
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrophosphination of boron–boron multiple bonds†
Tom E.
Stennett
ab,
Arumugam
Jayaraman
 ab,
Tobias
Brückner
ab,
Lea
Schneider
ab and
Holger
Braunschweig
*ab
ab,
Tobias
Brückner
ab,
Lea
Schneider
ab and
Holger
Braunschweig
*ab
aInstitut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 18th December 2019
Abstract
Five compounds containing boron–boron multiple bonds are shown to undergo hydrophosphination reactions with diphenylphosphine in the absence of a catalyst. With diborenes, the products obtained are highly dependent on the substitution pattern at the boron atoms, with both 1,1- and 1,2-hydrophosphinations observed. With a symmetrical diboryne, 1,2-hydrophosphination yields a hydro(phosphino)diborene. The different mechanistic pathways for the hydrophosphination of diborenes are rationalised with the aid of density functional theory calculations.
Introduction
Compounds containing covalent bonds between phosphorus and boron, while not enjoying the ubiquity of their nitrogen–boron analogues, display a rich chemistry.1–3 Phosphinoboranes – that is, compounds of the form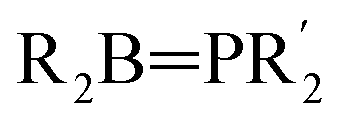
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 with a planar geometry at phosphorus and partial double bond character5 – can undergo addition reactions with such substrates as H2,6,7 ketones6 and dienes6 and coordinate in an η2 fashion to transition metals.8 Borylphosphines, in which the boron atom is not π-acidic (e.g. due to π-donor or carborane substituents) and phosphorus thus retains its lone pair and tetrahedral geometry, are highly electron rich η1 ligands for transition metal complexes.9–11 By far the most common route for the construction of P–B bonds is salt elimination12–19 using an alkali metal phosphide, MPR2, and a haloborane (Scheme 1A), although other routes are known, including nucleophilic addition of a boryl anion to a chlorophosphine (B),20 elimination of silyl halide (C),10,11,21 and cross-coupling of a B–I species with secondary phosphines using a palladium catalyst (D).9
4 with a planar geometry at phosphorus and partial double bond character5 – can undergo addition reactions with such substrates as H2,6,7 ketones6 and dienes6 and coordinate in an η2 fashion to transition metals.8 Borylphosphines, in which the boron atom is not π-acidic (e.g. due to π-donor or carborane substituents) and phosphorus thus retains its lone pair and tetrahedral geometry, are highly electron rich η1 ligands for transition metal complexes.9–11 By far the most common route for the construction of P–B bonds is salt elimination12–19 using an alkali metal phosphide, MPR2, and a haloborane (Scheme 1A), although other routes are known, including nucleophilic addition of a boryl anion to a chlorophosphine (B),20 elimination of silyl halide (C),10,11,21 and cross-coupling of a B–I species with secondary phosphines using a palladium catalyst (D).9
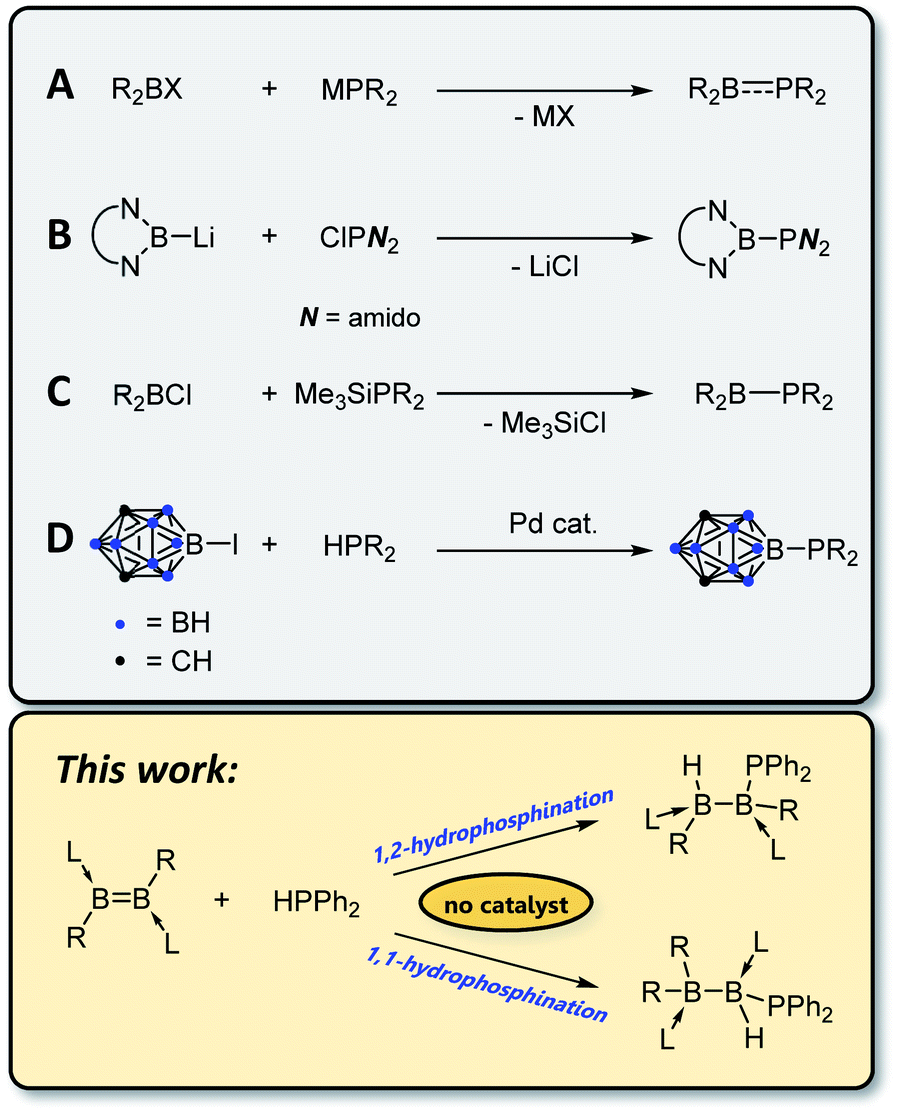 | ||
| Scheme 1 Reported methods for the construction of covalent boron–phosphorus bonds. | ||
Of the different synthetic options for the construction of phosphorus–carbon bonds, hydrophosphination of unsaturated carbon substrates has gained popularity in recent years.22–24 Advantages of this method over typical nucleophilic addition reactions include reduced waste (hydrophosphination is a 100% atom-economical process) and increased functional group tolerance due to the avoidance of highly reactive Grignard or organolithium reagents. However, these reactions typically require a catalyst in the form of a base or metal complex, or a radical initiator, and the scope is somewhat limited in terms of which secondary phosphines can be used. Whereas P–H bond activation at transition25 and main-group26 metal centres is well documented, to the best of our knowledge the sole reported example of hydrophosphination of a homonuclear double bond comprising elements other than carbon is the 2016 report by Wang and Wu of hydrophosphination of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in azobenzene with diphenylphosphine oxide.27 Recent studies in our laboratories have demonstrated the feasibility of adding H–H,28,29 B–H,30–32 B–B,33,34 S–S35,36 and Se–Se35 bonds across boron–boron multiple bonds. In this work, we show that uncatalysed hydrophosphination of both diborenes and diborynes can be used as a mild, selective and catalyst-free route to construct B–P bonds.
