
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic studies of cystobactamids as antibiotics and bacterial imaging carriers lead to compounds with high in vivo efficacy†
Giambattista
Testolin
a,
Katarina
Cirnski
bc,
Katharina
Rox
ab,
Hans
Prochnow
a,
Verena
Fetz
a,
Charlotte
Grandclaudon
ab,
Tim
Mollner
 a,
Alain
Baiyoumy
a,
Antje
Ritter
a,
Christian
Leitner
ab,
Jana
Krull
a,
Joop
van den Heuvel
d,
Aurelie
Vassort
e,
Sylvie
Sordello
f,
Mostafa M.
Hamed
c,
Walid A. M.
Elgaher
c,
Jennifer
Herrmann
bc,
Rolf W.
Hartmann
c,
Rolf
Müller
bc and
Mark
Brönstrup
*abg
a,
Alain
Baiyoumy
a,
Antje
Ritter
a,
Christian
Leitner
ab,
Jana
Krull
a,
Joop
van den Heuvel
d,
Aurelie
Vassort
e,
Sylvie
Sordello
f,
Mostafa M.
Hamed
c,
Walid A. M.
Elgaher
c,
Jennifer
Herrmann
bc,
Rolf W.
Hartmann
c,
Rolf
Müller
bc and
Mark
Brönstrup
*abg
aDepartment of Chemical Biology, Helmholtz Centre for Infection Research, Inhoffenstrasse 7, 38124 Braunschweig, Germany. E-mail: mark.broenstrup@helmholtz-hzi.de
bGerman Center for Infection Research (DZIF), Site Hannover-Braunschweig, Germany
cHelmholtz Institute for Pharmaceutical Research Saarland, Universitätscampus E8.1, 66123 Saarbrücken, Germany
dGroup Recombinant Protein Expression, Helmholtz Centre for Infection Research, Inhoffenstrasse 7, 38124 Braunschweig, Germany
eEvotec ID, 1541 Avenue Marcel Merieux, 69289 Marcy l'Etoile, France
fEvotec ID, Alderley Park, Cheshire SK10 4TG, UK
gCenter of Biomolecular Drug Research (BMWZ), Leibniz Universität, 30167 Hannover, Germany
First published on 10th December 2019
Abstract
There is an alarming scarcity of novel chemical matter with bioactivity against multidrug-resistant Gram-negative bacterial pathogens. Cystobactamids, recently discovered natural products from myxobacteria, are an exception to this trend. Their unusual chemical structure, composed of oligomeric para-aminobenzoic acid moieties, is associated with a high antibiotic activity through the inhibition of gyrase. In this study, structural determinants of cystobactamid's antibacterial potency were defined at five positions, which were varied using three different synthetic routes to the cystobactamid scaffold. The potency against Acinetobacter baumannii could be increased ten-fold to an MIC (minimum inhibitory concentration) of 0.06 μg mL−1, and the previously identified spectrum gap of Klebsiella pneumoniae could be closed compared to the natural products (MIC of 0.5 μg mL−1). Proteolytic degradation of cystobactamids by the resistance factor AlbD was prevented by an amide-triazole replacement. Conjugation of cystobactamid's N-terminal tetrapeptide to a Bodipy moiety induced the selective localization of the fluorophore for bacterial imaging purposes. Finally, a first in vivo proof of concept was obtained in an E. coli infection mouse model, where derivative 22 led to the reduction of bacterial loads (cfu, colony-forming units) in muscle, lung and kidneys by five orders of magnitude compared to vehicle-treated mice. These findings qualify cystobactamids as highly promising lead structures against infections caused by Gram-positive and Gram-negative bacterial pathogens.
