
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimetic O2 adsorption in an iron metal–organic framework for air separation†
Douglas A.
Reed
 a,
Dianne J.
Xiao
a,
Henry Z. H.
Jiang
a,
Khetpakorn
Chakarawet
a,
Julia
Oktawiec
a and
Jeffrey R.
Long
*abc
a,
Dianne J.
Xiao
a,
Henry Z. H.
Jiang
a,
Khetpakorn
Chakarawet
a,
Julia
Oktawiec
a and
Jeffrey R.
Long
*abc
aDepartment of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: jrlong@berkeley.edu
bDepartment of Chemical Engineering, University of California, Berkeley, CA 94720, USA
cMaterials Sciences Division, Lawrence Berkeley National Lab, Berkeley, CA 94720, USA
First published on 10th January 2020
Abstract
Bio-inspired motifs for gas binding and small molecule activation can be used to design more selective adsorbents for gas separation applications. Here, we report an iron metal–organic framework, Fe-BTTri (Fe3[(Fe4Cl)3(BTTri)8]2·18CH3OH, H3BTTri = 1,3,5-tris(1H-1,2,3-triazol-5-yl)benzene), that binds O2 in a manner similar to hemoglobin and therefore results in highly selective O2 binding. As confirmed by gas adsorption studies and Mössbauer and infrared spectroscopy data, the exposed iron sites in the framework reversibly adsorb substantial amounts of O2 at low temperatures by converting between high-spin, square-pyramidal Fe(II) centers in the activated material to low-spin, octahedral Fe(III)–superoxide sites upon gas binding. This change in both oxidation state and spin state observed in Fe-BTTri leads to selective and readily reversible O2 binding, with the highest reported O2/N2 selectivity for any iron-based framework.
Introduction
Metal–organic frameworks (MOFs) are a class of permanently porous solids built using organic linkers and inorganic nodes1–3 that have been studied for a wide variety of applications including catalysis4 and gas separations.5 The inherent tunability of metal–organic frameworks has made it possible to design structures with bio-inspired pores precisely engineered for applications in catalysis6,7 and cooperative substrate binding.8,9 The study of biomimetic gas binding to metal sites, a broad topic in the field of inorganic chemistry as a whole,10–14 can also be applied to gas adsorption and gas separations in porous frameworks.8,15–17 However, successful implementation of this strategy is somewhat challenging, as the highly specific metal–ligand bonds required for such model systems are difficult to engineer in sufficient densities to afford reasonable separation capacities, thus limiting their effectiveness as adsorbents. One process that would benefit from biomimetic gas binding is the industrial separation of air for the production of high-purity O2, which is widely used for chemical synthesis, chemical processing, and the implementation of oxyfuel combustion.18,19 Industrial air separation, dominated by the separation of O2 and N2, is primarily accomplished through energetically demanding cryogenic distillation; current adsorbent-based technologies, which are predominantly N2-selective, are not competitive at large scales.20 In contrast, biological systems perform air separations by selectively and reversibly binding O2 through various electronic transformations at transition metal binding sites, using carriers such as hemoglobin, hemocyanin, and hemerythrin.21 For example, the square pyramidal, high-spin Fe(II) heme centers in hemoglobin selectively bind O2 by undergoing both an oxidation and a spin state transition to form octahedral, low-spin Fe(III)–superoxo species. Replicating such selective binding mechanisms found in proteins is one intriguing strategy toward the design of more effective adsorbents for O2/N2 separations.Metal–organic frameworks bearing redox-active open metal sites have been investigated for the adsorption of O2,22–28 and the framework Fe2(dobdc) (dobdc4− = 2,5-dioxido-1,4-benzenedicarboxylate) reversibly binds O2 at low temperatures via charge transfer from exposed Fe(II) sites.23 However, below −50 °C the weak ligand field afforded by the framework linkers leads to only partial charge transfer to O2, generating high-spin iron sites with an oxidation state between Fe(II) and Fe(III). This Fe–O2 interaction is much weaker than that found in heme systems, and while the framework O2/N2 selectivities are much higher than traditional adsorbents studied for air separations,20 the selectivity could be further improved for practical applications. The framework PCN–224Fe, which features Fe(II) heme centers, can transform from an intermediate-spin, four-coordinate Fe(II) in the activated phase to a low-spin, five-coordinate superoxide-bound Fe(III) upon O2 adsorption, but the O2 capacity needs to be improved for separation purposes.15 A structurally flexible metal–organic framework with an electron-donating ligand environment would support the iron site electronic changes observed with O2 binding in hemoglobin, and combined with a high concentration of Fe(II) sites could consequently exhibit improved O2/N2 selectivities and capacities.
