
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptical monitoring of polymerizations in droplets with high temporal dynamic range†
Andrew C.
Cavell
a,
Veronica K.
Krasecki
a,
Guoping
Li
b,
Abhishek
Sharma
c,
Hao
Sun
 b,
Matthew P.
Thompson
b,
Christopher J.
Forman
b,
Si Yue
Guo
de,
Riley J.
Hickman
de,
Katherine A.
Parrish
a,
Alán
Aspuru-Guzik
defg,
Leroy
Cronin
c,
Nathan C.
Gianneschi
b and
Randall H.
Goldsmith
*a
b,
Matthew P.
Thompson
b,
Christopher J.
Forman
b,
Si Yue
Guo
de,
Riley J.
Hickman
de,
Katherine A.
Parrish
a,
Alán
Aspuru-Guzik
defg,
Leroy
Cronin
c,
Nathan C.
Gianneschi
b and
Randall H.
Goldsmith
*a
aDepartment of Chemistry, University of Wisconsin-Madison, 1101 University Avenue, Madison, WI 53706, USA. E-mail: rhg@chem.wisc.edu
bDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
cSchool of Chemistry, University of Glasgow, Joseph Black Building, University Avenue, Glasgow, Scotland G12 8QQ, UK
dDepartment of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada
eDepartment of Computer Science, University of Toronto, 40 St. George Street, Toronto, Ontario M5S 2E4, Canada
fCanadian Institute for Advanced Research (CIFAR) Senior Fellow, Toronto, Ontario M5S 1M1, Canada
gCIFAR Artificial Intelligence Chair, Vector Institute, Toronto, Ontario M5S 1M1, Canada
First published on 4th February 2020
Abstract
The ability to optically monitor a chemical reaction and generate an in situ readout is an important enabling technology, with applications ranging from the monitoring of reactions in flow, to the critical assessment step for combinatorial screening, to mechanistic studies on single reactant and catalyst molecules. Ideally, such a method would be applicable to many polymers and not require only a specific monomer for readout. It should also be applicable if the reactions are carried out in microdroplet chemical reactors, which offer a route to massive scalability in combinatorial searches. We describe a convenient optical method for monitoring polymerization reactions, fluorescence polarization anisotropy monitoring, and show that it can be applied in a robotically generated microdroplet. Further, we compare our method to an established optical reaction monitoring scheme, the use of Aggregation-Induced Emission (AIE) dyes, and find the two monitoring schemes offer sensitivity to different temporal regimes of the polymerization, meaning that the combination of the two provides an increased temporal dynamic range. Anisotropy is sensitive at early times, suggesting it will be useful for detecting new polymerization “hits” in searches for new reactivity, while the AIE dye responds at longer times, suggesting it will be useful for detecting reactions capable of reaching higher molecular weights.
Introduction
Chemical transformations frequently entail complex mixtures of relevant chemical species. In catalysed reactions, numerous intermediate states can be visited and catalyst speciation can contribute significantly to reaction dynamics.1–5 Polymerization reactions are natural producers of chemical heterogeneity, as growing chains of varying length and functionality can result in diverse and dynamic chemical environments that may influence reaction pathways and rates.6–9 As the number of processes developed for the synthesis of small molecules and polymeric materials continues to increase, so does the need for new, efficient techniques for gaining an understanding of their chemical composition and behaviour. The monitoring of these systems under synthetically relevant conditions is essential for further insight and progress in the informed development of new reactions. In particular, optical methods have the benefit of allowing for monitoring of reactions in real-time, with minimal disruption to the natural dynamics of the reactions under study.10–14 Photonic devices can act as multipliers for increasing sensitivity in optical spectroscopy of chemical reactions.15,16 Advancements in fluorescence microscopy over the last few decades have allowed for remarkable strides to be made in the study of chemical reactions.17–23 These techniques can even be employed at the single-molecule level to reveal unsynchronized dynamics of individual catalyst molecules.19,20,24–29 At the other extreme, the inherent scalability of optical methods makes them attractive readouts for massively parallel, high-throughput combinatorial testing of reaction conditions.30–35 Pairing this approach with machine-learning methods also enables accelerated discovery and screening of new functional materials.36–39 Ideally, such combinatorial searches will occur in chemical environments that strongly resemble the conditions that would be used in an industrial setting.Polymerization reactions performed in mixed phases and emulsions are used extensively in the industrial production of specialty materials.40 There is much interest in studying polymerizations carried out in dispersed media, as these processes form a toolkit for the facile production of complex polymeric and hybrid materials on large scales.41,42 Reactions in droplets are also highly relevant for microfluidic reactors, which can provide tight control of conditions in the continuous synthesis of complex structures,43–45 high-throughput screening of reaction conditions,45–47 and for applications in droplet-based assays.45,48,49 However, the inherent complexity and transient nature of these droplet systems makes for some difficulty in understanding their dynamics and behavior.50 For this reason, researchers have begun developing methods for in situ monitoring of this class of polymerizations.51,52 Indeed, while the confinement of reactions to small droplets can sometimes alter the rate of the reaction,53 it also plays to the advantage of optical monitoring strategies by allowing application of microscopy. Fluorescence-based methods are thus uniquely poised as powerful tools for the characterization and monitoring of polymerization catalysts in situ with high spatial resolution and signal-to-background.
One way of encoding useful chemical information into an optical signal is by relying on specific chemical transformations. Fluorogenic transformations rely on practically non-fluorescent reactants which yield strongly fluorescent products upon reaction, and have been used to monitor a variety of chemical transformations.24,26,54–60 Closely related to fluorogenic reactions, spectral shifts as a result of a change in functionality or extension of conjugation length have also been used to monitor chemical reactions.61,62 These methods generally require the use of reagents that have been specifically engineered to provide the measured response. These reagents, however, may display different reactivity than the desired substrate and so may lack generality. A more versatile approach would entail the simple introduction of an additive at ppm concentrations to allow monitoring of arbitrary reactions. This more generally applicable strategy would be particularly powerful for applications in combinatorial searches for new reactivity.
Fluorescence polarization anisotropy, which quantifies the rotational time scale of a molecule, provides one avenue for the study of polymer reaction dynamics that satisfies this need. By doping in a small amount of tracer fluorophore, the rotational dynamics of the tracer molecule can be used to report on the chemical evolution of its environment. Measurements of fluorophore rotational dynamics via fluorescence anisotropy have been used to investigate a broad variety of polymeric systems, including measuring the mobility of grafted polymer chains in microgels,63 observing the movement of small molecules in crosslinked polystyrene networks,64 following production of silica gels,65 photocuring of polymers,66 studying the self-assembly of block copolymers in water,67 quantifying distributions of conformers of intrinsically disordered proteins in solution,68,69 and studying dynamics within supported polymer thin films and glasses.70 There are very few studies that employ fluorescence polarization anisotropy measurements in droplets. One recent study measured the anisotropy of free rhodamine 6G dye molecules in microdroplets and examined the role of electrostatics at the droplet interface.71 Another recent study successfully determined protein–peptide dissociation constants by measuring the anisotropy of a series of droplets with systematically varying composition in a flow cell.72 To date, there are no examples of fluorescence anisotropy being used to monitor the progress of chemical reactions confined in droplets.
Another readout mode that satisfies this criterion, and can act through introduction of an additive, is aggregation-induced emission (AIE).73 AIE dyes typically possess internal rotational degrees of freedom which allow the dye to relax non-radiatively to the ground state upon excitation. As viscosity increases or aggregation of the dye occurs, these rotations are limited and radiative modes of relaxation are favoured, leading to an increase in fluorescence quantum yield and emission intensity. In this way, dyes that exhibit AIE or a viscosity-sensitive quantum yield can also be used to monitor the course of a polymerization reaction, either when the dye is covalently attached to a monomer74,75 or simply added to the solution,22,23 including in the dispersed phase,52 though this method has not been used at the level of single droplets. A priori, it is not evident whether AIE and fluorescence polarization anisotropy will reveal the same or complementary information about the polymerization.
Here, we demonstrate how fluorescence polarization anisotropy and AIE provide an information-rich readout of the state of a polymerization reaction in a single microdroplet. In particular, we show how an increase in fluorescence polarization anisotropy can be observed as a result of the incorporation of a fluorescent probe monomer into a growing polymer chain, and demonstrate its use to track the course of a polymerization. We will also describe how fluorescence polarization anisotropy and AIE can be used simultaneously. As will be shown below, the dynamic ranges of fluorescence polarization anisotropy and AIE are distinct and complementary.
