
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntra- and intermolecular interception of a photochemically generated terminal uranium nitride†
Munendra
Yadav
 *,
Alejandro
Metta-Magaña
and
Skye
Fortier
*
*,
Alejandro
Metta-Magaña
and
Skye
Fortier
*
Department of Chemistry and Biochemistry, University of Texas at El Paso, El Paso, TX 79968, USA. E-mail: asfortier@utep.edu
First published on 22nd January 2020
Abstract
The photochemically generated synthesis of a terminal uranium nitride species is here reported and an examination of its intra- and intermolecular chemistry is presented. Treatment of the U(III) complex LArUI(DME) ((LAr)2− = 2,2′′-bis(Dippanilide)-p-terphenyl; Dipp = 2,6-diisopropylphenyl) with LiNImDipp ((NImDipp)− = 1,3-bis(Dipp)-imidazolin-2-iminato) generates the sterically congested 3N-coordinate compound LArU(NImDipp) (1). Complex 1 reacts with 1 equiv. of Ph3CN3 to give the U(IV) azide LArU(N3)(NImDipp) (2). Structural analysis of 2 reveals inequivalent Nα–Nβ > Nβ–Nγ distances indicative of an activated azide moiety predisposed to N2 loss. Room-temperature photolysis of benzene solutions of 2 affords the U(IV) amide (N-LAr)U(NImDipp) (3) via intramolecular N-atom insertion into the benzylic C–H bond of a pendant isopropyl group of the (LAr)2− ligand. The formation of 3 occurs as a result of the intramolecular interception of the intermediately generated, terminal uranium nitride (LAr)U(N)(NImDipp) (3′). Evidence for the formation of 3′ is further bolstered by its intermolecular capture, accomplished by photolyzing solutions of 2 in the presence of an isocyanide or PMe3 to give (LAr)U[NCN(C6H3Me2)](NImDipp) (5) and (N,C-LAr*)U(N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PMe3)(NImDipp) (6), respectively. These results expand upon the limited reactivity studies of terminal uranium–nitride moieties and provide new insights into their chemical properties.
PMe3)(NImDipp) (6), respectively. These results expand upon the limited reactivity studies of terminal uranium–nitride moieties and provide new insights into their chemical properties.
Introduction
The study of metal–ligand multiple bonds has played a critical role in the understanding and development of d-block reactivity profiles1 and continues to be a widely studied area of interest. In comparison, the chemistry of actinide–ligand multiple bonding has lagged substantially behind. This is due in part to the poor radial extension of the actinide 5f valence orbitals, potential energetic mismatch between the metal and ligand bonding orbitals, and a higher degree of ionic bonding character,2 which can favour bridging and formation of higher nuclearity complexes. Despite these challenging factors, significant advances in early actinide–ligand multiple bonding have been achieved in recent years,3 from the synthesis of imido and heavier chalcogenide analogues of the pervasive uranyl cation, UO22+ (e.g., U(NtBu)2I2(THF)2);4 [Cp*2Co][U(E)(O)(NR2)3] (R = SiMe3; E = S, Se),5 to the synthesis of uranium tris- and tetrakis(imido) complexes and beyond.6Notably, special efforts have been dedicated over the years to the synthesis and isolation of uranium compounds possessing terminal nitride moieties. In contrast to the well-developed nitride chemistry of the transition metals,7 complexes featuring U![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds were once considered a so-called “holy grail” of actinide chemistry as results were long limited to matrix isolation experiments and systems containing bridging nitride moieties.8 The paucity of these compounds is surprising as the uranium–oxygen bonds of uranyl (UO22+) are remarkably robust and share an isolobal UO bonding relationship to UN functionalities. Moreover, complexes featuring terminal uranium-oxo bonds, once rare, are now fairly well-known.3,5,9
N bonds were once considered a so-called “holy grail” of actinide chemistry as results were long limited to matrix isolation experiments and systems containing bridging nitride moieties.8 The paucity of these compounds is surprising as the uranium–oxygen bonds of uranyl (UO22+) are remarkably robust and share an isolobal UO bonding relationship to UN functionalities. Moreover, complexes featuring terminal uranium-oxo bonds, once rare, are now fairly well-known.3,5,9