N bond in azobenzene with diphenylphosphine oxide.27 Recent studies in our laboratories have demonstrated the feasibility of adding H–H,28,29 B–H,30–32 B–B,33,34 S–S35,36 and Se–Se35 bonds across boron–boron multiple bonds. In this work, we show that uncatalysed hydrophosphination of both diborenes and diborynes can be used as a mild, selective and catalyst-free route to construct B–P bonds.
Results and discussion
Diborenes based on a chelating benzylphosphine group37 that holds the aryl substituent in the plane of the boron–boron π-bond have shown particularly high reactivity towards dienes38 and diboranes,33 so we chose these compounds as a starting point. The symmetrical diborene 1 was treated with diphenylphosphine at room temperature, leading to slow conversion to a compound with signals in the 11B NMR spectrum at −15.9 and −27.2 ppm, both in the region expected for four-coordinate boron atoms. Heating the mixture to 60 °C for 1 h resulted in the decolouration of the solution and full conversion to the product, which was isolated as a colourless solid after workup in 79% yield. X-ray diffraction on single crystals grown from a diethyl ether solution confirmed the structure as that of product 5, the result of a 1,2-hydrophosphination across the BB bond (Scheme 2 and Fig. 1). The PPh2 and hydrogen substituents display a gauche conformation, while the benzylphosphine units have reverted to a vicinal (1,2-) coordination mode; in contrast to other compounds bearing this substituent,38 no conversion to the geminal isomer was observed. At 2.051(2) Å, the B1–P1 bond is in the range of published base-stabilised borylphosphines,15 and markedly longer than both dative B–P bonds in the molecule (B1–P2 = 2.008(2), B2–P3 = 1.928(2) Å). As expected, P1 is highly pyramidal, with a sum of angles of 312.4°. In the 1H NMR spectrum, a multiplet centred at 3.08 ppm becomes visible upon 11B decoupling, corresponding to the newly formed B–H group. Two signals at 10.4 ppm and 6.1 ppm in the 31P NMR spectrum correspond to the now inequivalent benzylphosphine phosphorus atoms, whereas the PPh2 group is represented by a broad signal at −13.3 ppm.
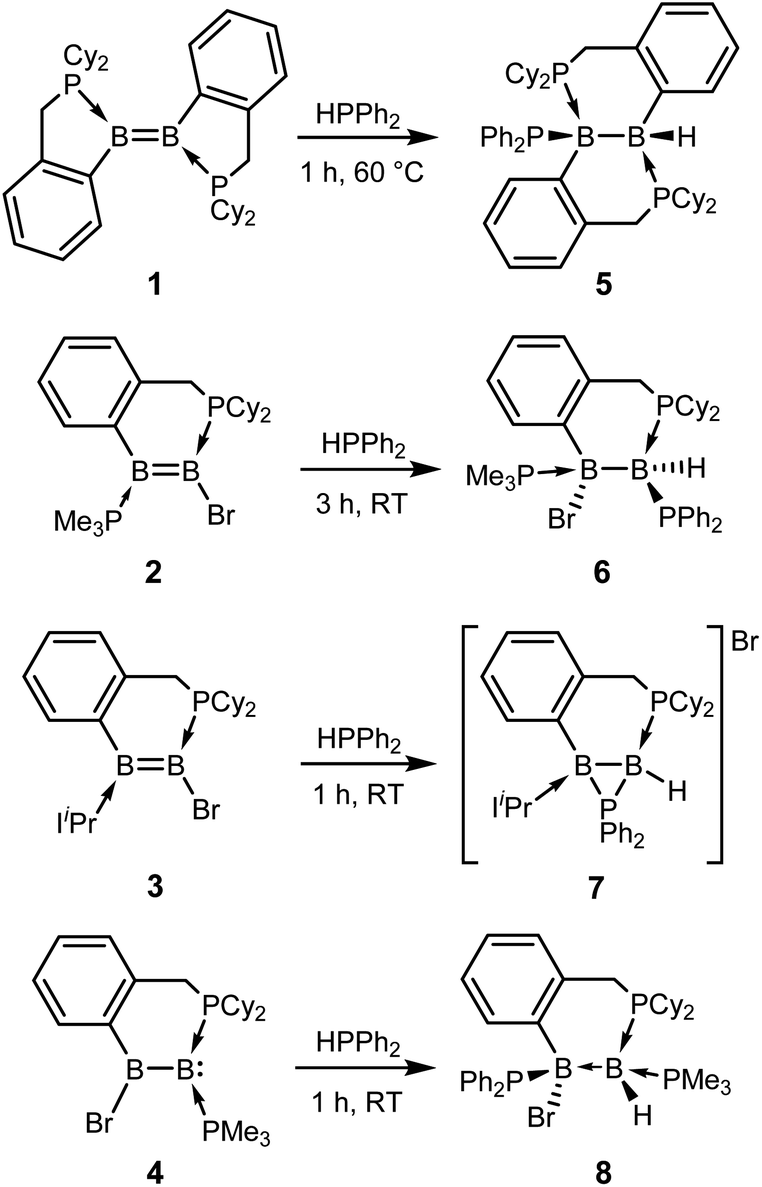 | ||
| Scheme 2 Hydrophosphination of diborenes and a borylborylene with diphenylphosphine. IiPr = 1,3-diisopropylimidazol-2-ylidene. | ||
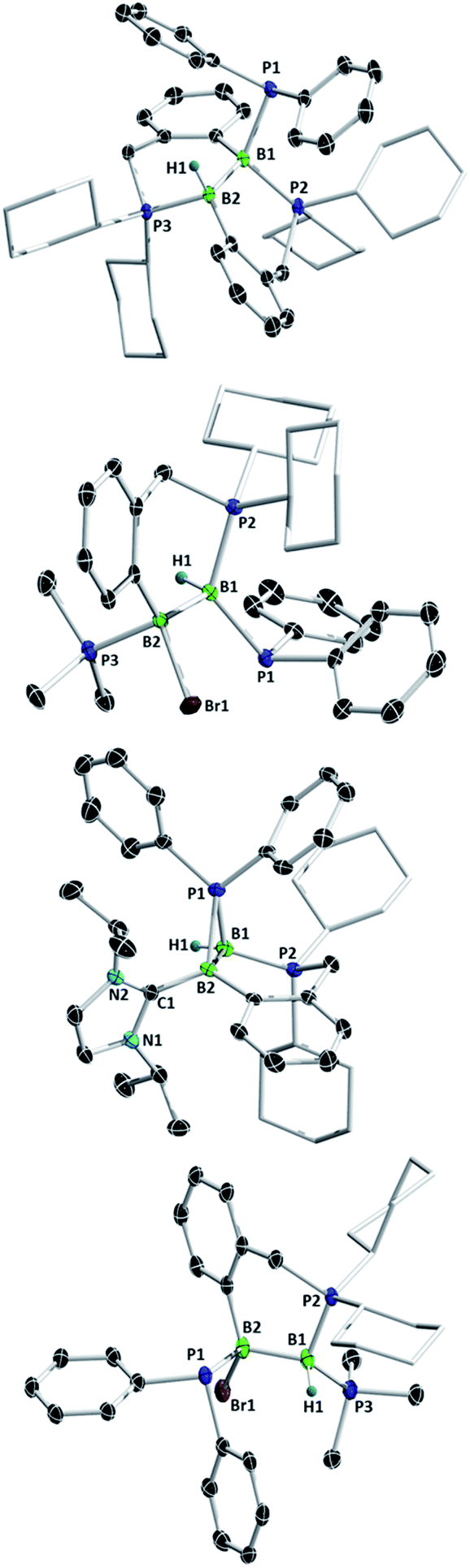 | ||