Ring-opening metathesis polymerization (ROMP) catalysed by the ruthenium-based Grubbs Generation II (GG2) catalyst76,77 was selected for the development of this monitoring strategy as it is relatively air and moisture-tolerant, easy to control, relatively well understood, and industrially relevant.78,79 ROMP polymers and their derivatives are widely represented in both industrial catalysis and the development of specialized materials.80 Diverse polymer morphologies and polyfunctional materials are now accessible via ROMP through reproducible means and under mild conditions.81 Many thousands of tons of polynorbornene polymers are produced industrially each year.78 Specifically, the ROMP of norbornene to form polynorbornene was chosen as the primary reaction under study to explore this readout method.
In our implementation, droplets of predefined compositions of organic reaction mixture are produced and deposited using a robotic platform, immobilized on fluorinated surfaces, and then monitored optically as the polymerization proceeds. The creation of large droplet arrays (from 2 × 2 up to 20 × 20 on a single coverslip) allows for investigation of multiple reaction conditions simultaneously, and even application of complementary bulk analysis tools. Rotational dynamics of a norbornene functionalized perylene diimide (PDI) dye82–90 molecule are monitored using fluorescence polarization anisotropy (Scheme 1). Simultaneously, the intensity of emission from a tetraphenylethylene (TPE)-labelled norbornene monomer91 which exhibits AIE is monitored. Together, these two methods provide a real-time readout of polymerization reaction progress with a greater temporal dynamic range than either method alone.
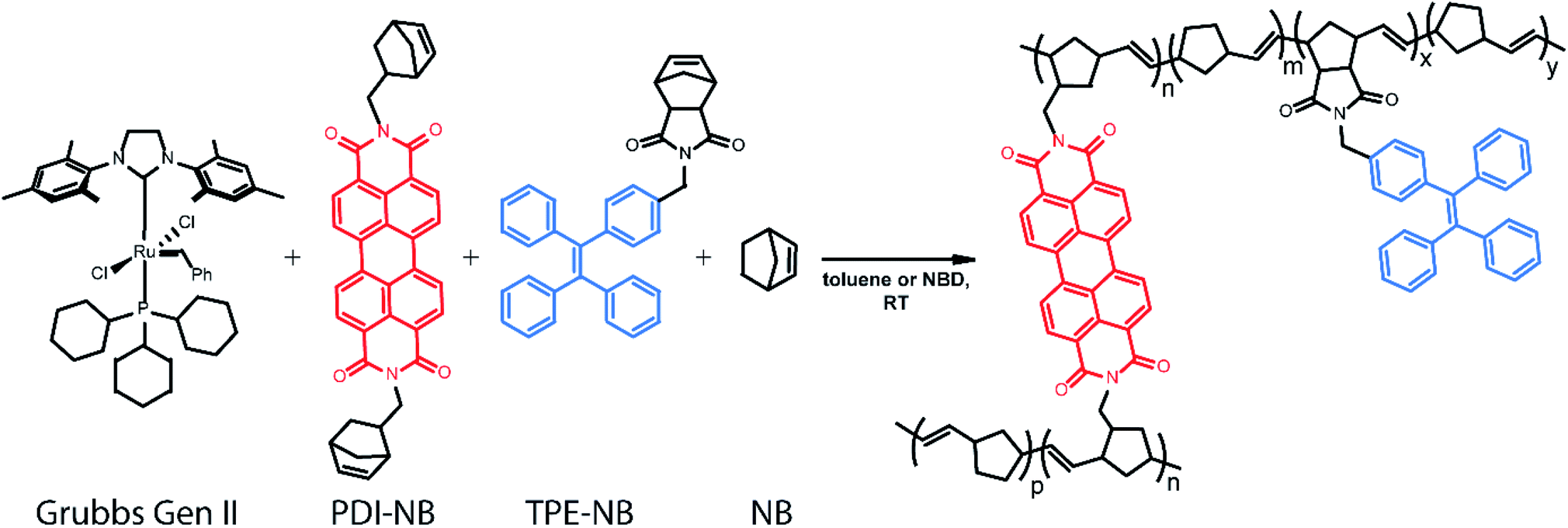 | ||
| Scheme 1 Grubbs Gen II-catalysed ROMP polymerization under study in this work, with TPE and PDI-based norbornene monomers as fluorescent probes. Probe monomers are present in low amounts (ppm for PDI and ppt for TPE) relative to unlabeled monomer (thus p, m, y ≫ n, x). | ||
Results and discussion
Surface functionalization for droplet immobilization
Measurements on individual reactions confined in droplets require that the droplets be immobilized for long-term imaging. We image through glass coverslips which are inherently hydrophilic, making it impractical to place an organic phase droplet onto the glass surface when water is present. A method was needed to functionalize the glass surfaces in a way that made them highly hydrophobic to repel the aqueous continuous phase but also lipophilic to immobilize the organic phase droplet. To this end, we functionalized pre-cleaned glass coverslips and all-glass reaction chambers92 using a solution-phase deposition of perfluorosilane, 1H,1H,2H,2H-perfluorodecyltriethoxysilane, to afford a hydrophobic and lipophilic fluorinated surface (see ESI† for details). Fluorinated surfaces are valuable as an immobilization technique,93,94 with applications in molecular catalysis95 and for immobilization of molecules in microarrays for biological assays.96 While perfluorinated compounds are typically hydrophobic, certain fluorinated compounds are more lipophilic than others depending on fluorinated chain length.97 By using a longer fluorinated chain to functionalize the glass surface, a surface is created where droplets of organic solvent placed in contact with these coverslips under an aqueous continuous phase will stick to the fluorinated surface and will stay immobilized (Fig. 1). The surfaces are not readily wetted by the continuous phase and so the organic droplet phase stays in contact with the glass despite its low density relative to the surrounding water. Importantly, more typical alkylated hydrophobic/lipophilic surfaces (such as those made with octylsilane) did not result in as robust droplet phase immobilization as did the fluorinated surfaces.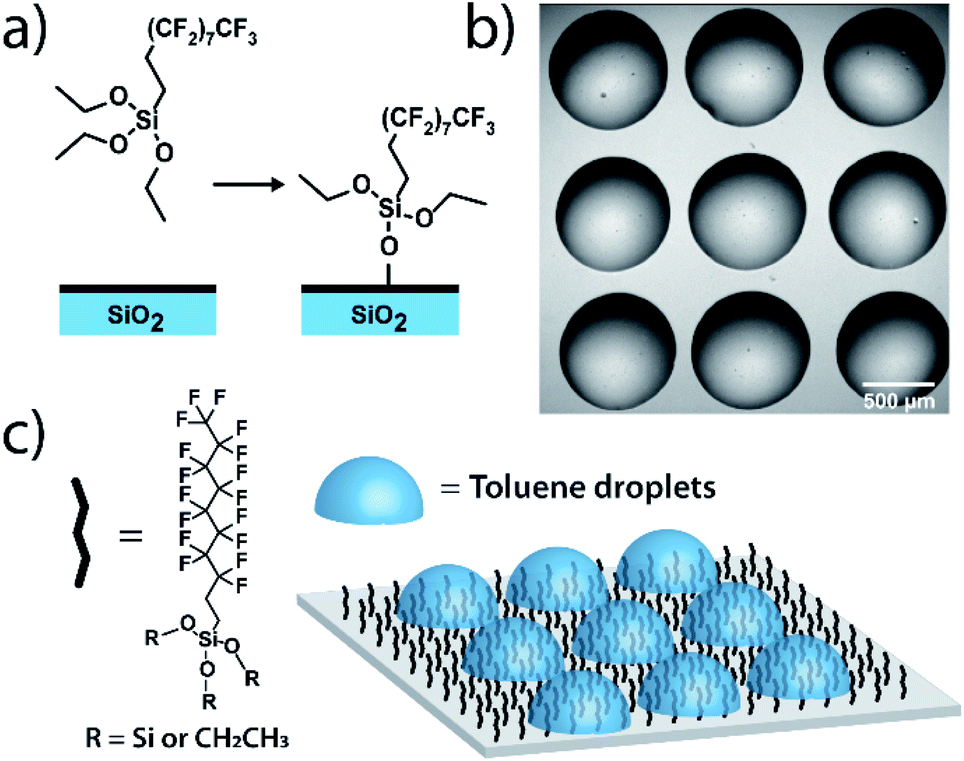 | ||
| Fig. 1 Surface functionalization for droplet immobilization: (a) deposition of perfluorodecyltriethoxysilane. (b) Bright field image of a 3 × 3 array of toluene droplets immobilized on a glass coverslip, surrounded by an aqueous continuous phase. (c) The prepared hydrophobic fluorinated glass surface has a high relative affinity for organic solvent, allowing droplet array immobilization. | ||
Droplet placement with a robotic platform