It bears mentioning that the study of bridged actinide nitrides of the type AnNAn has proven a to be a fruitful area of investigation with rich chemistry.6e,10 This has included such novel transformations as the cleavage of CO by [Cs{[U(OSi(OtBu)3)3]2(μ-N)}] to give the bridged cyanide complex [Cs{[U(OSi(OtBu)3)3]2(μ-CN)(μ-O)}]10c as well as reversible dihydrogen activation by the same bridged nitride complex to give the hydride imide [Cs{U(OSi(OtBu)3)3}2(μ-H)(μ-NH)].10g
In 2010, a major milestone was reached when Kiplinger and co-workers provided evidence for the intermediate formation of a terminal uranium–nitride species generated through the photolysis of the U(IV) azide Cp*2U[N(SiMe3)2](N3) to give the intramolecularly C–H activated product Cp*(η5-C5Me4CH2NH)U[N(SiMe3)2].11 This result demonstrated that terminal uranium nitrides are photochemically accessible but suggested an inherent instability. A few years later, in a landmark result, Liddle and co-workers established that not only could terminal uranium nitrides be synthesized but could also be isolated when using an appropriate ligand architecture under specific reaction conditions. Namely, they showed that treatment of the bulky U(III) complex U(TrenTIPS) (TrenTIPS = [N(CH2CH2NSiiPr3)3]3−) with NaN3 gives the mononuclear U(V) nitride [Na(12-crown-4)2][U(N)(TrenTIPS)] upon workup and addition of 12-crown-4 polyether, though this compound is sensitive and rapidly decomposes in ethereal solutions.12 A follow-up study showed that oxidation with molecular iodine affords access to the hexavalent, terminal nitride U(N)(TrenTIPS).13 On the heels of this, Cloke et al., using a bulky U(III) metallocene platform treated with NaN3, reported the synthesis and structural characterization of the sodium-capped U(V) nitride [C8H6(SiiPr3)2]Cp*U(N)Na(OEt2)2.14
Critically, these nitride complexes provide valuable new insights into the area of actinide metal–ligand multiple bonding; however, given the rarity of these species, much remains to be learned regarding the reactivity of UN bonds with studies thus far largely limited to Liddle's system. For instance, [Na(12-crown-4)2][U(N)(TrenTIPS)] reacts with Me3SiCl to give the imide U(NSiMe3)(TrenTIPS),12 while [U(N)(TrenTIPS)]n− (n = 0, 1) is reactive towards CE2 (E = O, S) heteroallenes leading to the formation of U(O)(TrenTIPS) and U-NCE products through multiple bond metathesis and complex redox pathways.9l On this note, [U(N)(TrenTIPS)]n− (n = 0, 1) is susceptible to reductive carbonylation upon addition of CO, leading to the formation of U-NCO isocyanates.15
Furthermore, evidence is beginning to mount which shows the reactivity of mononuclear uranium nitrides to be particularly distinct from other uranium–ligand multiple bonds as they appear to show a propensity for C–H bond activation chemistry. As in the case of the putative Cp*2U(N)[N(SiMe3)2],11 photolysis of U(N)(TrenTIPS) gives rise to the U(IV) N-atom inserted amide U[N(CH2CH2NSiiPr3)2(CH2CH2NSiiPr2CHMeCH2N)].13 More recently, it was shown that the one-electron reduction of the U(IV) bisazide (PN)2U(N3)2 (PN = N(2-iPr2P-4-MeC6H3)(2,4,6-Me3C6H2)) does not lead to the formation of a U(V) nitride but instead generates the potassium-capped parent imido complex [K(THF)3]{(PN)U(NH)[iPr2P(C6H3Me)N(C6H2Me2CH2)]} by way of intramolecular C–H bond addition of a pendant methyl group.16
In our laboratory, we have been exploring the chemistry of uranium supported by a bis(anilide) terphenyl ligand ((LAr)2−). In the complex LArUI(DME) (A) (Scheme 1), the uranium is straddled between two nitrogen atoms in a nearly linear fashion with the metal situated above the central ring of the terphenyl moiety with steric protection afforded by encumbering 2,6-diisopropylphenyl (Dipp) groups.17 This platform has been shown able to support a rare U–Fe actinide–metal bond,17 and we reasoned that the [LArU] system may be suitable for accessing other unique bonding motifs with uranium, such as UN bonds. Here, we present our synthesis of a uranium nitride complex and present its intra- and intermolecular reactivity, thus expanding the limited portfolio of terminal actinide–nitride chemistry.
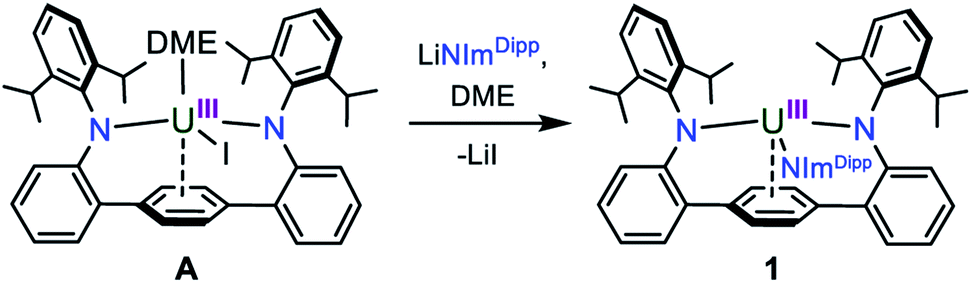 | ||
| Scheme 1 Synthesis of 1. | ||
Results and discussion
