-type bis(amido)argentates for directed ortho argentation†
Noriyuki
Tezuka
 *ab,
Keiichi
Hirano
*ab,
Andrew J.
Peel
c,
Andrew E. H.
Wheatley
c,
Kazunori
Miyamoto
ab and
Masanobu
Uchiyama
*abd
*ab,
Keiichi
Hirano
*ab,
Andrew J.
Peel
c,
Andrew E. H.
Wheatley
c,
Kazunori
Miyamoto
ab and
Masanobu
Uchiyama
*abd
aGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: tezuka-noriyuki@g.ecc.u-tokyo.ac.jp; k1hirano@mol.f.u-tokyo.ac.jp; uchiyama@mol.f.u-tokyo.ac.jp
bCluster of Pioneering Research (CPR), Advanced Elements Chemistry Laboratory, RIKEN, 2-1 Hirosawa, Wako-shi, Saitama 351-0198, Japan
cDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK
dResearch Initiative for Supra-Materials (RISM), Shinshu University, 3-15-1 Tokida, Ueda, Nagano 386-8567, Japan
First published on 7th January 2020
Abstract
Bis(amido)argentate (TMP)2Ag(CN)Li2 (3, TMP-Ag-ate; TMP = 2,2,6,6-tetramethylpiperidido) was designed as a tool for chemoselective aromatic functionalization via unprecedented directed ortho argentation (DoAg). X-Ray crystallographic analysis showed that 3 takes a structure analogous to that of the corresponding Lipshutz cuprate. DoAg with this TMP-Ag-ate afforded multifunctional aromatics in high yields in processes that exhibited high chemoselectivity and compatibility with a wide range of functional groups. These included organometallics- and transition metal-susceptible substituents such as methyl ester, aldehyde, vinyl, iodo, (trifluoromethanesulfonyl)oxy and nitro groups. The arylargentates displayed good reactivity with various electrophiles. Chalcogen (S, Se, and Te) installation and azo coupling reactions also proceeded efficiently.
Introduction
Copper and silver are categorized as congeneric elements, being vertically adjacent in the periodic table. They have many similarities, including electronegativity (Cu 1.9 vs. Ag 1.9)1 and coordination geometry. However, whereas organocopper species have opened up many new avenues in organic synthesis,2 the silver analogues are quite poorly characterized,3 partly due to a lack of efficient preparative methods (Fig. 1a). For example, organocopper (R–Cu(I)) compounds are commonly prepared by the oxidative cupration of organic halides with Cu(0).4 Unfortunately, this method cannot be applied to the preparation of organosilver (R–Ag(I)) compounds, because the Ag(0 → I) redox potential is 0.8 V (Fig. 1b).5,6 Neither hydro (or carbo)-metalation nor halogen–metal exchange, which are also effective methods for the preparation of organocopper compounds, have been effectively applied to organosilver chemistry to date.7 Transmetalation from organometallics to an Ag(I) source is another route offering potential access to the corresponding organosilver species.7b,c,8 However, the high reactivity (including wrt silver salts) of commonly used organolithiums and organomagnesiums severely restricts the employment of functionalized aromatic compounds as substrates. Directed ortho metalation (DoM) is another synthetic option for the regioselective preparation of aryl metal compounds9,10 and could offer a route to aromatic silver complexes of a type that have recently received attention as potential reaction intermediates.8l–n Here we present an unprecedented directed ortho argentation (DoAg) reaction using a newly designed silver ate base, and demonstrate its applicability to the derivatization of a range of functionalized aromatics.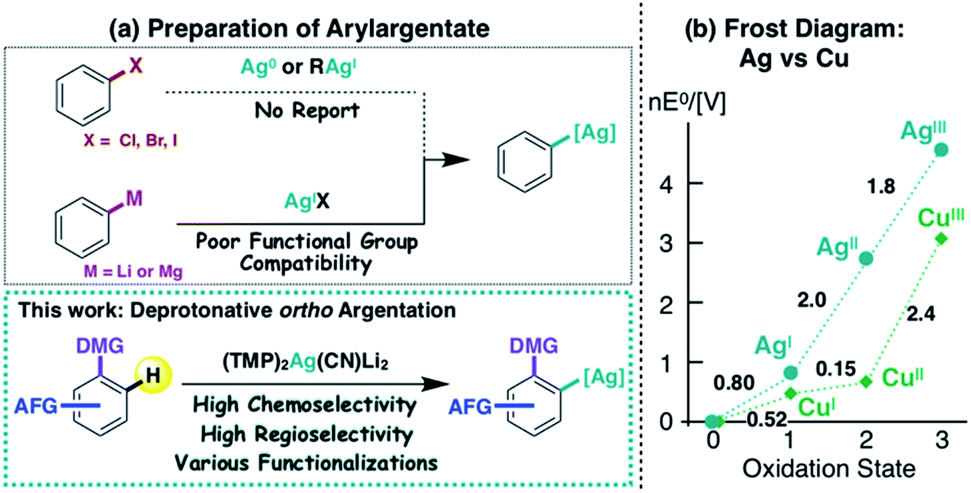 | ||
