
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBase induced isomerisation of a phosphaethynolato-borane: mechanistic insights into boryl migration and decarbonylation to afford a triplet phosphinidene†
Daniel W. N.
Wilson
 a,
Mauricio P.
Franco
a,
William K.
Myers
b,
John E.
McGrady
a and
Jose M.
Goicoechea
*a
a,
Mauricio P.
Franco
a,
William K.
Myers
b,
John E.
McGrady
a and
Jose M.
Goicoechea
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK
bDepartment of Chemistry, University of Oxford, Centre for Advanced ESR, Inorganic Chemistry Laboratory, South Parks Road, Oxford, OX1 3QR, UK. E-mail: jose.goicoechea@chem.ox.ac.uk
First published on 2nd December 2019
Abstract
We report on the (tert-butyl)isocyanide-catalysed isomersation of a phosphaethynolato-borane, [B]OCP ([B] = N,N′-bis(2,6-diisopropylphenyl)-2,3-dihydro-1H-1,3,2-diazaboryl), to its linkage isomer, a phosphaketenyl-borane, [B]PCO. Mechanistic insight into this unusual isomerisation was gained through a series of stoichiometric reactions of [B]OCP with isocyanides and theoretical calculations at the Density Functional Theory (DFT) level. [B]PCO decarbonylates under photolytic conditions to afford a novel boryl-substituted diphosphene, [B]P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P[B]. This reaction proceeds via a transient triplet phosphinidene which we have been able to observe spectroscopically by Electron Paramagnetic Resonance (EPR) spectroscopy.
P[B]. This reaction proceeds via a transient triplet phosphinidene which we have been able to observe spectroscopically by Electron Paramagnetic Resonance (EPR) spectroscopy.
1. Introduction
The 2-phosphaethynolate anion, PCO−, is an ambidentate pseudo-halide that exhibits comparable nucleophilicity to its lighter, and more thoroughly studied congener, the cyanate ion.1 Over the course of the last eight years, the chemistry of PCO− has been studied extensively by a number of research groups worldwide. One of the most notable empirical observations from the research carried out thus far concerns the binding preferences of the PCO− ion, revealing that phosphorus-bonded phosphaketenes, [E]–PCO,2 far outnumber their linkage isomers, oxygen-bonded phosphaethynolates, [E]–O–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P.3–7 In fact, this latter family of compounds is largely the purview of the s- and f-block elements, and to date, only three structurally authenticated phosphaethynolate compounds have been reported for the elements of the d- and p-blocks.5–7 This is perhaps unsurprising considering that coordination via the oxygen atom, particularly in systems that exhibit a significant degree of covalency, generates a species with a localised CP π bond at the expense of a much stronger CO double bond. Consequently, it is only in highly ionic compounds, or in complexes with strong [E]–O bonds, that bonding via the oxygen atom is observed.
P.3–7 In fact, this latter family of compounds is largely the purview of the s- and f-block elements, and to date, only three structurally authenticated phosphaethynolate compounds have been reported for the elements of the d- and p-blocks.5–7 This is perhaps unsurprising considering that coordination via the oxygen atom, particularly in systems that exhibit a significant degree of covalency, generates a species with a localised CP π bond at the expense of a much stronger CO double bond. Consequently, it is only in highly ionic compounds, or in complexes with strong [E]–O bonds, that bonding via the oxygen atom is observed.
We recently reported an example of an oxygen-bonded PCO− ion in the form of an isolable phosphaethynolato-borane, [B]–O–CP (1; [B] = N,N′-bis(2,6-diisopropylphenyl)-2,3-dihydro-1H-1,3,2-diazaboryl; pictured in Scheme 1).6 At the time we noted that the phosphaethynolate could be isolated despite the fact that the phosphorus-bonded isomer, the phosphaketene ([B]–PCO), was thermodynamically more stable by 7 kcal mol−1. However heating a toluene solution of [B]OCP to 140 °C showed no evidence of thermal isomerization even over the course of several weeks. In this report we show that strong Lewis bases such as (tert-butyl)isocyanide (tBuNC) can be used to catalytically induce the isomerisation of [B]OCP to [B]PCO, and that when employed in stoichiometric quantities, isonitriles can trap a series of low valent phosphorus compounds, including base-stabilised phosphinidenes. We also demonstrate that photolysis of [B]PCO generates a triplet phosphinidene which could be observed by EPR spectroscopy, but rapidly dimerises to afford a diphosphene, [B]PP[B].
 | ||
| Scheme 1 Reactivity of 1 towards isocyanides. Dipp = 2,6-diisopropylphenyl. | ||
2. Results and discussion
2.1 Reactivity of 1 with isocyanides
For clarity, prior to discussing our catalytic studies, we will focus on reactions of 1 with stoichiometric amounts of isocyanides as they provide some indication of the mechanism of isomerization. Given their greater abundance, the reactivity of phosphaketene compounds, [E]–PCO, towards Lewis bases (e.g. carbenes) has been extensively explored, and shown to induce isomerisation or decarbonylation.2i,8,9 By contrast, the reactivity of phosphaethynolate species toward such reagents is entirely unexplored. Reaction of 1 with one equivalent of MesNC (Mes = 2,4,6-trimethylphenyl) cleanly affords a single product after 24 h with a singlet 31P NMR resonance at −195.2 ppm (shifted to a notably higher frequency from known phosphaethynolato or phosphaketene compounds). Pale orange crystals of the resulting product ([B]C(PCO)NMes; 2a) could be obtained in good yield (74%) by cooling a concentrated hexane solution of the reaction mixture.‡
The crystal structure of 2a (Fig. 1) reveals migration of the boryl functionality from the oxygen atom of 1 to the carbon atom of the isocyanide, and the presence of a phosphaketene also associated with the isocyanide carbon. Bond metric data for the phosphaketene moiety reveal a linear ion (P1–C1–O1 173.6(2)°) with a P–C bond of 1.670(2) Å and a C–O bond of 1.155(3)Å which compare well with previously isolated compounds of this type.