In an effort to enhance the relative electron richness of our [LArU] platform, while adding increased steric protection to disfavour possible U–N–U cluster formation, the 1,3-bis(Dipp)imidazolin-2-iminato (ImDippN−) ligand was reacted with A to give the 3N-coordinated U(III) complex LArU(NImDipp) (1) (Scheme 1). Complex 1 is isolated in 36% yield as dark brown crystals from the storage of concentrated DME solutions at −22 °C for 2 days.The solid-state molecular structure of 1·DME reveals a highly congested uranium centre, which is effectively illustrated through its space-filling model (Fig. 1). As compared to the uranium–anilide distances of A (U–N = 2.524(4)–2.558(3) Å), the U1–N1 = 2.429(3) Å and U1–N2 = 2.369(3) Å bonds are significantly shorter, while the N1–U1–N2 = 128.8(1)° bond angle is now nearly thirty degrees more acute. Of note, the distance between the uranium and the central ring of the terphenyl ligand (U–Ccentroid = 2.38 Å) in 1·DME also contracts substantially as compared to A (U–Ccentroid = 2.56 Å). We ascribe the significant structural changes within the [LArU] core from A to 1·DME as a consequence of having to accommodate the sterically imposing ImDippN− ligand.
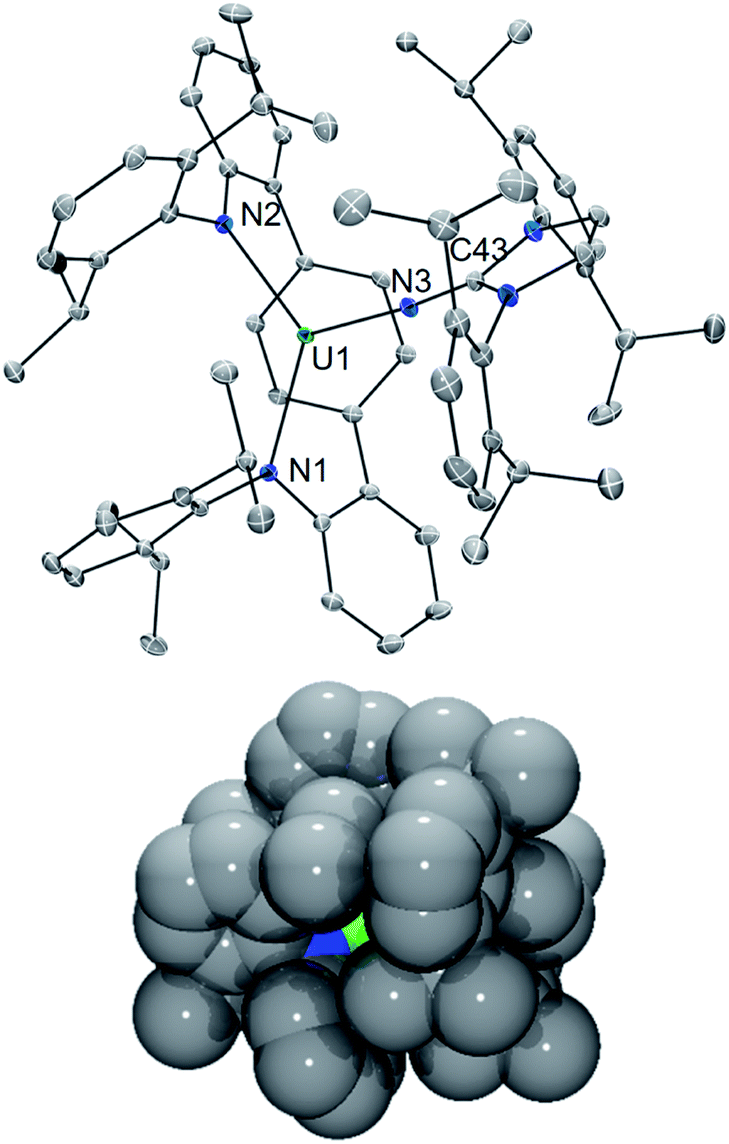 | ||
| Fig. 1 Solid-state molecular structure of 1·DME (top). Space filling model of 1 showing uranium (green), nitrogen (blue), and carbon (gray) atom spheres (bottom). | ||
It should be noted that the coordination of iminic N-donor ligands to U(III) is rare, and inspection of the U–NIm bond length in 1·DME (U1–N3 = 2.169(3) Å) reveals it to be much shorter than the non-bridging uranium–ketimide bond in the inverted sandwich complex [Na2(Et2O)][UN(tBu)(Mes)]2(μ:η6-C7H8) (U–N = 2.24(1) Å).18 However, the U–NIm bonding metrics (U1–N3 = 2.169(3) Å; U1–N3–C43 = 170.4(3)°) are similar to that reported for tetravalent U(NImDipp)[N(SiMe3)2]3 (U–N = 2.137(6) Å; U–N–C = 169.5(5)°).19
With 1 in hand, addition of 1 equiv. of Me3SiN3 in DME at room temperature leads to the formation of the U(IV) complex LArU(N3)(NImDipp) (2). Compound 2 is produced in low yield and is accompanied by the formation of (Me3Si)2LAr. As the reaction appears to proceed via the one-electron reduction of Me3SiN3, we reasoned that Ph3CN3 would be a viable azide source as reduction would lead to the formation of Gomberg's dimer which should avoid back-reactions with the ligand. As such, use of Ph3CN3 with U[N(SiMe3)2]3 generates the U(IV) azide U(N3)[N(SiMe3)2]3.9i Accordingly, treatment of 1 with Ph3CN3 in C6D6 at room temperature leads to the quantitative formation of 2 as observed by 1H NMR spectroscopy. The reaction can be performed on a preparative scale at room temperature in solutions of toluene with crystals of 2 generated from the diffusion of hexanes into concentrated toluene/benzene solutions stored at −22 °C, providing 2 in 57% isolated yield (Scheme 2).
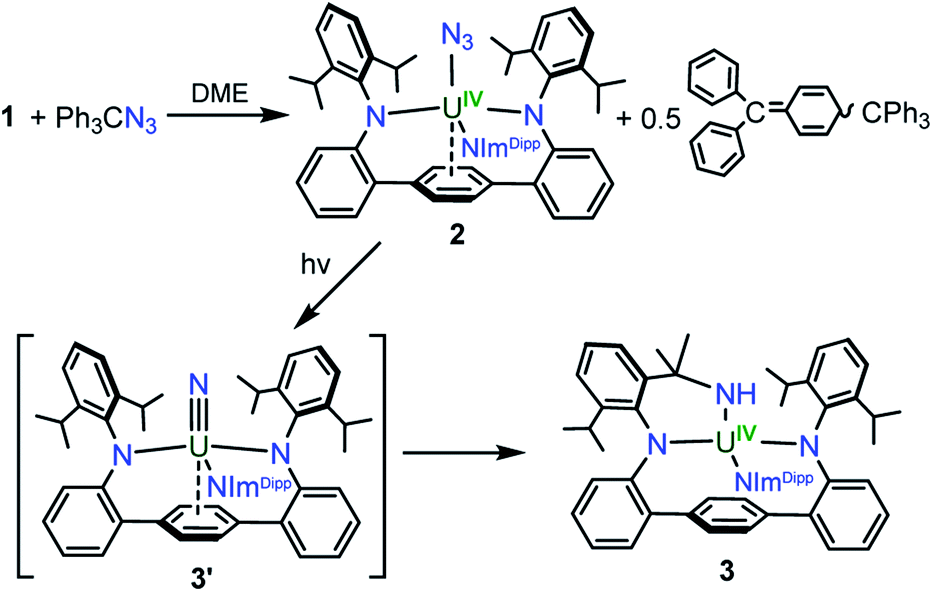 | ||
| Scheme 2 Synthesis of 2 and proposed formation of 3via terminal nitride 3′. | ||
The solid-state molecular structure of 2·C6H6 is shown in Fig. 2. Interestingly, oxidation to U(IV) leads to an elongation of the U–LAr bonds (U1–N1 = 2.455(5) Å; U1–N2 = 2.414(5) Å; U–Ccentroid = 2.58 Å). On the other hand, the U–NIm bond is observed to undergo contraction (U1–N3 = 2.135(4) Å). Of particular note, the U–N3 distance (U1–N6 = 2.142(5) Å) is on the shorter end of the range reported for tetravalent uranium azides (2.22–2.56 Å).20 Moreover, the azide unit exhibits inequivalent nitrogen–nitrogen distances (N6–N7 = 1.24(1) Å, N7–N8 = 1.13(2) Å) which indicates activation of the azide moiety. Together, the short U–N3 bond and azide activation is characteristic of what has been described in the literature as a covalent metal–azide interaction.21
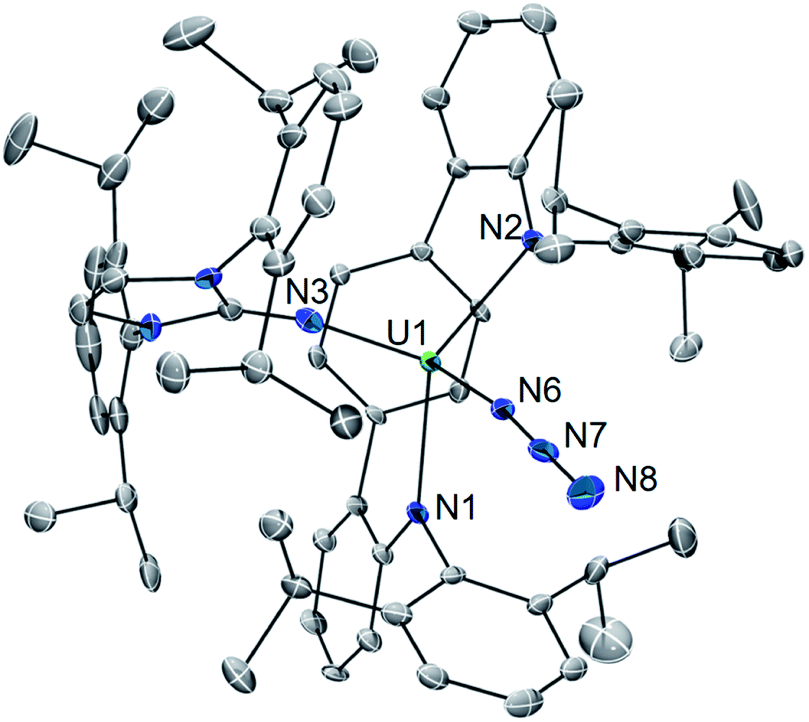 | ||
| Fig. 2 Solid-state molecular structure of 2·C6H6. | ||
Complex 2 is soluble in ethereal solvents while possessing moderate solubility in aromatic solvents such as benzene and toluene. The 1H NMR spectrum of 2 in toleuene-d8 is complicated, giving rise to 50 distinct resonances appearing from −22.3 to 28.9 ppm (see ESI Fig. SI3 and SI4†), which we attribute to increased rotational restriction owing to occupation of the axial position by the azide unit. Accordingly, each of the 16 methyl groups of the isopropyl functionalities in 2 gives rise to a unique resonance integrating to three protons each.