| Fig. 1 (a) Preparative strategies for arylargentates, and (b) comparison of electrochemical properties of copper and silver. | ||
Results and discussion
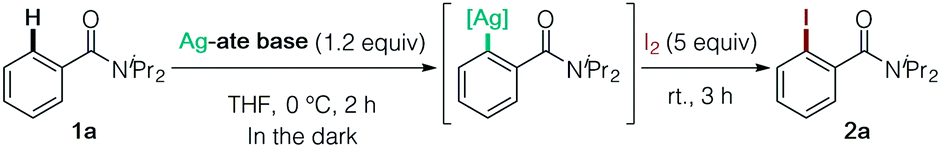
We initially screened Ag-ate bases prepared by combining a range of silver salts and amido ligands for DoAg using N,N-diisopropylbenzamide (1a) as a model substrate, followed by trapping with iodine (Table 1). The use of silver(I) salts with equimolar TMPLi failed to provide the corresponding iodinated product 2a (entries 1 and 2). We next assessed the reactivity of TMP-argentates prepared from several silver sources using a two-fold excess of TMPLi (entries 3–6). (TMP)2Ag(CN)Li23, generated from silver cyanide, uniquely gave the desired product 2a,11 whereas other TMP-argentates were totally unproductive (entry 6 vs. entries 3–5). Moreover, 3 was capable of deprotonating two equivalents of substrate 1a (entry 7). From the viewpoint of practical use, it is important to emphasize that AgCN is comparable to CuCN in price.12 Next, we examined amido ligands and solvents. The HMDS-argentate failed to give 2a, presumably due to its low basicity (entry 8). Meanwhile, in contrast to the poor yield obtained with (iPr2N)2Ag(CN)Li2,132a was obtained quantitatively by using (Cy2N)2Ag(CN)Li2 (entries 9 vs. 10). This result is practically of great importance in terms of cost reduction.14 The heteroleptic amidoargentate Me(TMP)Ag(CN)Li2 proved unproductive (entry 11).15 Lastly, THF was found to be a particularly effective solvent for DoAg (entry 6 vs. entries 12–15).16 It is worth noting that neither 3 nor the putative intermediate arylargentate generated was sensitive to light, meaning that all DoAg operations can be conducted without shading tools (entry 16). Based on these data, all subsequent experiments were conducted without special precautions to exclude light.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Ag-ate base | Solvent | Yield (%) | Entry | Ag-ate base | Solvent | Yield (%) |
| a NMR yields based on mesitylene as an internal standard. Isolated yield in parentheses. ND: not detected. b Ag-ate base (0.5 equiv.). c Deprotonation conducted at room temperature. d Exposed to light. TMP: 2,2,6,6-tetramethylpiperidido. TfO: trifluoromethanesulfonato. Cy: cyclohexyl. HMDS: 1,1,1,3,3,3-hexamethyldisilazido. | |||||||
| 1 | (TMP)Ag(NO3)Li | THF | ND | 9 | (iPr2N)2Ag(CN)Li2 | THF | 11 |
| 2 | (TMP)Ag(CN)Li | THF | ND | 10 | (Cy2N)2Ag(CN)Li2 | THF | 99 |
| 3 | (TMP)2Ag(NO3)Li2 | THF | ND | 11 | Me(TMP)Ag(CN)Li2 | THF | ND |
| 4 | (TMP)2Ag(1/2·CO3)Li2 | THF | ND | 12 | (TMP)2Ag(CN)Li2 | Et2O | 31 |
| 5 | (TMP)2Ag(OTf)Li2 | THF | ND | 13c | (TMP)2Ag(CN)Li2 | Dioxane | ND |
| 6 | (TMP)2Ag(CN)Li2 | THF | 99 (92) | 14 | (TMP)2Ag(CN)Li2 | Benzene | 49 |
| 7b | (TMP)2Ag(CN)Li2 | THF | 93 | 15 | (TMP)2Ag(CN)Li2 | Hexane | 46 |
| 8 | (HMDS)2Ag(CN)Li2 | THF | ND | 16d | (TMP)2Ag(CN)Li2 | THF | 99 |
The structure of the THF-solvate of (TMP)2Ag(CN)Li23 was obtained by X-ray crystallography and corroborated by DFT calculations (Fig. 2). Single crystals were obtained from a hexane/toluene solution of 3 prepared from AgCN and TMPLi in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.17 Complex 3(THF) has an almost linear –Ag– geometry (176.01(8)°) and the two lithium centers are bridged by cyanide. The resulting lower-order structure mirrors that seen in the corresponding Lipshutz cuprate.15 However, since the related and yet relatively inert Gilman cuprate18 is also based on