 | ||
| Fig. 1 Molecular structure of 2a. Anisotropic displacement ellipsoids set at 50% probability. Hydrogen atoms have been omitted for clarity. Atoms of the Dipp and Mes groups are pictured as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: P1–C1 1.670(2), C1–O1 1.155(3), C2–P1 1.870(2), C2–B1 1.580(2), C2–N1 1.270(2); P1–C1–O1 173.6(2), C2–P1–C1 96.67(9), B1–C2–P1 122.97(12), B1–C2–N1 120.34(15), P1–C2–N1 116.66(13). | ||
Photolysis of a solution of 2a affords a new species with a singlet resonance in its 31P NMR spectrum at −262.6 ppm accompanied by an additional, unidentified product at 8.6 ppm. Separation of the former species by fractional crystallization allowed us to identify it as an isocyanide-stabilized phosphinidene ([B]PCNMes, 3a). An analogous species can also be obtained under non-photolytic conditions when 1 is reacted with a stoichiometric excess (4 equivalents) of the smaller isocyanide tBuNC. The 31P NMR spectrum of this reaction mixture reveals the quantitative formation of a single product over 4 days with a singlet resonance at −256.2 ppm which can be assigned to [B]PCNtBu (3b). Two intermediates could be observed in the reaction mixture by 31P NMR spectroscopy (at −207.8 and 192.5 ppm), one of which corresponds to 2b (−207.8 ppm; computed resonance: −196 ppm; see ESI for details†). These two intermediates convert to a single species with a 31P NMR chemical shift of −337.1 ppm (vide infra) which ultimately affords 3b over the course of several days. It is interesting to note that while decarbonylation of 2a to afford 3a requires photolytic treatment, 2b decarbonylates much more readily, presumably a result of the greater inductive character of the tBu functionality. Due to the π-acidic character of the boryl group, the chemical shifts observed for 3a and 3b (−262.6 and −256.2 ppm, respectively) differ from related species such as AdNCP{P[N(Dipp)(CH2)2]2} which exhibits a 31P NMR resonance at −128.7 ppm.9a The single crystal X-ray structure of 3a is shown in Fig. 2. Structurally 3a and 3b are very similar, with a PCN core that is essentially co-planar with the boryl ring (mean deviation from plane: 0.16 (3a) and 0.14 Å (3b)). P–B and P–C bonds are 1.912(2)/1.916(2) and 1.674(2)/1.661(3) Å for 3a/3b, respectively, the latter being in the expected range for PC double bonds (1.69 Å).10
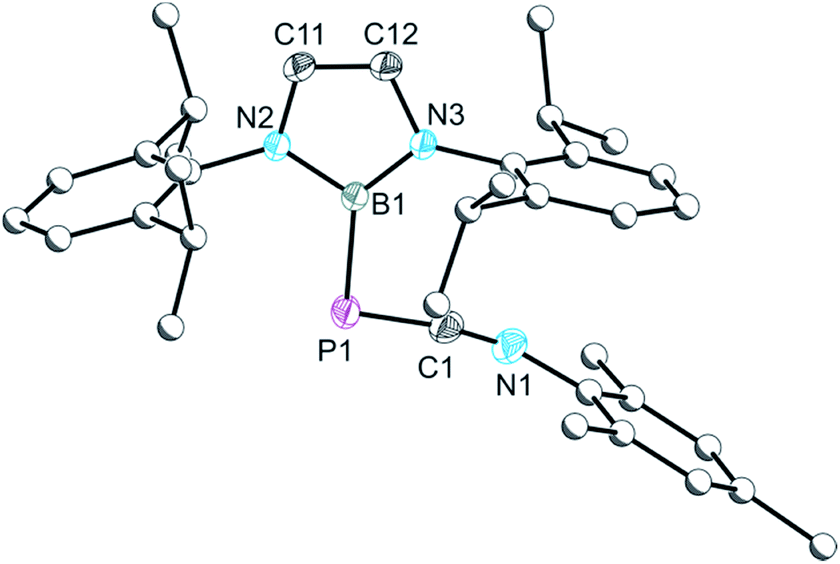 | ||
| Fig. 2 Molecular structure of 3a. Anisotropic displacement ellipsoids set at 50% probability. Hydrogen atoms have been omitted for clarity. Atoms of the Dipp and Mes groups are pictured as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: P1–C1 1.674(2), C1–N1 1.195(2), P1–B1 1.912(2); P1–C1–N1 167.50(13), C1–P1–B1 101.81(7). | ||
In the presence of 1.2 equivalents of MesNC, compound 2a loses CO to afford a novel compound [B]C(PCNMes)NMes, 4a, where the carbon monoxide is displaced by a valence isoelectronic isocyanide. Related decarbonylation reactions of phosphaketenes have been reported by Bertrand and co-workers revealing that the phosphorus atom in phosphaketenes can act as a Lewis acid, leading to ligand displacement reactions.9 The structure of 4a (Fig. 3) is closely related to that of 2a, with a C1–P1 bond length of 1.841(2) Å compared to 1.870(2) Å in 2a. Similarly, the B–C1 bond length between the boryl functionality and the isocyanide carbon in 4a, 1.580(2) Å, is identical to that observed in 2a.
 | ||
| Fig. 3 Molecular structure of 4a. Anisotropic displacement ellipsoids set at 50% probability. Hydrogen atoms have been omitted for clarity. Atoms of the Dipp and Mes groups are pictured as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: P1–C1 1.841(2), C1–N1 1.286(2), C1–B1 1.580(2), P1–C11 1.707(2); C1–P1–C11 105.51(5), P1–C1–N1 130.90(9), P1–C1–B1 113.71(7), B1–C1–N1 115.30(10). | ||
The aforementioned reactions prompted us to explore the role that catalytic amounts of these bases play in the reaction chemistry. When a solution of 1 with 10 mol% of tBuNC was monitored by 31P NMR spectroscopy, a new product, [B]PCO (5), was detected after 48 hours. 5 exhibits a singlet resonance at −337.1 ppm (cf. −285.9 ppm for 1) in its 31P NMR spectrum.