Introduction
The public health threat posed by bacterial infections has increased to an alarming extent, because bacterial resistance against common antibiotics has risen over the last two decades while, at the same time, an innovation gap in antibiotic discovery led to a shriveled pipeline of novel drugs.1 This is particularly true for Gram-negative pathogens: since infections with carbapenem-resistant Enterobacteriaceae are associated with mortality rates of 40–50%, these species as well as drug-resistant Acinetobacter baumannii and Pseudomonas aeruginosa have been classified as ‘critical’ by the WHO.2 The main scientific problem that hampers the discovery of novel antibiotics against Gram-negative species concerns the lack of knowledge on how to design molecules that can achieve sufficient intracellular concentrations by overcoming the highly effective penetration barriers imposed by the inner and outer membranes on the one hand, and by avoiding efflux-mediated export on the other hand.3,4 Recent progress was achieved by establishing structural guidelines for synthetic small molecules,5 or by antibiotic conjugation to actively transported moieties (the ‘Trojan Horse’ principle).6 However, studying natural products arguably remains the most fruitful strategy to discover novel antibiotic lead structures against Gram-negative bacteria.1,7 This is exemplified by the recently discovered cystobactamids, myxobacterial natural products that exhibit potent, broad-spectrum antibacterial properties through the inhibition of bacterial gyrase A.8 Cystobactamids, their derivatives coralmycin9 and the closely related albicidin10 are featured by an unusual aromatic oligopeptide structure, composed of five para-aminobenzoic acid (PABA)-derived moieties and a central aliphatic amino acid (Scheme 1). An in-depth isolation study from Myxococcus sp. resulted in the discovery of derivative 861-2 as the most potent cystobactamid analog.11 Soon after their first isolation, total syntheses of albicidin (1) and the cystobactamids 861-2 (2) and 919-2 (3) were reported,11–15 all highlighting the hidden challenges behind the chemistry of these natural products, such as low reactivity in aromatic amine couplings combined to solubility problems. First variations of the albicidin structure at various positions led to mostly equal antibacterial activity compared to the natural product.16–19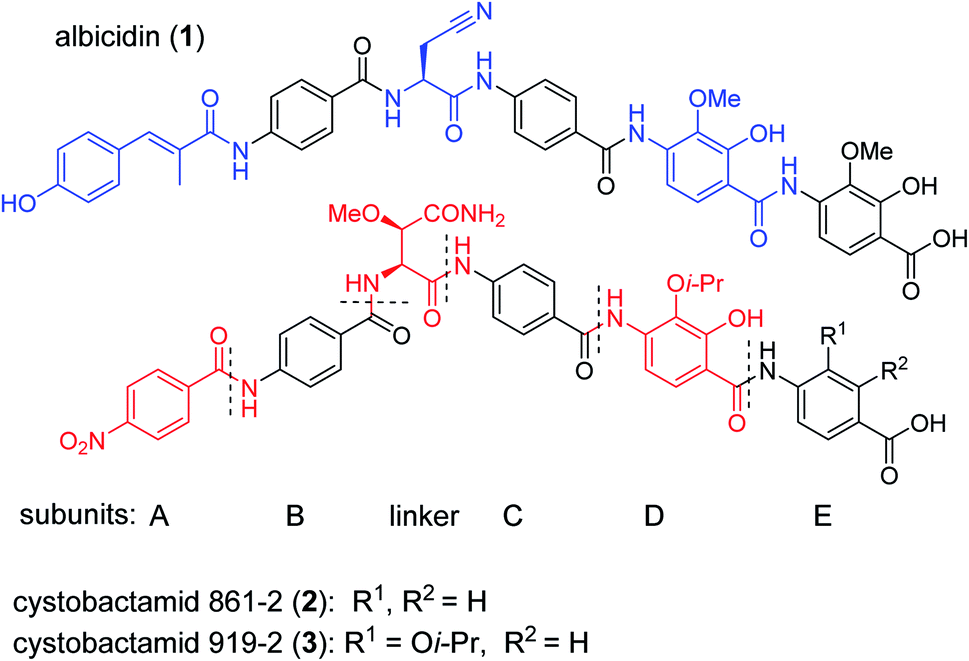 | ||
| Scheme 1 Chemical structures of albicidin and cystobactamids 861-2 and 919-2. Characteristic structural features of the classes are highlighted in blue for albicidin and in red for cytobactamids. | ||
In this paper, we report the establishment of three different synthetic routes for a systematic study of cystobactamid's structure–activity relationships (SAR), in order to optimize the activity profile across a larger panel of bacterial pathogens. We obtained compounds with significantly improved antibacterial activity, and a rescaffolding of the C-ring led to the circumvention of AlbD-mediated resistance, an endopeptidase that site-specifically cleaves albicidin in two inactive fragments. Furthermore, a proof of principle that the cystobactamid scaffold can be used as a targeting moiety for bacterial imaging is provided. Finally, the pharmacokinetic properties as well as a first in vivo proof of concept of derivative 22 are presented.