Recently, we reported the material Fe-BTTri (Fe3[(Fe4Cl)3(BTTri)8]2·18CH3OH, H3BTTri = 1,3,5-tris(1H-1,2,3-triazol-5-yl)benzene), which features Fe(II) centers bound in an environment similar to hemoglobin iron sites.29 This framework has been shown to accommodate both high- and low-spin iron centers, making it an excellent candidate to study the various electronic transformations required in biomimetic O2 binding. Here, we demonstrate that Fe-BTTri can indeed undergo an electronic transformation similar to that of hemoglobin upon binding oxygen, resulting in O2/N2 selectivities that are substantially higher than other iron-based adsorbents.
Results and discussion
The framework Fe-BTTri was synthesized and activated according to a previously reported procedure.29 Similar to hemoglobin, the material features five-coordinate iron centers in a square pyramidal geometry, with equatorial sites occupied by N-atom heterocyclic donor ligands and an open coordination site for O2 binding (Fig. 1). Previous structural, spectroscopic, and magnetic studies of Fe-BTTri have shown that the high-spin Fe(II) sites in the activated phase are present in sufficiently high density to allow for large gas adsorption capacities.29 The material is irreversibly oxidized upon exposure to air, but similar to Fe2(dobdc), the O2 adsorption is selective and reversible under more controlled, anhydrous conditions.23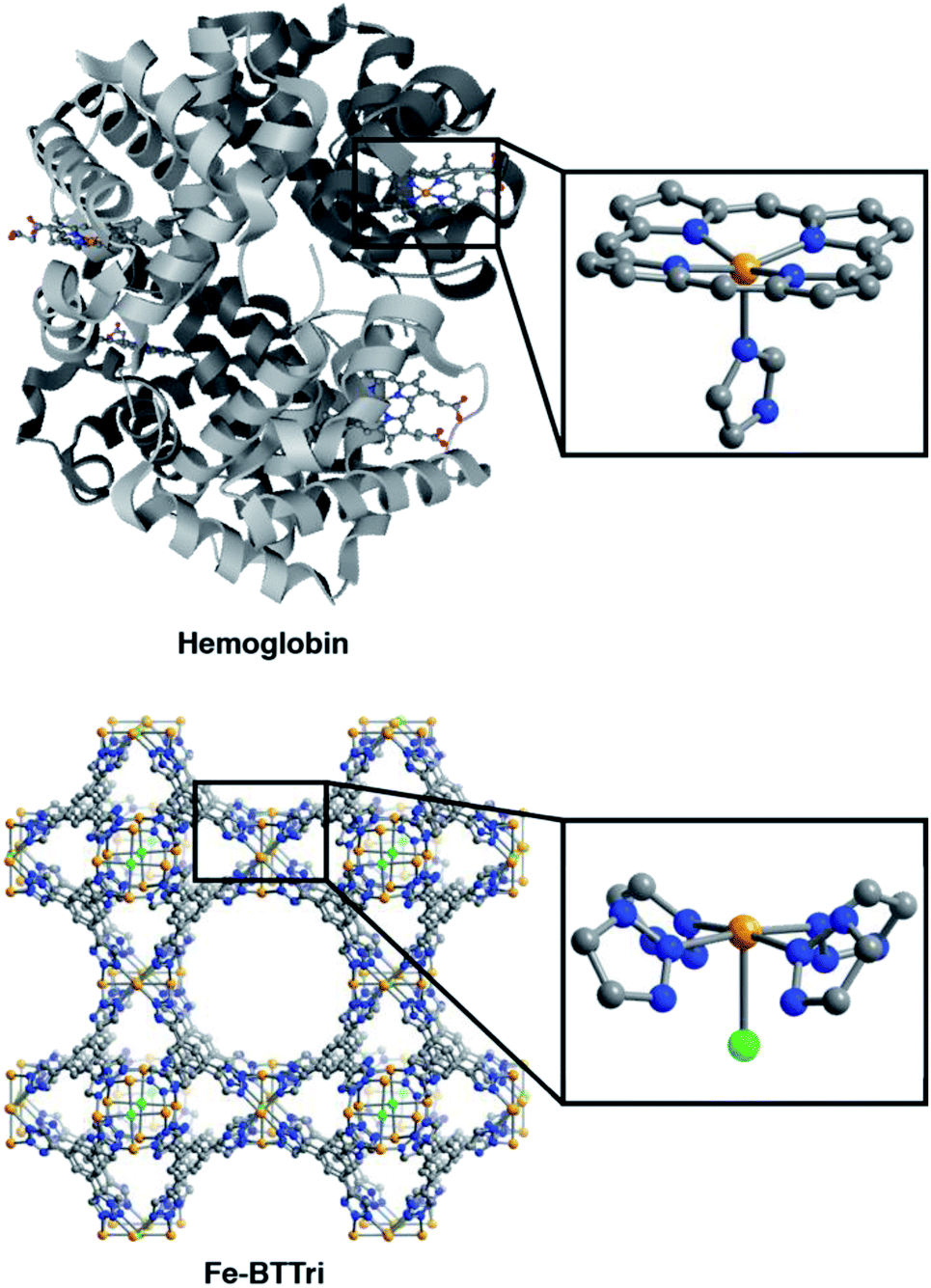 | ||
| Fig. 1 Structures of hemoglobin (top)42 and Fe-BTTri (bottom),29 with expanded views of the individual iron coordination environments in each material. Orange, grey, blue, red, and green spheres represent Fe, C, N, O, and Cl atoms, respectively; H-atoms and solvent molecules are omitted for clarity. | ||