As noted above, a species with the same 31P NMR fingerprint (−337.1 ppm) was also observed as an intermediate in the reactions which led to the isolation of 3b. Indeed, we find that 3b can also be accessed by reaction of an isolated sample of 5 with tBuNC, confirming it as the intermediate. Crystals of 5 suitable for X-ray diffraction were obtained from a cooled hexane solution, and the resulting structure is shown in Fig. 4. The X-ray structure confirms the presence of a B–P bond and a “side-on” interaction of the PCO− moiety with the boryl group (C–P–B: 98.92(6)°), both of which are co-planar (mean deviation from plane: 0.27 Å). Notably, there is an elongation of the P–C bond of 5 relative to the isomeric phosphaethynolato-borane (1.652(2) vs. 1.545(2) Å, respectively) and a concomitant shortening of the C–O bond (1.153(2) vs. 1.269(2) Å). These two molecules (1 and 5) constitute the first pair of linkage isomers of the PCO− ion that has been structurally characterised to date. It is worth noting that when the bulkier isocyanide MesNC is employed, no catalytic activity is observed, and only unreacted [B]OCP and small amounts of 2a are observed in the reaction mixtures.
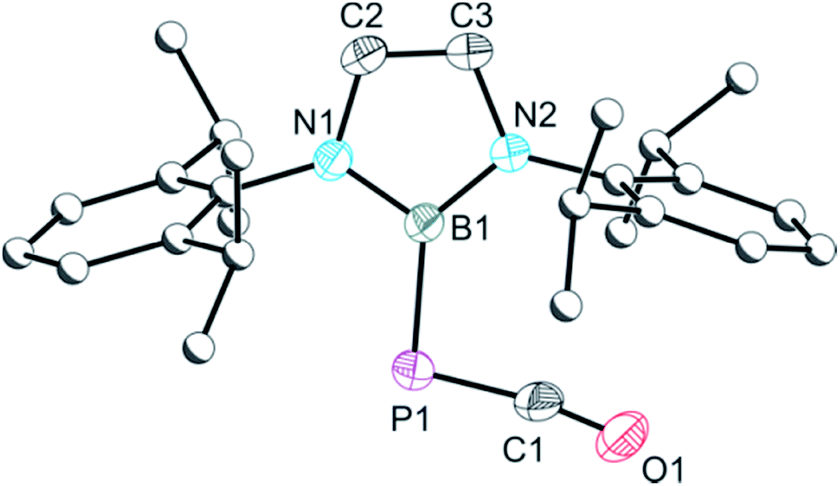 | ||
| Fig. 4 Molecular structure of 5. Anisotropic displacement ellipsoids set at 50% probability. Hydrogen atoms have been omitted for clarity. Atoms of the Dipp groups are pictured as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: P1–C1 1.652(2), C1–O1 1.153(2), P1–B1 1.913(2); P1–C1–O1 170.91(13), C1–P1–B1 98.92(6). | ||
2.2. Density functional theory
In an attempt to shed further light on this rather complex reaction chemistry, we turned to density functional theory to map out important regions of the free-energy hypersurface. In the first instance, the structures of 1, 2a and 5 were optimised (M06-L). In all three cases, the key structural parameters are in excellent agreement with their crystallographic counterparts (ESI, Fig. S37†). Of the two PCO linkage isomers, 1 and 5, the latter is the more stable by 10 kcal mol−1, consistent with our previous calculations performed at the PBE1PBE level.6 The free energy surface summarising the various pathways connecting the minima is presented in Fig. 5, where in all cases a model isonitrile, HNC, has been used. We have located a transition state, TS1, that directly connects the two linkage isomers without participation of the isonitrile (red pathway in Fig. 4). In the transition structure, the B–O bond is substantially lengthened compared to 1 (1.84 Å vs. 1.41 Å) while the central carbon atom of the OCP unit approaches within bonding distance of the boron centre (1.72 Å vs. 2.40 Å in 1 and 2.70 Å in 5) to offset the loss of electron density from the breaking B–O bond. However, TS1 lies 33 kcal mol−1 above 1 and the very high barrier is immediately consistent with the lack of isomerisation, even at 140 °C, in the absence of base. In our search for a viable base-catalysed pathway, we have considered various trajectories for the approach of the isonitrile ligand, HNC. The lowest energy of these (black pathway in Fig. 5) connects the reactants to the product 2via a barrier of 16.4 kcal mol−1 (TS2). In the transition structure, TS2, the HNC ligand is weakly coordinated to the boron centre (B–C = 1.61 Å), which is approximately tetrahedral, and, just like in TS1, the B–O bond is substantially elongated compared to the reactant (1.71 Å vs. 1.41 Å). Unlike TS1, the central carbon atom of the PCO unit remains far from the boron centre (2.46 Å), suggesting that it is the HNC nucleophile, rather than the carbon atom of the PCO ligand, that fulfils the function of offsetting the loss of electron density from the B–O bond. After the transition structure, the phosphorus terminus of the PCO ligand moves towards the bound carbon of the isonitrile, while the C–P–C and P–C–O angles open up to expel the oxygen from the boron coordination sphere, generating product 2. From 2, the HNC unit can then be extruded to generate 5 and HNC viaTS3 in a reaction that is slightly exergonic (ΔG = +9 kcal mol−1). The transition structure, TS3, is almost isoenergetic with TS2. TS2 and TS3 are also rather similar structurally, in so much as the boron centre is