
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium-initiated polymerization of lactide: a route to remarkable cellular uptake for photodynamic therapy of cancer†
Nancy
Soliman
 ab,
Luke K.
McKenzie
bc,
Johannes
Karges
b,
Emilie
Bertrand
ab,
Mickaël
Tharaud
d,
Marta
Jakubaszek
be,
Vincent
Guérineau
f,
Bruno
Goud
e,
Marcel
Hollenstein
c,
Gilles
Gasser
*b and
Christophe M.
Thomas
*a
ab,
Luke K.
McKenzie
bc,
Johannes
Karges
b,
Emilie
Bertrand
ab,
Mickaël
Tharaud
d,
Marta
Jakubaszek
be,
Vincent
Guérineau
f,
Bruno
Goud
e,
Marcel
Hollenstein
c,
Gilles
Gasser
*b and
Christophe M.
Thomas
*a
aChimie ParisTech, PSL University, CNRS, Institut de Recherche de Chimie Paris, 75005 Paris, France. E-mail: christophe.thomas@chimie-paristech.fr
bChimie ParisTech, PSL University, CNRS, Institute of Chemistry for Life and Health Sciences, Laboratory for Inorganic Chemical Biology, 75005 Paris, France. E-mail: gilles.gasser@chimieparistech.psl.eu
cInstitut Pasteur, Department of Structural Biology and Chemistry, Laboratory for Bioorganic Chemistry of Nucleic Acids, CNRS UMR 3523, 75015 Paris, France
dUniversité de Paris, Institut de Physique du Globe de Paris, CNRS, 75005 Paris, France
eInstitut Curie, PSL University, CNRS UMR 144, 75005 Paris, France
fInstitut de Chimie des Substances Naturelles, CNRS UPR2301, Université Paris-Sud, Université Paris-Saclay, Avenue de la Terrasse, 91198 Gif-sur-Yvette Cedex, France
First published on 30th January 2020
Abstract
Ruthenium complexes have attracted a lot of attention as potential photosensitizers (PSs) for photodynamic therapy (PDT). However, some of these PSs are unsuitable for PDT applications due to their low cellular uptake, which is possibly the consequence of their relatively low degree of lipophilicity, which prevents them from penetrating into tumor cells. Here, we report the simple one-pot synthesis of ruthenium-containing nanoconjugates from a non-cell-penetrating, non-phototoxic ruthenium(II) polypyridyl complex (RuOH), by a drug-initiated ring-opening polymerization of lactide through the formation of a zinc initiator. These conjugates were then formulated into nanoparticles by nanoprecipitation and characterized by means of nuclear magnetic resonance spectroscopy (NMR), matrix-assisted laser desorption/ionization – time of flight mass spectrometry (MALDI-TOF MS) and dynamic light scattering (DLS). Finally, their photo-therapeutic activity (λexc = 480 nm, 3.21 J cm−2) in cancerous human cervical carcinoma (HeLa) and non-cancerous retinal pigment epithelium (RPE-1) cells was tested alongside that of RuOH and their cellular uptake in HeLa cells was assessed by confocal microscopy and inductively coupled plasma – mass spectrometry (ICP-MS). All nanoparticles showed improved photophysical properties including luminescence and singlet oxygen generation, enhanced cellular uptake and, capitalizing on this, an improved photo-toxicity. Overall, this study demonstrates how it is possible to transform a non-phototoxic PDT PS into an active PS using an easy, versatile polymerization technique.
Introduction
In recent years, photodynamic therapy (PDT) has gained considerable attention as a complementary/alternative tool in cancer treatment. It consists of a two-stage procedure involving the local or systemic administration of a photosensitizer (PS) followed by local irradiation with light at a specific wavelength. Irradiation leads to the formation of the activated photosensitizer (PS*), which subsequently interacts with surrounding cellular molecular oxygen 3O2 to generate cytotoxic reactive oxygen species (ROS) including singlet oxygen 1O2.1,2 Most clinically approved PSs are based on cyclic tetrapyrrole structures such as porphyrins, chlorins or phthalocyanines. However, most of these PSs suffer from several drawbacks including (i) poor water solubility, (ii) lack of cancer selectivity and (iii) slow clearance from the body leading to prolonged photosensitivity.3 Numerous efforts have therefore been made to develop new non-tetrapyrrole-based PSs. Among them, transition metal complexes and in particular ruthenium(II) polypyridyl complexes were found to be extremely promising,4–10 with one example starting phase II clinical trials in Canada for the treatment of bladder cancer.11,12 While a multitude of ruthenium(II) polypyridyl complexes have been reported in the literature, the compound [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2 (RuOH) with bipy = 2,2′-bipyridine and dppz = dipyrido[3,2-a:2′,3′-c]phenazine was previously found to have no photo-toxicity, due to a lack of cellular uptake owing to its low degree of lipophilicity (Fig. 1).13 However, its photophysical properties were ideal for PDT applications.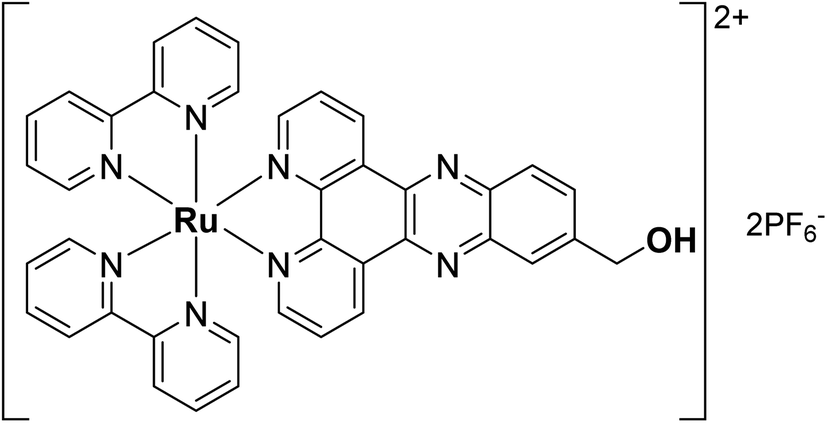 | ||