| Fig. 1 Molecular structures of (top to bottom) 5, 6, 7 and 8, with selected atomic displacement ellipsoids at the 50% probability level. Hydrogen atoms except for those bound to boron, a bromide counterion (compound 7) and co-crystallised solvent molecules are omitted for clarity. Selected bond lengths (Å), angles and torsions (°): compound 5: B1–B2 1.780(2), B1–P1 2.051(2), B1–P2 2.008(2), B2–P3 1.928 (2), P1–B1–B2–H1 24.7(9); compound 6: B1–B2 1.777(3), B1–P1 1.974(2), B1–P2 1.932(2), B2–P3 1.953(2); compound 7: B1–B2 1.772(5), B1–P1 1.918(4), B2–P1 1.948(4), B1–P2 1.885(4), B2–C1 1.601(5), B1–P1–B2 54.6(2) P2–B1–B2–C1 141.1(3); compound 8: B1–B2 1.789(4), B2–P1 1.986(4), B1–P2 1.938(3), B1–P3 1.935(4). | ||
Unsymmetrically-substituted diboron compounds are known to display higher reactivity than their symmetrical counterparts.39,40 We have previously shown that unsymmetrical diborenes 2 and 3 (Scheme 2) react more quickly than 1 in diboration reactions with B2cat2.33 Treatment of compound 2 with HPPh2 resulted in complete conversion to a new compound within 3 h at room temperature. Signals in the 11B NMR spectrum at −7.7 and −34.2 ppm again indicated the presence of inequivalent, four-coordinate boron atoms. Three broad signals in the 31P NMR spectrum at 23.1, 13.8 and −6.9 ppm also suggested three boron-bound phosphorus environments. Measurement of a 1H NMR spectrum with selective 11B decoupling (δ11B offset = 34 ppm) again revealed a complex multiplet for a B–H moiety at 2.15 ppm. X-ray diffraction confirmed the product as 6, the result of a formal 1,1-hydrophosphination rather than the expected 1,2-addition across the double bond (Fig. 1). The B1–B2 distance (1.777(3) Å) and B–P distances (B1–P1 = 1.974(2), B1–P2 = 1.932(2), B2–P3 = 1.953(2) Å) lie in the respective expected ranges. N-Heterocyclic-carbene (NHC) supported diborene 3 also undergoes a rapid reaction with HPPh2 at room temperature to give a compound with 11B NMR signals at −21.6 and −37.6 ppm. In this case however, a sharp doublet in the 31P NMR spectrum at −70.9 ppm alongside a broader signal at 8.9 ppm suggested a different outcome. The insolubility of the compound in benzene and tetrahydrofuran also led us to suspect an ion-separated structure. Single crystals obtained from a dichloromethane/pentane solution allowed determination of the structure by X-ray diffraction (Fig. 1), which confirmed the compound to be cationic diborane(5) 7, bearing a Br− counterion. Here, the PPh2 unit bridges the two boron atoms almost symmetrically (B1–P1 = 1.948(4) Å, B2–P1 = 1.918(4) Å). Such three-membered rings are far from common – the only structurally characterised examples of non-cluster B2P rings are the butterfly-shaped B2P2 diradicals reported by Bertrand and co-workers.41–43