The O2 adsorption isotherm for Fe-BTTri collected at −78 °C features an initial steep rise to a capacity of 1.2 mmol g−1 at just 100 μbar followed by a more gradual increase to a capacity of 5.9 mmol g−1 at 1 bar (Fig. 2). Importantly, at 210 mbar, corresponding to the partial pressure of O2 in air, the adsorption capacity is a substantial 3.3 mmol g−1, or approximately 10 wt% of O2, highlighting the potential of this material for practical separations. This capacity corresponds to adsorption of O2 at 80% of the framework iron sites (excluding charge balancing extra-framework Fe(II) cations). At this temperature, powder crystalline samples of the material remain highly crystalline when exposed to O2 (Fig. S6†). Moreover, the powder X-ray diffraction (PXRD) data show that the unit cell volume decreases relative to the bare framework, indicative of a structural contraction upon O2 adsorption (Fig. S6†).
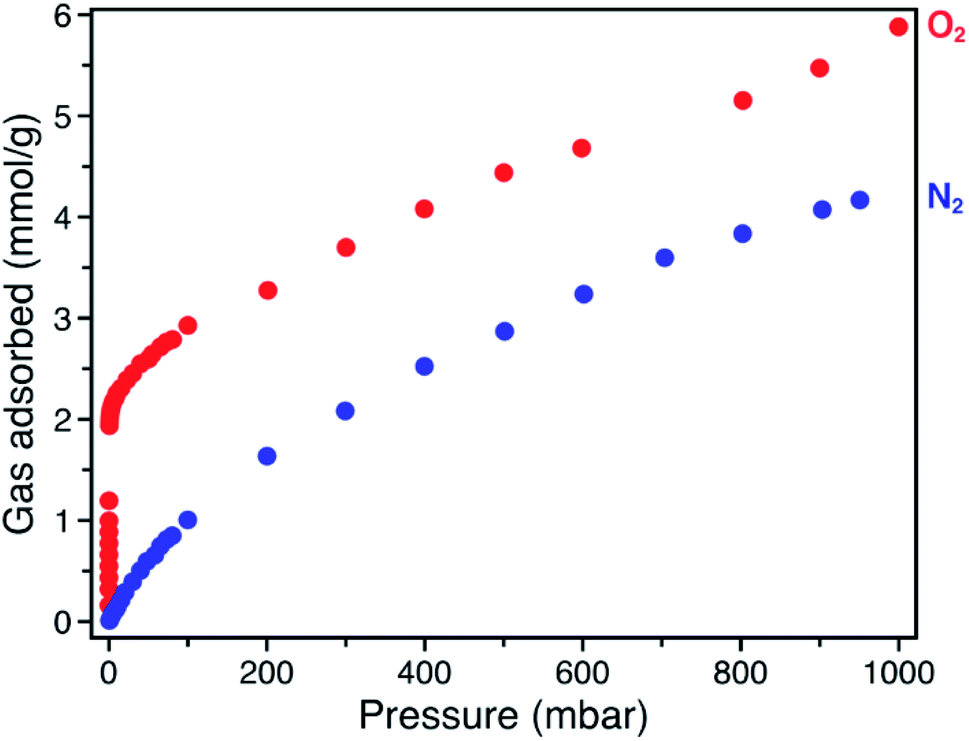 | ||
| Fig. 2 Gas adsorption isotherms of O2 (red) and N2 (blue), collected at −78 °C for Fe-BTTri. | ||
We first turned to Mössbauer spectroscopy to investigate a possible iron redox or spin state transition following exposure of Fe-BTTri to O2. Previous studies of the activated framework yielded isomer shift values (δ) ranging from 1.05–1.07 mm s−1 and quadrupole splitting values (ΔEQ) from 1.80–3.06 mm s−1.29 The slightly different iron environments giving rise to these values can be ascribed to the presence of disordered, charge-balancing extra-framework cations as well as residual solvent molecules remaining upon activation, which has also been seen in the structurally analogous framework Fe-BTT (Fe3[(Fe4Cl)3(BTT)8(CH3OH)4]2, BTT3− = 1,3,5-benzenetristetrazolate).30 Dosing activated Fe-BTTri with 210 mbar of O2 at −78 °C resulted in the appearance of several new Mössbauer spectral features (Fig. 3). The dominant feature (δ = 0.309(2) mm s−1, ΔEQ = 2.370(3) mm s−1) comprises 50.8(9)% of the total absorption area and is consistent with low-spin, octahedrally-coordinated Fe(III), which typically exhibit quadrupole splitting values >2 mm s−1.31 This result is the first example of an iron site in a metal–organic framework converting from high-spin Fe(II) to low-spin Fe(III) upon adsorbate binding. Importantly, these