A home-built robotic platform was constructed to allow for precise 2D placement of droplets of varying compositions.98,99 The robot consists of two linear actuators mounted on rails above a sample preparation stage, topped with a stepper motor for precision z positioning of a custom 3D-printed syringe module. The linear actuators allow for x–y control of the position of the syringe head, which can then be lowered to the sample stage for taking up each sample and placing droplets. The syringe head assembly contains a second stepper motor that controls the position of the syringe plunger to dispense small volumes of solution to create droplets. The configuration used for these experiments consisted of a 10 μL Hamilton Gastight syringe held by the stepper motor in a 3D-printed assembly allowing for precise actuation of the plunger down to sub-nanoliter steps. The syringe was fitted with a 27-gauge blunt-tip needle. The use of a blunt tip is essential, as the angled sharp tip on common laboratory needles allows for the organic phase reaction mixture to flow out the side of the needle aperture and float to the surface of the continuous phase. A blunt needle forces the organic phase to contact the coverslip surface directly and stick in place. This computer-controlled robotic platform is modular in design, allowing for simple modification of the sample stage and easy interchange of different syringe types and volumes. While a simple syringe module was used for this work, the head could be swapped with a variety of alternate dispensing modules or microfluidic devices to scale droplet production and immobilization for the needs of future applications.Tracer dye design and synthesis
Ensuring that polymerization will result in a measurable change in fluorescence polarization anisotropy necessitates the use of a fluorophore that changes its rotational dynamics significantly upon polymerization. To this end, we designed and synthesized (see ESI†) a perylene diimide-based fluorescent crosslinking monomer (PDI-NB, Scheme 1), which is expected to experience a significant loss of rotational freedom upon incorporation into the growing polymer chain. The ability of this monomer to undergo a second incorporation (i.e. to cross-link) should additionally limit its rotation as the reaction proceeds, yielding a significant overall increase in the monomer's fluorescence anisotropy.In addition to the rotational probe, a monomer labelled with tetraphenylethylene (TPE-NB, Scheme 1) was incorporated into the polymerizations for use in a complementary readout strategy. TPE exhibits AIE, wherein a restriction of intramolecular rotation yields an increase in fluorescence quantum yield. The chosen TPE-based ROMP monomer has been utilized previously in the synthesis of ion-and-pH-sensing fluorescent polymers100,101 and in the preparation of fluorescent nano-objects.91 Based on recent reports exploring the incorporation of TPE dyes in RAFT polymerizations,74,75 we hypothesized that the fluorescence turn-on exhibited by the TPE-NB dye upon aggregation should also be observable during polymerization.
Polymerization reactions
The polymerization reactions in our experiments consist of a high concentration of unlabelled norbornene in toluene (typically 7 M unless otherwise noted) with lower concentrations of TPE-NB monomer (2 mM) and our PDI-NB monomer (∼1 μM). The tracer dyes are kept at low concentrations to limit their effect on product polymer morphology and make any potential influence on reaction kinetics negligible. Use of low concentrations also prevents aggregation of the dyes, which would otherwise result in artificially low anisotropy values or high initial AIE intensities. Absorption spectra of PDI-NB taken in 7 M norbornene confirm the absence of dye aggregation (see Fig. S15†).102 Reactions were also explored using norbornadiene as a solvent and comonomer, with the unlabelled norbornene and norbornadiene present at 2 M and 7 M, respectively. Stock solutions of GG2 catalyst were freshly prepared before each experiment and added to each reaction mixture immediately before droplet production. The droplet arrays were assembled onto fluorinated surfaces underneath a continuous phase of water using the robotic platform described above. In experiments monitoring multiple reaction conditions simultaneously, the robot placed droplets of each composition in order, automatically rinsing the syringe in between samples. Immediately following droplet placement, these prepared arrays were placed on a fluorescence microscopy setup for imaging (Fig. 2).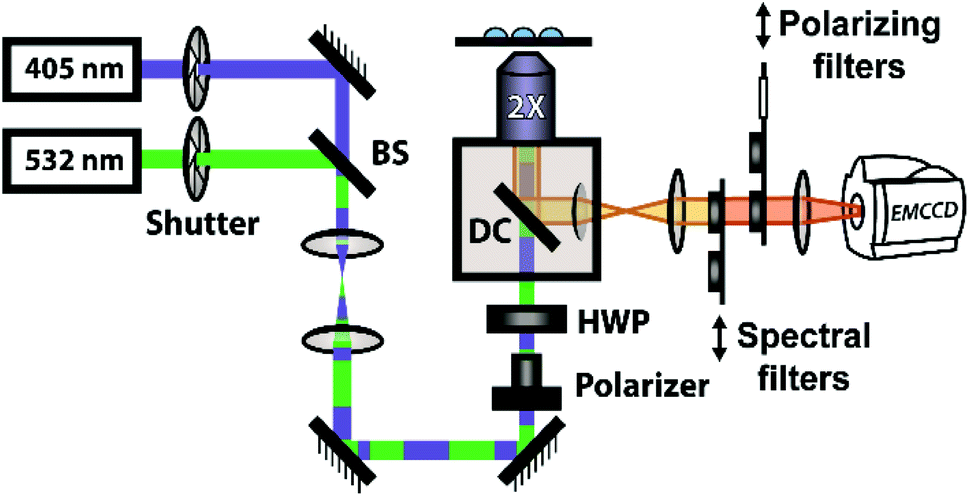 | ||
| Fig. 2 Optical setup for fluorescence measurements. Synchronized shutters and sliding mounts allow for alternating excitation with 405 nm and 532 nm light for the two probe monomers and collection through appropriate spectral and polarizing filters. BS: beamsplitter; HWP: half-wave plate; DC: dichroic. | ||
Fluorescence anisotropy measurements
The steady-state fluorescence anisotropy of the PDI-based fluorescent monomer was measured throughout each polymerization reaction using a home-built fluorescence microscopy setup (Fig. 2). The droplets containing PDI dye were excited in a widefield geometry using a vertically polarized 532 nm laser. Emission from the PDI fluorophores was isolated from the excitation light via a multi-edge band-pass dichroic and subsequent 532 nm long-pass filter. Images of the parallel and perpendicular components (I‖ and I⊥) of the emission from the PDI monomers were recorded sequentially every 5 minutes using a single camera (EMCCD, Andor Ixon) by taking alternating frames through two orthogonally oriented polarizing filters mounted in a computer-controlled sliding mount. The anisotropy (r) was calculated using eqn (1):103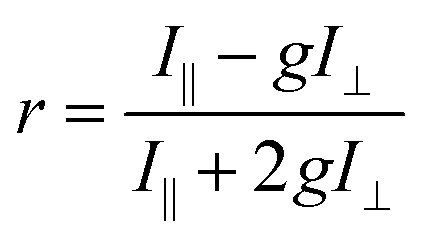 | (1) |