Results
Cystobactamids do not obey the guidelines for physicochemical properties that are important for activity against Gram-negative bacteria: according to O'Shea and Moser, a low molecular weight (cutoff ca. 600 Da) and high polarity are important features of Gram-negative antibiotics to facilitate transport through porin channels.20 In addition, the so-called “eNTRy” rules proposed by Richter et al. state that accumulation in Gram-negative bacteria is more likely to occur if a molecule possesses basic nitrogens (primary amines being optimal), low globularity and high rigidity.5 In contrast, cystobactamids have no basic nitrogen, their molecular weight (>850 Da) is far beyond the cutoff, they are highly nonpolar (clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 4.19), and 15 (principally) rotatable bonds limit their rigidity (ESI Section 2†). Because the potent activity of cystobactamids against Gram-negative pathogens is achieved with atypical physicochemical properties, we did not apply such parameters as filters for compound design. In the absence of structural information on the binding to gyrase and on uptake determinants, the synthesis of novel compounds was driven by their antibacterial activity and their ability to inhibit gyrase.
P = 4.19), and 15 (principally) rotatable bonds limit their rigidity (ESI Section 2†). Because the potent activity of cystobactamids against Gram-negative pathogens is achieved with atypical physicochemical properties, we did not apply such parameters as filters for compound design. In the absence of structural information on the binding to gyrase and on uptake determinants, the synthesis of novel compounds was driven by their antibacterial activity and their ability to inhibit gyrase.
In order to understand the minimal structural requirement for high antibiotic activity, a simplification of the cystobactamid structure was targeted, starting with the most potent analog 2. We first decided to investigate the relevance of the methoxy group in the central linker region, as the high activity of albicidin suggested that a substituent at position C-3 of the aliphatic amino acid may not be mandatory. Therefore, the des-methoxy analog 4 was synthesized following the retrosynthetic approach that we recently reported11 for the total synthesis of 2 (Scheme 2), with the main difference that the central fragment 6 bears L-Asn instead of the (2S,3R)-β-methoxy-Asn moiety of 2.
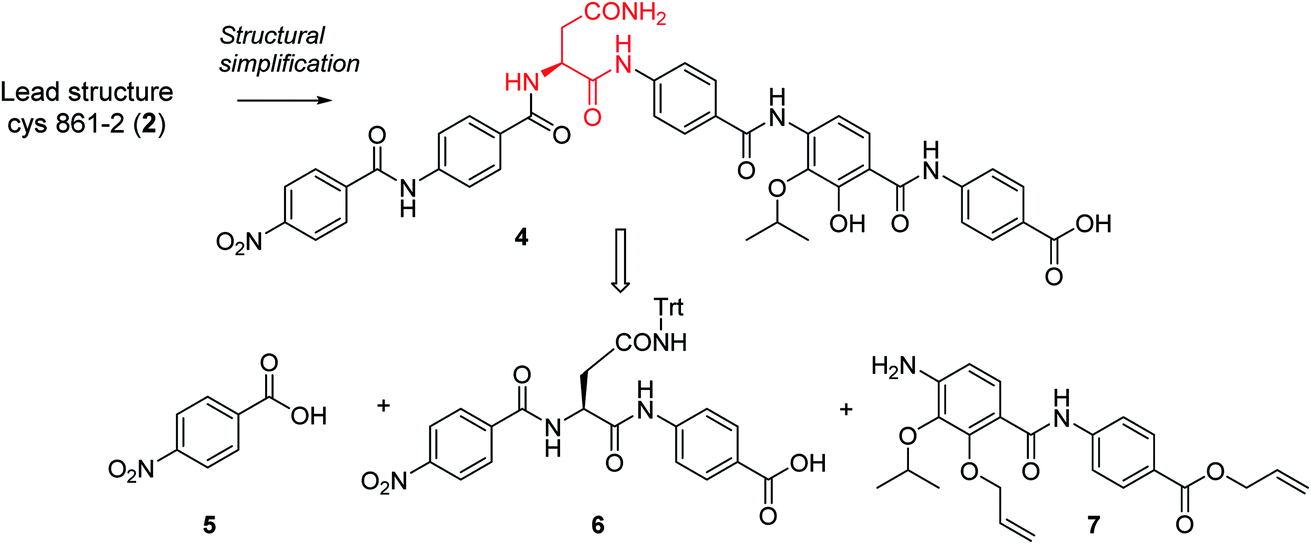 | ||