| Fig. 1 Structure of the ruthenium complex RuOH. | ||
The lipophilicity of drugs has long been established as a crucial physicochemical parameter in determining their potency and toxicity.50,51 Numerous studies have shown a strong correlation between the increased lipophilicity of Ru(II) polypyridyl complexes and their biological activity, explained in most cases by an increased cellular uptake (hence, increased intracellular concentrations of the complexes).49 Increased lipophilicity of Ru(II) polypyridyl complexes can be performed either by substitution of the bipy ancillary ligands46,47 or by enlargement of the aromatic ring system of the dppz moiety, using the π-expansive ligand dppn (dppn = benzo[i]dipyrido[3,2-a:2′,3′-h]quinoxaline) instead.48,52,53 However, an increased lipophilicity also raises concern in the way of administration as the drug may have to be dissolved in an organic solvent such as DMSO or DMF, which may lead to serious side effects. To avoid using potentially toxic excipients, encapsulation into nanoparticles with hydrophobic properties may be a better strategy.21
Nanoparticles including polymeric nanoparticles offer, as a drug delivery platform, the possibility to improve accumulation at the tumor site by taking advantage of the abnormalities in cancer cells through the enhanced permeability and retention (EPR) effect,14 therefore improving treatment specificity and reducing side effects. So far, drug delivery systems using the [Ru(bipy)2(dppz)]2+ scaffold as nanobody-conjugation15 and mesoporous silica nanoparticle formulation16 showed only limited use due to a lack of ROS generation inside the cell and loading limitations.
Biodegradable aliphatic polyester based nanoparticles have been widely used for encapsulation of drugs with an aim to harmlessly deliver them in a controlled and triggered fashion.4,17,18 Generally, therapeutic agents are incorporated into the polymer matrix through physical interaction. However, this type of entrapment suffers from strong limitations that hamper intravenous administration such as (i) significant “burst release” effect, (ii) uncontrolled and low encapsulation efficiency and (iii) poor drug loading (generally less than 10%).19 To overcome these constraints, Kricheldorf was the first to report the preparation of drug–polyester conjugates by ring-opening polymerization (ROP) of cyclic esters via the formation of reactive initiators by mixing triethylaluminium with hydroxyl containing bioactive molecules.20 Inspired by this pioneering work, several research groups developed drug-initiated polymerizations of different monomers.21,25–29 Although the synthetic covalent approaches are interesting, the relationship between the properties of the polymer and the properties of the final polymer-encapsulated complex has rarely been clarified or linked to the physical properties of these conjugates. Therefore, physical encapsulation remains the most common strategy to deliver metal complexes because it is often considered simpler and faster than covalent conjugation.
We hypothesized that covalent incorporation of RuOH into aforementioned nanoparticles would allow access to the promising photophysical properties of the molecule by improving cellular penetration. Herein, we report the conjugation of RuOH to polylactide (PLA), a well-established and FDA-approved biodegradable and biocompatible aliphatic polyester for drug delivery applications,22–24via a simple and straightforward drug-initiated method. We prepared active and easy-to-prepare nanoconjugates from two metal-based precursors for the synthesis of hydrophobic nanoparticles. Polymers of different molecular weights were synthesized from D,L-lactide, L-lactide and D-lactide yielding respectively, atactic and isotactic polymers that could be formulated into nanoparticles by nanoprecipitation. The influence of molecular weight, tacticity and nanostructure on the photophysical properties, phototherapeutic activity, cellular uptake and photosensitizer release kinetics was evaluated.