This anisotropy value (r) gives a measure of the depolarization of the emission relative to the excitation polarization. Said another way, this value relates the amount of rotational displacement over the emission lifetime of the dye. The calculated anisotropy should increase from r ≈ 0 to r ≈ 0.4 as the dye transitions from a state of fast rotation to one with highly limited rotation. This calculation was carried out for each pixel of interest in the recorded image. The constant g (the “g-factor”) is a correction factor to account for differences in collection efficiency between the parallel and perpendicular channels and was calculated and applied on a per-pixel basis (see ESI† for details). The anisotropy value reported for each droplet is an average taken over all pixels within the droplet. This value is recorded for every droplet in the array at every frame to produce plots of anisotropy over time (Fig. 3), while the per-pixel anisotropy values are used to create colorscale images of the anisotropy across the droplet arrays at every frame (as in Fig. 4). The use of larger arrays of droplets accelerates data acquisition by allowing the monitoring of multiple reactions simultaneously, enabling a direct visual comparison between varied reaction conditions and internal controls in parallel. Movies of the monitoring of multiple droplets are available in the online ESI.†
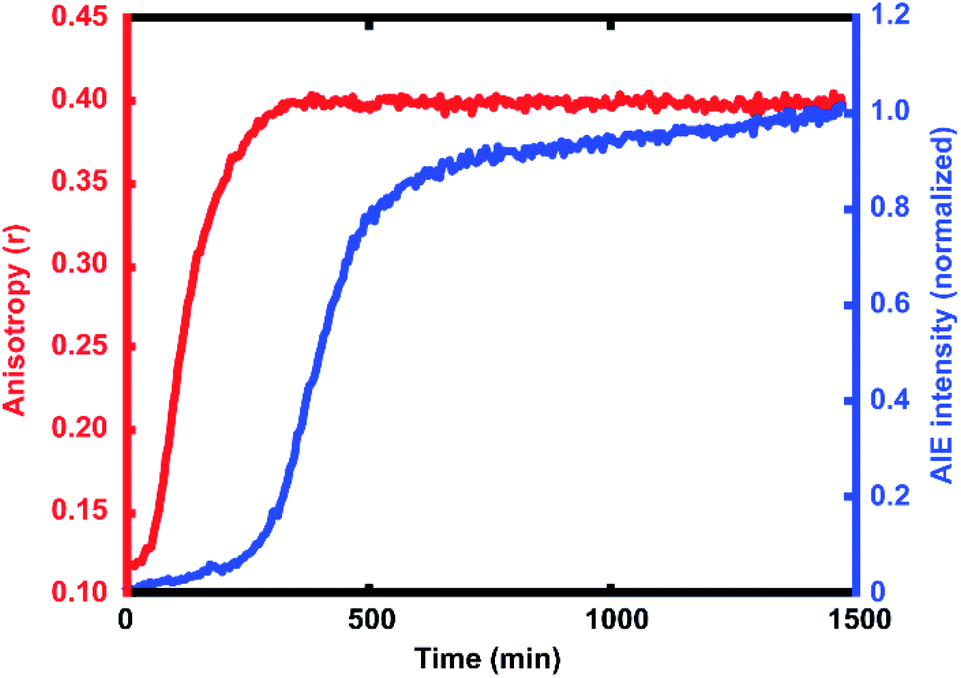 | ||
| Fig. 3 Average anisotropy vs. time (red) and average AIE intensity vs. time (blue), showing the additional dynamic range given by the offset response of the AIE signal relative to the increase in anisotropy. | ||
 | ||
| Fig. 4 Monitoring the polymerization progress in a single droplet. (a) Colorscale image of anisotropy values in droplets over time. As the reaction progresses, the droplet increases in anisotropy. (b) Emission intensity from the AIE monomer probe in a droplet over time. Droplet shown is the same as in the anisotropy images above. Each frame corresponds to the same timepoint as above (scale bar 250 μm). | ||
For every polymerization reaction, we observe an increase in the measured steady-state anisotropy of the reaction mixture droplet. A non-zero starting value of r ≈ 0.1 is seen in each case, due to the relatively high initial viscosity of the unpolymerized reaction mixture as compared to toluene alone (where r = 0.05, see ESI†). As the reactions continue, the anisotropy increases until at later times it saturates near the theoretical maximum value (r = 0.4). Experiments with arrays containing varying catalyst concentrations show a shift in this response to later times for droplets with less catalyst, and earlier times for droplets with more catalyst, providing further evidence that the anisotropy increase depends on the rate of the polymerization reaction (Fig. 5).
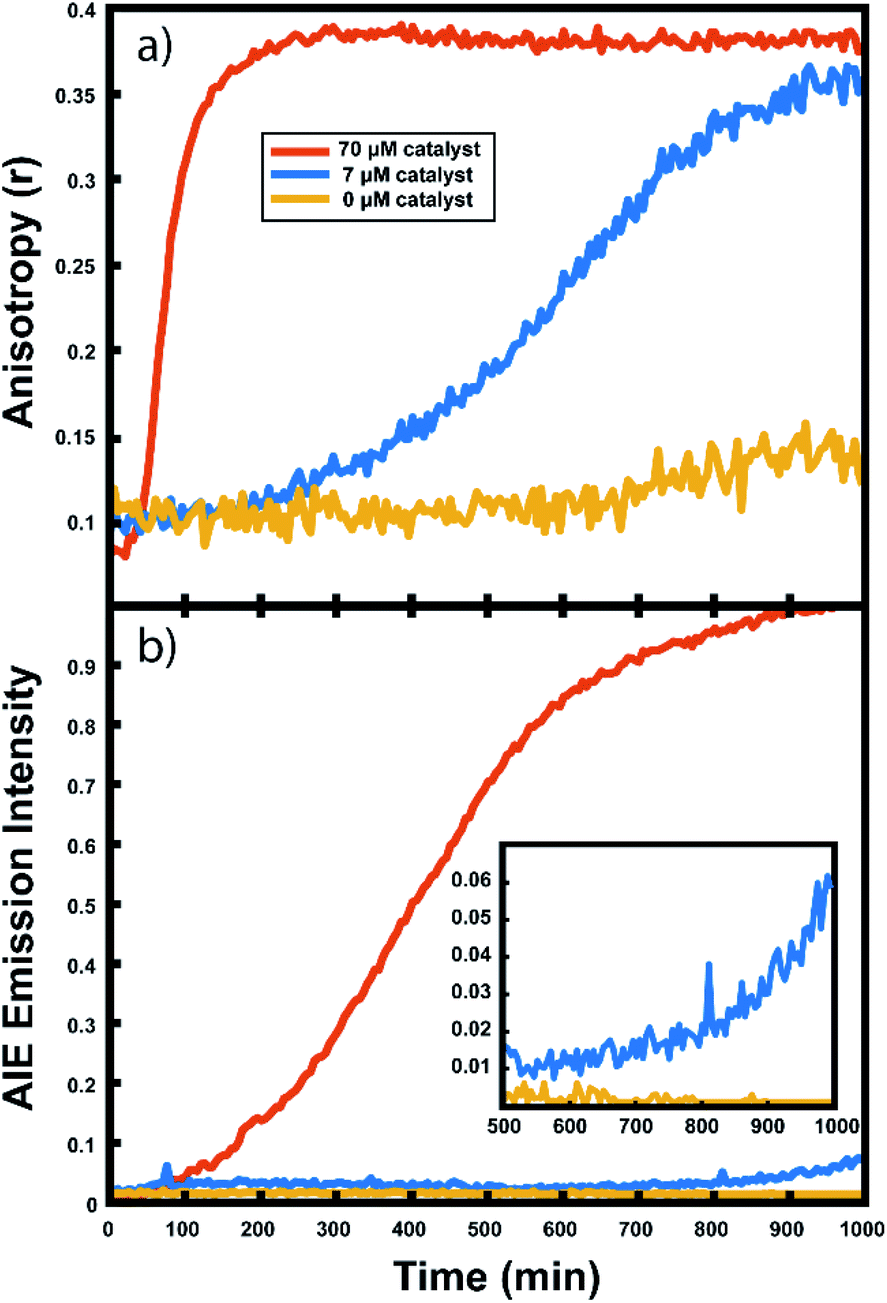 | ||
| Fig. 5 Monitoring norbornene/toluene droplets at different catalyst concentrations: (a) anisotropy response curves and (b) AIE response curves in droplets containing 70 μM (red), 7 μM (blue), and 0 μM (yellow) Grubbs Gen II catalyst. Inset shows late time intensity dynamics. | ||
Multiple competing phenomena could potentially contribute to a restricted degree of rotation for the fluorescent probe, potentially leading to an increase in fluorescence anisotropy that does not accurately track polymerization reaction progress. One potential cause of a false positive increase in anisotropy is lack of droplet stability resulting in loss of solvent to the surrounding continuous phase. If the droplet phase dissolves into the continuous phase too quickly, then an observed increase in anisotropy could be due to changes in freedom of motion resulting from a decrease in droplet volume (and consequent change in viscosity). In our experiments, we see that the immobilized droplets are stable over long periods of time, preventing this effect from being dominant on the timescale of the polymerizations. Still, some droplet contraction is seen at longer times, causing the slight rise seen in the yellow trace in Fig. 5, and when left for many hours, the organic droplets are observed to disappear entirely, eventually dissolving into the surrounding aqueous continuous phase. Notably, the droplets containing polymer are more stable at these later times and are seen to shrink much more slowly than the catalyst-free droplets. Our attempts to use various surfactants to increase their stability led to an even faster contraction of the droplets, likely due to the solubilizing effect of the surfactants. Control experiments comparing droplets with and without catalyst present show that the anisotropy increases observed in droplets containing active polymerizations occur well before any changes in anisotropy occur due to solvent loss in inactive droplets (see ESI†). Further confirmation of the correlation between anisotropy and molecular weight will be given below.