Mössbauer parameters also closely align with those obtained for oxyhemoglobin and biomimetic iron porphyrin–O2 compounds.32,33 The remaining spectral features are attributed to residual high-spin Fe(II) in extra-framework and framework sites, which is expected based on the gas sorption data. A small amount of high-spin Fe(III) impurity is also observed due to sample warming (see the Discussion in the ESI† for additional details).
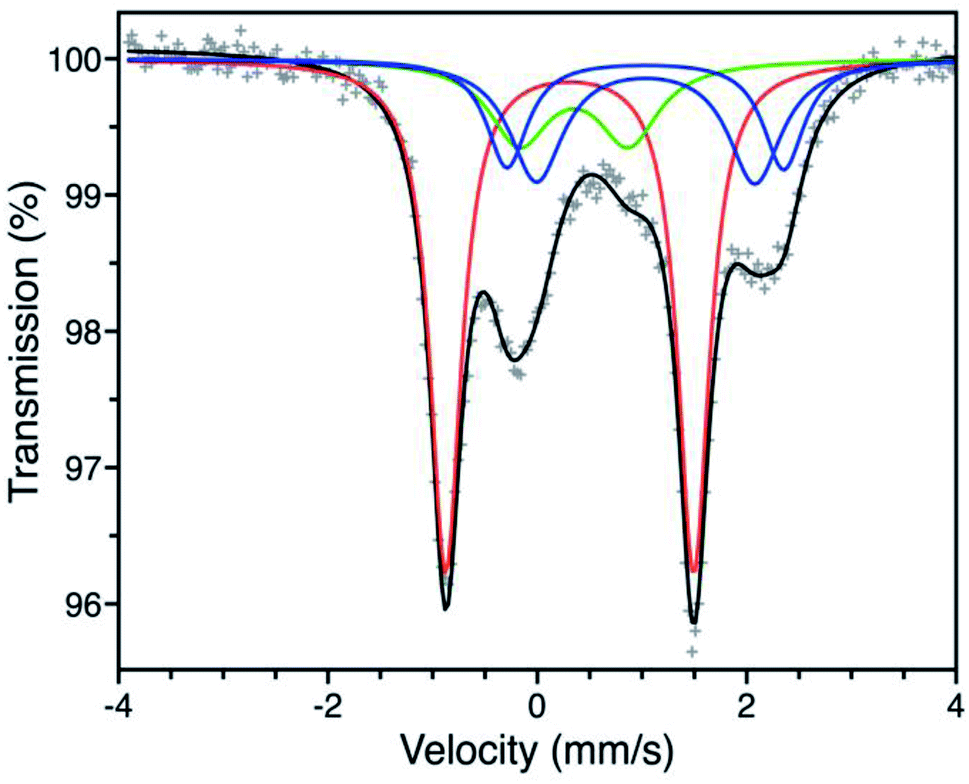 | ||
| Fig. 3 Mössbauer spectrum of Fe-BTTri collected at −78 °C under a 210 mbar atmosphere of O2. Experimental data are shown as grey plusses and the total fit is shown in black. The red component is assigned to low-spin Fe(III) centers bound to O2−, the green component to a high-spin Fe(III) decomposition product, and the blue components to high-spin Fe(II) species at framework and extra-framework sites. The ESI† contains spectral parameters for all sites (Table S3†), along with supplemental discussion of the green and blue components. | ||
The nature of the bound dioxygen molecule was also investigated via in situ infrared spectroscopy, using a custom-built DRIFTS setup (Fig. S7†). Cooling a sample of Fe-BTTri to −78 °C and dosing with 15 mbar of 16O2 resulted in the appearance of a new stretch at 1199 cm−1 that shifts to 1136 cm−1 with isotopically labeled 18O2. Given that several of the framework peaks change upon O2 binding, the spectrum of the 18O2-dosed material was subtracted from that of the 16O2-dosed framework to isolate the O2-based vibrations. The difference spectrum confirms that the peaks at 1136 and 1199 cm−1 arise from the O–O stretch. The resonance at 1199 cm−1 is characteristic of a superoxo (O2−) species, which typically show infrared stretching frequencies between 1070–1200 cm−1,34 and is similar to stretching frequencies observed for O2 in oxyhemoglobin and other model systems.35,36