As reported recently, compound 2 rearranges upon heating to its more thermodynamically stable isomer, 4, which possesses a polarised boron–boron bond and a borylborylene resonance form.37 Whereas one might expect such a compound to undergo 1,1-addition reactions to the “borylene” boron atom, both 4 (ref. 33) and a related compound reported by Kinjo's group44 give 1,2-addition products with B2cat2. Compound 4 reacts instantaneously with HPPh2 upon solvation in C6D6 to give different NMR signals to those seen in the corresponding reaction with 2. In the 11B NMR spectrum, two signals at 1.5 and −33.4 ppm are observed, whereas the 31P NMR spectrum reveals two broad signals at 16.4 and −7.8 ppm and a pseudo-triplet at −20.5 ppm. Reduction of the volume of the reaction mixture gave single crystals of the product, which were shown by X-ray diffraction to be 1,2-hydrophosphination product 8 (Fig. 1). As with the diboration product of 4, compound 8 can be drawn as a borylene–borane adduct. However, it is notable that the B–B distance (1.789(4) Å) and the B–P distances (B2–P1 = 1.986(4), B1–P2 = 1.938(3), B1–P3 = 1.935(4) Å) are statistically indistinguishable from those in its constitutional isomer 6, indicating that the electronic situation at the B2 units may not be as different as these formalisms suggest.
To gain mechanistic insight into how different hydrophosphination outcomes were obtained using the diboron compounds 1–4, DFT computations at the ωB97XD/6-31++g(d,p)/SMD//ωB97XD/6-31g(d,p) level of theory were performed. For computations, truncated models of diborenes, A1–D1, in which the cyclohexyl substituents of the chelating phosphines were replaced with methyl groups, were examined. The 1,1- and 1,2-hydrophosphination reactions observed are significantly exergonic, with ΔG values ranging from −23.9 to −38.4 kcal mol−1 (Fig. 2 and 3). For the 1,2-hydrophosphination of diborene 1, yielding product 5, the likely mechanism is shown in Fig. 2a. Initially, coordination of HPPh2 to one of the boron atoms affords adduct A2. This is followed by a 1,3-H shift from the coordinated HPPh2 to the distal boron atom, leading to the formation of a 1,2-hydrophosphinated intermediate A3, with the PPh2 and H units located syn to one another. This intermediate subsequently rearranges via phosphine dissociation/association to form the product A6. For this reaction, the 1,3-H shift step was found to be rate limiting and has a barrier of 33.7 kcal mol−1 (TSA(2–3)). The calculated barrier for this pathway is seemingly high for a room-temperature reaction, and we attribute this to the use of methyl groups rather than cyclohexyl groups in the calculations. The energy reduction due to the London dispersion effect45 from the bulky cyclohexyl groups is lost, which affects the crowded transition state more than the starting diborene. Two other reasonable mechanistic possibilities for this reaction were also explored, both of which gave higher barriers (see ESI†).