approximately tetrahedral in both cases. It is, however, now the B–P bond that is formed via a 3-membered transition state (2.98 Å in 2, 2.08 Å in TS3, 1.91 Å in 5) rather than the 5-membered transition structure involved in TS2. Overall, then, the black section of the free energy profile indicates that the isomerisation of 1 and 5 occurs via the isolated intermediate 2, which connects reactants to products via two transition states, TS2 and TS3. The barrier for the first step in the catalysed reaction (viaTS2) is only 16.4 kcal mol−1 but the second (viaTS3) is 32 kcal mol−1, only marginally lower than the uncatalysed process viaTS1. The region of the free energy surface connecting 2 to 5 is, however, likely to be sensitive to the substituents on the isonitrile, and so we have re-optimised the structures of 2, TS3 and RNC using both MesNC (2a and TS3a) and tBuNC (2b and TS3b) The structures of the stationary points are very similar to those reported for the model reactions with HNC, but the presence of bulkier substituents has the effect of destabilising the intermediates, 2a/b, relative to both the product (5 + RNC) and the intervening transition structure (TS3a/b). For tBuNC, the overall reaction 2b → 5 + tBuNC is now only very slightly endergonic (+1 kcal mol−1vs. 9 kcal mol−1 for the HNC model) while the barrier (2b → TS3b) is reduced to 27 kcal mol−1. The very delicate energetic balance between 2b and 5 + tBuNC will render the reaction very sensitive to concentration, with excess amounts of tBuNC affording 2a which can react with more tBuNC to give 3b, while lower stoichiometric loadings, or indeed catalytic amounts, favour 5 (+tBuNC). For MesNC the relative destabilisation of 2a is less dramatic and it remains 5 kcal mol−1 below 5 + MesNC, with a barrier of 35 kcal mol−1. The much higher barrier for MesNC is consistent with the fact that 5 cannot be formed from 2a + MesNC.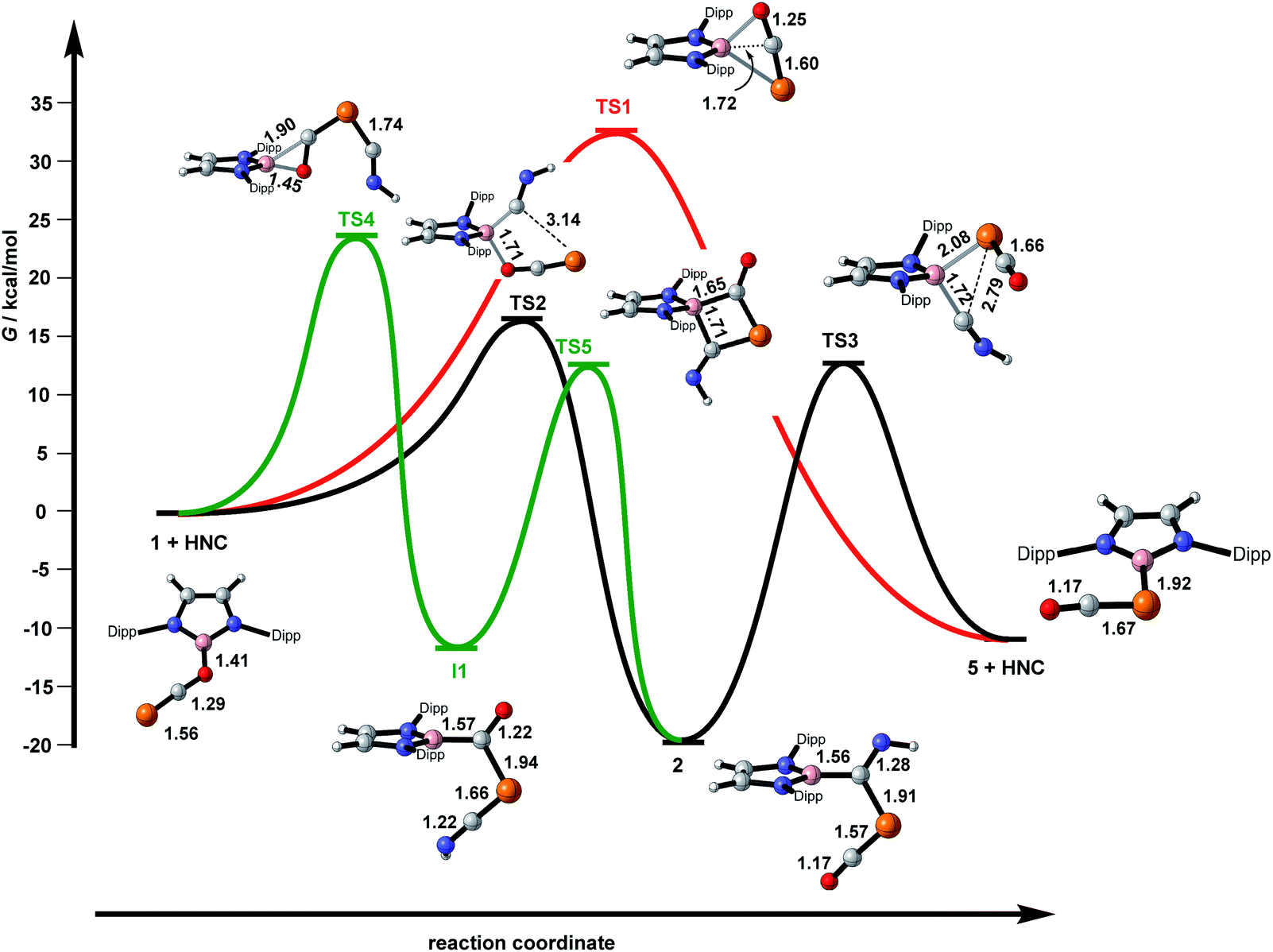 | ||
| Fig. 5 Free energy surface for the reactions of [B]-OCP with the model isonitrile, HNC. | ||
It is worth emphasising the point that although the model isonitrile ligand is clearly bound to the boron centre in both transition states, TS2 and TS3, we have never located a stable minimum where it remains bound, nor have we found any evidence for isonitrile binding in the NMR spectra. The catalytic role of the base is therefore to labilise the B–O bond via binding to the boron centre only in the transition structures. The central importance of binding of the nucleophile to the boron centre contrasts starkly with our previous reports of the reactions of the anionic nucleophiles Mes− and [Cp*Fe(CO)2]−, where direct attack at the phosphorus centre leads to isolated enolate intermediates where the carbon centre of the PCO unit, rather than the oxygen, is bound to the boryl group.6,11 We do not locate any such stationary points in reactions with a neutral nucleophile such as RNC, simply because they would require a very substantial separation of charge. The presence of a mobile cation is therefore vital to the stabilisation of the enolates. When we do allow the HNC nucleophile to approach the terminal phosphorus centre of 1, we locate a further intermediate, I1, with a ketonic HNCP–C(O)– ligand. This species has not been detected in any of our experiments but the enolate intermediates referred to above react with sources of [(PR3)Au]+, to generate species with a (Nu)(Au–PR3)P–C(O)– ligand. The intervening transition state, TS4, features an η2-coordination mode for the C–O unit of the PCO ligand, and the barrier is 25 kcal mol−1, similar to that for the extrusion of RCN from 2. Given that I1 is marginally more stable than the 5 + HCN asymptote, it is not immediately obvious why we have not observed I1 in any of our reactions. However, we note that just like 1 and 5, I1 and 2 also constitute a pair of linkage isomers, and they can be interconverted via a transition state, TS4, featuring a 4-membered ring where the HNC unit is again bound to the boron. For the model HNC ligand this is a relatively facile process (barrier of 24 kcal mol−1) and so any I1 formed through remote attack of the nucleophile at the phosphorus centre will rearrange rapidly to form the more stable intermediate 2.