To further probe the interaction between the iron centers and O2, gas adsorption isotherms were collected at various temperatures and analyzed to determine the Fe–O2 binding enthalpy, or isosteric heat of adsorption. Isotherm data collected at −78, −61, and −49 °C were fit to a dual-site Langmuir–Freundlich equation (Fig. S1†), and the Clausius–Clapeyron relation was used to calculate an isosteric heat of adsorption of −51 kJ mol−1 (Fig. S4†). This value is similar to those reported for O2 binding in heme systems, which typically range from −52 to −62 kJ mol−1.10,37 Additionally, when compared to the value of −41 kJ mol−1 obtained for O2 binding in Fe2(dobdc),23 it is clear the different binding mechanism in Fe-BTTri facilitates substantially stronger iron–O2 interactions. Indeed, among frameworks exhibiting fully reversible O2 adsorption,15,23,24,26 Fe-BTTri shows the highest binding affinity for O2.
The similar O2 binding in Fe-BTTri and heme systems suggests that Fe-BTTri should perform selective and reversible O2 binding. Low-temperature cycling experiments revealed that O2 binding in Fe-BTTri is fully reversible, and the material can be completely regenerated under mild conditions with very small temperature swings (Fig. 4). Heating O2-bound Fe-BTTri to −40 °C under vacuum for just 30 minutes fully regenerates the material, leading to an identical O2 uptake at 210 mbar compared to that of the pristine material over five cycles. Variable-temperature Mössbauer spectra collected under 210 mbar of O2 similarly show that >80% of the O2 bound to low-spin Fe(III) is released upon warming the sample from −78 to −28 °C (Fig. S8†). Finally, variable-temperature PXRD data collected on a framework sample dosed with 7 mbar of O2 show fully reversible unit cell volume contractions and expansions over multiple cycles between −80 and −30 °C, consistent with the reversible binding and release of O2 (Fig. S6†). We note that above −15 °C, adsorption of O2 becomes irreversible. Ultimately, the combined oxidation and spin state transition observed for Fe-BTTri enables highly selective and reversible low-temperature adsorption of O2.