Attempts to reduce 2 with external reductants such as KC8 or [Li(18-crown-6)][biphenyl]22 inevitably led to the formation of alkali metal salts of the (LAr)2− ligand. Additionally, treatment of 2 with strong Lewis acids (e.g. Sm(OTf)3) also failed to initiate N2 loss. Furthermore, thermolysis of C6D6 solutions of 2 up to refluxing temperatures does not lead to any change within the 1H NMR spectrum.
Gratifyingly, photolyzing solutions of 2 with UV light (365 nm) for at least 24 hours does lead to new reactivity. This generates a paramagnetic species with a 1H NMR spectrum that features 50 resonances within the range of −56.6 to 97.0 ppm (see ESI Fig. SI6†). Removal of the solvent and recrystallisation from hexanes provides crystals of the C1 symmetric U(IV) N-atom inserted product (N-LAr)U(NImDipp) (3·C6H14) (Scheme 2), and its solid-state molecular structure is depicted in Fig. 3.
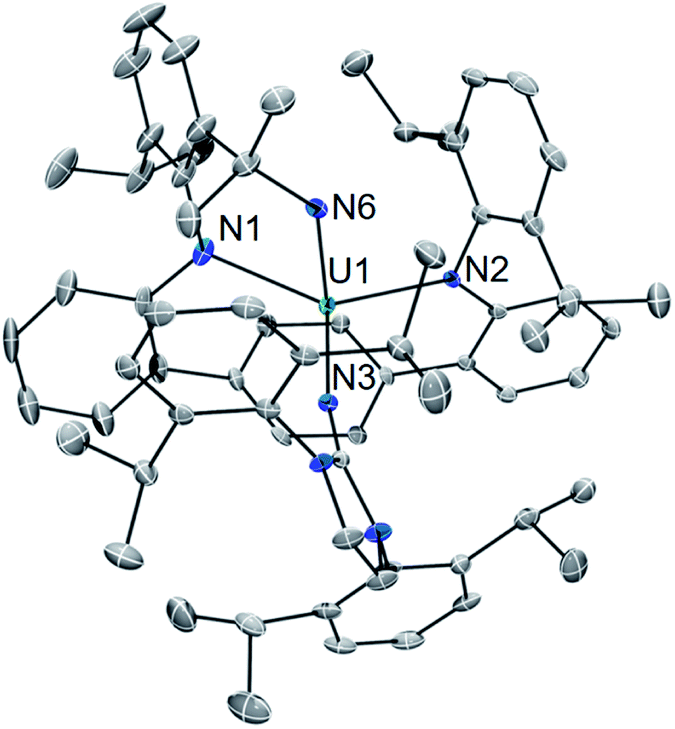 | ||
| Fig. 3 Solid-state molecular structure of 3·C6H14. | ||
Overall, the structural changes as compared to 2·C6H6 are modest (3·C6H14: U1–N1 = 2.415(3) Å, U1–N2 = 2.388(3) Å, U1–N3 = 2.159(3) Å); yet, the uranium atom does become offset from the central terphenyl ring moving from a nominal η6 interaction to a η4 orientation. By far, the most salient feature of 3·C6H14 is the formation of a new U–amide bond (U1–N6 = 2.167(3) Å) from insertion of a nitrogen atom into a pendant isopropyl group of the (LAr)2− ligand. This result is strongly reminiscent of the chemistry observed in the photolysis of Cp*2U[N(SiMe3)2](N3) and U(N)(TrenTIPS),11,13 providing strong evidence for the passing formation of (LAr)U(N)(NImDipp) (3′) possessing a terminal uranium nitride bond (Scheme 2). And, as with these systems, the ligand in 2 acts to intercept the nitride through intramolecular trapping.
The formation of 3′ begs the question of the formal oxidation state assignment of its uranium centre. The elimination of N2 from the azide unit is typically a two-electron process, which would render a U(VI) assignment for a diamagnetic 3′. Yet, formation of radical or nitrene character at the nitride moiety clouds canonical oxidation state assignments. Following the photoreaction by 1H NMR and UV-vis spectroscopies (see ESI Fig. SI5 and SI13†) shows the conversion of 2 to 3 without the apparent formation of any intermediate species. Furthermore, following the low temperature photolysis of 2 in toluene-d8 at −10 °C by 1H NMR spectroscopy also fails to trap or detect a fleeting species. This suggests that the nitride intermediate formed in the reaction is highly reactive and undergoes rapid reactivity with the ligand.