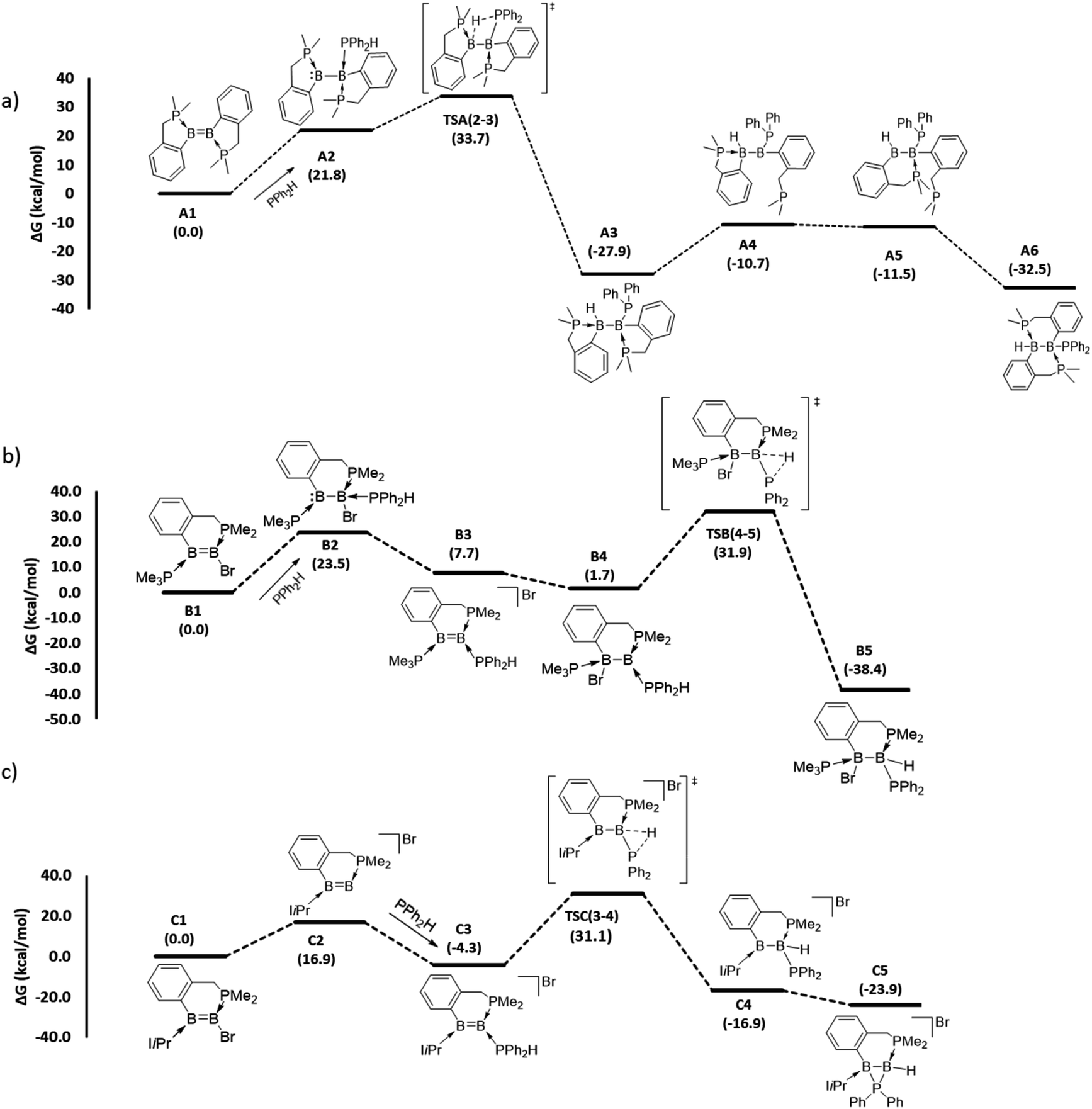 | ||
| Fig. 2 DFT-computed mechanistic pathways for (a) 1,2-hydrophosphination of diborene A1 (model of 1), (b) 1,1-hydrophosphination of diborene B1 (model of 2) and (c) 1,1-hydrophosphination of diborene C1 (model of 3). Note: only the transition states for the rate limiting P–H bond activation steps are shown; selected representative transition states for phosphine association/dissociation steps were calculated and found to have energies marginally higher than the higher energy intermediate (see ESI†). | ||
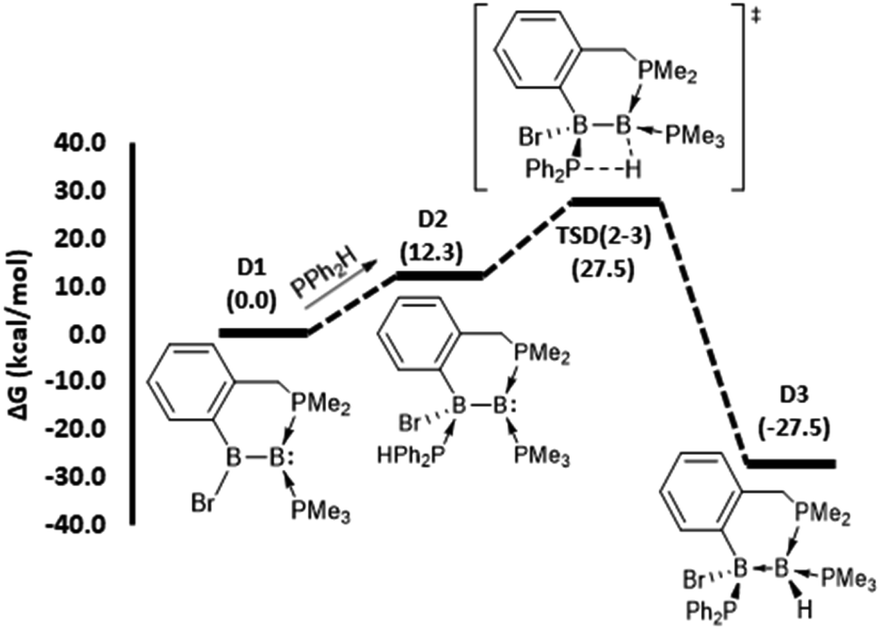 | ||
| Fig. 3 DFT-computed mechanistic pathways for 1,2-hydrophosphination of borylborylene D1 (model of 4). Note: the transition state between D1 and D2 was found to have a very low barrier (14.3 kcal mol−1) and is not shown. | ||
For the conversion of diborene 2 into 6, the proposed mechanism (Fig. 2b) involves (i) an initial coordination of HPPh2 to the boron centre that bears the electronegative bromide substituent and the chelating phosphine ligand, (ii) stepwise transfer (dissociation and association) of the bromide to the adjacent boron atom and (iii) 1,2-H shift from the coordinated HPPh2 to the proximal boron atom, leading to the formation of 1,1-hydrophosphinated product B5. Here, the rate limiting step is the 1,2-H shift, which has a barrier of 31.9 kcal mol−1. Our efforts to locate a transition state for a direct bromide shift from B2 to B4, or to find the minimum energy structure of a bromide-bridged intermediate, were unsuccessful. All transition state optimization efforts led to dissociation of either bromide or HPPh2. Minimization attempts to find the bromide-bridged intermediate led to convergence to one of the non-bridging intermediates B2 and B4. Use of a highly truncated model with hydrogen substituents at all phosphorus atoms produced a single transition state for a direct bromide shift, but the free energy barrier of 66.8 kcal mol−1 is too high to represent a feasible mechanism (see ESI†). Thus, a dissociative pathway was proposed. Other additional mechanistic pathways were examined, but were found to have higher barriers than that described above. We also investigated the corresponding 1,2-hydrophosphination of B1, which was confirmed to have a slightly higher overall barrier than the 1,1-addition.