linear, lower-order Cu (like the cyanide-free argentate, see ESI Fig. S2†), the geometry of the group 11 element cannot alone explain the high reactivity of 3(THF) in DoAg; the inclusion of LiCN must play a vital role. Overall, the bond lengths and angles in the solid-state structure of 3(THF) are in good agreement with those obtained by DFT calculation. Meanwhile, preliminary attempts to prove the formation of aryl silver species during our proposed DoAg reactions have been initiated. With this in mind, equimolar reaction between 1a and 3 has provided evidence for the 1:1 complex of a diarylargentate and bis(TMP)argentate, presumably the result of a disproportionation (Fig. 2d). Further evidence that Gilman-type diarylargentates can form came from the 2:1 reaction of 1a with 3 (Fig. 2e). Though work is ongoing, the ability of aryl silver species to form in these systems is clear. Moreover, Ag–aryl bond lengths in our initial structures are comparable to those in diarylargentates previously reported by van Koten.8d
:2.17 Complex 3(THF) has an almost linear –Ag– geometry (176.01(8)°) and the two lithium centers are bridged by cyanide. The resulting lower-order structure mirrors that seen in the corresponding Lipshutz cuprate.15 However, since the related and yet relatively inert Gilman cuprate18 is also based on linear, lower-order Cu (like the cyanide-free argentate, see ESI Fig. S2†), the geometry of the group 11 element cannot alone explain the high reactivity of 3(THF) in DoAg; the inclusion of LiCN must play a vital role. Overall, the bond lengths and angles in the solid-state structure of 3(THF) are in good agreement with those obtained by DFT calculation. Meanwhile, preliminary attempts to prove the formation of aryl silver species during our proposed DoAg reactions have been initiated. With this in mind, equimolar reaction between 1a and 3 has provided evidence for the 1:1 complex of a diarylargentate and bis(TMP)argentate, presumably the result of a disproportionation (Fig. 2d). Further evidence that Gilman-type diarylargentates can form came from the 2:1 reaction of 1a with 3 (Fig. 2e). Though work is ongoing, the ability of aryl silver species to form in these systems is clear. Moreover, Ag–aryl bond lengths in our initial structures are comparable to those in diarylargentates previously reported by van Koten.8d
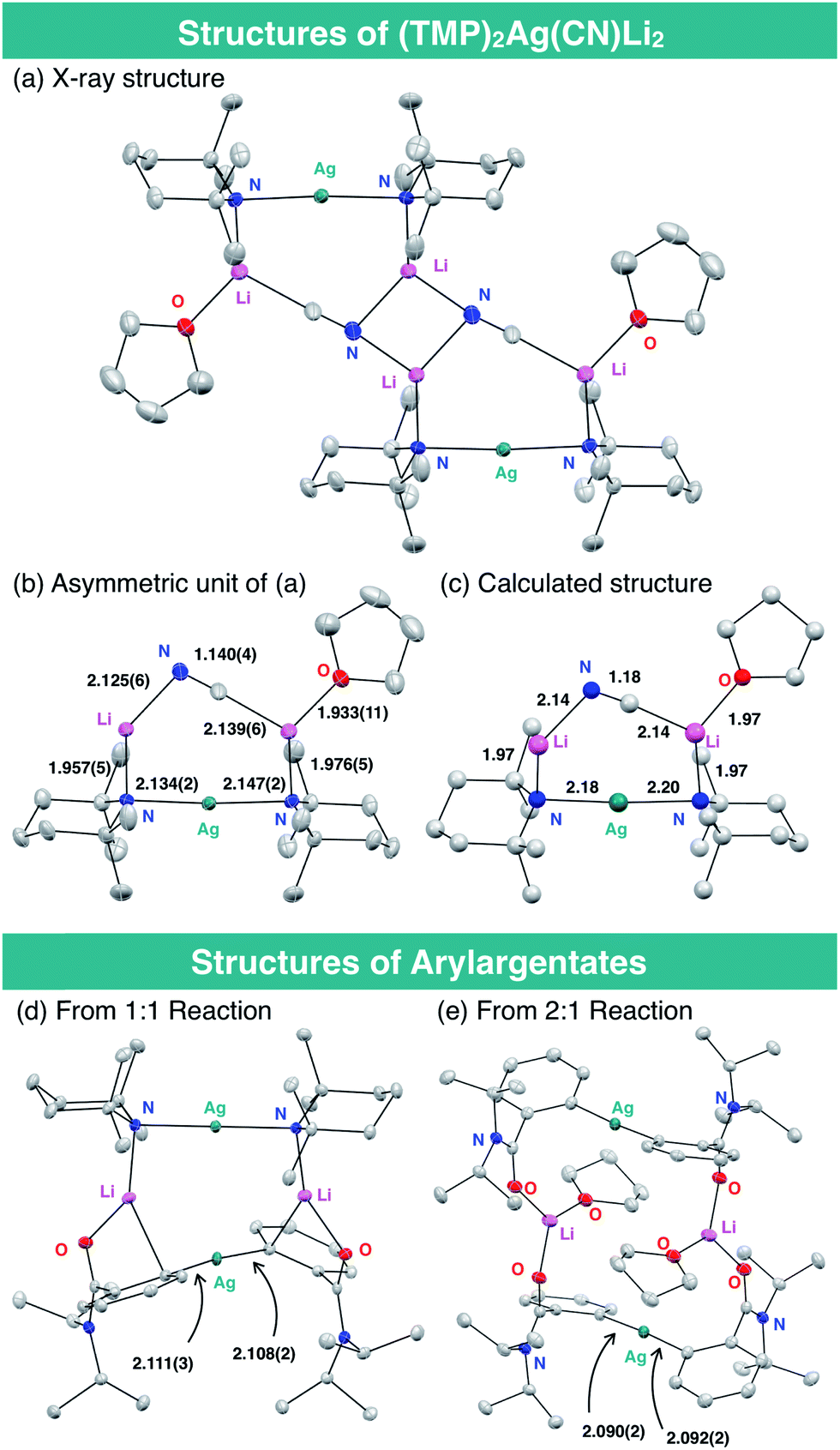 | ||
| Fig. 2 X-ray and calculated structures of argentates. (a) Crystal structure of the dimer of (TMP)2Ag(CN)Li2(THF) [3(THF)]2; (b) the asymmetric unit (monomer) from (a); (c) the asymmetric unit extracted from the structure of [3(THF)]2 calculated at the M06/6-31+G*&LanL2DZ(Ag) level; (d) crystal structure of a diarylargentate adduct obtained by 1:1 reaction of 1a and 3 and (e) of a diarylargentate dimer from the 2:1 reaction of 1a and 3. All atomic displacement parameters in crystal structures shown at 30% probability, with H atoms (and THF disorder in (a and b)) omitted. Selected bond lengths in Å. | ||
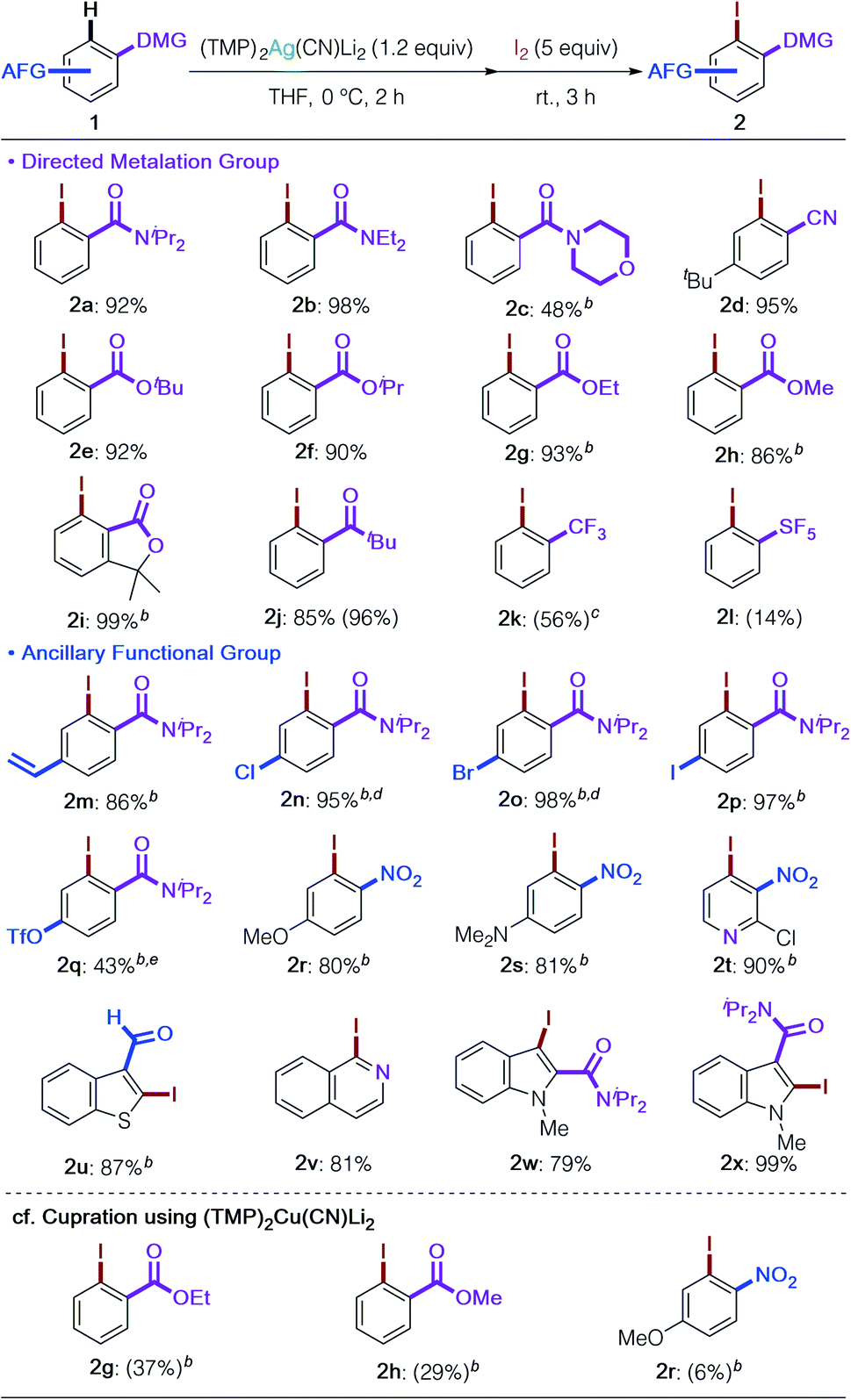