2.3 Reactions of a phosphinidene intermediate
Phosphaketenes such as 5 have previously been employed to generate transient phosphindenes (RP),12 and in one case, an isolable singlet phosphinidene.13 There has been a long-running interest in the isolation of such species which are often generated by photolytic or thermal dissociation of phospha-Wittig reagents or ring-strained compounds such as phosphiranes and phosphirenes.14,15In an attempt to isolate a terminal phosphinidene, [B]–P, 5 was photolyzed in a C6D6 solution (Scheme 2, top). The 31P NMR spectrum of the reaction mixture reveals the formation of a single product with a singlet resonance at 596.0 ppm, while the 11B and 1H NMR spectra are consistent with the presence of a single boryl functionality. The 31P NMR shift of the resulting product is in the expected region for a diphosphene ([B]–PP–[B]; 6). A closely related system, featuring a saturated boryl functionality, has been previously reported by Yamashita and co-workers.16 Orange crystals of the resulting product suitable for single crystal X-ray diffraction could be isolated in good yield (68%) by cooling a concentrated hexane solution of the product to −35 °C.
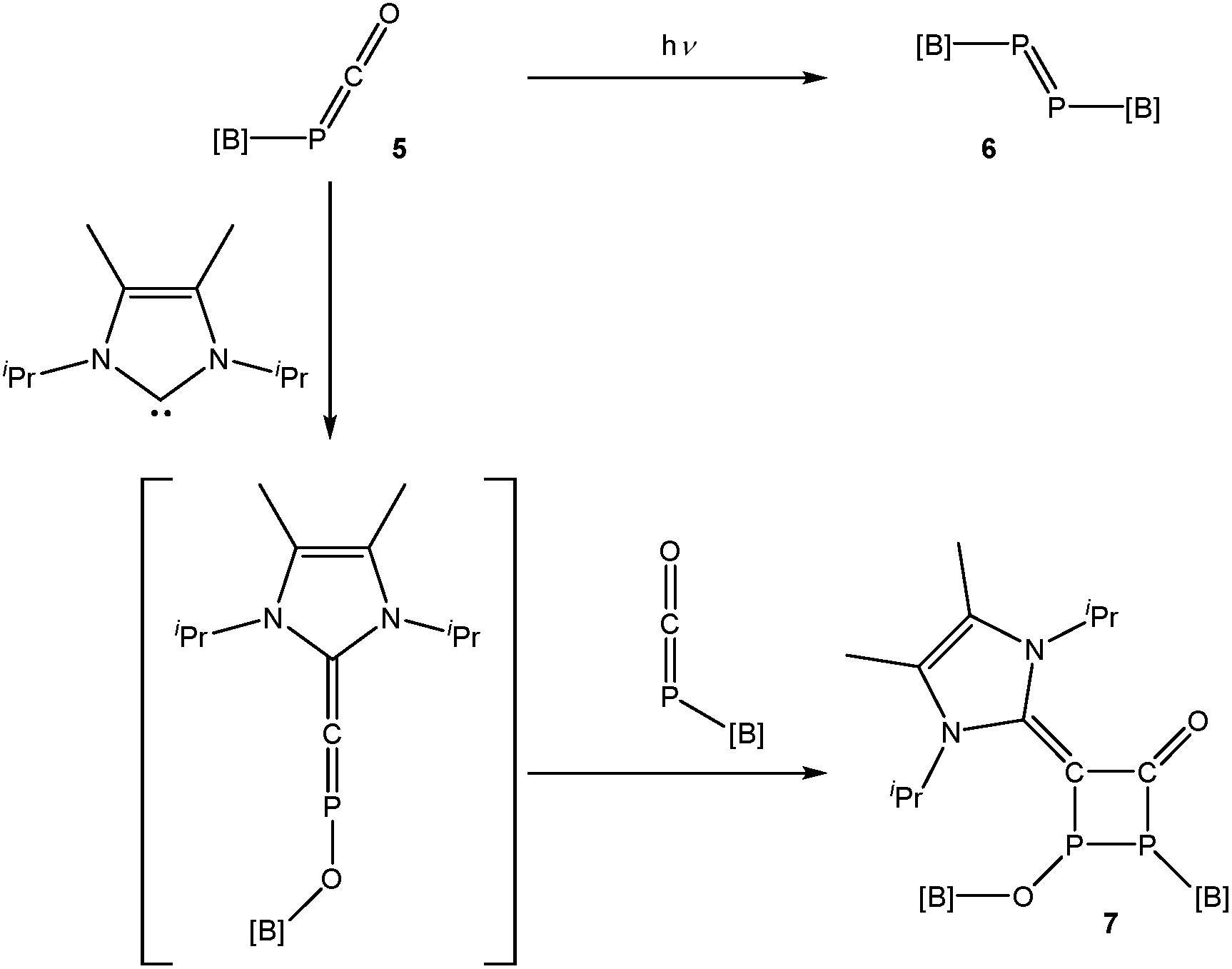 | ||
| Scheme 2 Photolysis of 5 and reactivity towards an N-heterocyclic carbene. | ||
The single crystal X-ray structure of 6 shown in Fig. 6 reveals a diphosphene with a relatively short P–P bond distance of 2.027(1) Å, which is in line with the expected distance for a PP double bond (2.04 Å) and comparable to other literature reported diphosphenes, e.g. [PB{N(Dipp)(CH2)2}2]2 (2.066(2) Å) and [P(C6H2-2,4,6-tBu3)]2 (2.034(2) Å).16,17
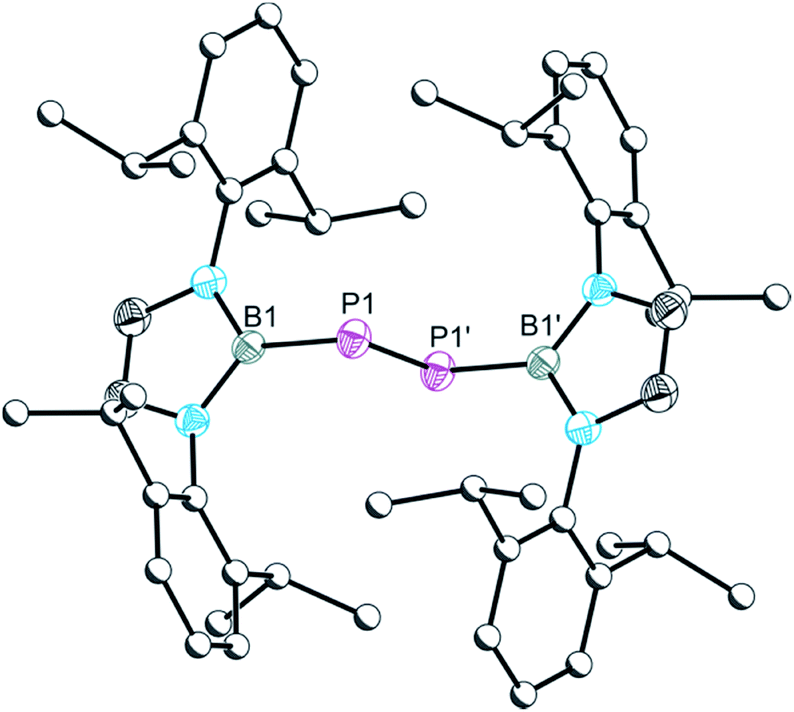 | ||
| Fig. 6 Molecular structure of 6. Anisotropic displacement ellipsoids set at 50% probability. Hydrogen atoms have been omitted for clarity. Atoms of the Dipp groups are pictured as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: B1–P1 1.926(2), P1–P1′ 2.026(1); B1–P1–P1′ 97.02(4). Symmetry operation′: −x, 1 year, −z. | ||
Formation of 6 is postulated to proceed via a short-lived triplet phosphinidene. Efforts to observe this transient intermediate were made using EPR spectroscopy. Generation of [B]–P from photolysis of 5 led to an electron paramagnetic resonance (EPR) signal at X-band (∼9.4 GHz) in the 1.15 to 1.25 Tesla field range characteristic of molecular oxygen. As shown in Fig. 7, the signal may be simulated with an axial zero-field splitting parameter, D, of 4 cm−1 and an orthorhombic parameter, E, of 3.7 × 10−3 cm−1 and hyperfine coupling of the electrons and 31P nucleus, A(31P)iso = 300 MHz. These values are in close agreement with a prior report of zero-field splitting parameters D = 4.116 cm−1, E = 4 × 10−3 cm−1, and hyperfine interactions Ax, Ay(31P) = 269, 356 MHz, respectively, for the related triplet mesitylphosphinidene.18 Several g ≈ 2 EPR signals are observed during photolysis at room temperature with a molecular concentrations ≥5 mM. With the total EPR radical concentration being less than 0.6% of molecular concentration, meaningful fits of the kinetic rates on an A → [B] → C intermediate detection scheme were not possible; however, inclusion of three components greatly improved the fit over one component. At one fifth concentration (5 mM) a sample did yield a single prominent component that fits well with a tricarbontriphosphide tricyclic radical as previously reported by Grützmacher by scaling the DFT hyperfine values by a factor of two (see ESI† for further details).19