Aggregation-induced emission measurements
In addition to the anisotropy measurements described above, emission intensity from an aggregation-induced emission dye, TPE-NB, was measured as a complementary readout of reaction progress. With progressing polymerization, the measured fluorescence from the TPE dye should increase both from incorporation into the growing polymer and consequent steric hindrance, as well as from increasing viscosity of the surrounding chemical environment. These effects will both contribute to a restriction of intramolecular rotation, suppressing non-radiative decay from the excited state, and thereby increasing its emission intensity. The TPE-NB monomer was excited at 405 nm for the AIE measurements. These measurements were made at the same time as the anisotropy measurements described above. The 405 nm laser necessary for the AIE readout was coaligned with the 532 nm anisotropy beam (Fig. 2), and computer-controlled shutters and sliding mounts were used to excite and collect the fluorescence from each probe monomer individually at each timepoint (additional details in ESI†). In this way, time-lapse videos were created of the changing emission from each of the droplets.Complementary measurements
The collected AIE videos reveal increases in emission intensity over time for the polymerizing droplets, followed by eventual saturation of the response. As with the anisotropy experiments, the time at which the increases occur is sensitive to different reaction conditions and tracks with changes in catalyst concentration. Importantly, the AIE response is seen to occur after the anisotropy response in all cases. The onset of the AIE response is delayed relative to the onset of the anisotropy increase, and it continues to increase well after the anisotropy measurement has saturated at its highest value. This separation reveals the ongoing polymerization continuing for hours after the anisotropy response has run out of dynamic range. At the same time, the anisotropy response is sensitive to the developing polymerization at shorter timescales that the AIE measurement misses. The combination of these two readout methods yields a significant extension to the temporal dynamic range, as the measurement of fluorescence anisotropy adds the ability to observe polymerization reactions at much earlier times (and lower Mw) than the AIE response would allow for on its own. Fluorescence polarization anisotropy may be better suited for assessing if a polymerization has occurred at all (i.e. for differentiating a small amount of polymerization from no polymerization), as would be valuable in identifying a “hit” in a combinatorial screen, while AIE may be more suited to determine if a large molecule weight has been reached (though it may be insensitive to small degrees of polymer formation).This separation of observed dynamic ranges is not entirely unexpected, as these different measurements are probing different phenomena. Previous work examining the AIE of growing polymers shows that, for some reaction conditions, there is not an appreciable increase in AIE until a critical molecular weight has been reached, after which the emission intensity scales with increasing conversion.74 The difference in observed dynamic ranges can be explained by the different mechanisms that give rise to each observed increase – in the anisotropy measurement, the rotational correlation time of the fluorescent monomer must only be slowed relative to its emission lifetime to see an increase in r. As the PDI dye is incorporated into the large polymer chain, its rotation is slowed dramatically. Though the TPE dye's global rotation is also slowing at these earlier timepoints (as it is also being incorporated), it is the intramolecular rotations of the phenyl rotors that must be slowed to turn on the AIE response. Judging from the observed separation in time scales, slowing this process requires more time for the reaction to reach a much higher effective solution viscosity before increased emission intensity can be observed. In some cases, it may be that intermolecular interactions between multiple TPE molecules or TPE molecules with other pi systems may be required to reduce these internal degrees of freedom.73
Experiments in neat monomer
Many important polymerizations are carried out in neat monomer. To investigate the influence of the presence of solvent on our reaction readout, we performed experiments in which we replaced the toluene component of the reaction mixture with norbornadiene (NBD). Norbornadiene may also act as a monomer in the polymerization along with norbornene, but is a liquid at room temperature, allowing for its use as a solvent for the polymerization reaction. Importantly, norbornadiene's reactivity differs slightly from that of norbornene due to differences in ring strain energies of the two monomers, and it may undergo some crosslinking as the second double bond is opened, though this second ring opening is relatively unfavorable.104,105Fluorescence anisotropy and AIE were used to monitor the progression of the ring opening metathesis polymerization of NB/NBD to form a statistical copolymer of polynorbornene with lightly crosslinked norbornadiene units. The data collected from droplets under these conditions show increases in anisotropy and AIE as before (Fig. 6). These experiments showed that both the anisotropy and the AIE readout methods are capable of tracking polymerizations in reactions of neat monomer, a common condition for industrial polymerizations.
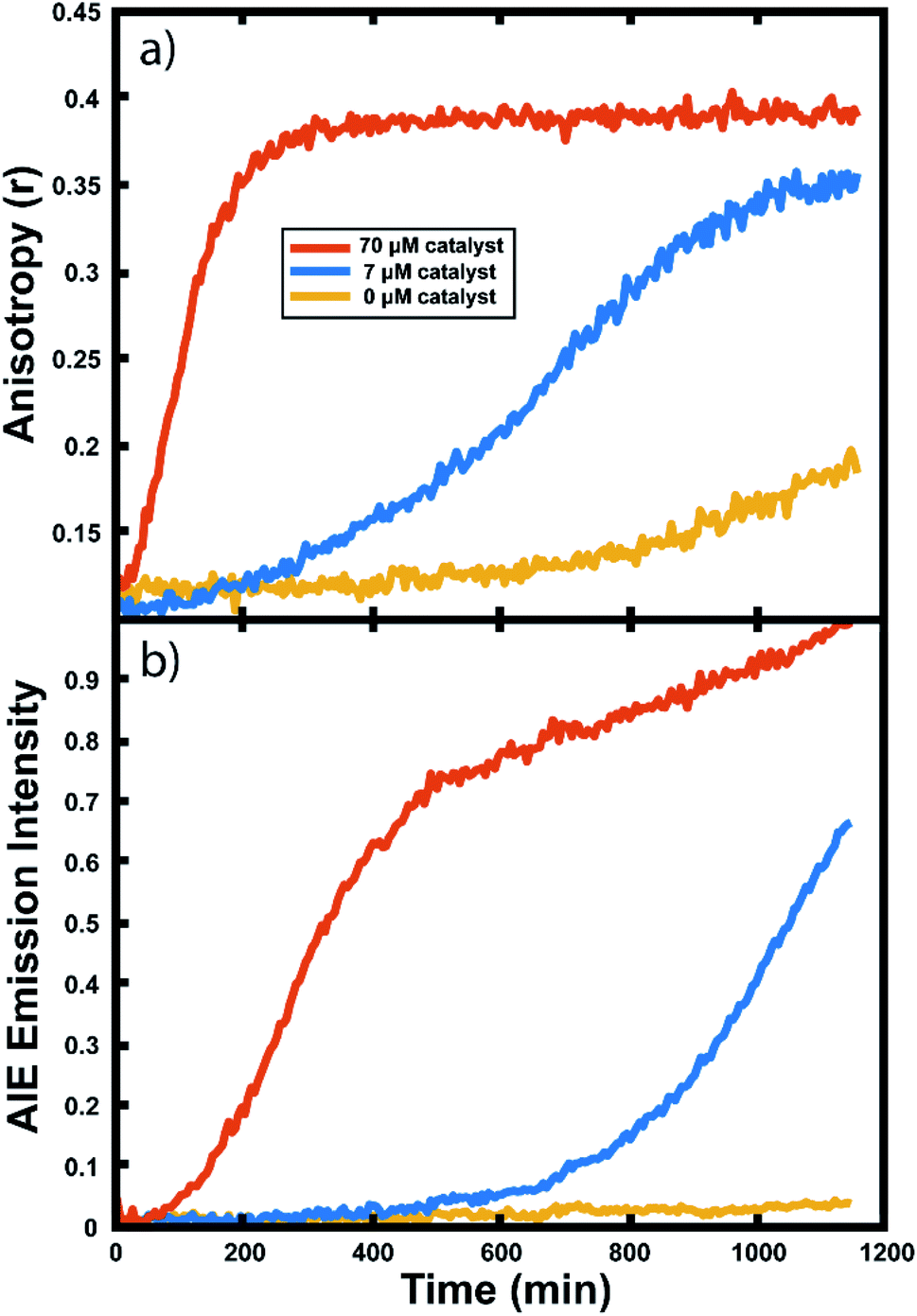 | ||
| Fig. 6 Monitoring norbornene/norbornadiene droplets at different catalyst concentrations: (a) anisotropy response curves and (b) AIE response curves in droplets containing 70 μM (red), 7 μM (blue), and 0 μM (yellow) Grubbs Gen II catalyst. | ||
Calibration of anisotropy readout with molecular weight
In order to confirm that the observed increases in anisotropy correspond to increasing polymer molecular weight, GPC-MALS (Gel Permeation Chromatography with Multiple Angle Light Scattering) measurements were taken of polymers formed in reactions in immobilized droplet arrays. To accomplish this, larger scale experiments (consisting of >1000 droplets each) were performed under the same conditions as those detailed above. For each run, a smaller droplet array was produced as usual for optical monitoring, while 3 additional 20 × 20 arrays of the same size droplets were allowed to polymerize simultaneously. We carried out these reactions with 25 μM GG2 and ended them at varying times from 30–1200 minutes. At the desired end timepoint, the anisotropy value was recorded and the reactions in the larger arrays were quenched by addition of ethyl vinyl ether. In this way, large enough samples could be produced for GPC-MALS without altering the reaction conditions used in earlier optical experiments (details in ESI†).The resulting relationship between Mw and steady state anisotropy, shown in Fig. 7, reveals that the shorter time reactions which produced smaller molecular weight polymers correspond to lower measured anisotropy values, while the samples which consisted of higher molecular weight polymers are those where high anisotropy values were measured, as expected. This relationship saturates at around r = 0.4, the maximum possible steady state anisotropy value, and so it appears that for this reaction system all polymers greater than ∼300 kDa would be expected to yield this limiting anisotropy value. The scatter in the data likely results from variability in how quickly the polymerization was quenched after measuring each sample's anisotropy. This observed correlation of anisotropy with molecular weight further confirms the viability of this measurement technique for tracking polymerization reactions in situ, particularly at early times where AIE is insensitive.