Results and discussion
Preparation of Ru–PLA nanoconjugates
ROP of lactide using metal-coordination initiators including metal alkoxides (MORs) is arguably the most efficient method to prepare well-controlled polylactides (PLAs).30 The MORs – synthesized prior to polymerization by mixing a hydroxyl-containing compound with an active metal catalyst – can initiate and control the ROP of LA leading to quantitative insertion of the alcohol into the PLA chain-end.31 The commercially available stannous octoate Sn(Oct)2 has been the most utilized ROP metal catalyst so far. However, it is difficult to be removed entirely from the polymer, raising concerns as the amount of residual tin in polymers for biomedical applications should remain low according to the Food and Drug Administration (FDA).32 Therefore, in view of pharmaceutical applications, special interest has been devoted to non-toxic and biocompatible metal centers such as zinc.33,34 As RuOH bears a hydroxyl group on the dppz ligand, it can react with Zn(N(SiMe3)2)2 to form an active zinc alkoxide to be used for the initiation and control of LA polymerization (Scheme 1).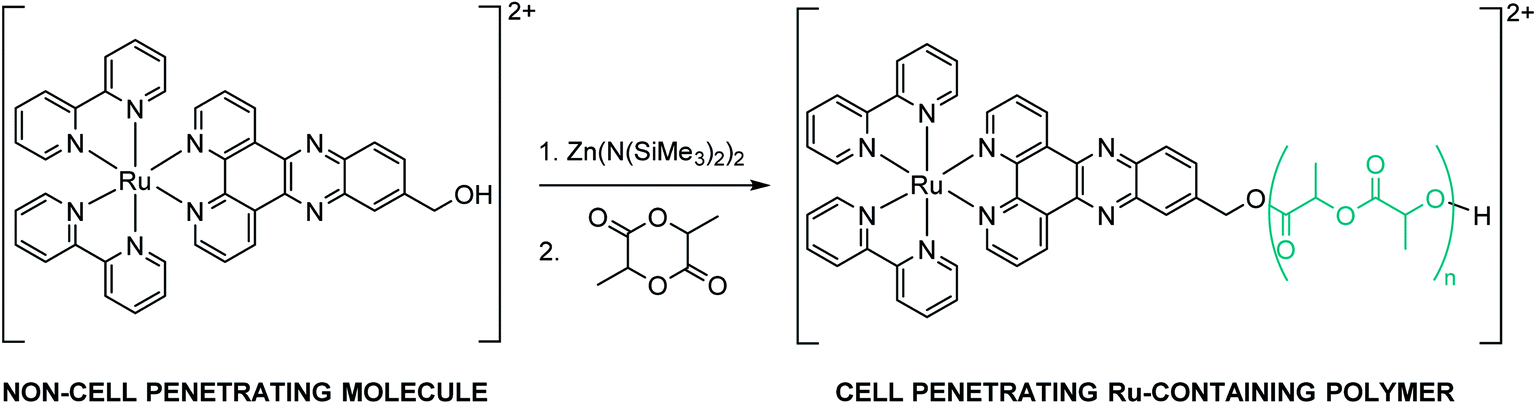 | ||
| Scheme 1 Synthesis of RuPLA. | ||
RuOH, as a PF6 salt, was only readily soluble in acetonitrile limiting reactions in this solvent system. Anion exchange from PF6− to OTf−, BF4− or BPh4− did not improve its solubility in non-polar solvents such as toluene or in polar organic solvents such as THF or CH2Cl2. The reaction between equimolar amounts of RuOH and Zn(N(SiMe3)2)2 resulted in a Schlenk equilibrium that strongly favors the formation of zinc bis(alkoxide) over time, as characterized by 1H NMR (Fig. S1†). As zinc bis(alkoxide) is the most thermodynamically stable species, it was used in the ROP of LA by directly adding two equivalents of RuOH to a solution of Zn(N(SiMe3)2)2 in acetonitrile. Polymerizations of racemic (D,L-lactide) or enantiopure lactide (D-lactide or L-lactide) were performed using the formed zinc alkoxide initiator with a monomer concentration of 3.0 M at 60 °C for 1 h. Conversion was measured by 1H NMR in CDCl3 using the signals from the methine protons of the unreacted monomer (q, 5.04 ppm) and the ones of the polymer (5.18 ppm). After 1 h of polymerization, the resulting ruthenium–polylactide (Ru–PLA) conjugates were simply isolated by precipitation to remove unreacted monomer (Table 1). Polymers P1, P2 and P5, synthesized from D,L-lactide, yielded amorphous polymers as a result of random sequence of D- and L-units along the polymer backbone. For polymers P3 and P4, derived from the enantiopure monomers, we obtained semi-crystalline polymers as the sequence of the absolute configuration is the same along the polymer backbone. The quantitative conjugation of RuOH to PLA through an ester linkage was shown by 1H and 13C NMR spectroscopy as the peak integrating for the –CH2O– on the dppz ligand of RuOH has shifted from 4.98 to 5.54 ppm in the 1H NMR spectrum and from 64.11 to 66.89 ppm in the 13C NMR spectrum after polymerization (Fig. S2–S4†). In addition, experimental number-average molecular weights (Mn, calculated by NMR end-group analysis) were close to the theoretical ones, which indicates that the polymerization proceeded in a controlled fashion (i.e., without any significant side reactions), as confirmed by MALDI-TOF mass spectrometry (Fig. S5†). The MALDI-TOF MS analysis reveals two distribution of peaks, one spaced by Δm/z = 144.03 corresponding to one lactide unit and the second spaced by Δm/z = 72.02 attributed to the presence of transesterification reactions which is not surprising with this type of catalyst. Besides, the isotopic pattern of each peak clearly shows the presence of ruthenium. Of note, these polymers could not be analyzed by size exclusion chromatography (SEC) with refractive index detector (RID) and THF or DMF as eluent since the characterization of bipyridine ruthenium complex-containing polymers by SEC is rather difficult. As already reported by Schubert and co-workers,35 this can be explained by the interaction of the metal ions and nitrogen atoms with the SEC column material, which is a highly cross-linked polystyrene divinylbenzene (PS-DVB) gel. Once these Ru–PLA conjugates were synthesized and fully characterized, they were formulated into nanoparticles NPs by a nanoprecipitation method.36Ru–PLA was dissolved in THF, a water–miscible organic solvent and the resulting solution was added dropwise to water, a non-solvent containing 0.3% w/v of poly(vinyl alcohol) (PVA). The instantaneous diffusion of the organic solvent into the aqueous solution resulted, after solvent