 | ||
| Fig. 7 X-band EPR of 5 under two photolytic conditions with EPR simulations shown in red and the data is in black. In panel (a), the temperature was 30 K with a flame-sealed sample and a pulsed LASER was used, the microwave power was 0.635 mW and frequency was 9.693 GHz with a modulation amplitude of 0.3 mT. In panel (b), the temperature was 5 K with a J. Young sample tube and a Xenon arc lamp was used, the microwave power was 1 mW and frequency was 9.391 GHz with a modulation amplitude of 1.5 mT. | ||
In additional attempts to chemically induce decarbonylation of 5, a toluene solution of this compound was reacted with one equivalent of 1,3-diisopropyl-4,5-dimethyl-1,3-dihydro-2λ2-imidazole (Me2IPr). The 31P NMR spectrum of the reaction reveals that the outcome is not consistent with a simple displacement of carbon monoxide by the N-heterocyclic carbene. Rather, the reaction give rise to a product exhibiting two doublet resonances in its 31P NMR spectrum at 90.6 and −22.5 ppm (1JP–P = 191.0 Hz). The 11B and 1H NMR spectra are consistent with the presence of two distinct boryl functionalities which suggests dimerization. Crystallization of the resulting product from a concentrated hexane solution afforded single crystals suitable for X-ray diffraction (Fig. 8). The structure of 7 can be rationalized as the [2 + 2] cycloaddition product of 5 with [B]–O–PCMe2IPr (Scheme 2, bottom). Such phosphorus-containing fulminate analogues are not without precedent, and related NHC-induced isomerisation reactions involving phosphaketenes (R–PCO) have previously been reported by Grützmacher and co-workers by reaction of, for example, Ph3SiPCO with IPr.20 The cyclic four-membered core of 7 is highly unusual albeit related to a diphosphacyclobutenonyl ligand previously reported by Scheer and co-workers.21
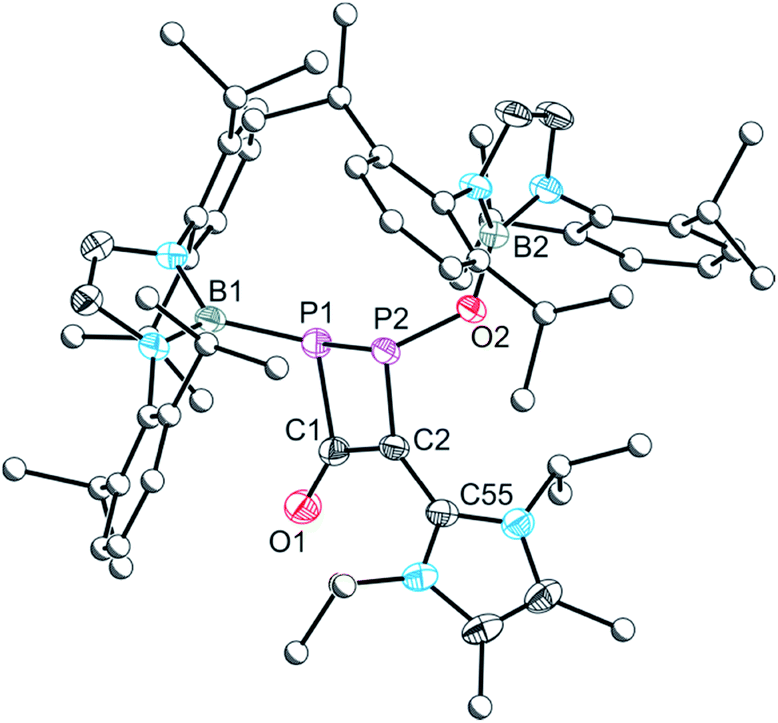 | ||
| Fig. 8 Molecular structure of 7. Anisotropic displacement ellipsoids set at 50% probability. Hydrogen atoms have been omitted for clarity. Atoms of the Dipp, iPr and Me groups are pictured as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: P1–P2 2.2369(6), P1–C1 1.8874(17), C1–O1 1.239(2), C1–C2 1.407(2), C2–C55 1.449(2), C2–P2 1.7872(16), P1–B1 1.915(2), O2–B2 1.371(2); B1–P1–P2 108.05(6), B1–P1–C1 109.32(8), P1–C1–O1 128.70(13), P1–C1–C2 99.50(11), O1–C1–C2 131.76(16), C1–C2–P2 103.95(11), C1–C2–C55 123.93(15), C2–P2–P1 77.15(5), C2–P2–O2 103.38(7), P2–O2–B2 125.73(11). | ||
Conclusions
To conclude, we have shown that (tert-butyl)isocyanide, tBuNC, can induce the isomerization of a phosphaethynolato-borane, [B]OCP, to its linkage isomer [B]PCO. In doing so we have isolated and fully characterized for the first time two linkage isomers of the same PCO−-containing species. The isomerization was probed through a series of stoichiometric reactions with isocyanides and DFT studies. [B]PCO can be photolyzed to afford a transient triplet phosphinidene [B]P which we were able to observe spectroscopically, but that ultimately dimerizes to afford a bis(boryl)diphosphene. These reactions help shine light on the versatile chemistry of the 2-phosphaethynolate anion and its utility in accessing novel low valent phosphorus containing compounds. Studies probing the reactivity of [B]PCO as a phosphinidene transfer agent are currently on-going.Experimental
Computational methods