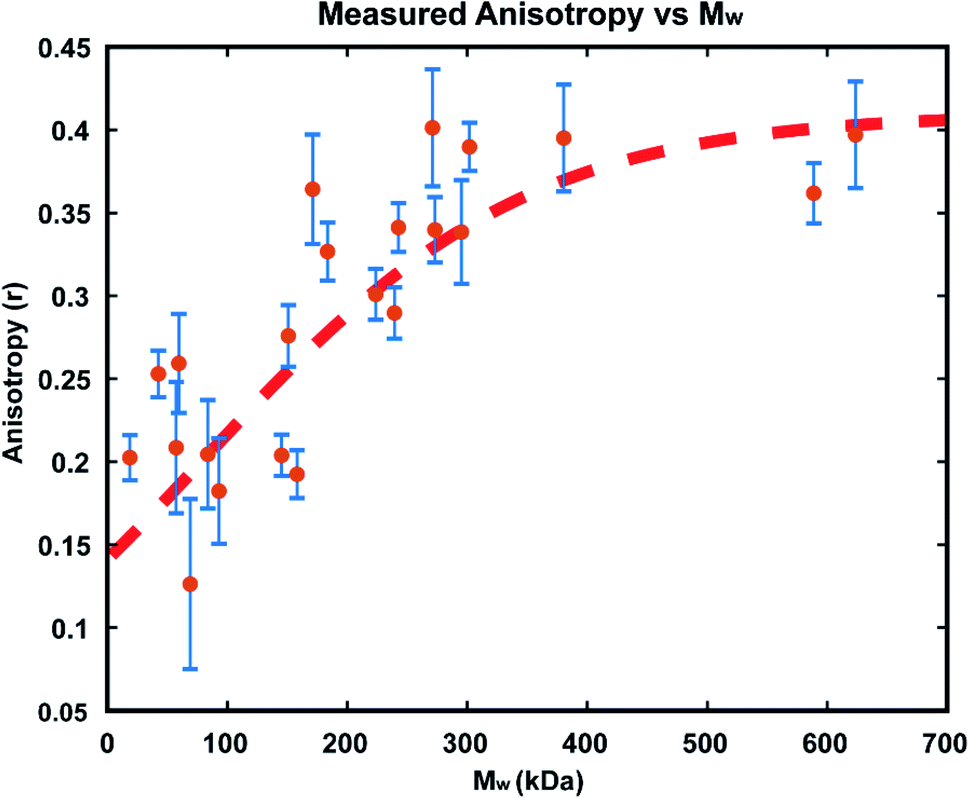 | ||
| Fig. 7 Calibration of measured anisotropy values with molecular weight, as measured by GPC-MALS. A correlation of measured anisotropy is seen with increasing molecular weight until the maximum value of 0.4 is reached. Red dashed line is a sigmoidal fit of the data added as a visual guide and is not meant as a quantitative model. | ||
Conclusions
We have demonstrated a method for optical monitoring of polymerization reaction progress in single droplets based on two complementary approaches. As the reaction progresses, increases in molecular weight of the growing ROMP polymers are accompanied by an increase in the fluorescence polarization anisotropy of a PDI tracer molecule. This method is informative at early reaction times for polymerizations of varying compositions and morphologies, suggesting utility for combinatorial searches for new polymerization reactions. Following the saturation of the anisotropy response, the fluorescence intensity of an AIEgen-based monomer is seen to increase at later times, suggesting utility for monitoring production of high molecular weight polymers. The combination of these two optically orthogonal readouts allows for a larger temporal dynamic range than either of the methods could provide on its own. Both methods can be used to monitor the reactions in droplets at the single-droplet level, with a flourous surface providing convenient immobilization. Confinement of the reactions under study to precisely positioned sub-mm droplets of organic phase, coupled with the use of widefield fluorescence microscopy to monitor their optical response, allows for measurement of many reaction conditions in parallel, with simple incorporation of simultaneous internal controls with many replicates. The advantages afforded by a computer-controlled robotic platform for precision droplet placement lays the groundwork for the future development of multi-droplet computing paradigms.106 Simple scaling to larger droplet arrays (>20 × 20) and the use of automated image processing opens a path to massively high throughput, combinatorial testing of reaction chemistries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Defense Advanced Research Projects Agency (DARPA) for funding this project under award number W911NF-18-2-0036 from the Molecular Informatics program. A. A.-G. is grateful to Anders G. Fröseth's generous support. The authors gratefully acknowledge use of facilities and instrumentation at the UW-Madison Wisconsin Centers for Nanoscale Technology (wcnt.wisc.edu) partially supported by the NSF through the University of Wisconsin Materials Research Science and Engineering Center (DMR-1720415).Notes and references
- D. B. Eremin and V. P. Ananikov, Coord. Chem. Rev., 2017, 346, 2–19 CrossRef.
- S. Shekhar, P. Ryberg, J. F. Hartwig, J. S. Mathew, D. G. Blackmond, E. R. Strieter and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 3584–3591 CrossRef PubMed.
- M. T. Reetz and E. Westermann, Angew. Chem., Int. Ed., 2000, 39, 165 CrossRef PubMed.
- N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef.
- K. P. Sullivan, M. Wieliczko, M. Kim, Q. S. Yin, D. L. Collins-Wildman, A. K. Mehta, J. Bacsa, X. L. Lu, Y. V. Geletii and C. L. Hill, ACS Catal., 2018, 8, 11952–11959 CrossRef.
- Q. T. Easter, V. Trauschke and S. A. Blum, ACS Catal., 2015, 5, 2290–2295 CrossRef.
- C. M. Liu, K. R. Kubo, E. D. Wang, K. S. Han, F. Yang, G. Q. Chen, F. A. Escobedo, G. W. Coates and P. Chen, Science, 2017, 358, 352–355 CrossRef PubMed.
- A. Pandey, Y. Champouret and S. Rastogi, Macromolecules, 2011, 44, 4952–4960 CrossRef.