For the cyclic product 7, obtained from the reaction between HPPh2 and the unsymmetrical, NHC-substituted diborene 3, a similar mechanistic pathway to that of B1 was investigated (Fig. 2c). In contrast to the B1 case, complexation of HPPh2 to diborene C1 only occurs after initial dissociation of the bromide ion. This difference can be primarily attributed to the large steric hindrance exerted by the two isopropyl peripheries of the NHC ligand as well as ample charge flow from the NHC to the distal boron centre. Unlike in B1, the dissociated bromide cannot feasibly bind to the adjacent boron in cationic intermediate C3, again due to the strong electron donation and steric hindrance of the NHC ligand. As a result, C3 is forced to undergo a 1,2-proton shift from the coordinated HPPh2, leading to the formation of 1,1-hydrophosphinated cationic intermediate C4, with the positive charge predominantly localised at the NHC-coordinated boron centre. As a means to alleviate the electron deficiency at this boron centre, in the subsequent step the phosphinyl substituent establishes a dative interaction, leading to the observed product C5. For this reaction, an overall barrier of 31.1 kcal mol−1 was found for the 1,2-H shift step.
The mechanistic pathway for the 1,2-hydrophosphination of borylborylene 4 to form 8 can be described straightforwardly (Fig. 3). Given that the boron atom of the boryl unit holds a partial positive charge (vide infra), it can easily bind the incoming phosphine. The resulting loss of π-delocalisation of the borylene lone pair leaves it strongly localised, and in the ensuing step a protic 1,3-H shift from the HPPh2 unit to the borylene boron atom occurs, affording the product D3. This rate limiting step has a barrier of only 27.5 kcal mol−1, which is the lowest among the reactions investigated. Notably, the inverse addition of the P–H bond across the diborene is strongly disfavoured (see ESI†), supporting the selectivity to the observed product.
To understand why diborene 1 and borylborylene 4 prefer 1,2-hydrophosphination, while diborenes 2 and 3 undergo 1,1-hydrophosphination, a selection of NPA partial charges were analysed (Table 1). The symmetrical diborene A1 displays partial negative charge on both boron atoms (entry 2, parentheses), as expected for diborenes.46 In line with our previous analysis of the Hirshfeld charges of the unsymmetrical species 2–4,37 the NPA partial charges of the model diborenes B1 and C1 and borylborylene D1 (parentheses of entries 3–5) displayed a very similar trend for the charge accumulation and the course of charge flow on the boron atoms. Upon coordination of HPPh2 to the electron-rich diborene C1, the negative charges on both boron atoms increase further, with a relatively high negative charge localised on BDIST (the boron atoms are denoted as BPROX and BDIST, respectively, for those proximal and distal to the incoming HPPh2). Simultaneously, the partial positive charge at the HPPh2 phosphorus atom increases, confirming the charge donation from the incoming phosphine. Another intriguing fact about the coordinated HPPh2 is that its hydrogen substituent obtains a partial positive charge in contrast to its free form, which is marginally negative at hydrogen (entry 1). Thus, it is apparent that the hydrogen transfer from phosphorus to boron during hydrophosphination is a proton transfer, not a hydride transfer. As anticipated, the HPPh2 coordination A1–D1 is endergonic due to addition of a weak phosphine nucleophile to an electron-rich diborene. As the initially formed phosphine adducts of diborene A1 and borylborylene D1 are the immediate precursors to the transition states that involve a proton shift step, the partial charges at both boron atoms of these intermediates (A2 and D2) were analyzed. The adduct A2 shows a high negative charge build-up on the boron atom distal to the coordinated HPPh2 (entry 2). As a result, BDIST abstracts the proton from the coordinated HPPh2. Likewise, the localized high negative charge on BDIST in intermediate D2 (entry 5) drives the proton shift to this distal boron. For the reaction coordinates of diborenes B1 and C1, the immediate precursors to the proton transfer transition state are B4 and C3, respectively. In these two intermediates (entries 3 and 4), a high negative charge build-up was found on BPROX (bound to HPPh2), leading in both cases to 1,1-hydrophosphination.
| Entry | Species | q (H) | q (P(PPh2H)) | q (BPROX) | q (BDIST) |
|---|---|---|---|---|---|
| a BPROX refers to the PPh2H-coordinated boron atom, whereas BDIST refers to the other boron atom. The charges in parentheses are of the boron atoms of starting diborenes. | |||||
| 1 | PPh2H | −0.02 | 0.613 | — | — |
| 2 | A2 | 0.041 | 1.138 | −0.468 (−0.324) | −0.611 (−0.324) |
| 3 | B4 | 0.044 | 1.155 | −1.230 (−0.451) | −0.113 (−0.453) |
| 4 | C3 | 0.017 | 1.197 | −1.061 (−0.546) | 0.222 (−0.102) |
| 5 | D2 | 0.005 | 1.117 | −0.055 (0.298) | −1.238 (−1.205) |
Overall, these computations show that the hydrophosphination of diborenes begins with an initial coordination of the substrate HPPh2 to the boron atoms of electron-rich diborenes, followed by a proton shift from the coordinated HPPh2 to the proximal or distal boron, depending on which has a higher localised negative partial charge, dictating whether the 1,1- or 1,2-hydrophosphinated product is obtained.
Recent success in adding B–B and B–H bonds across boron–boron triple bonds32,34 led us to consider the hydrophosphination of diborynes. We chose the least sterically encumbered diboryne currently available, B2(SIDep)2 (9, SIDep = 1,3-bis(2,6-diethylphenyl)imidazolin-2-ylidene),28 in the hope that this would allow the approach of the relatively bulky substrate. Treatment of 9 with excess HPPh2 at −78 °C in toluene (Scheme 3) resulted in an immediate colour change from red to purple. After warming to room temperature, analysis by NMR spectroscopy revealed clean conversion to a new species with a doublet signal at −25.9 ppm (J = 46 Hz) in the 31P NMR spectrum. Two broad resonances were observed at 38.3 and 18.0 ppm in the 11B NMR spectrum, shifted significantly from that of 9 (δ = 55.9 ppm) and in the expected region for diborenes. The 1H{11B} NMR spectrum exhibited a conspicuous broadened doublet at 4.19 ppm, indicating a boron-bound proton coupled to the phosphorus nucleus (3JPH = 46 Hz). Crystals obtained from a pentane solution allowed determination of the structure of the compound (Fig. 4). The product, diborene 10, is the result of a 1,2-hydrophosphination across the boron–boron triple bond. Compound 10 is the first example of a phosphinodiborane. The B–B bond distance of 1.567(3) Å is somewhat shorter than in the related hydro(boryl)diborene SIDipMes(H)BB(Bcat)SIDipMes (SIDipMes = 1-(2,4,6-trimethylphenyl)-3-(2,6-diisopropylphenyl)-imidazolin-2-ylidene, BB = 1.609(2) Å),32 while the B1–P1 distance is shorter than that in phosphinodiboranes 5–8, at 1.945(2) Å, on account of the sp2 hybridisation at boron.