With the optimized reaction conditions and an understanding of the nature of the base in hand (Table 1, entry 16 and Fig. 2), we next examined the substrate scope of DoAg (Table 2). We first looked at the choice of DMG. It proved possible to use diethylamide 1b and morpholine amide 1c, both of which are chemically more labile than diisopropylamide 1a. Regioselective argentation of benzonitrile (1d) proceeded smoothly in 95% yield, leaving the cyano group intact. Various esters, including CO2tBu, –iPr, and –Et, were compatible with the Ag-ate base (1e–1i). Of considerable importance given that the mechanism of deprotonation in reactions involving the application of 3 is the subject of ongoing study, even methyl ester was tolerated, giving the corresponding iodinated product in high yield (2h).19 Furthermore, traditionally base-susceptible lactones remained intact (2i: 99%). The carbonyl group of a ketone also worked as DMG to give the desired product 2j without any observable side reactions. Benzotrifluoride was also usable; the corresponding iodobenzotrifluoride 2k was obtained in 56% yield with moderate ortho selectivity (78:15:7 for ortho:meta:para).20 PhSF5 was also subjected to the DoAg process and argentation took place with perfect ortho selectivity (2l). Though this reaction needs further optimization, it is remarkable that the metalation proceeded, and this strongly suggests that DoAg could find a role in the diverse functionalization of pharmaceutically important pentafluorosulfanylarenes.21,22 High ancillary functional group (AFG) tolerance is a key feature of the present argentation reaction. Styrene-type substrate 1m could be employed without observable polymerization as a side reaction. Halogens and pseudo-halogens also survived (2n–2q). Especially remarkable was the successful retention of the (trifluoromethanesulfonyl)oxy group in 2q. To our knowledge, this is the first example of this AFG surviving a deprotonative metalation reaction. The highlight of the current development of DoAg is the outstanding chemoselectivity revealed by the first directed ortho metalation of a nitroarene by an amidometal ate base. Deprotonative metalation of nitroarenes has been acknowledged for many years to be especially difficult due to incompatibility between the nitro group and strong bases as well as the instability of generated arylmetals.23 Thus, until now only specific substrates have enabled this chemistry.24 In contrast, our argentate successfully achieved the conversion of both electron-rich and electron-deficient nitro(hetero)arenes in high yields with perfect regiocontrol next to the nitro group in the presence of other potential DMGs (2r–2t). Our work has also demonstrated that an aldehyde function is tolerant to the use of our Ag-ate (2u).19,24b,c,25 Heteroarenes such as isoquinoline and indoles, with DMGs at the 2- or 3-positions, were also transformed into the desired products in high yields (2v–2x). As expected, (TMP)2Ag(CN)Li23 exhibited a stark difference in chemoselectivity in comparison to its cuprous counterpart, (TMP)2Cu(CN)Li2.15 Upon cupration, sterically unshielded methyl and ethyl esters suffered from uncontrollable side reactions (2g and 2h) and the nitroarene gave only trace amounts of the iodined product 2r due to the redox activity of Cu.
We next investigated the synthetic use of the putative arylargentate 4a in aromatic functionalization (Scheme 1). Reactions with benzoyl chloride (5), allyl bromide (6), D2O (7), trimethylsilyl chloride (TMSCl: 8)26 and N-chlorophthalimide (NCPI: 9) proceeded well. The reactivity with NCPI was particularly noteworthy, since the alternative use of redox active (TMP)2Cu(CN)Li2 led to significantly less of the desired product on account of undesired oxidative biaryl formation (9:60% and biaryl: 35% NMR yields). Unfortunately, Pd- and Ni-catalyzed cross-coupling reactions8l–n have not yet proved successful.