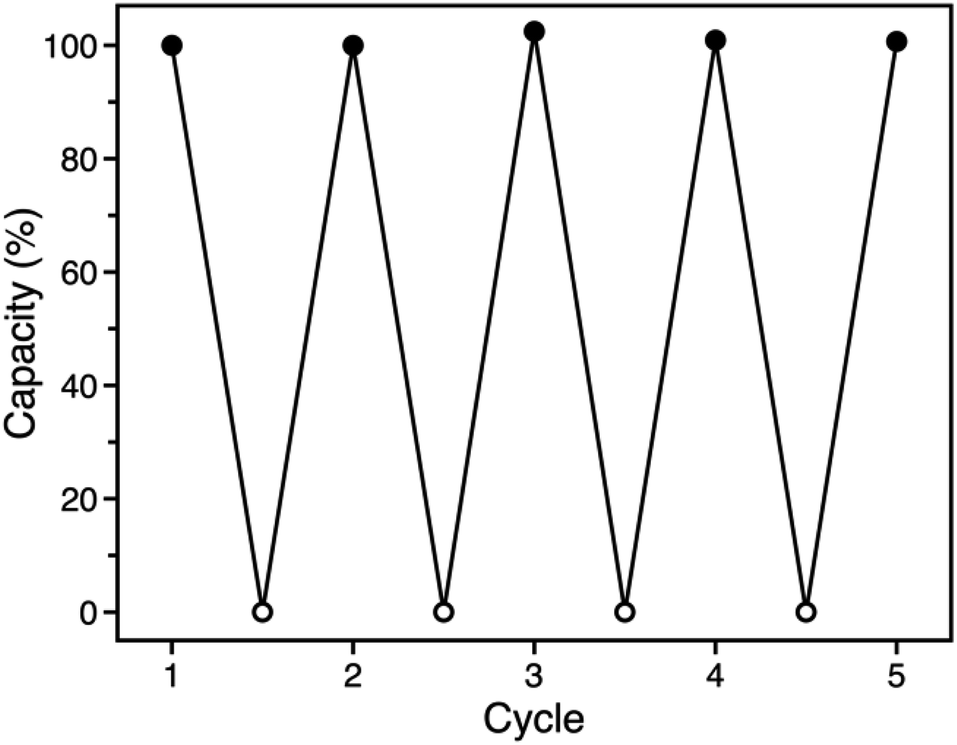 | ||
| Fig. 4 Cycling data for successive adsorption and desorption of O2 in Fe-BTTri, expressed as a percentage of the capacity measured for cycle 1. Adsorption (solid symbols) occurred within 10 minutes upon dosing O2 at 210 mbar and −78 °C, and complete desorption (open symbols) was accomplished by warming the sample to −40 °C under dynamic vacuum for 30 minutes. | ||
The O2/N2 selectivity of Fe-BTTri for air separation applications was assessed from analysis of O2 and N2 gas adsorption data (Fig. 2). In comparison to that of O2, the N2 adsorption curve rises much more gradually, and at all measured pressures Fe-BTTri adsorbs significantly more O2 than N2. Furthermore, the isosteric heat of adsorption for N2 is only −14 kJ mol−1, substantially smaller in magnitude than that measured for O2 (Fig. S4†). Ideal Adsorbed Solution Theory (IAST) was used to quantify the framework O2/N2 selectivity from the single component isotherms. For 21% O2 in N2 at a total pressure of 1 bar, which approximates the composition of air, Fe-BTTri shows an IAST selectivity value of 27 at −78 °C, which corresponds to 88% pure O2 in the adsorbed phase. This value is the highest reported for any iron-based framework. For comparison, Fe2(dobdc) displays an IAST selectivity value of approximately 11 under similar conditions.23 Additionally, the binding enthalpy of all other gaseous components of air, such as CO2, are significantly smaller in magnitude than that of O2 and should not affect the O2/N2 selectivity.29 This is another important contrast to Fe2(dobdc), where CO2 adsorbs with a similar binding affinity to that of O2 and may impact the O2 selectivity performance in air separations.38
Interestingly, the O2/N2 IAST selectivity values for Fe-BTTri remain relatively constant over a wide temperature range. For the 21% O2 in N2 mixture, the selectivity increases to 31 when the temperature is increased to −61 °C, and even at −49 °C the selectivity remains high at 28. An increase in selectivity with temperature has been found for other separations involving adsorbate-induced spin state transitions,29 but this result is in stark contrast to that for other O2 adsorbents, which exhibit diminished selectivity with increasing temperature.23,24 For example, the selectivity of Fe2(dobdc) decreases by over 30%, from 11 to 7.5, over a much smaller temperature range of −72 to −59 °C.23 The ability of redox and spin state flexible adsorbents to remain selective at higher temperatures is another advantage of this binding mechanism. To the best of our knowledge, this is the first example of temperature-independent selectivity in O2-selective adsorbents.
Lastly, we examined the potential utility of this material for O2-scrubbing applications, as O2 is a potential poison for several N2 fixation catalysts.39 The O2 capacity of Fe-BTTri is approximately 1.2 mmol g−1 at just 100 ppm, corresponding to a removal capacity of nearly 4 wt% at low O2 partial pressures (Fig. S3†). In comparison, the adsorption capacity of Fe2(dobdc) at these temperatures and pressures is approximately 0.2 mmol g−1.23 Importantly, due to its steep O2 adsorption isotherm profile, Fe-BTTri is also highly selective for O2 at low O2 partial pressures (Fig. S5†). Analysis of the IAST selectivity values for varying O2/N2 compositions at 1 bar total pressure and −78 °C revealed that the selectivity increases at lower O2 concentrations, reaching a value of 68 for a 5% O2 in N2 mixture. This result is again in contrast to other materials,23 for which the selectivity remains constant or even decreases at low O2 concentrations, and demonstrates that Fe-BTTri is promising for both air separation and O2-scrubbing applications.