removal under reduced pressure, in the formation of narrowly dispersed polymeric NPs as characterized by their intensity-average diameter Dz with a polydispersity index (PdI) lower than 0.3 (Table 1). Interestingly, the two polymers synthesized from the enantiopure lactide could not be formulated into NPs using these conditions. However, NPs could be obtained by mixing these two at equimolar ratio giving stereocomplex nanoparticles NPs-3. The stereocomplex formation was confirmed by differential scanning calorimetry (DSC) showing, for the stereocomplex, a melting temperature (Tm) 60 °C higher than the one of the enantiopure parent polymers. This result is in accordance with what has already been reported. It is, indeed, well documented in the literature that stereocomplexation between poly(D-lactide) and poly(L-lactide) can take place in solution.37,38
| Entry | LA/Zn | LA | Conv.b (%) | M n,NMR (kDa) | DPb | M n,theo (kDa) | % RuOHd (wt%) | NPs | D z ± SDe (nm) | PdI ± SDe |
|---|---|---|---|---|---|---|---|---|---|---|
| a All reactions were performed at 60 °C for 1 h with [LA]0 = 3 M unless otherwise stated. b Conversion of monomers, degree of polymerization (DP) and Mn,NMR were calculated by 1H NMR spectroscopy in CD3CN. c Calculated according to Mn,theo = ((LA/Zn) × conv. × M(LA))/2 + M(RuOH) with M(LA) = 144.13 g mol−1 and M(RuOH) = 1015.7 g mol−1. d Calculated according to (M(RuOH)/Mn,NMR) × 100. e Determined by dynamic light scattering (DLS) as an average of three measurements, values given with standard deviation (SD). f This reaction was performed with [LA]0 = 2 M. | ||||||||||
| P1 | 11 | D,L-LA | 75 | 1.9 | 5 | 1.7 | 53 | NPs-1 | 309.7 ± 1.815 | 0.198 ± 0.022 |
| P2 | 41 | D,L-LA | 92 | 4.0 | 20 | 3.7 | 25 | NPs-2 | 119.6 ± 0.406 | 0.236 ± 0.003 |
| P3 | 41 | D-LA | 91 | 4.0 | 20 | 3.7 | 25 | NPs-3 | 174.1 ± 1.429 | 0.192 ± 0.006 |
| P4 | 41 | L-LA | 91 | 3.7 | 18 | 3.7 | 27 | |||
| P5 | 70 | D,L-LA | 95 | 7.0 | 41 | 5.8 | 15 | NPs-4 | 248.8 ± 1.601 | 0.100 ± 0.011 |
Photophysical properties
After formulation into NPs, their photophysical properties were investigated to determine whether the attachment of a polymer chain or the nanoparticle formulation influenced these properties. As expected, the absorption (Fig. S6†) and emission spectra (Fig. S7†) did not show a significant difference. Strikingly, NPs showed a highly improved luminescence in H2O (Table S1†) in comparison to RuOH which was only measurable on the detection limit of our apparatus (emission quantum yield Φem,NPs-2,3,4 = 1.3–1.4%, Φem,RuOH < 0.1%). Following this, the excited state lifetimes in a degassed and air saturated aqueous solution were investigated. The obtained values were found in the nanosecond range (Table S1 and Fig. S8–S11†) as for other published ruthenium(II) polypyridyl complexes.42,43 Importantly, the lifetime drastically decreases in the presence of air indicating that the excited triplet state of the ruthenium(II) polypyridyl complex can interact with 3O2 in the air to produce 1O2. To validate this and quantify the amount of 1O2 generated upon irradiation at 450 nm, singlet oxygen quantum yields Φ1O2 were determined by two complementary techniques: (1) direct by measuring the phosphorescence of 1O2 at 1270 nm, (2) indirect by measuring the change in absorbance of a 1O2 scavenger.44 Interestingly, NPs showed a highly improved singlet oxygen production in H2O (Table S2†) in comparison to RuOH (Φ1O2,NPs-2,3,4 = 11–12%, Φ1O2,RuOH = 3%). These results are a direct consequence of the prevention of quenching effects in water attributed to hydrogen bonding of water to the nitrogen atoms of the phenazine moiety,39–41 which leads to a drastic improvement of the photophysical properties of NPs in comparison to RuOH.Photosensitizer release kinetic studies
As the ruthenium complex is linked to PLA by an ester bond, its release from the resulting nanoconjugates is mainly governed by hydrolysis. The RuOH release goes with the degradation of PLA into its shorter oligomers and eventually, lactic acid. The latter is a known, non-toxic byproduct of several metabolic pathways. There is no systemic toxicity associated with the use of this type of nanoparticles.21 Also, the longer the polymer chain is, the slower the release is (Fig. S12 and S13†). It is difficult to establish a rational correlation between the release kinetics of RuOH and the nanoparticles' cytotoxicities as the RuPLA nanoconjugates does not act as a prodrug which becomes active when the linkage between the drug and the polymer gets cleaved. However, as shown by the photophysical studies, the nanoconjugate formulation improves the overall photophysical properties of RuOH.Biological evaluation