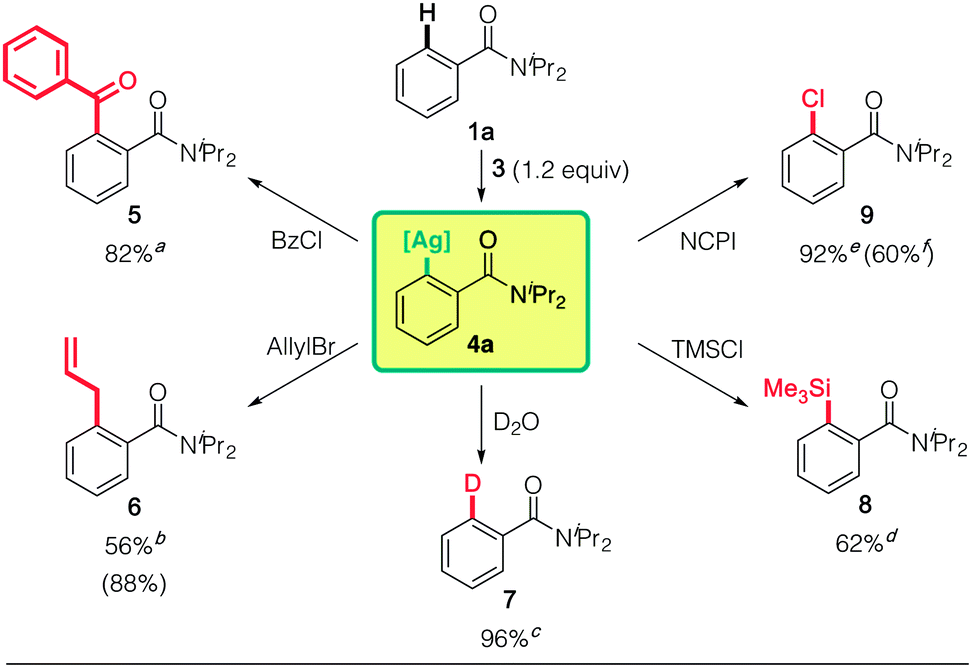 | ||
| Scheme 1 Reaction scope of arylargentate 4a. Isolated yields. DoAg was conducted with (TMP)2Ag(CN)Li2 (3) (1.2 equiv.), 0 °C, 2 h. aBzCl (3.5 equiv.), 80 °C, 16 h. bAllylBr (5.0 equiv.), 80 °C, 16 h.; NMR yield in parentheses. cD2O (55 equiv.), rt., 16 h.; D/H = 97/3. dTMSCl (5.0 equiv.), 80 °C, 16 h. eDoAg for 0.5 h.; NCPI (3.0 equiv.), rt., 1 h. fNMR yield when using (TMP)2Cu(CN)Li2 instead of 3. Bz: benzoyl. TMS: trimethylsilyl. NCPI: N-chlorophthalimide. | ||
Interestingly, putative 4a is reluctant to react with some typical electrophiles, such as methyl iodide and benzyl bromide (see ESI Table S1†). This can be attributed to the high Ag(I → III) redox potential, and results in distinctly different reactivity from the SN2 processes of organocopper reagents, where the positive charge of the Cu center first increases and then decreases as it goes from Cu(I) to Cu(III) and then back to Cu(I).27
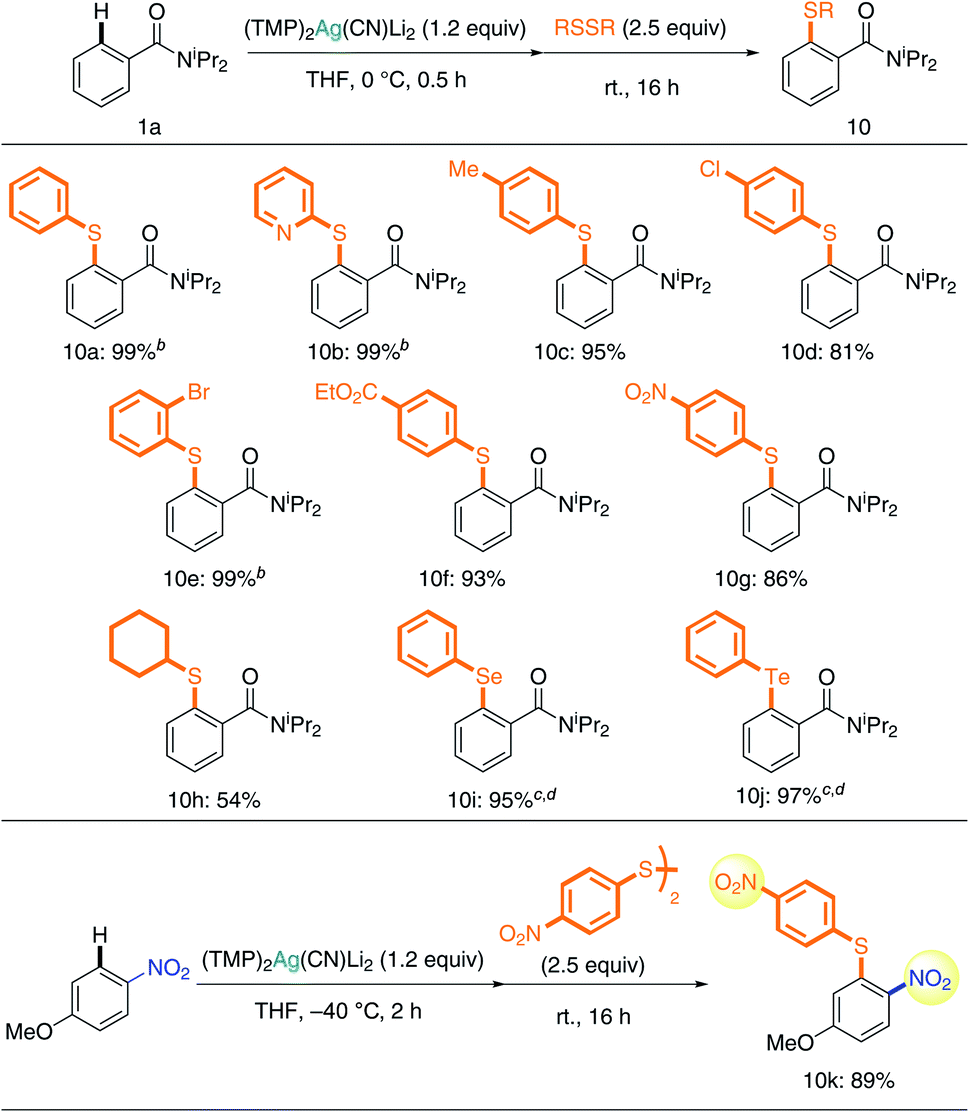
A study of chalcogen introduction reactions showed that disulfides are excellent substrates for TMP-Ag-ate reaction (Table 3). Unsymmetrically (hetero)arylated sulfides 10a and 10b were obtained quantitatively. Excellent chemoselectivity was also observed during reactions with disulfide; benzyl (10c), chloro (10d), bromo (10e), ester (10f), and even nitro groups (10g) were tolerated, and the corresponding sulfides were obtained in high yields. Taking advantage of the unusual tolerance of the nitro group to our Ag-ate (vide supra), the synthesis of diaryl sulfide 10k with nitro groups on both aryl rings was achieved. In the case of an alkyl substituent, dicyclohexyl disulfide, 10h was obtained in 54% yield. Diaryl sulfides are important chemical units in medicinal chemistry research.28 Though the reaction of aryllithium or -magnesium reagents with disulfides can be used to access diaryl sulfides, the yields are variable and the AFG compatibility is low, due to the nature of these organometallics.29,30 In contrast, our more tolerant argentate-based methodology is a powerful new tool for obtaining diverse diaryl sulfides. Moreover, it allows chalcogen introduction to be extended to the preparation of diaryl selenides and tellurides31 (10i and 10j).