Conclusions
The results shown here demonstrate that materials exhibiting new mechanisms for metal-site gas binding can be achieved using inspiration from biology. In particular, the framework Fe-BTTri, featuring iron sites structurally similar to heme iron, shows high selectivities for O2 over N2 for a range of temperatures and pressures. The highly tunable nature of metal–organic frameworks should enable the design and installation of other bio-inspired motifs, such as pendant hydrogen bond donors via secondary coordination sphere modification,40 bimetallic O2 binding sites using neighboring metal centers capable of synergistic binding,7,41 and cooperative adsorption sites.9 These approaches may afford new means of binding and activating gaseous substrates and serve as models for the continued development of advanced adsorbents.Conflicts of interest
The authors declare the following competing financial interests: J. R. L. has a financial interest in Mosaic Materials, Inc., a startup company working to commercialize metal–organic frameworks for gas separations. The University of California, Berkeley has applied for a patent on some of the materials discussed herein, on which J. R. L. and D. A. R. are listed as inventors.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award DE-SC0019992. Powder X-ray diffraction data were collected at Beamline 17-BM at the Advanced Photon Source, a DOE Office of Science User Facility, operated by Argonne National Laboratory under contract DE-AC02-06CH11357. We thank the National Science Foundation for graduate fellowship support of D. A. R., D. J. X., and J. O., and thank Dr K. R. Meihaus for editorial assistance.Notes and references
- O. M. Yaghi, H. Li, M. Eddaoudi and M. O'Keefe, Nature, 1999, 402, 276 CrossRef.
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. R. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- Z.-Y. Gu, J. Park, A. Raiff, Z. Wei and H.-C. Zhou, ChemCatChem, 2014, 6, 67 CrossRef CAS.
- D. Y. Osadchii, A. I. Olivos-Suarez, Á. Szécsényi, G. Li, M. A. Nasalevich, I. A. Dugulan, P. S. Crespo, E. J. M. Hensen, S. L. Veber, M. V. Fedin, G. Sankar, E. A. Pidko and J. Gascon, ACS Catal., 2018, 8, 5542 CrossRef CAS.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303 CrossRef CAS PubMed.
- D. A. Reed, B. K. Keitz, J. Oktawiec, J. A. Mason, T. Runčevski, D. J. Xiao, L. E. Darago, V. Crocella, S. Bordiga and J. R. Long, Nature, 2017, 550, 96 CrossRef CAS.
- J. P. Collman, J. I. Brauman and K. S. Suslick, J. Am. Chem. Soc., 1975, 97, 7185 CrossRef CAS.
- J. P. Collman, J. I. Brauman, E. Rose and K. S. Suslick, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 1052 CrossRef CAS PubMed.
- C. Citek, S. Herres-Pawlis and T. D. P. Stack, Acc. Chem. Res., 2015, 48, 2424 CrossRef CAS PubMed.
- W. B. Tolman, Acc. Chem. Res., 1997, 30, 227 CrossRef CAS.
- T. J. Mizoguchi and S. J. Lippard, J. Am. Chem. Soc., 1998, 120, 11022 CrossRef CAS.
- J. S. Anderson, A. T. Gallagher, J. A. Mason and T. D. Harris, J. Am. Chem. Soc., 2014, 136, 16489 CrossRef CAS PubMed.
- A. M. Wright, Z. Wu, G. Zhang, J. L. Mancuso, R. J. Comito, R. W. Day, C. H. Hendon, J. T. Miller and M. Dinca, Chem, 2018, 4, 2894 CAS.
- C. E. Bien, K. K. Chen, S.-C. Chien, B. R. Riener, L.-C. Lin, C. R. Wade and W. S. W. Ho, J. Am. Chem. Soc., 2018, 140, 12662 CrossRef CAS PubMed.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Butterworth Heinemann, Burlington, MA, 2nd edn, 2002 Search PubMed.
- M. J. Kirschner, A. Alekseev, S. Dowy, M. Grahl, L. Jansson, P. Keil, G. Lauermann, M. Meilinger, W. Schmehl, H. Weckler and C. Windmeier, Oxygen, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2017 Search PubMed.
- A. R. Smith and J. Klosek, Fuel Process. Technol., 2001, 70, 115 CrossRef CAS.
- K. Imai, Oxygen-Binding Proteins, in Encyclopedia of Molecular Biology, John Wiley and Sons, 2002 Search PubMed.