The dark and light cytotoxicities of NPs were tested in human cervical carcinoma (HeLa) and non-cancerous retinal pigment epithelium (RPE-1) cells alongside RuOH following a 4, 24 and 48 h incubations (Tables 2 and S3†). All compounds were non-cytotoxic following a 4 h incubation in the dark (IC50 > 100 μM for NPs; >500 μM for RuOH). Low dose light irradiation (λexc = 480 nm, 10 min, 3.21 J cm−2) yielded photosensitization with NPs in HeLa cells with a phototoxicity index (PI) ranging from >2.5 to >6, while no photosensitization was observed for NPs-2 and NPs-3 in RPE-1 cells (IC50 > 100 μM). Notably we chose 480 nm as irradiation wavelength since it is the optimal wavelength for this PDT PS. RuOH remained non-cytotoxic following irradiation (IC50 > 500 μM). Following a 24 h incubation, the compounds remained non-cytotoxic in the dark (IC50s 81.3 μM, >100 μM and >500 μM for NPs-4, NPs-1,2,3 and RuOH, respectively) while an increase in photosensitization was observed for NPs (PI range >5.9 to >11.25) and extremely modest photosensitization close to no photosensitization was observed with RuOH in HeLa cells (IC50 274.4 μM). Following a 48 h incubation, all compounds were somewhat cytotoxic in the dark in HeLa cells (IC50s 31.8–62.8 μM and 99.1 μM for NPs and RuOH respectively) while their light cytotoxicities remained relatively stable. Worthy of note, the blue light irradiation at 480 nm is not toxic to the two cell lines at a fluence of 3.21 J cm−2. The cytotoxicity of NPs and RuOH in HeLa cells was correlated to their cellular uptake which was investigated by ICP-MS following 4, 24 and 48 h incubations (Fig. 2). As expected, RuOH had limited cellular uptake (0.0019–0.0019 pg per cell) while a variation in cellular uptakes was observed for NPs. Relatively low cellular uptake was measured for NPs-1 (0.038–0.064 pg per cell), which is expected due to the short polymer chain length and lower hydrophobicity. NPs-2,3 were moderately more cell-penetrating (0.174–0.230 pg per cell and 0.101–0.408 pg per cell for NPs-2 and NPs-3, respectively) with a cellular uptake twice higher after 48 h incubation for NPs-3 compared to NPs-2 justifying the interest in forming stereocomplex nanoparticles. As predicted from the higher chain length and hence hydrophobicity (hence implied lipophilicity), NPs-4 was observed to be the most cell accumulating (0.256–2.448 pg per cell) with a 9.5× increase in cellular ruthenium content from the 4 h timepoint to the 48 h timepoint. The trend in cellular uptake fits with the hydrophobicity of the compounds.| 4 h | 24 h | 48 h | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Light | Dark | PIb | Light | Dark | PIb | Light | Dark | PIb | |
| a IC50 values were an average of three measurements. b PI refers to the phototoxicity index, which is the ratio between the IC50 values in the dark and the ones upon light irradiation. | |||||||||
| NPs-1 | 28.0 ± 3.2 | >100 | 3.6 | 18.7 ± 3.6 | >100 | 5.9 | 12.7 ± 3.3 | 43.4 ± 17.8 | 3.4 |
| NPs-2 | 34.2 ± 17.4 | >100 | 2.9 | 14.5 ± 6.3 | >100 | 6.9 | 23.4 ± 3.8 | 61.4 ± 17.9 | 2.6 |
| NPs-3 | 41.3 ± 4.5 | >100 | 2.5 | 9.5 ± 1.1 | >100 | 11.25 | 8.4 ± 4.3 | 62.9 ± 13.4 | 7.5 |
| NPs-4 | 16.7 ± 4.3 | >100 | 6 | 7.8 ± 7.7 | 81.3 ± 10.9 | 10.9 | 4.4 ± 0.8 | 31.8 ± 7.1 | 7.5 |
| RuOH | >500 | >500 | 274.4 ± 70.1 | >500 | 1.8 | 99.1 ± 12.7 | 248.6 ± 37.7 | 2.5 | |
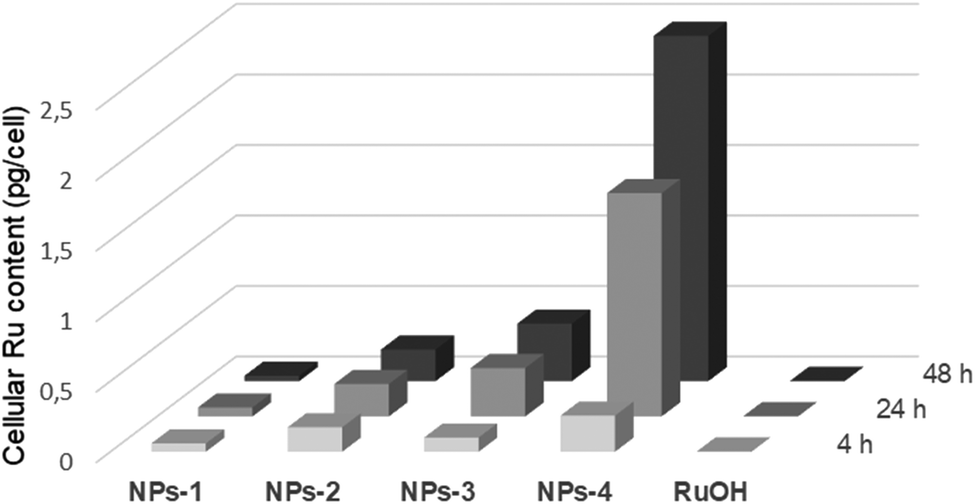 | ||
| Fig. 2 Cellular uptake in HeLa cells as measured by ICP-MS. Values given in pg per cell. | ||
The subcellular localization of the compounds was investigated using confocal microscopy in HeLa cells (Fig. 3 and S14†). Cellular localization of NPs can be observed by confocal microscopy (λexc = 480 nm, λem = 650–750 nm) while no luminescence was observed for RuOH. Similar staining patterns were observed for NPs with punctuate cytoplasmic signals dominating. Costaining with the anti-LAMP lysosomal antibody indicated no signal overlap (Fig. S14†). Endocytosis is implicated in the cellular uptake of nanoparticles45 and may be, at least, the partial source of the punctate staining. While localization varies slightly between NPs, no significant change in subcellular localization is observed with increased incubation time, suggesting that the increase in photosensitizing effect is not due to a change in localization. These data clearly indicate an increase in cellular uptake, and photosensitization, in cells by the polymer encapsulated ruthenium complex as compared to its ‘free’ counterpart, RuOH. This encapsulation may be suitable for use with small complexes allowing cellular access.