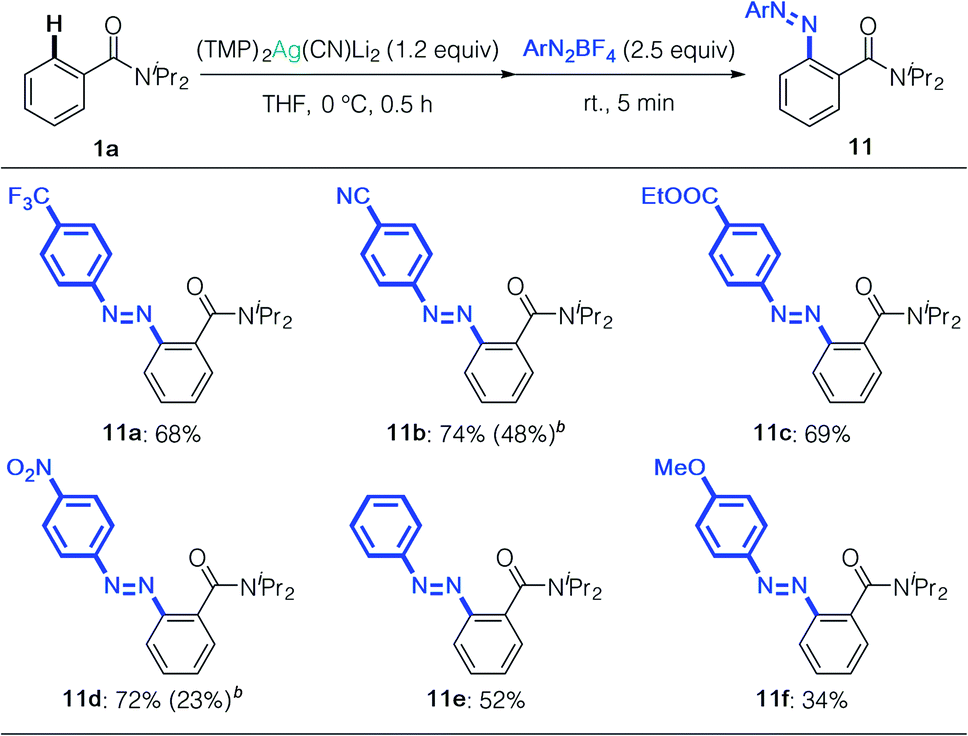
We next focused on the synthesis of azo compounds from diazonium salts. Arylmetals generally react with diazoniums in SNAr or SET fashion (Sandmeyer-type reaction) and electron-neutral or electron-deficient arenes do not react with diazonium salts by themselves.32 Minakata's oxidative aniline cross-coupling represents a remarkable advance in this field, albeit with unavoidable formation of the homo-coupled product.33 We treated 4a with phenyldiazonium tetrafluoroborate, and obtained the corresponding azo compound 11a in 68% yield (Table 4). This approach selectively produced heterocoupling products, and cyano, ester, and even nitro groups remained intact during the reaction, affording the corresponding azo compounds with two electron-deficient arenes in high yields (11b–11d). Aryldiazonium salts without electron-withdrawing groups reacted selectively to give 11e and 11f. As expected, reference experiments showed that the arylcuprate prepared from 1a and (TMP)2Cu(CN)Li2 formed biaryl by-products, which strongly suggests that the oxidation of copper competed in this case – something we were able to obviate by using silver.
Conclusions
In summary, we have developed a method for the highly chemo- and regioselective deprotonative argentation of a range of (hetero)arenes by designing TMP-Ag-ate base 3, the structure of which is consistent with its deprotonation efficacy. The resultant putative arylargentate 4a exhibited excellent reactivity towards electrophiles, including disulfide, diselenide, and ditelluride, with high ancillary functional group tolerance. Deprotonative argentation was also found to be efficient for preparing unsymmetrically substituted azo compounds in combination with diazonium tetrafluoroborates. Further theoretical and spectroscopic studies to expand the reaction scope and to investigate the physicochemical properties of these unique aromatic silver compounds are underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by JSPS Grant-in-Aid for Scientific Research on Innovative Areas (No. 17H05430), JSPS KAKENHI (S) (No. 17H06173), NAGASE Science Technology Foundation, Sumitomo Foundation (to M. U.), JSPS Grant-in-Aid for Young Scientists (A) (No. 16H06214), Challenging Research (Exploratory) (No. 18K19390), the Uehara Memorial Foundation, and the Naito Foundation (to K. H.), and Grant-in-Aid for JSPS Fellows (No. 18J12045) and Graduate Program for Leaders in Life Innovation, The University of Tokyo Life Innovation Leading Graduate School from MEXT, Japan (to N. T.). Support also came from the U.K. EPSRC through grants EP/J500380/1 and EP/N509620/1 (to A. J. P.) and also EP/K039520/1. RIKEN Integrated Cluster of Clusters (RICC), HOKUSAI-GreatWave and HOKUSAI-BigWaterfall provided the computer resources for the DFT calculations.Notes and references
- (a) L. Pauling, J. Am. Chem. Soc., 1932, 54, 3570–3582 CrossRef CAS; (b) Handbook of Chemistry: Pure Chemistry, ed. Chem. Soc. Japan, Maruzen, Tokyo, 1966 Search PubMed.
- For a review on organocopper chemistry, see: (a) Modern Organocopper Chemistry, ed. N. Krause, Wiley-VCH Verlag GmbH, Weinheim, 2002 Search PubMed. Conjugate addition is a key feature of organocuprates, see: (b) N. Yoshikai and E. Nakamura, Chem. Rev., 2012, 112, 2339–2372 CrossRef CAS PubMed; (c) S. Woodward, Chem. Soc. Rev., 2000, 29, 393–401 RSC. The reactions between organocoppers and multiple bonds have been investigated in detail, see: (d) H. Yoshida, ACS Catal., 2016, 6, 1799–1811 CrossRef CAS; (e) Y. Shimizu and M. Kanai, Tetrahedron Lett., 2014, 55, 3727–3737 CrossRef CAS.
- The numbers of reports on “Argentate” vs. “Cuprate”: 3760 vs. 97269, SciFinder®, Jan 1st 2020.