In an effort to chemically intercept 3′, CO was added to a C6D6 solution of 2. Upon addition, the immediate appearance of a complex series of resonances is observed in the 1H NMR spectrum concomitant with partial consumption of the starting material. The reaction is complete in 12 hours at room temperature. Workup of the reaction mixture and crystallisation from a hexanes/Et2O solution provides small yellow crystals of the U(IV) compound (CO-LAr)U(N3)(NImDipp) (4) in low yield, formed from insertion of CO into a uranium–anilide bond (Scheme 3). Photolysis of 2 in the presence of a CO atmosphere also leads to the formation of 4 as the only isolable product. While such reactivity is not well-documented among 5f-element amide compounds, it has been previously reported in the reactions of Cp*2M(NR2)2 (M = Th, U, R = CH3; M = U, R = C2H5) with CO to give Cp*2M(η2-CO-NR2)2.23
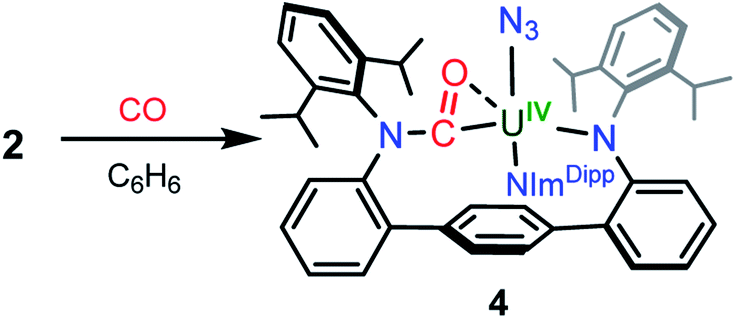 | ||
| Scheme 3 Synthesis of 4. | ||
Weak X-ray diffraction data for 4 precludes an in-depth structural analysis, but connectivity is definitively established (see ESI Fig. SI1†) and shows the carbonyl moiety binds to the uranium in an η2-fashion. Photolysis of solutions of 4 does not result in any observed reactivity, signalling that the U–N3 interaction is highly sensitive to the coordination environment of the uranium metal centre.
Using a bulky isocyanide to prevent ligand insertion, room temperature photolysis of 2 in the presence of CN(C6H3Me2) in C6D6 results in the formation of a new paramagnetic species as indicated by 1H NMR spectroscopy (see ESI Fig. SI8†). Removal of the solvent and extraction into a mixture of diethyl ether and hexanes followed by storage at −22 °C for 4 days affords dark brown-red crystals in 44% yield. Analysis by single crystal X-ray diffraction revealed the formation of the U(IV) carbodiiminate complex (LAr)U[NCN(C6H3Me2)](NImDipp) (5·Et2O) (Scheme 4) and (Fig. 4).
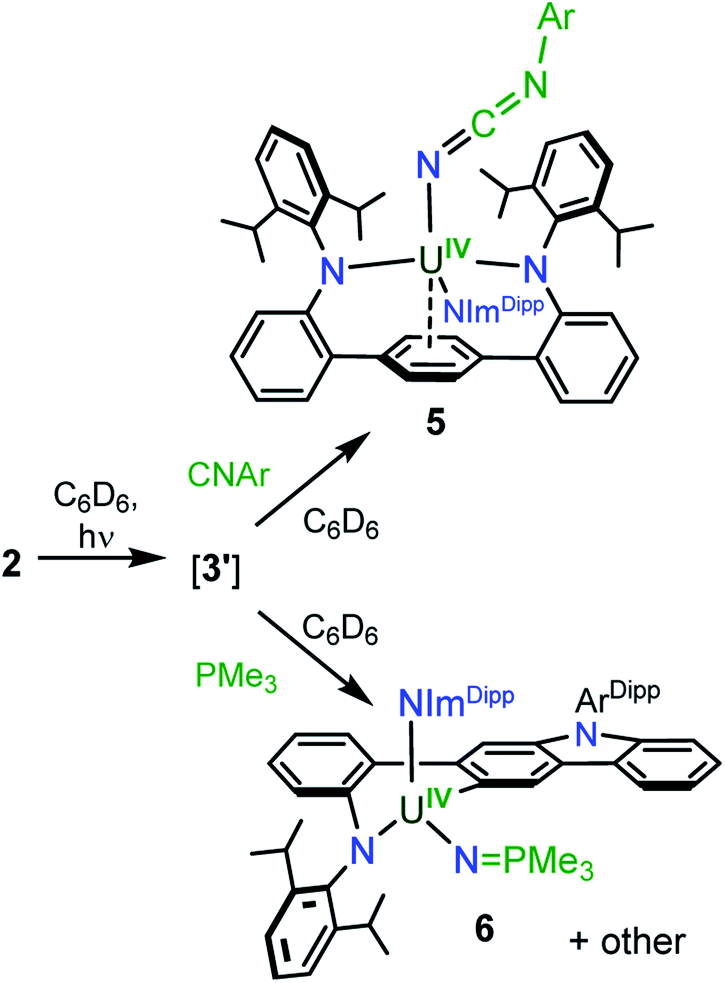 | ||
| Scheme 4 Synthesis of 5 and 6. | ||
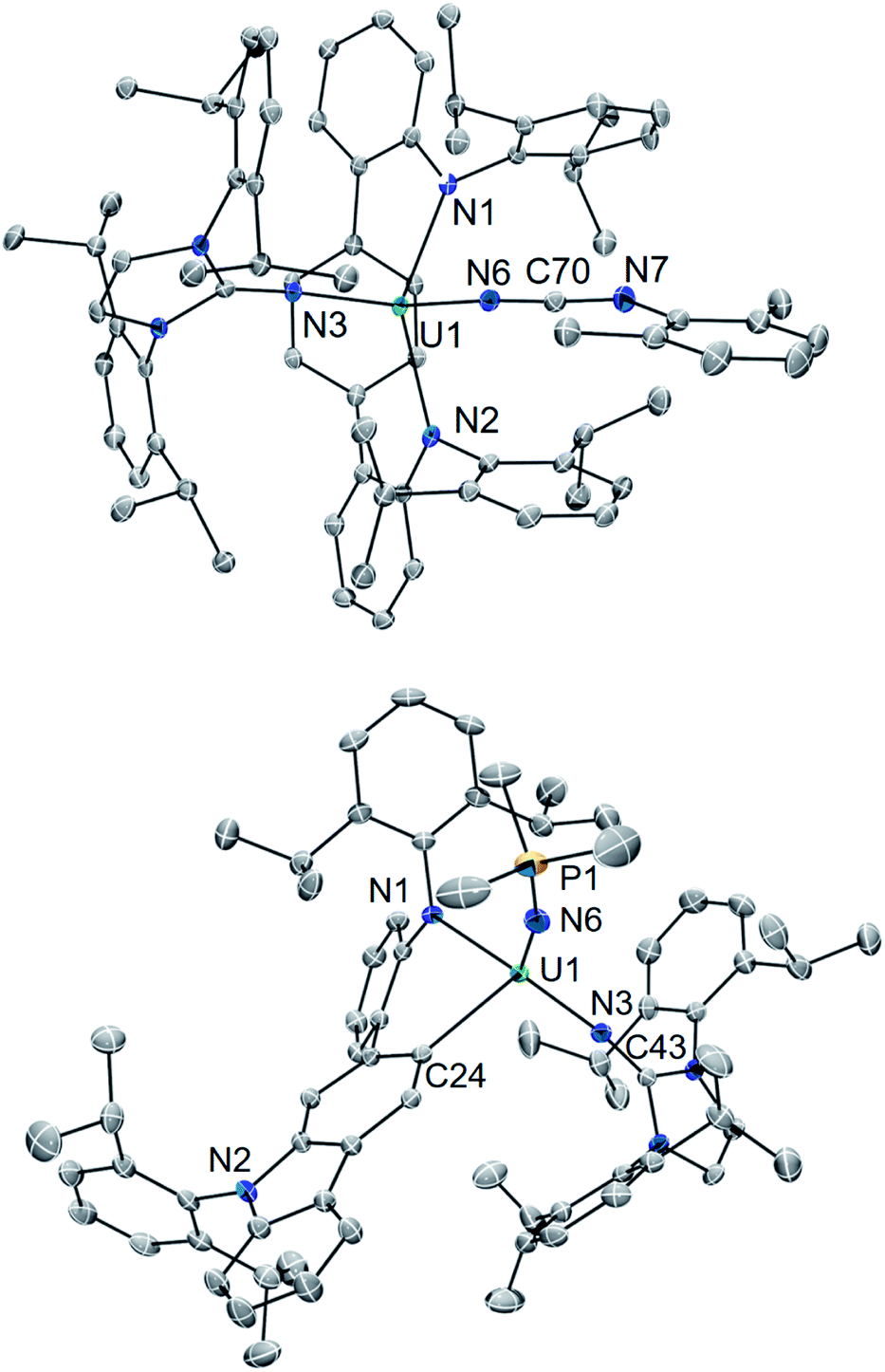 | ||
| Fig. 4 Solid-state molecular structures of 5·Et2O (top) and 6·Et2O (bottom). | ||
The core [(LAr)U(NImDipp)]+ structure of 5·Et2O (U1–N1 = 2.408(3) Å, U1–N2 = 2.450(3) Å, U1–N3 = 2.136(3) Å, U1–Ccentroid = 2.58 Å, N1–U1–N2 = 142.6(1)°) is similar to that found for 2·C6H6. Moreover, the uranium–carbodiiminate bond (U1–N6 = 2.235(3) Å) is longer in length than the U–N3 bond in 2·C6H6 but much shorter than the U–Ncarbo bond found in the U(IV) triazacyclononane tris(aryloxide) complex [(tBuArO)3tacn]U(NCNMe) (U–N = 2.327(3) Å), formed by a multiple-bond metathesis reaction that proceeds via nitrene transfer between [(tBuArO)3tacn]UNSiMe3 and CNMe.24
Furthermore, photolyzing benzene solutions of 2 with a slight excess of PMe3 leads to a highly complicated mixture of products as shown through 1H NMR spectroscopy. From these mixtures, one major product is reproducibly isolated in approximately 15% yield as yellow crystals, though separation of pure material has been hampered by co-crystallisation of other as-of-yet unidentified products. The 1H NMR spectrum of the major product in C6D6 is suggestive of C1 symmetry in solution as it exhibits 51 distinct resonances with 19 peaks attributable to the three protons of each methyl group (see ESI Fig. SI10†). Moreover, only one resonance in the 31P{1H} NMR spectrum is observed in the reaction mixture, appearing at 12.1 ppm (see ESI Fig. SI11†).