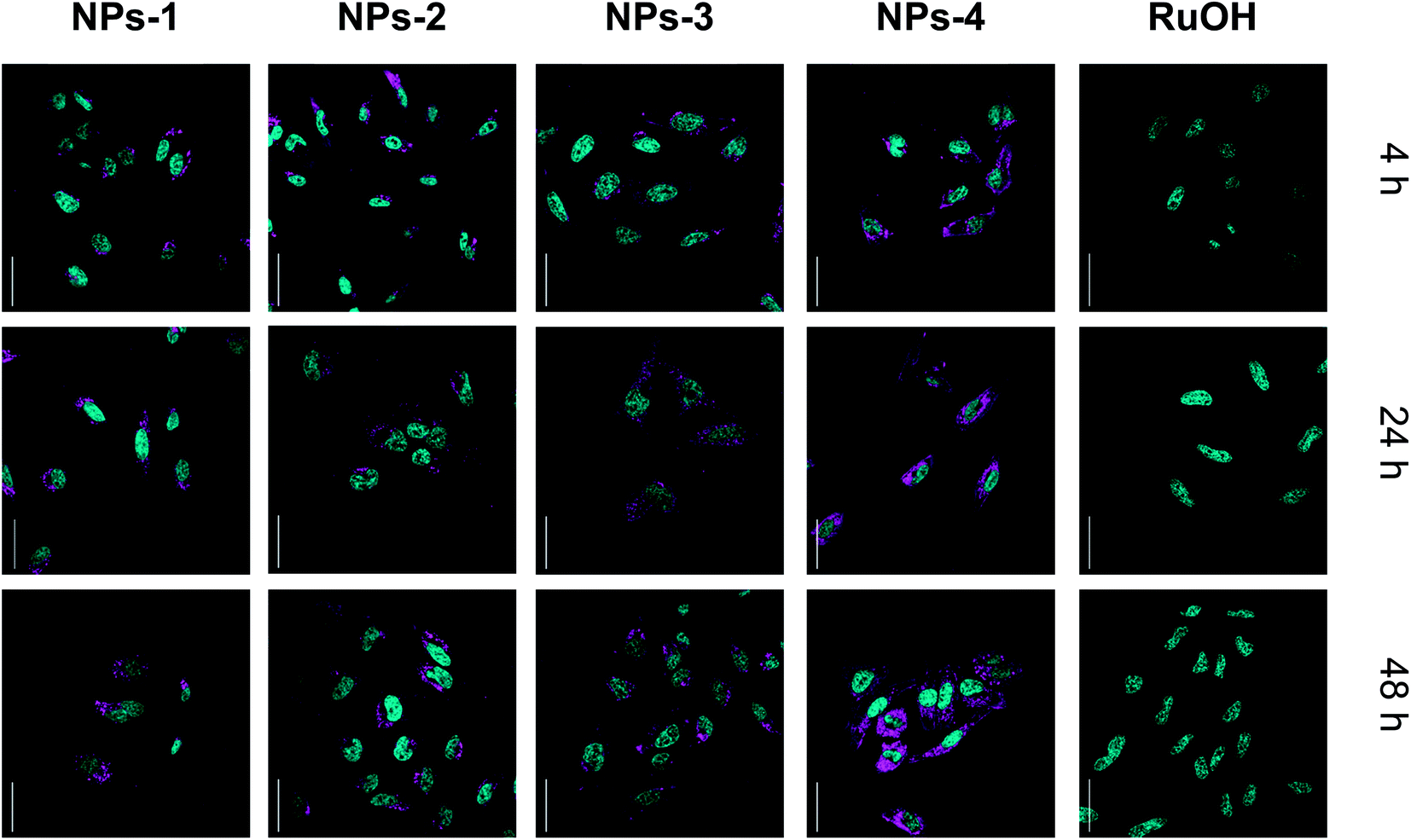 | ||
| Fig. 3 Confocal microscopy images of HeLa cells incubated with NPs and RuOH (50 μM, 37 °C, 4, 24, 48 h, magenta). NPs co-stained with the nuclear stain, NucBlue (cyan), scale bar 50 μm. | ||
Conclusion
A series of ruthenium polylactide RuPLA conjugates with different degrees of polymerization, tacticity and a drug loading up to 53% has been successfully prepared by the drug-initiated ROP of lactide using a bimetallic initiator formed from RuOH. This one-pot synthetic strategy prevents the use of the cost-effective and time-consuming preparative size exclusion chromatography currently applied for purification of ruthenium–polymer conjugates prepared from chelation of a polymeric macroligand to the metal salt, as the only purification step here is precipitation. These conjugates were then formulated into narrowly dispersed nanoparticles with superior photophysical properties including their luminescence and singlet oxygen generation due to lower amount of quenching effects in H2O. Capitalizing on this, the particles were biologically tested on HeLa cervical cancer cells, showing an enhanced cellular uptake of RuOH overtime and hence, an improved phototherapeutic activity overtime. Owing to its simplicity, this strategy can be expanded and applied to a broad range of ruthenium complexes. This opens new avenues in PDT treatment in which patients could be treated over several days using a single injection.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
CNRS, ENSCP and French Ministry of Research and Higher Education are thanked for financial support of this work. This work was also financially supported by an ERC Consolidator Grant PhotoMedMet to G. G. (GA 681679) and has received support under the program Investissements d'Avenir launched by the French Government and implemented by the ANR with the reference ANR-10-IDEX-0001-02 PSL (G. G.). We thank Dr Philippe Goldner for access to state-of-the-art laser apparatus. M. H. acknowledges funding from Institut Pasteur. N. S. gratefully acknowledges financial support from Cancéropôle Île-de-France for her PhD scholarship. L. K. M. acknowledges a fellowship from the ARC Foundation for Cancer Research (grant number: S-FB18006). Île-de-France Region is gratefully acknowledged for financial support of 500 MHz NMR spectrometer of Chimie ParisTech in the framework of the SESAME equipment project. We acknowledge the loan of Agilent's equipment to Chimie ParisTech. Part of this work was supported by IPGP multidisciplinary program PARI and by Region Île-de-France SESAME Grant no. 12015908. The authors would like to thank Purac for a generous loan of D,L-lactide. C. M. T. is grateful to the Institut Universitaire de France.References