- R. D. Rieke, D. E. Stack, B. T. Dawson and T.-C. Wu, J. Org. Chem., 1993, 58, 2483–2491 CrossRef CAS.
- P. Vanýsek, in CRC Handbook of Chemistry and Physics, ed. D. W. H. Lide, CRC Press, Boca Raton, 89th edn, 2005, pp. 8-20–8-29 Search PubMed.
- A. A. Frost, J. Am. Chem. Soc., 1951, 73, 2680–2682 CrossRef CAS.
- While hydroboration of alkynes via borylargentation has been developed by Yoshida and coworkers, this reaction mode is still rare for silver. See: (a) H. Yoshida, I. Kageyuki and K. Takaki, Org. Lett., 2014, 16, 3512–3515 CrossRef CAS PubMed. Tyrra and Naumann revealed fluoroargentation and halogen-metal exchange of extremely electron deficient substrates, see: (b) W. Tyrra and D. Naumann, J. Fluorine Chem., 2004, 125, 823–830 CrossRef CAS. Seminal work on carboargentation across conjugated triple bonds was reported by Vermeer, see: (c) H. Kleijn, M. Tigchelaar, J. Meijer and P. Vermeer, Recl. Trav. Chim. Pays-Bas, 1981, 100, 337–341 CrossRef CAS and references therein.
- For the representative reports on the synthesis of organosilvers via transmetalation, see: (a) S. Weske, R. A. Hardin, T. Auth, R. A. J. O'Hair, K. Koszinowski and C. A. Ogle, Chem. Commun., 2018, 54, 5086–5089 RSC; (b) D. Joven-Sancho, M. Baya, A. Martín and B. Menjón, Chem.–Eur. J., 2018, 24, 13098–13101 CrossRef CAS PubMed; (c) S. Martínez de Salinas, Á. L. Mudarra, J. Benet-Buchholz, T. Parella, F. Maseras and M. H. Pérez-Temprano, Chem.–Eur. J., 2018, 24, 11895–11898 CrossRef PubMed; (d) C. M. P. Kronenburg, J. T. B. H. Jastrzebski, J. Boersma, M. Lutz, A. L. Spek and G. van Koten, J. Am. Chem. Soc., 2002, 124, 11675–11683 CrossRef CAS PubMed; (e) C.-S. Hwang and P. P. Power, J. Organomet. Chem., 1999, 589, 234–238 CrossRef CAS; (f) T. Kauffmann, C. Neiteler and S. Robbe, Chem. Ber., 1992, 125, 2409–2418 CrossRef CAS; (g) C. Eaborn, P. B. Hitchcock, J. D. Smith and A. C. Sullivan, J. Chem. Soc., Chem. Commun., 1984, 870–871 RSC; (h) J. Blenkers, H. K. Hofstee, J. Boersma and G. J. M. van der Kerk, J. Organomet. Chem., 1979, 168, 251–258 CrossRef CAS; (i) H. K. Hofstee, J. Boersma and G. J. M. van der Kerk, J. Organomet. Chem., 1978, 168, 241–249 CrossRef; (j) E. Krause and M. Schmitz, Ber. Dtsch. Chem. Ges., 1919, 52, 2150–2164 CrossRef. Decarboxylative preparation of diphenyl argentate have been reported as well, see: (k) M. I. S. Röhr, J. Petersen, C. Brunet, R. Antoine, M. Broyer, P. Dugourd, V. Bonačić-Koutecký, R. A. J. O'Hair and R. Mitrić, J. Phys. Chem. Lett., 2012, 3, 1197–1201 CrossRef PubMed. Deprotonative argentation in Pd-catalyzed cross-coupling system is also proposed, see: (l) M. D. Lotz, N. M. Camasso, A. J. Canty and M. S. Sanford, Organometallics, 2017, 36, 165–171 CrossRef CAS; (m) S. Y. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 15278–15284 CrossRef CAS PubMed; (n) D. Whitaker, J. Burés and I. Larrosa, J. Am. Chem. Soc., 2016, 138, 8384–8387 CrossRef CAS PubMed.
- For a comprehensive review on DoM reactions, see: (a) J. Board, J. L. Cosman, T. Rantanen, S. P. Singh and V. Snieckus, Platinum Met. Rev., 2013, 57, 234–258 CrossRef; (b) V. Snieckus and T. Macklin, in Handbook of C–H Transformations: Applications in Organic Synthesis, ed. G. Dyker, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2008, vol. 1, pp. 106–115 Search PubMed; (c) V. Snieckus, Chem. Rev., 1990, 90, 879–933 CrossRef CAS; (d) P. Beak and V. Snieckus, Acc. Chem. Res., 1982, 15, 306–312 CrossRef CAS. For stereo- and regiocontrolled deprotonative metalation of various types of C–H bonds by utilizing complex-induced proximity effects, see: (e) P. Beak and A. I. Meyers, Acc. Chem. Res., 1986, 19, 356–363 CrossRef CAS.
- For a review on deprotonative metalation using metal amide bases, see: (a) F. Chevallier, F. Mongin, R. Takita and M. Uchiyama, in Arene Chemistry, ed. J. Mortier, John Wiley & Sons, Inc, Hoboken, New Jersey, 2015, pp. 777–812 Search PubMed; (b) R. E. Mulvey, F. Mongin, M. Uchiyama and Y. Kondo, Angew. Chem., Int. Ed., 2007, 46, 3802–3824 CrossRef CAS PubMed.
- Cyanide anion should play an important roll in the deprotonation step rather than the iodination step (Scheme S1†).
- CuCN 0.84 USD per mmol (47.00 USD per 5 g) vs. AgCN 0.85 USD per mmol (63.80 USD per 10 g) at Sigma-Aldrich, Nov. 27th 2019.
- Hydride elimination from iPr2N anion might account for the low reactivity of the corresponding amidoargentate, see: B.-A. Feit, S. Shapira and A. Herbst, Tetrahedron, 1995, 51, 317–328 CrossRef CAS.
- Cy2NH (0.051 USD per mmol; 27.90 USD per 100 g) is less expensive than TMPH (0.72 USD per mmol; 128.00 USD per 25 g) at Sigma-Aldrich, Nov. 27th 2019. Knochel discussed this issue: (a) M. R. Becker and P. Knochel, Org. Lett., 2016, 18, 1462–1465 CrossRef CAS PubMed; (b) M. R. Becker, M. A. Ganiek and P. Knochel, Chem. Sci., 2015, 6, 6649–6653 RSC.