| Scheme 2 Retrosynthetic disconnection of 4. | ||
In brief, Fmoc-Asn(Trt)-OH and methyl-p-aminobenzoate were coupled efficiently by means of POCl3 at 0 °C. Keeping the temperature low was important in order to avoid racemization of the aliphatic amino acid (Scheme 3).21 After deprotection of the Fmoc group, the second PABA unit was installed using HBTU and para-nitrobenzoic acid (PNBA) to give 94 in 78% yield. Because the ester hydrolysis under standard hydrolytic conditions (LiOH, water/THF) led to complete racemization, the carboxylic acid 6 was obtained using an excess of LiI in ethyl acetate under refluxing conditions.22
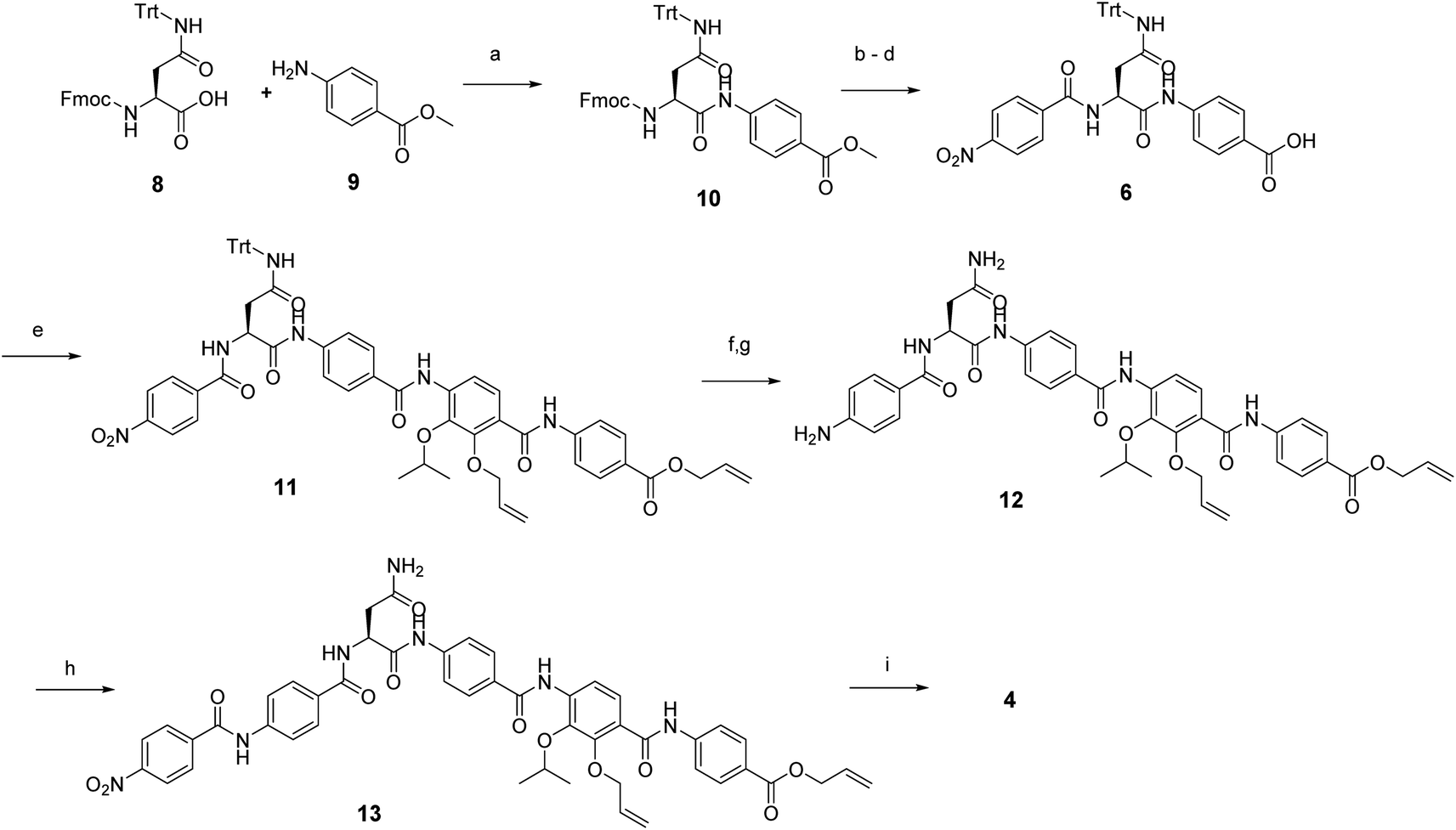 | ||
| Scheme 3 Reagent and conditions: (a) POCl3, TEA, DCM, 0 °C, 2 h (96%); (b) 20% Et2NH in CH3CN, rt, 30 min; (c) HBTU, DiPEA, CH3CN, rt, 3 h (78% 2 steps); (d) LiI, EtOAc, 90 °C, 5 d (86%); (e) amine 7, POCl3, DiPEA, THF/DCM, rt, 6 h (54%); (f) Zn dust, 10% AcOH, THF/EtOH, rt, 5 h, (g) TFA, Tips, DCM (94% 2 steps); (h) 4-nitrobenzoic acid, BTC, collidine, DiPEA, THF, rt, 4 h (80%); (i) Pd(PPh3)4; PhSiH3, THF, rt, overnight (21%). | ||
Fragment 7, synthesized as reported previously,11 was coupled to 6 using POCl3 to obtain the pentapeptide 11. The reduction of the nitro group and the removal of the trityl protecting group afforded 12. The N-terminal PNBA moiety was activated as an acyl chloride and coupled to 12. After the removal of the allyl protecting groups and a final purification by preparative reversed phase (RP) HPLC, 4 was obtained as an ammonium salt. A Marfey analysis,23–25 used to assess the enantiomeric ratio, revealed that less than 5% of racemization occurred during the synthesis (see ESI†). The overall synthesis had 12 steps in the longest linear sequence and an overall yield of 1.9%.
The minimal inhibitory concentrations (MICs) of all compounds were determined against a small panel of three clinically important pathogens comprising Escherichia coli, Pseudomonas aeruginosa and Staphylococcus aureus. For the two former species, mutants with impaired efflux (E. coli ΔtolC and P. aeruginosa ΔmexAB) were tested in order to assess transport-mediated effects. In addition, a functional gyrase supercoiling assay served to probe the target-specific activity of the cystobactamids. Compound 4 showed high antibacterial activities that were comparable to those of 2, with the notable exception of a strongly decreased activity against P. aeruginosa (Table 1). The overall profile confirmed the assumption of the β-methoxy group in the central amino acid linker is not essential. Because 4 was slightly more potent than 2 in a target-based gyrase supercoiling assay (IC50 values of 0.11 μM vs. 0.22 μM), the additional methoxy moiety of 2 seems to be beneficial for intracellular compound accumulation in P. aeruginosa WT.