In line with this, the solid-state molecular structure of the yellow crystals obtained through X-ray diffraction analysis revealed the formation of the asymmetric, arylated U(IV) phosphinimide (N,C-LAr*)U(NPMe3)(NImDipp) (6) (Scheme 4). As seen in Fig. 4, a number of structural and chemical changes occur including ligand cyclization to give a carbazole functionality accompanied by deprotonation of the central terphenyl ring to give a U–Caryl bond (U1–C24). Nonetheless, clearly present in 6·Et2O is the formation of a –NPMe3 group (U1–N6 = 2.115(2) Å; U1–N6–P1 = 161.7(2)°), with metrical parameters comparable to its U–NIm interaction (U1–N3 = 2.141(2) Å; U–N3–C43 = 168.8(2)°) and the U–NPPh3 bond in Cp3UNPPh3 (U–N = 2.07(2) Å; U–N–P = 172(1)°).25 We reason that upon formation of the phosphinimide, steric congestion forces structural rearrangement, consequently initiating a cascade of chemical events that gives the complicated product mixture containing 6. Photolysis in the presence of the more encumbered phosphine, PMe2Ph, leads exclusively to the formation of 3.
Importantly, complex 2 does not react with CN(C6H3Me2) or PMe3 in the absence of light, and the formation of 5 and 6 occur exclusively under photolysis. This proves that 3′ is a key intermediate and a sufficiently long-lived species such that it is susceptible to chemical trapping, bypassing competitive intramolecular C–H insertion pathways. Moreover, the formation of these complexes represents the first examples of intermolecular chemistry for a photochemically generated uranium nitride.
The reactivity of 3′ with nucleophiles is akin to that observed for the reductive carbonylation of UVI(N)(TrenTIPS) with CO to give UIV(NCO)(TrenTIPS),15 and while 3′ is a putative terminal nitride, its reactivity is further reminiscent of the chemistry between dinuclear {[U(NtBuAr)3]2(μ-N)}+ (Ar = 3,5-dimethylphenyl) and CN− which generates the bridged carbodiiminate [U(NtBuAr)3]2(μ-NCN).26 Overall, the formation of 3, 5, and 6 through partial N-atom transfer concomitant with reduction of the metal centre is, from first principles, consistent with classic electrophilic nitride character. While we favour this particular perspective, we acknowledge it may be an oversimplification as uranium nitrides have been demonstrated to exhibit ambiphilic character. For instance, DFT analysis indicates that the reaction of U(N)(TrenTIPS) with CO proceeds through donation of the nitride lone-pair into the CO π*-orbital, signalling initiation via nucleophilic attack of the nitride onto CO.14 This is not unlike the reactivity profile known for some terminal iron nitrides which have also been described as possessing a dual-nature transition state, where the nitride simultaneously exhibits both nucleophilic and electrophilic character in the reactions of PhB(MesIm)3FeN (PhB(MesIm)3 = tris(mesitylcarbene)borate)27 and [LAr*]FeN(py) (LAr* = (tBu2CN)C(Ar*N)2, Ar* = 2,6-bis(diphenylmethyl)-4-tert-butylphenyl)28 with phosphines to give [Fe–NPMe3] products. As such, this reveals an interesting reactivity pattern for uranium nitrides, and further study of 3′ is underway to better elucidate the reactivity character of its nitride group.