All calculations were performed with Gaussian09 package, version D.01. Geometry optimizations were performed using the M06L functional,22 and a 6-31+G(d,p) basis for P, O, B, N, and the carbon atoms of the isonitrile and PCO moieties.23 A 6-31G basis set was used for all other C and H atoms. Intermediates and transition state (TS) were identified by the absence of imaginary frequencies and the presence of a single imaginary frequency, respectively. Transition states were connected to reactants and products by calculation of the Intrinsic Reaction Coordinates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC and the University of Oxford for financial support of this research (DTA studentship D.W.N.W.) and the University of Oxford for access to Chemical Crystallography and Advanced Research Computing (ARC) facilities (DOI: 10.5281/zenodo.22558). MPF acknowledges a CAPES (Coordination for the Improvement of Higher Education Personnel) scholarship that supported his stay in Oxford (88881.188450/2018-01). The Centre for Advanced ESR (CAESR) is supported by UK EPSRC (EP/L011972/1).Notes and references
- For recent reviews on the chemistry of the 2-phosphaethynolate anions see: (a) J. M. Goicoechea and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 CrossRef CAS; (b) L. Weber, Eur. J. Inorg. Chem., 2018, 2175–2227 CrossRef CAS.
- (a) S. Alidori, D. Heift, G. Santiso-Quinones, Z. B. H. Grützmacher, M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem.–Eur. J., 2012, 18, 14805–14811 CrossRef CAS; (b) D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 43, 5920–5928 RSC; (c) Z. Li, X. Chen, M. Bergeler, M. Reiher, C.-Y. Su and H. Grützmacher, Dalton Trans., 2015, 44, 6431–6438 RSC; (d) A. R. Jupp, M. B. Geeson, J. E. McGrady and J. M. Goicoechea, Eur. J. Inorg. Chem., 2016, 639–648 CrossRef CAS; (e) L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CrossRef CAS; (f) L. Liu, D. A. Ruiz, F. Dahcheh, G. Bertrand, R. Suter, A. M. Tondreauc and H. Grützmacher, Chem. Sci., 2016, 7, 2335–2341 RSC; (g) N. Del Rio, A. Baceiredo, N. Saffon-Merceron, D. Hashizume, D. Lutters, T. Müller and T. Kato, Angew. Chem., Int. Ed., 2016, 55, 4753–4758 CrossRef CAS; (h) S. Yao, Y. Xiong, T. Szilvási, H. Grützmacher and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 4781–4785 CrossRef CAS; (i) Z. Li, X. Chen, Z. Benkő, L. Liu, D. A. Ruiz, J. L. Peltier, G. Bertrand, C.-Y. Su and H. Grützmacher, Angew. Chem., Int. Ed., 2016, 55, 6018–6022 CrossRef CAS; (j) Y. Wu, L. Liu, J. Su, J. Zhu, Z. Ji and Y. Zhao, Organometallics, 2016, 35, 1593–1596 CrossRef CAS; (k) Y. Xiong, S. Yao, T. Szilvási, E. Ballestero-Martínez, H. Grützmacher and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 4333–4336 CrossRef CAS; (l) A. Hinz and J. M. Goicoechea, Chem. - Eur. J., 2018, 24, 7358–7363 CrossRef CAS; (m) L. N. Grant, J. Krzystek, B. Pinter, J. Telser, H. Grützmacher and D. J. Mindiola, Chem. Commun., 2019, 55, 5966–5969 RSC.
- For s-block compounds see: (a) G. Becker, W. Schwarz, N. Seidler and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 612, 72–82 CrossRef CAS; (b) M. Westerhausen, S. Schneiderbauer, H. Piotrowski, M. Suter and H. Nöth, J. Organomet. Chem., 2002, 643–644, 189–193 CrossRef CAS; (c) F. F. Puschmann, D. Stein, D. Heift, C. Hendriksen, Z. A. Gal, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2011, 50, 8420–8423 CrossRef CAS; (d) A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064–10067 CrossRef CAS; (e) R. J. Gilliard Jr., D. Heift, Z. Benkő, J. M. Keiser, A. L. Rheingold, H. Grützmacher and J. D. Protasiewicz, Dalton Trans., 2018, 47, 666–669 RSC; (f) F. Hennersdorf, J. Frötschel and J. J. Weigand, J. Am. Chem. Soc., 2017, 139, 14592–14604 CrossRef CAS.