- M. S. Sanford, J. A. Love and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 6543–6554 CrossRef PubMed.
- M. A. Banares, Adv. Mater., 2011, 23, 5293–5301 CrossRef PubMed.
- S. J. Tinnemans, J. G. Mesu, K. Kervinen, T. Visser, T. A. Nijhuis, A. M. Beale, D. E. Keller, A. M. J. van der Eerden and B. M. Weckhuysen, Catal. Today, 2006, 113, 3–15 CrossRef.
- B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2003, 5, 4351–4360 RSC.
- P. Muller, S. P. Burt, A. M. Love, W. P. McDermott, P. Wolf and I. Hermans, ACS Catal., 2016, 6, 6823–6832 CrossRef.
- P. Muller and L. Hermans, Ind. Eng. Chem. Res., 2017, 56, 1123–1136 CrossRef.
- K. D. Heylman, K. A. Knapper, E. H. Horak, M. T. Rea, S. K. Vanga and R. H. Goldsmith, Adv. Mater., 2017, 29, 1700037 CrossRef.
- A. M. Cubillas, S. Unterkofler, T. G. Euser, B. J. M. Etzold, A. C. Jones, P. J. Sadler, P. Wasserscheid and P. S. Russell, Chem. Soc. Rev., 2013, 42, 8629–8648 RSC.
- M. B. J. Roeffaers, J. Hofkens, G. De Cremer, F. C. De Schryver, P. A. Jacobs, D. E. De Vos and B. F. Sels, Catal. Today, 2007, 126, 44–53 CrossRef.
- G. De Cremer, B. F. Sels, D. E. De Vos, J. Hofkens and M. B. J. Roeffaers, Chem. Soc. Rev., 2010, 39, 4703–4717 RSC.
- K. Kitagawa and S. A. Blum, ACS Catal., 2017, 7, 3786–3791 CrossRef.
- M. B. J. Roeffaers, G. De Cremer, H. Uji-i, B. Muls, B. F. Sels, P. A. Jacobs, F. C. De Schryver, D. E. De Vos and J. Hofkens, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12603–12609 CrossRef.
- O. Valdesaguilera, C. P. Pathak and D. C. Neckers, Macromolecules, 1990, 23, 689–692 CrossRef.
- F. Mikes, F. Gonzalez-Benito, B. Serrano, J. Bravo and J. Baselga, Polymer, 2002, 43, 4331–4339 CrossRef.
- R. O. Loutfy, J. Polym. Sci., Polym. Phys. Ed., 1982, 20, 825–835 CrossRef.
- W. L. Xu, J. S. Kong, Y. T. E. Yeh and P. Chen, Nat. Mater., 2008, 7, 992–996 CrossRef CAS.
- J. D. Ng, S. P. Upadhyay, A. N. Marquard, K. M. Lupo, D. A. Hinton, N. A. Padilla, D. M. Bates and R. H. Goldsmith, J. Am. Chem. Soc., 2016, 138, 3876–3883 CrossRef CAS.
- B. P. English, W. Min, A. M. van Oijen, K. T. Lee, G. B. Luo, H. Y. Sun, B. J. Cherayil, S. C. Kou and S. N. Xie, Nat. Chem. Biol., 2006, 2, 168 CrossRef CAS.
- Q. T. Easter, A. Garcia and S. A. Blum, ACS Catal., 2019, 9, 3375–3383 CrossRef CAS.
- Q. T. Easter and S. A. Blum, Angew. Chem., Int. Ed., 2018, 57, 1572–1575 CrossRef CAS.
- Q. T. Easter and S. A. Blum, Angew. Chem., Int. Ed., 2017, 56, 13772–13775 CrossRef CAS.
- Z. T. Allen, J. R. Sackey-Addo, M. P. Hopps, D. Tahseen, J. T. Anderson, T. A. Graf and C. B. Cooley, Chem. Sci., 2019, 10, 1017–1022 RSC.
- M. N. Hopkinson, A. Gomez-Suarez, M. Teders, B. Sahoo and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 4361–4366 CrossRef CAS.
- J. A. Friest, S. Broussy, W. J. Chung and D. B. Berkowitz, Angew. Chem., Int. Ed., 2011, 50, 8895–8899 CrossRef CAS.
- B. T. Herrera, S. R. Moor, M. McVeigh, E. K. Roesner, F. Marini and E. V. Anslyn, J. Am. Chem. Soc., 2019, 141, 11151–11160 CrossRef CAS.
- J. P. Stambuli, S. R. Stauffer, K. H. Shaughnessy and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 2677–2678 CrossRef CAS.
- S. R. Stauffer and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 6977–6985 CrossRef CAS.
- V. Dragone, V. Sans, A. B. Henson, J. M. Granda and L. Cronin, Nat. Commun., 2017, 8, 15733 CrossRef.
- P. S. Gromski, A. B. Henson, J. M. Granda and L. Cronin, Nat. Rev. Chem., 2019, 3, 119–128 CrossRef.
- R. Gomez-Bombarelli, J. Aguilera-Iparraguirre, T. D. Hirzel, D. Duvenaud, D. Maclaurin, M. A. Blood-Forsythe, H. S. Chae, M. Einzinger, D. G. Ha, T. Wu, G. Markopoulos, S. Jeon, H. Kang, H. Miyazaki, M. Numata, S. Kim, W. L. Huang, S. I. Hong, M. Baldo, R. P. Adams and A. Aspuru-Guzik, Nat. Mater., 2016, 15, 1120 CrossRef CAS.
- D. P. Tabor, L. M. Roch, S. K. Saikin, C. Kreisbeck, D. Sheberla, J. H. Montoya, S. Dwaraknath, M. Aykol, C. Ortiz, H. Tribukait, C. Amador-Bedolla, C. J. Brabec, B. Maruyama, K. A. Persson and A. Aspuru-Guzik, Nat. Rev. Mater., 2018, 3, 5–20 CrossRef CAS.
- J. M. Asua, Adv. Polym. Sci., 2018, 281, 1–22 CAS.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS.
- S. C. Thickett and G. H. Teo, Polym. Chem., 2019, 10, 2906–2924 RSC.
- W. J. Jeong, J. Y. Kim, J. Choo, E. K. Lee, C. S. Han, D. J. Beebe, G. H. Seong and S. H. Lee, Langmuir, 2005, 21, 3738–3741 CrossRef CAS.
- T. Brugarolas, F. Q. Tu and D. Lee, Soft Matter, 2013, 9, 9046–9058 RSC.
- S. Mashaghi, A. Abbaspourrad, D. A. Weitz and A. M. van Oijen, TrAC, Trends Anal. Chem., 2016, 82, 118–125 CrossRef CAS.
- G. S. Du, Q. Fang and J. M. J. den Toonder, Anal. Chim. Acta, 2016, 903, 36–50 CrossRef CAS.
- K. Churski, P. Korczyk and P. Garstecki, Lab Chip, 2010, 10, 816–818 RSC.
- E. Brouzes, M. Medkova, N. Savenelli, D. Marran, M. Twardowski, J. B. Hutchison, J. M. Rothberg, D. R. Link, N. Perrimon and M. L. Samuels, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14195–14200 CrossRef CAS.
- K. Churski, T. S. Kaminski, S. Jakiela, W. Kamysz, W. Baranska-Rybak, D. B. Weibel and P. Garstecki, Lab Chip, 2012, 12, 1629–1637 RSC.
- S. C. Thickett and R. G. Gilbert, Polymer, 2007, 48, 6965–6991 CrossRef CAS.
- C. Frochot, M. Mascherin, A. Haumont, M. L. Viriot and E. Marie, J. Appl. Polym. Sci., 2011, 119, 219–224 CrossRef CAS.
- X. G. Qiao, H. H. Ma, Z. Zhou, Y. L. Shi, X. C. Pang and S. Z. Zhou, Dyes Pigm., 2020, 172, 107796 CrossRef CAS.
- D. C. Crans, B. Baruah, A. Ross and N. E. Levinger, Coord. Chem. Rev., 2009, 253, 2178–2185 CrossRef CAS.
- B. Rotman and B. W. Papermaster, Proc. Natl. Acad. Sci. U. S. A., 1966, 55, 134 CrossRef CAS.