Conclusion
In closing, while the landmark isolation of monouranium nitrides have been achieved through the synthesis of [U(N)(TrenTIPS)]n− (n = 0, 1) and [C8H6(SiiPr3)2]Cp*U(N)Na(OEt2)2 by the groups of Liddle and Cloke, respectively,12–14 much remains to be learned regarding the character of UN bonds. Here, we have demonstrated that LArU(N3)(NImDipp) (2) provides an accessible platform for testing such reactivity as its photolysis generates the (LAr)U(N)(NImDipp) (3′) intermediate which can be intra- and intermolecularly intercepted. In the latter case, addition of the nucleophiles CN(C6H3Me2) and PMe3 to 3′ gives the U(IV)–imine products (LAr)U[NCN(C6H3Me2)](NImDipp) (5) and (N,C-LAr*)U(NPMe3)(NImDipp) (6), respectively, produced as a result of nitride capture and partial N-atom transfer. Thus, for the first time, we demonstrate that accessing intermolecular uranium–nitride chemistry under photochemical conditions is possible. These reactions can be classically described as proceeding through an electrophilic nitride, though the potential for nitride ambiphilicity cannot be ruled out at this stage and a thorough reaction survey of 3′ with other substrates is ongoing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Welch Foundation (AH-1922-20170325) and the NSF PREM Program (DMR-1827745) for financial support of this work. S. F. is an Alfred P. Sloan Foundation research fellow and is thankful for their support. We also wish to acknowledge the NSF-MRI program (CHE-1827875) for providing funding for the purchase of an X-ray diffractometer.Notes and references
- W. A. Nugent and J. M. Mayer, Metal-Ligand Multiple Bonds, John Wiley & Sons, New York, 1988 Search PubMed.
- (a) S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed; (b) M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS; (c) R. Beekmeyer and A. Kerridge, Inorganics, 2015, 3, 482–499 CrossRef CAS; (d) W. W. Lukens, M. Speldrich, P. Yang, T. J. Duignan, J. Autschbach and P. Kogerler, Dalton Trans., 2016, 45, 11508–11521 RSC.
- (a) T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC; (b) T. W. Hayton, Dalton Trans., 2010, 39, 1145–1158 RSC.
- T. W. Hayton, J. M. Boncella, B. L. Scott, P. D. Palmer, E. R. Batista and P. J. Hay, Science, 2005, 310, 1941–1943 CrossRef CAS PubMed.
- J. L. Brown, S. Fortier, G. Wu, N. Kaltsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2013, 135, 5352–5355 CrossRef CAS PubMed.
- (a) N. H. Anderson, S. O. Odoh, Y. Y. Yao, U. J. Williams, B. A. Schaefer, J. J. Kiernicki, A. J. Lewis, M. D. Goshert, P. E. Fanwick, E. J. Schelter, J. R. Walensky, L. Gagliardi and S. C. Bart, Nat. Chem., 2014, 6, 919–926 CrossRef CAS PubMed; (b) N. H. Anderson, J. Xie, D. Ray, M. Zeller, L. Gagliardi and S. C. Bart, Nat. Chem., 2017, 9, 850–855 CrossRef CAS PubMed; (c) B. M. Gardner, G. Balazs, M. Scheer, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2015, 7, 582–590 CrossRef CAS PubMed; (d) E. P. Wildman, G. Balazs, A. J. Wooles, M. Scheer and S. T. Liddle, Nat. Commun., 2017, 8, 14769 CrossRef CAS PubMed; (e) S. L. Staun, D. C. Sergentu, G. Wu, J. Autschbach and T. W. Hayton, Chem. Sci., 2019, 10, 6431–6436 RSC; (f) W. S. Ren, G. F. Zi, D. C. Fang and M. D. Walter, J. Am. Chem. Soc., 2011, 133, 13183–13196 CrossRef CAS PubMed; (g) N. L. Bell, L. Maron and P. L. Arnold, J. Am. Chem. Soc., 2015, 137, 10492–10495 CrossRef CAS PubMed; (h) C. C. Zhang, G. H. Hou, G. F. Zi and M. D. Walter, Dalton Trans., 2019, 48, 2377–2387 RSC.
- (a) J. M. Smith, Prog. Inorg. Chem., 2014, 58, 417–470 CAS; (b) J. F. Berry, Comments Inorg. Chem., 2009, 30, 28–66 CrossRef CAS; (c) K. Dehnicke and J. Strahle, Angew. Chem., Int. Ed., 1992, 31, 955–978 CrossRef.
- D. M. King and S. T. Liddle, Coord. Chem. Rev., 2014, 266–267, 2–15 CrossRef CAS.
- (a) A. J. Lewis, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 13185–13192 CrossRef CAS PubMed; (b) A. J. Lewis, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 511–518 CrossRef CAS PubMed; (c) C. J. Hoerger, H. S. La Pierre, L. Maron, A. Scheurer, F. W. Heinemann and K. Meyer, Chem. Commun., 2016, 52, 10854–10857 RSC; (d) N. S. Settineri, A. A. Shiau and J. Arnold, Chem. Commun., 2018, 54, 10913–10916 RSC; (e) S. M. Franke, B. L. Tran, F. W. Heinemann, W. Hieringer, D. J. Mindiola and K. Meyer, Inorg. Chem., 2013, 52, 10552–10558 CrossRef CAS PubMed; (f) O. Cooper, C. Camp, J. Pecaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716–6723 CrossRef CAS PubMed; (g) A. C. Schmidt, F. W. Heinemann, W. W. Lukens and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef CAS PubMed; (h) D. M. King, F. Tuna, J. McMaster, W. Lewis, A. J. Blake, E. J. L. McInnes and S. T. Liddle, Angew. Chem., Int. Ed., 2013, 52, 4921–4924 CrossRef CAS PubMed; (i) K. C. Mullane, A. J. Lewis, H. Yin, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2014, 53, 9129–9139 CrossRef CAS PubMed; (j) D. P. Halter, F. W. Heinemann, J. Bachmann and K. Meyer, Nature, 2016, 530, 317 CrossRef CAS PubMed; (k) K. C. Mullane, T. Cheisson, E. Nakamaru-Ogiso, B. C. Manor, P. J. Carroll and E. J. Schelter, Chem.–Eur. J., 2018, 24, 826–837 CrossRef CAS PubMed; (l) P. A. Cleaves, C. E. Kefalidis, B. M. Gardner, F. Tuna, E. J. L. McInnes, W. Lewis, L. Maron and S. T. Liddle, Chem.–Eur. J., 2017, 23, 2950–2959 CrossRef CAS PubMed; (m) S. J. Kraft, J. Walensky, P. E. Fanwick, M. B. Hall and S. C. Bart, Inorg. Chem., 2010, 49, 7620–7622 CrossRef CAS PubMed.