- For f-block compounds see: (a) C. Camp, N. Settineri, J. Lefèvre, A. R. Jupp, J. M. Goicoechea, L. Maron and J. Arnold, Chem. Sci., 2015, 6, 6379–6384 RSC; (b) C. J. Hoerger, F. W. Heinemann, E. Louyriac, L. Maron, H. Grützmacher and K. Meyer, Organometallics, 2017, 36, 4351–4354 CrossRef CAS; (c) S. Bestgen, Q. Chen, N. H. Rees and J. M. Goicoechea, Dalton Trans., 2018, 47, 13016–13024 RSC.
- L. N. Grant, B. Pinter, B. C. Manor, H. Grützmacher and D. J. Mindiola, Angew. Chem., Int. Ed., 2018, 57, 1049–1052 CrossRef CAS.
- D. W. N. Wilson, A. Hinz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 2188–2193 CrossRef CAS.
- Y. Mei, J. E. Borger, D.-J. Wuab and H. Grützmacher, Dalton Trans., 2019, 48, 4370–4374 RSC.
- (a) Z. Li, X. Chen, Y. Li, C.-Y. Su and H. Grützmacher, Chem. Commun., 2016, 52, 11343–11346 RSC; (b) Z. Li, X. Chen, D. M. Andrada, G. Frenking, Z. Benkő, Y. Li, J. R. Harmer, C.-Y. Su and H. Grützmacher, Angew. Chem., Int. Ed., 2017, 56, 5744–5749 CrossRef CAS; (c) Z. Li, Y. Hou, Y. Li, A. Hinz, J. R. Harmer, C.-Y. Su, G. Bertrand and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 198–202 CrossRef CAS.
- (a) M. M. Hansmann, R. Jazzar and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 8356–8359 CrossRef CAS; (b) M. M. Hansmann and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 15885–15888 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef.
- D. W. N. Wilson and J. M. Goicoechea, Chem. Commun., 2019, 55, 6842–6845 RSC.
- (a) L. Liu, D. A. Ruiz, F. Dahcheh, G. Bertrand, R. Suter, A. M. Tondreau and H. Grützmacher, Chem. Sci., 2016, 7, 2335–2341 RSC; (b) Y. Xiong, S. Yao, T. Szilvási, E. Ballestero-Martínez, H. Grützmacher and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 4333–4336 CrossRef CAS; (c) T. Krachko, A. W. Ehlers, M. Nieger, M. Lutz and J. C. Slootweg, Angew. Chem., Int. Ed., 2018, 57, 1683–1687 CrossRef CAS.
- L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CAS.
- S. Shah, M. C. Simpson, R. C. Smith and J. D. Protasiewicz, J. Am. Chem. Soc., 2001, 123, 6925–6926 CrossRef CAS.
- (a) F. Mathey, Angew. Chem., Int. Ed. Engl., 1987, 26, 275–286 CrossRef; (b) X. Li, D. Lei, M. Y. Chiang and P. P. Gaspar, J. Am. Chem. Soc., 1992, 114, 8526–8531 CrossRef CAS; (c) X. Li, S. I. Weissman, T.-S. Lin, P. P. Gaspar, A. H. Cowley and A. I. Smirnov, J. Am. Chem. Soc., 1994, 116, 7899–7900 CrossRef CAS; (d) M.-A. Courtemanche, W. J. Transue and C. C. Cummins, J. Am. Chem. Soc., 2016, 138, 16220–16223 CrossRef CAS; (e) W. J. Transue, A. Velian, M. Nava, C. García-Iriepa, M. Temprado and C. C. Cummins, J. Am. Chem. Soc., 2017, 139, 10822–10831 CrossRef CAS; (f) A. Mardyukov and D. Niedek, Chem. Commun., 2018, 54, 13694 RSC; (g) W. J. Transue, M. Nava, M. W. Terban, J. Yang, M. W. Greenberg, G. Wu, E. S. Foreman, C. L. Mustoe, P. Kennepohl, J. S. Owen, S. J. L. Billinge, H. J. Kulik and C. C. Cummins, J. Am. Chem. Soc., 2019, 141, 431–440 CrossRef CAS; (h) M. B. Geeson, W. J. Transue and C. C. Cummins, J. Am. Chem. Soc., 2019, 141, 13336–13340 CrossRef CAS; (i) K. M. Szkop, M. B. Geeson, D. W. Stephan and C. C. Cummins, Chem. Sci., 2019, 10, 3627 RSC.
- S.-s. Asami, M. Okamoto, K. Suzuki and M. Yamashita, Angew. Chem., Int. Ed., 2016, 55, 12827–12831 CrossRef CAS.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS.
- A. V. Akimov, Y. S. Ganushevich, D. V. Korchagin, V. A. Miluykov and E. Y. Misochko, Angew. Chem., Int. Ed., 2017, 56, 7944–7947 CrossRef CAS.
- Z. Li, Y. Hou, Y. Li, A. Hinz, J. R. Harmer, C. Y. Su, G. Bertrand and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 198–202 CrossRef CAS.
- (a) Z. Li, X. Chen, Y. Li, C.-Y. Su and H. Grützmacher, Chem. Commun., 2016, 52, 11343–11346 RSC; (b) Z. Li, X. Chen, Z. Benkő, L. Liu, D. A. Ruiz, J. L. Peltier, G. Bertrand, C.-Y. Su and H. Grützmacher, Angew. Chem., Int. Ed., 2016, 55, 6018–6022 CrossRef CAS.
- M. Scheer, D. Himmel, B. P. Johnson, C. Kuntz and M. Schiffer, Angew. Chem., Int. Ed., 2007, 46, 3971–3975 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101–194118 CrossRef.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental (including spectra and X-ray data) and computational details. CCDC 1963000–1963006. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05969e |
| ‡ Full experimental details (including spectra) can be found in the ESI.† |
| This journal is © The Royal Society of Chemistry 2020 |