- (a) S. Usui, Y. Hashimoto, J. V. Morey, A. E. H. Wheatley and M. Uchiyama, J. Am. Chem. Soc., 2007, 129, 15102–15103 CrossRef CAS PubMed; (b) N. Tezuka, K. Shimojo, K. Hirano, S. Komagawa, K. Yoshida, C. Wang, K. Miyamoto, T. Saito, R. Takita and M. Uchiyama, J. Am. Chem. Soc., 2016, 138, 9166–9171 CrossRef CAS.
- The inability to metalate 1a in dioxane might be attributed to undesired reaction of the putative argentate with dioxane. For related work, see: J. García-Álvarez, E. Hevia, A. R. Kennedy, J. Klett and R. E. Mulvey, Chem. Commun., 2007, 2402–2404 RSC.
- THF was included while the crude 3 was processed in THF prior to recrystallization from hexane/toluene. For details, see ESI.†.
- A. J. Peel, N. Tezuka, J. M. D'Rozario, M. Uchiyama and A. E. H. Wheatley, Chem. Sci., 2019, 10, 3385–3400 RSC.
- The excellent compatibility of DoAg with nucleophile-sensitive AFGs such as ethyl and methyl esters and aldehyde (Table 2, 2g, 2h and 2u) rules out the formation of arylllithium. For the condensation of ortho-lithiated benzoates, see: (a) T. D. Krizan and J. C. Martin, J. Am. Chem. Soc., 1983, 105, 6155–6157 CrossRef CAS; (b) C. J. Upton and P. Beak, J. Org. Chem., 1975, 40, 1094–1098 CrossRef CAS; (c) W. E. Parham and Y. A. Sayed, J. Org. Chem., 1974, 39, 2053–2056 CrossRef CAS.
- J. A. Garden, D. R. Armstrong, W. Clegg, J. García-Alvarez, E. Hevia, A. R. Kennedy, R. E. Mulvey, S. D. Robertson and L. Russo, Organometallics, 2013, 32, 5481–5490 CrossRef CAS.
- SF5 (super trifluoromethyl) is an attractive functional group. Its chemical and physical properties are receiving increasing attention in the pharmaceutical sciences, see: Fluorine in Pharmaceutical and Medicinal Chemistry; Molecular Medicine and Medicinal Chemistry, ed. V. Gouverneur and K. Müller, Imperial College Press, Covent Garden, London, 2012 Search PubMed.
- NBO analysis by modeled DFT calculation estimates that the lithium argentate derived from PhSF5 has a strong interaction between Li and F (10.2 kcal mol−1), while PhCF3 has a moderate Li–F interaction (6.1 kcal mol−1) (see ESI†).
- (a) G. Bartoli, R. Dalpozzo and M. Nardi, Chem. Soc. Rev., 2014, 43, 4728–4750 RSC; (b) M. Mąkosza, Chem.–Eur. J., 2014, 20, 5536–5545 CrossRef PubMed.
- (a) W. C. Black, B. Guay and F. Scheuermeyer, J. Org. Chem., 1997, 62, 758–760 CrossRef CAS PubMed; (b) S. H. Wunderlich and P. Knochel, Angew. Chem., Int. Ed., 2007, 46, 7685–7688 CrossRef CAS PubMed; (c) T. Bresser, G. Monzon, M. Mosrin and P. Knochel, Org. Process Res. Dev., 2010, 14, 1299–1303 CrossRef CAS.
- S. P. Shahi and K. Koide, Angew. Chem., Int. Ed., 2004, 43, 2525–2527 CrossRef CAS PubMed.
- K. Murakami, K. Hirano, H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2008, 47, 5833–5835 CrossRef CAS PubMed.
- W. Nakanishi, M. Yamanaka and E. Nakamura, J. Am. Chem. Soc., 2005, 127, 1446–1453 CrossRef CAS PubMed.
- (a) G. De Martino, G. La Regina, A. Coluccia, M. C. Edler, M. C. Barbera, A. Brancale, E. Wilcox, E. Hamel, M. Artico and R. Silvestri, J. Med. Chem., 2004, 47, 6120–6123 CrossRef CAS PubMed; (b) G. Liu, J. R. Huth, E. T. Olejniczak, R. Mendoza, P. DeVries, S. Leitza, E. B. Reilly, G. F. Okasinski, S. W. Fesik and T. W. von Geldern, J. Med. Chem., 2001, 44, 1202–1210 CrossRef CAS PubMed; (c) A. Marcincal-Lefebvre, J. C. Gesquiere, C. Lemer and B. Dupuis, J. Med. Chem., 1981, 24, 889–893 CrossRef CAS.
- Several diaryl disulfide syntheses via the reaction of diaryl disulfide and either aryl lithium or aryl Grignard reagents have been reported, see: Li: (a) G. D. Figuly and J. C. Martin, J. Org. Chem., 1980, 45, 3728–3729 CrossRef CAS; (b) J. M. Muchowski and M. C. Venuti, J. Org. Chem., 1980, 45, 4798–4801 CrossRef CAS. Mg: (c) B.-X. Du, Z.-J. Quan, Y.-X. Da, Z. Zhang and X.-C. Wang, Adv. Synth. Catal., 2015, 357, 1270–1276 CrossRef CAS.
- Knochel and co-workers recently reported diaryl sulfide synthesis utilizing aromatic zinc reagents, see; Z.-B. Dong, M. Balkenhohl, E. Tan and P. Knochel, Org. Lett., 2018, 20, 7581–7584 CrossRef CAS PubMed.
- H. A. Stefani, J. M. Pena, F. Manarin, R. A. Ando, D. M. Leal and N. Petragnani, Tetrahedron Lett., 2011, 52, 4398–4401 CrossRef CAS.
- Diazo Chemistry I, ed. H. Zollinger, VCH Verlagsgesellschaft mbH, Weinheim, 1994 Search PubMed.
- (a) Y. Takeda, S. Okumura and S. Minakata, Synthesis, 2013, 45, 1029–1033 CrossRef CAS; (b) Y. Takeda, S. Okumura and S. Minakata, Angew. Chem., Int. Ed., 2012, 51, 7804–7808 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1919739, 1919740, 1957572 and 1960037. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06060j |
| This journal is © The Royal Society of Chemistry 2020 |