- (a) L. Chatelain, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2016, 138, 1784–1787 CrossRef CAS PubMed; (b) M. Falcone, L. Chatelain and M. Mazzanti, Angew. Chem., Int. Ed., 2016, 55, 4074–4078 CrossRef CAS PubMed; (c) M. Falcone, C. E. Kefalidis, R. Scopelliti, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2016, 55, 12290–12294 CrossRef CAS PubMed; (d) M. Falcone, L. Chatelain, R. Scopelliti and M. Mazzanti, Chimia, 2017, 71, 209–212 CrossRef CAS PubMed; (e) M. Falcone, L. Chatelain, R. Scopelliti, I. Zivkovic and M. Mazzanti, Nature, 2017, 547, 332–335 CrossRef CAS PubMed; (f) M. Falcone and M. Mazzanti, Chimia, 2018, 72, 199–202 CrossRef CAS PubMed; (g) M. Falcone, L. N. Poon, F. F. Tirani and M. Mazzanti, Angew. Chem., Int. Ed., 2018, 57, 3697–3700 CrossRef CAS PubMed; (h) L. Barluzzi, L. Chatelain, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, Chem. Sci., 2019, 10, 3543–3555 RSC; (i) M. Falcone, L. Barluzzi, J. Andrez, F. F. Tirani, I. Zivkovic, A. Fabrizio, C. Corminboeuf, K. Severin and M. Mazzanti, Nat. Chem., 2019, 11, 154–160 CrossRef CAS PubMed; (j) C. T. Palumbo, L. Barluzzi, R. Scopelliti, I. Zivkovic, A. Fabrizio, C. Corminboeuf and M. Mazzanti, Chem. Sci., 2019, 10, 8840–8849 RSC; (k) J. Z. Du, C. Alvarez-Lamsfus, E. P. Wildman, A. J. Wooles, L. Maron and S. T. Liddle, Nat. Commun., 2019, 10, 4203 CrossRef PubMed; (l) J. Z. Du, D. M. King, L. Chatelain, E. L. Lu, F. Tuna, E. J. L. McInnes, A. J. Wooles, L. Maron and S. T. Liddle, Chem. Sci., 2019, 10, 3738–3745 RSC.
- R. K. Thomson, T. Cantat, B. L. Scott, D. E. Morris, E. R. Batista and J. L. Kiplinger, Nat. Chem., 2010, 2, 723–729 CrossRef CAS PubMed.
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS PubMed.
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482–488 CrossRef CAS PubMed.
- N. Tsoureas, A. F. R. Kilpatrick, C. J. Inman and F. G. N. Cloke, Chem. Sci., 2016, 7, 4624–4632 RSC.
- P. A. Cleaves, D. M. King, C. E. Kefalidis, L. Maron, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 10412–10415 CrossRef CAS PubMed.
- K. C. Mullane, H. Ryu, T. Cheisson, L. N. Grant, J. Y. Park, B. C. Manor, P. J. Carroll, M.-H. Baik, D. J. Mindiola and E. J. Schelter, J. Am. Chem. Soc., 2018, 140, 11335–11340 CrossRef CAS PubMed.
- S. Fortier, J. R. Aguilar-Calderon, B. Vlaisavljevich, A. J. Metta-Magana, A. G. Goos and C. E. Botez, Organometallics, 2017, 36, 4591–4599 CrossRef CAS.
- P. L. Diaconescu and C. C. Cummins, Inorg. Chem., 2012, 51, 2902–2916 CrossRef CAS PubMed.
- I. S. R. Karmel, N. Fridman, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2014, 136, 17180–17192 CrossRef CAS PubMed.
- (a) S. Fortier, G. Wu and T. W. Hayton, Dalton Trans., 2010, 39, 352–354 RSC; (b) O. Bénaud, J.-C. Berthet, P. Thuéry and M. Ephritikhine, Inorg. Chem., 2011, 50, 12204–12214 CrossRef PubMed.
- I. C. Tornieporthoetting and T. M. Klapotke, Angew. Chem., Int. Ed., 1995, 34, 511–520 CrossRef CAS.
- M. Castillo, A. J. Metta-Magana and S. Fortier, New J. Chem., 2016, 40, 1923–1926 RSC.
- P. J. Fagan, J. M. Manriquez, S. H. Vollmer, C. S. Day, V. W. Day and T. J. Marks, J. Am. Chem. Soc., 1981, 103, 2206–2220 CrossRef CAS.
- I. Castro-Rodriguez, H. Nakai and K. Meyer, Angew. Chem., Int. Ed., 2006, 45, 2389–2392 CrossRef CAS PubMed.
- R. E. Cramer, F. Edelmann, A. L. Mori, S. Roth, J. W. Gilje, K. Tatsumi and A. Nakamura, Organometallics, 1988, 7, 841–849 CrossRef CAS.
- A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250–3251 CrossRef CAS PubMed.
- J. J. Scepaniak, C. G. Margarit, J. N. Harvey and J. M. Smith, Inorg. Chem., 2011, 50, 9508–9517 CrossRef CAS PubMed.
- A. K. Maity, J. Murillo, A. J. Metta-Magana, B. Pinter and S. Fortier, J. Am. Chem. Soc., 2017, 139, 15691–15700 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and procedures, additional crystallographic information, and NMR spectral data. CCDC 1958453, 1958456, 1958640, 1958626 and 1958455. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05992j |
| This journal is © The Royal Society of Chemistry 2020 |