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS.
- D. van Straten, V. Mashayekhi, H. S. de Bruijn, S. Oliveira and D. J. Robinson, Cancers, 2017, 9, 19 CrossRef PubMed.
- A. E. O'Connor, W. M. Gallagher and A. T. Byrne, Photochem. Photobiol., 2009, 85, 1053–1074 CrossRef PubMed.
- (a) L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC; (b) J. D. Knoll, B. A. Albani and C. Turro, Acc. Chem. Res., 2015, 48, 2280–2287 CrossRef CAS; (c) M. A. Sgambellone, A. David, R. N. Garner, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2013, 135, 11274–11282 CrossRef CAS; (d) K. Arora, M. Herroon, M. H. Al-Afyouni, N. P. Toupin, T. N. Rohrabaugh Jr, L. M. Loftus, I. Podgorski, C. Turro and J. J. Kodanko, J. Am. Chem. Soc., 2018, 140, 14367–14380 CrossRef CAS PubMed.
- J. Liu, C. Zhang, T. W. Rees, L. Ke, L. Ji and H. Chao, Coord. Chem. Rev., 2018, 363, 17–28 CrossRef CAS.
- L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2018, 379, 2–29 CrossRef.
- M. Jakubaszek, B. Goud, S. Ferrari and G. Gasser, Chem. Commun., 2018, 54, 13040–13059 RSC.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS.
- S. A. McFarland, A. Mandel, R. Dumoulin-White and G. Gasser, Curr. Opin. Chem. Biol., 2020, 56, 23–27 CrossRef CAS PubMed.
- P. S. Felder, S. Keller and G. Gasser, Adv. Ther., 2019, 1900139 Search PubMed.
- Theralase Annouces First Patient Treated in Phase II Non-Muscle Invasive Bladder Cancer Clinical Study, https://www.streetinsider.com/dr/news.php?id=15879950%26gfv=1, accessed September 24, 2019.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS.
- C. Mari, V. Pierroz, R. Rubbiani, M. Patra, J. Hess, B. Spingler, L. Oehninger, J. Schur, I. Ott, L. Salassa, S. Ferrari and G. Gasser, Chem.–Eur. J., 2014, 20, 14421–14436 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- J. Karges, M. Jakubaszek, C. Mari, K. Zarschler, B. Goud, H. Stephan and G. Gasser, ChemBioChem, 2019 DOI:10.1002/cbic.201900419 , accepted.
- Y. Ellahioui, M. Patra, C. Mari, R. Kaabi, J. Karges, G. Gasser and S. Gómez-Ruiz, Dalton Trans., 2019, 48, 5940–5951 RSC.
- E. Villemin, Y. C. Ong, C. M. Thomas and G. Gasser, Nat. Rev. Chem., 2019, 3, 261–282 CrossRef CAS.
- D. K. Chatterjee, L. S. Fong and Y. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1627–1637 CrossRef CAS PubMed.