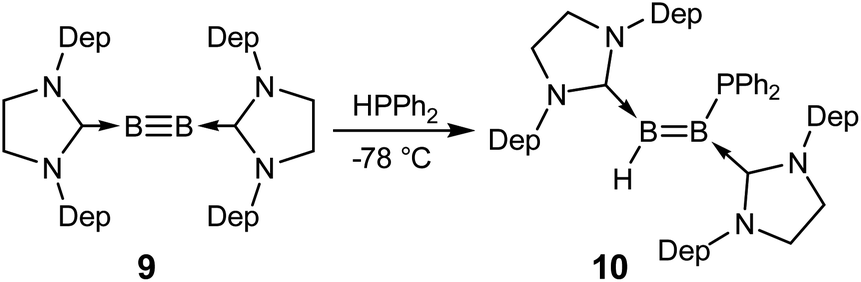 | ||
| Scheme 3 Hydrophosphination of the boron–boron triple bond in a diboryne. Dep = 2,6-diethylphenyl. | ||
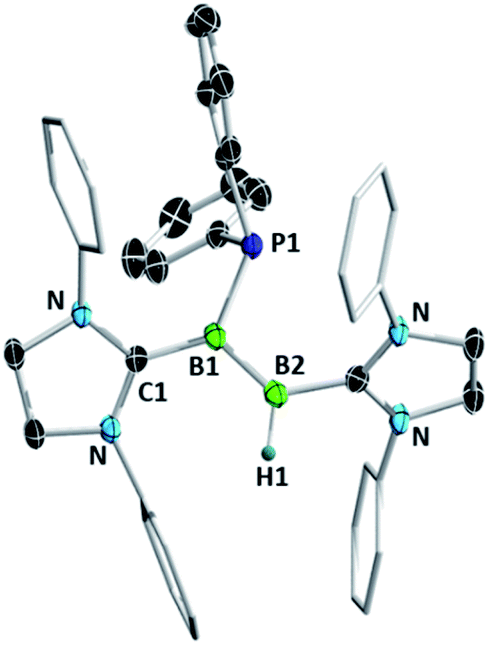 | ||
| Fig. 4 Molecular structure of compound 10 with selected atomic displacement ellipsoids at the 50% probability level. Hydrogen atoms except for H1, ethyl groups and a co-crystallised hexane molecule are omitted for clarity. Selected bond lengths (Å) and torsions (°): B1–B2 1.567(3), B1–P1 1.945(2), B1–C1 1.553(3), B2–C2 1.583(3), C1–B1–B2–B1 177.1(2). | ||
No further reaction of 10 with HPPh2 could be observed, even after heating the sample to 100 °C in toluene solution, in stark contrast to the behaviour of compounds 1–4. We ascribe this lack of reactivity to the large steric bulk of the flanking NHC ligands when compared to the accessible boron–boron bonds of the benzylphosphine-supported species.
Conclusion
In summary, we have demonstrated that diborenes undergo catalyst-free hydrophosphination reactions with diphenylphosphine under mild conditions. The substituents at the diborene dictate whether the reaction proceeds as a 1,1- or 1,2-addition. The major influencing factor for the regiochemistry appears to be the relative partial charges at the boron atoms in the initial adduct. We have also reported 1,2-hydrophosphination of a diboryne, yielding an unsymmetrical hydro(phosphino)diborene. Our current efforts are focussed on exploring more challenging σ-bond activations at these highly reactive boron–boron multiple bonds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the European Research Council for funding under the European Union Horizon 2020 Research and Innovation Program (grant agreement no. 669054). A. J. thanks the government of Canada for an NSERC postdoctoral fellowship.Notes and references
- J. A. Bailey and P. G. Pringle, Coord. Chem. Rev., 2015, 297, 77–90 CrossRef.
- D. C. Pestana and P. P. Power, J. Am. Chem. Soc., 1991, 113, 8426–8437 CrossRef CAS.
- R. T. Paine and H. Nöth, Chem. Rev., 1995, 95, 343–379 CrossRef.
- X. D. Feng, M. M. Olmstead and P. P. Power, Inorg. Chem., 1986, 25, 4615–4616 CrossRef CAS.
- For the sake of consistency, we have used the definitions of Bailey and Pringle (ref. 1) to make the distinction between the two structural extremes of monomeric P–B compounds..
- J. M. Breunig, A. Hubner, M. Bolte, M. Wagner and H. W. Lerner, Organometallics, 2013, 32, 6792–6799 CrossRef CAS.
- S. J. Geier, T. M. Gilbert and D. W. Stephan, J. Am. Chem. Soc., 2008, 130, 12632–12633 CrossRef CAS PubMed.
- A. Amgoune, S. Ladeira, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2012, 134, 6560–6563 CrossRef CAS PubMed.
- A. M. Spokoyny, C. D. Lewis, G. Teverovskiy and S. L. Buchwald, Organometallics, 2012, 31, 8478–8481 CrossRef CAS PubMed.
- J. A. Bailey, M. F. Haddow and P. G. Pringle, Chem. Commun., 2014, 50, 1432–1434 RSC.
- J. A. Bailey, M. Ploeger and P. G. Pringle, Inorg. Chem., 2014, 53, 7763–7769 CrossRef CAS PubMed.