- R. V. Rozhkov, V. J. Davisson and D. E. Bergstrom, Adv. Synth. Catal., 2008, 350, 71–75 CrossRef CAS.
- L. E. Greene, R. Lincoln, K. Krumova and G. Cosa, ACS Omega, 2017, 2, 8618–8624 CrossRef CAS.
- M. B. J. Roeffaers, B. F. Sels, H. Uji-i, F. C. De Schryver, P. A. Jacobs, D. E. De Vos and J. Hofkens, Nature, 2006, 439, 572–575 CrossRef CAS.
- X. C. Zhou, W. L. Xu, G. K. Liu, D. Panda and P. Chen, J. Am. Chem. Soc., 2010, 132, 138–146 CrossRef CAS.
- J. B. Sambur, T. Y. Chen, E. Choudhary, G. Q. Chen, E. J. Nissen, E. M. Thomas, N. M. Zou and P. Chen, Nature, 2016, 530, 77 CrossRef CAS PubMed.
- H. P. Lu, L. Y. Xun and X. S. Xie, Science, 1998, 282, 1877–1882 CrossRef CAS.
- A. Rybina, C. Lang, M. Wirtz, K. Grussmayer, A. Kurz, F. Maier, A. Schmitt, O. Trapp, G. Jung and D. P. Herten, Angew. Chem., Int. Ed., 2013, 52, 6322–6325 CrossRef CAS PubMed.
- G. De Cremer, M. B. J. Roeffaers, E. Bartholomeeusen, K. F. Lin, P. Dedecker, P. P. Pescarmona, P. A. Jacobs, D. E. De Vos, J. Hofkens and B. F. Sels, Angew. Chem., Int. Ed., 2010, 49, 908–911 CrossRef CAS PubMed.
- Y. Chen, P. Sun, Y. Zhang and Y. Ye, J. Appl. Polym. Sci., 2018, 135, 46742 CrossRef.
- M. Levitus, J. L. Bourdelande, G. Marques and P. F. Aramendia, J. Photochem. Photobiol., A, 1999, 126, 77–82 CrossRef CAS.
- C. D. Geddes, J. Karolin and D. J. S. Birch, J. Phys. Chem. B, 2002, 106, 3835–3841 CrossRef CAS.
- S. F. Scarlata and J. A. Ors, Polym. Commun., 1986, 27, 41–42 CAS.
- M. Beija, A. Fedorov, M. T. Charreyre and J. M. G. Martinho, J. Phys. Chem. B, 2010, 114, 9977–9986 CrossRef CAS.
- A. K. Foote, L. H. Manger, M. R. Holden, M. Margittai and R. H. Goldsmith, Phys. Chem. Chem. Phys., 2019, 21, 1863–1871 RSC.
- L. H. Manger, A. K. Foote, S. L. Wood, M. R. Holden, K. D. Heylman, M. Margittai and R. H. Goldsmith, Angew. Chem., Int. Ed., 2017, 56, 15584–15588 CrossRef CAS.
- K. Paeng, R. Richert and M. D. Ediger, Soft Matter, 2012, 8, 819–826 RSC.
- Z. P. Zhou, X. Yan, Y. H. Lai and R. N. Zare, J. Phys. Chem. Lett., 2018, 9, 2928–2932 CrossRef CAS PubMed.
- F. Gielen, M. Butz, E. J. Rees, M. Erdelyi, T. Moschetti, M. Hyvonen, J. B. Edel, C. F. Kaminski and F. Hollfelder, Anal. Chem., 2017, 89, 1092–1101 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- S. J. Liu, Y. H. Cheng, H. K. Zhang, Z. J. Qiu, R. T. K. Kwok, J. W. Y. Lam and B. Tang, Angew. Chem., Int. Ed., 2018, 57, 6274–6278 CrossRef CAS PubMed.
- C. P. Ma, Q. Q. Ling, S. D. Xu, H. N. Zhu, G. Zhang, X. Zhou, Z. G. Chi, S. W. Liu, Y. Zhang and J. R. Xu, Macromol. Biosci., 2014, 14, 235–243 CrossRef CAS.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Angew. Chem., Int. Ed., 2000, 39, 2903–2906 CrossRef CAS PubMed.
- L. Delaude and A. F. Noels, Kirk-Othmer Encyclopedia of Chemical Technology, 2005 DOI:10.1002/0471238961.metanoel.a01.
- C. Pariya, K. N. Jayaprakash and A. Sarkar, Coord. Chem. Rev., 1998, 168, 1–48 CrossRef CAS.
- A. Leitgeb, J. Wappel and C. Slugovc, Polymer, 2010, 51, 2927–2946 CrossRef CAS.
- D. B. Wright, M. A. Touve, M. P. Thompson and N. C. Gianneschi, ACS Macro Lett., 2018, 7, 401–405 CrossRef CAS.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed.
- H. Langhals, O. Krotz, K. Polborn and P. Mayer, Angew. Chem., Int. Ed., 2005, 44, 2427–2428 CrossRef CAS PubMed.
- X. W. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- R. H. Goldsmith, L. E. Sinks, R. F. Kelley, L. J. Betzen, W. H. Liu, E. A. Weiss, M. A. Ratner and M. R. Wasielewski, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3540–3545 CrossRef CAS.
- M. J. Ahrens, L. E. Sinks, B. Rybtchinski, W. H. Liu, B. A. Jones, J. M. Giaimo, A. V. Gusev, A. J. Goshe, D. M. Tiede and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 8284–8294 CrossRef CAS.
- E. A. Weiss, M. J. Ahrens, L. E. Sinks, A. V. Gusev, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 5577–5584 CrossRef CAS PubMed.
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed.
- B. Rybtchinski, L. E. Sinks and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 12268–12269 CrossRef CAS PubMed.
- S. Yagai, T. Seki, T. Karatsu, A. Kitamura and F. Wurthner, Angew. Chem., Int. Ed., 2008, 47, 3367–3371 CrossRef CAS PubMed.
- Y. Zhao, Y. Wu, G. Yan and K. Zhang, RSC Adv., 2014, 4, 51194–51200 RSC.
- S. P. Upadhyay, K. M. Lupo, A. N. Marquard, J. D. Ng, D. M. Bates and R. H. Goldsmith, J. Phys. Chem. C, 2015, 119, 19703–19714 CrossRef CAS.
- C. M. Santos, A. Kumar, W. Zhang and C. Z. Cai, Chem. Commun., 2009, 2854–2856, 10.1039/b821148e.
- J. M. Vincent, Chem. Commun., 2012, 48, 11382–11391 RSC.
- C. C. Tzschucke, C. Markert, H. Glatz and W. Bannwarth, Angew. Chem., Int. Ed., 2002, 41, 4500–4503 CrossRef CAS PubMed.
- K. S. Ko, F. A. Jaipuri and N. L. Pohl, J. Am. Chem. Soc., 2005, 127, 13162–13163 CrossRef CAS PubMed.
- T. Darmanin and F. Guittard, Soft Matter, 2013, 9, 5982–5990 RSC.
- J. M. P. Gutierrez, T. Hinkley, J. W. Taylor, K. Yanev and L. Cronin, Nat. Commun., 2014, 5, 5571 CrossRef CAS.
- J. Grizou, L. J. Points, A. Sharma and L. Cronin, arXiv:1904.12635 [cond-mat.soft], 2019.
- Y. M. Zhao, W. Zhu, L. X. Ren and K. Zhang, Polym. Chem., 2016, 7, 5386–5395 RSC.
- Y. M. Zhao, W. Zhu, Y. Wu, L. Qu, Z. P. Liu and K. Zhang, Polym. Chem., 2016, 7, 6513–6520 RSC.
- M. J. Ahrens, R. F. Kelley, Z. E. X. Dance and M. R. Wasielewski, Phys. Chem. Chem. Phys., 2007, 9, 1469–1478 RSC.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, Berlin, 3rd edn, 2006 Search PubMed.
- J. Howell, J. D. Goddard and W. Tam, Tetrahedron, 2009, 65, 4562–4568 CrossRef CAS.
- P. R. Khoury, J. D. Goddard and W. Tam, Tetrahedron, 2004, 60, 8103–8112 CrossRef CAS.
- G. Escuela, G. Gruenert and P. Dittrich, Nat. Comput., 2014, 13, 247–256 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05559b |
| This journal is © The Royal Society of Chemistry 2020 |