- S. Shen, Y. Wu, Y. Liu and D. Wu, Int. J. Nanomed., 2017, 12, 4085–4109 CrossRef CAS PubMed.
- H. R. Kricheldorf and I. Kreiser-Saunders, Polymer, 1994, 35, 4175–4180 CrossRef CAS.
- J. Nicolas, Chem. Mater., 2016, 28, 1591–1606 CrossRef CAS PubMed.
- B. Tyler, D. Gullotti, A. Mangraviti, T. Utsuki and H. Brem, Adv. Drug Delivery Rev., 2016, 107, 163–175 CrossRef CAS PubMed.
- S. Farah, D. G. Anderson and R. Langer, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS PubMed.
- R. James, Adv. Drug Delivery Rev., 2016, 107, 277–288 CrossRef CAS PubMed.
- R. Tong and J. Cheng, Angew. Chem., Int. Ed., 2008, 47, 4830–4834 CrossRef CAS PubMed.
- R. Tong and J. Cheng, J. Am. Chem. Soc., 2009, 131, 4744–4754 CrossRef CAS.
- R. Tong and J. Cheng, Bioconjugate Chem., 2010, 21, 111–121 CrossRef CAS PubMed.
- R. Tong and J. Cheng, Macromolecules, 2012, 45, 2225–2232 CrossRef CAS.
- R. M. Johnson and C. L. Fraser, Biomacromolecules, 2004, 5, 580–588 CrossRef CAS.
- C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC.
- The MORs can also synthesized by nucleophilic attack of the metal-based chloride precursor on an epoxide, see for instance: C. Robert, T. E. Schmidt, V. Richard, P. Haquette, S. K. Raman, M.-N. Rager, R. M. Gauvin, Y. Morin, X. Trivelli, V. Guérineau, I. del Rosal, L. Maron and C. M. Thomas, J. Am. Chem. Soc., 2017, 139, 6217–6225 CrossRef CAS.
- J. Guo, P. Haquette, J. Martin, K. Salim and C. M. Thomas, Angew. Chem., Int. Ed., 2013, 52, 13584–13587 CrossRef CAS PubMed.
- P. Marin, M. J.-L. Tschan, P. Haquette, T. Roisnel, I. del Rosal, L. Maron and C. M. Thomas, Eur. Polym. J., 2019, 120, 109208 CrossRef CAS.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS.
- M. A. R. Meier, B. G. G. Lohmeijer and U. S. Schubert, Macromol. Rapid Commun., 2003, 24, 852–857 CrossRef CAS.
- C. J. Martínez Rivas, M. Tarhini, W. Badri, K. Miladi, H. Greige-Gerges, Q. A. Nazari, S. A. Galindo Rodríguez, R. Á. Román, H. Fessi and A. Elaissari, Int. J. Pharm., 2017, 532, 66–81 CrossRef PubMed.
- P. Marin, M. J.-L. Tschan, F. Isnard, C. Robert, P. Haquette, X. Trivelli, L.-M. Chamoreau, V. Guérineau, I. del Rosal, L. Maron, V. Venditto and C. M. Thomas, Angew. Chem., Int. Ed., 2019, 58, 12585–12589 CrossRef CAS.
- H. Tsuji, Adv. Drug Delivery Rev., 2016, 107, 97–135 CrossRef CAS.
- A. E. Friedman, J.-C. Chambron, J.-P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960–4962 CrossRef CAS.
- C. Turro, S. H. Bossmann, Y. Jenkins, J. K. Barton and N. J. Turro, J. Am. Chem. Soc., 1995, 117, 9026–9032 CrossRef CAS.
- J. Olofsson, B. Önfelt and P. Lincoln, J. Phys. Chem. A, 2004, 108, 4391–4398 CrossRef CAS.
- M. J. Cook, A. P. Lewis, G. S. G. McAuliffe, V. Skarda, A. J. Thomson, J. L. Glasper and D. J. Robbins, J. Chem. Soc., Perkin Trans. 2, 1984, 1293–1301 RSC.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- J. Karges, O. Blacque, P. Goldner, H. Chao and G. Gasser, Eur. J. Inorg. Chem., 2019, 2019, 3704–3712 CrossRef CAS.
- S. Zhang, H. Gao and G. Bao, ACS Nano, 2015, 9, 8655–8671 CrossRef CAS.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2007, 129, 46–47 CrossRef CAS.
- C. A. Puckett and J. K. Barton, Biochemistry, 2008, 47, 11711–11716 CrossRef CAS PubMed.
- U. Schatzschneider, J. Niesel, I. Ott, R. Gust, H. Alborzinia and S. Wölfl, ChemMedChem, 2008, 3, 1104–1109 CrossRef CAS PubMed.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–775s6 RSC.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef CAS PubMed.
- J. A. Arnott and S. L. Planey, Expert Opin. Drug Discovery, 2012, 7, 863–875 CrossRef CAS PubMed.
- H. Yin, M. Stephenson, J. Gibson, E. Sampson, G. Shi, T. Sainuddin, S. Monro and S. A. McFarland, Inorg. Chem., 2014, 53, 4548–4559 CrossRef CAS.
- T. Sainuddin, J. McCain, M. Pinto, H. Yin, J. Gibson, M. Hetu and S. A. McFarland, Inorg. Chem., 2016, 55, 83–95 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05976h |
| This journal is © The Royal Society of Chemistry 2020 |