- D. Dou, G. W. Linti, T. Q. Chen, E. N. Duesler, R. T. Paine and H. Nöth, Inorg. Chem., 1996, 35, 3626–3634 CrossRef CAS.
- T. Q. Chen, E. N. Duesler, R. T. Paine and H. Nöth, Inorg. Chem., 1999, 38, 4993–4999 CrossRef CAS PubMed.
- T. Q. Chen, E. N. Duesler, H. Nöth and R. T. Paine, J. Organomet. Chem., 2000, 614, 99–106 CrossRef.
- S. J. Geier, T. M. Gilbert and D. W. Stephan, Inorg. Chem., 2011, 50, 336–344 CrossRef CAS PubMed.
- A. Lik, D. Kargin, S. Isenberg, Z. Kelemen, R. Pietschnig and H. Helten, Chem. Commun., 2018, 54, 2471–2474 RSC.
- M. Kaaz, C. Baucker, M. Deimling, S. König, S. H. Schlindwein, J. Bender, M. Nieger and D. Gudat, Eur. J. Inorg. Chem., 2017, 2017, 4525–4532 CrossRef CAS.
- A. Ordyszewska, N. Szynkiewicz, E. Perzanowski, J. Chojnacki, A. Wiśniewska and R. Grubba, Dalton Trans., 2019, 48, 12482–12495 RSC.
- G. E. Coates and J. G. Livingstone, J. Chem. Soc., 1961, 1000–1008 RSC.
- M. Kaaz, J. Bender, D. Forster, W. Frey, M. Nieger and D. Gudat, Dalton Trans., 2014, 43, 680–689 RSC.
- A. D. Gorman, J. A. Bailey, N. Fey, T. A. Young, H. A. Sparkes and P. G. Pringle, Angew. Chem., Int. Ed., 2018, 57, 15802–15806 CrossRef CAS PubMed.
- O. Delacroix and A. C. Gaumont, Curr. Org. Chem., 2005, 9, 1851–1882 CrossRef CAS.
- D. S. Glueck, Top. Organomet. Chem., 2010, 31, 65–100 CrossRef CAS.
- I. Wauters, W. Debrouwer and C. V. Stevens, Beilstein J. Org. Chem., 2014, 10, 1064–1096 CrossRef PubMed.
- S. Greenberg and D. W. Stephan, Chem. Soc. Rev., 2008, 37, 1482–1489 RSC.
- S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed.
- G. Hong, X. Y. Zhu, C. Hu, A. N. Arum, S. Y. Wu and L. M. Wang, J. Org. Chem., 2016, 81, 6867–6874 CrossRef CAS PubMed.
- M. Arrowsmith, J. Böhnke, H. Braunschweig, M. A. Celik, T. Dellermann and K. Hammond, Chem.–Eur. J., 2016, 22, 17169–17172 CrossRef CAS PubMed.
- J. H. Muessig, M. Thaler, R. D. Dewhurst, V. Paprocki, J. Seufert, J. D. Mattock, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 4405–4409 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst, C. Hörl, A. K. Phukan, F. Pinzner and S. Ullrich, Angew. Chem., Int. Ed., 2014, 53, 3241–3244 CrossRef CAS PubMed.
- H. Braunschweig and C. Hörl, Chem. Commun., 2014, 50, 10983–10985 RSC.
- T. Brückner, T. E. Stennett, M. Heß and H. Braunschweig, J. Am. Chem. Soc., 2019, 141, 14898–14903 CrossRef PubMed.
- T. E. Stennett, R. Bertermann and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 15896–15901 CrossRef CAS PubMed.
- T. Brückner, R. D. Dewhurst, T. Dellermann, M. Müller and H. Braunschweig, Chem. Sci., 2019, 10, 7375–7378 RSC.
- H. Braunschweig, P. Constantinidis, T. Dellermann, W. C. Ewing, I. Fischer, M. Hess, F. R. Knight, A. Rempel, C. Schneider, S. Ullrich, A. Vargas and J. D. Woollins, Angew. Chem., Int. Ed., 2016, 55, 5606–5609 CrossRef CAS PubMed.
- J. Böhnke, T. Dellermann, M. A. Celik, I. Krummenacher, R. D. Dewhurst, S. Demeshko, W. C. Ewing, K. Hammond, M. Heß, E. Bill, E. Welz, M. I. Rohr, R. Mitric, B. Engels, F. Meyer and H. Braunschweig, Nat. Commun., 2018, 9, 1197 CrossRef PubMed.
- T. E. Stennett, J. D. Mattock, I. Vollert, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 4098–4102 CrossRef CAS PubMed.
- T. E. Stennett, J. D. Mattock, L. Pentecost, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 15276–15281 CrossRef CAS PubMed.
- R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594–9607 RSC.
- J. Böhnke, M. Arrowsmith and H. Braunschweig, J. Am. Chem. Soc., 2018, 140, 10368–10373 CrossRef PubMed.
- J. B. Bourg, A. Rodriguez, D. Scheschkewitz, H. Gornitzka, D. Bourissou and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 5741–5745 CrossRef CAS PubMed.
- A. Rodriguez, F. S. Tham, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed., 2004, 43, 4876–4880 CrossRef CAS PubMed.
- A. Rodriguez, G. Fuks, J. B. Bourg, D. Bourissou, F. S. Tham and G. Bertrand, Dalton Trans., 2008, 4482–4487, 10.1039/b718631b.
- W. Lu, K. Xu, Y. X. Li, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2018, 57, 15691–15695 CrossRef CAS PubMed.
- J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed., 2015, 54, 12274–12296 CrossRef CAS PubMed.
- M. Arrowsmith, H. Braunschweig and T. E. Stennett, Angew. Chem., Int. Ed., 2017, 56, 96–115 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1954176–1954180. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05908c |
| This journal is © The Royal Society of Chemistry 2020 |