- L. J. Murray, M. Dincă, J. Yano, S. Chavan, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7856 CrossRef CAS PubMed.
- E. D. Bloch, L. J. Murray, W. L. Queen, S. Chavan, S. N. Maximoff, J. P. Bigi, R. Krishna, V. K. Peterson, F. Grandjean, G. J. Long, B. Smit, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14814 CrossRef CAS PubMed.
- D. J. Xiao, M. I. Gonzalez, L. E. Darago, K. D. Vogiatzis, E. Haldoupis, L. Gagliardi and J. R. Long, J. Am. Chem. Soc., 2016, 138, 7161 CrossRef CAS.
- E. D. Bloch, W. L. Queen, M. R. Hudson, J. A. Mason, D. J. Xiao, L. J. Murray, R. Flacau, C. M. Brown and J. R. Long, Angew. Chem., Int. Ed., 2016, 55, 8605 CrossRef CAS PubMed.
- A. T. Gallagher, J. Y. Lee, V. Kathiresan, J. S. Anderson, B. M. Hoffman and T. D. Harris, Chem. Sci., 2018, 9, 1596 RSC.
- A. T. Gallagher, M. L. Kelty, J. G. Park, J. S. Anderson, J. A. Mason, J. P. S. Walsh, S. L. Collins and T. D. Harris, Inorg. Chem. Front., 2016, 3, 536 RSC.
- D. Denysenko, M. Grzywa, J. Jelic, K. Reuter and D. Volkmer, Angew. Chem., Int. Ed., 2014, 53, 5832 CrossRef CAS PubMed.
- D. A. Reed, D. J. Xiao, M. I. Gonzalez, L. E. Darago, Z. R. Herm, F. Grandjean and J. R. Long, J. Am. Chem. Soc., 2016, 138, 5594 CrossRef CAS PubMed.
- K. Sumida, S. Horike, S. S. Kaye, Z. R. Herm, W. L. Queen, C. M. Brown, F. Grandjean, G. J. Long, A. Dailly and J. R. Long, Chem. Sci., 2010, 1, 184 RSC.
- P. Gütlich, E. Bill and A. X. Trautwein, Mossbauer Spectroscopy and Transition Metal Chemistry, Springer-Verlag, Berlin, 2007 Search PubMed.
- Y. Maeda, T. Harami, Y. Morita, A. Trautwein and U. Gonser, J. Chem. Phys., 1981, 75, 36 CrossRef CAS.
- K. Spartalian, G. Lang, J. P. Collman, R. R. Gagne and C. A. Reed, J. Chem. Phys., 1975, 63, 5375 CrossRef CAS.
- R. D. Jones, D. A. Summerville and F. Basolo, Chem. Rev., 1979, 79, 139 CrossRef CAS.
- C. H. Barlow, J. C. Maxwell, W. J. Wallace and W. S. Caughey, Biochem. Biophys. Res. Commun., 1973, 55, 91 CrossRef CAS.
- C. A. Reed and S. K. Cheung, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 1780 CrossRef CAS PubMed.
- E. C. Neiderhoffer, J. H. Timmons and A. E. Martell, Chem. Rev., 1984, 84, 137 CrossRef.
- W. L. Queen, M. R. Hudson, E. D. Bloch, J. A. Mason, M. I. Gonzalez, J. S. Lee, D. Gygi, J. D. Howe, K. Lee, T. A. Darwish, M. James, V. K. Peterson, S. J. Teat, B. Smit, J. B. Neaton, J. R. Long and C. M. Brown, Chem. Sci., 2014, 5, 4569 RSC.
- K. C. Waugh, D. Butler and B. E. Harden, Catal. Lett., 1994, 24, 197 CrossRef CAS.
- D. J. Xiao, J. Oktawiec, P. J. Milner and J. R. Long, J. Am. Chem. Soc., 2016, 138, 14371 CrossRef CAS PubMed.
- M. I. Gonzalez, M. T. Kapelewski, E. D. Bloch, P. J. Milner, D. A. Reed, M. R. Hudson, J. A. Mason, G. Barin, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2018, 140, 3412 CrossRef CAS PubMed.
- S.-Y. Park, T. Yokoyama, N. Shibayama, Y. Shiro and J. R. H. Tame, J. Mol. Biol., 2006, 360, 690 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, additional gas adsorption data and analysis, additional characterization data and analysis (Mössbauer spectroscopy, infrared spectroscopy, powder X-ray diffraction), and supplementary discussion. See DOI: 10.1039/c9sc06047b |
| This journal is © The Royal Society of Chemistry 2020 |