As the synthesis of 4 was more amenable to scale up compared to that of 2, a series of cystobactamid analogs bearing L-asparagine as a central element was designed that carry alterations in the N-terminal (ring A) and C-terminal (rings D–E) regions of the molecule.
A-ring variations
For A-ring variations, the advanced intermediate 12 was coupled to different carboxylic acids that varied in the nature, number and position of substituents (ESI Scheme S1†). The acids were activated as acyl chlorides by means of BTC and collidine12 or oxalyl chloride. Alternatively, when the substituents required a milder activation strategy, a combination of EDC and HOAt was chosen. Most derivatives were purified by preparative RP-HPLC with a water/acetonitrile gradient using 10 mM NH4HCO3 as a modifier and obtained as ammonium salts.The antibacterial properties of all analogs were determined subsequently (Table 2). The electronic properties and the position of the N-terminal substituent turned out to be essential, as the replacement of the nitro group by a hydrogen atom in 15 was associated with a drop of activity against E. coli from 1 μg mL−1 to >64 μg mL−1. Also its shift from the para-to meta- and ortho-positions led to MICs of >64 μg mL−1 and 8 μg mL−1, respectively. Likewise, reduced analogs bearing an amino group (17) or an N-acetylamino group (14) had lower activities of 2 μg mL−1 and 16 μg mL−1, respectively. An ureido substitution as in 21 resulted in equipotent compounds against E. coli, but reduced activity against S. aureus or P. aeruginosa. Also the installation of a photoactivatable trifluoromethyl-diazirine moiety at the para-position led to pronounced drops in gyrase inhibition and antibacterial activity, suggesting that 28 is not ideally suited for the mapping of the gyrase binding site in cross-linking experiments.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound |
|
MIC [μg mL−1] | E. coli gyrase IC50 [μM] | ||||||
| E. coli ΔtolCa | E. coli WTb | P. ae. ΔmexABa | P. ae. WTc | S. aureus | |||||
| R | |||||||||
| o | m | p | |||||||
| a Strain provided by collaborators, see ESI. b DSM-1116. c PA14. d ATCC29213. e nd = not determined, P. ae. = P. aeruginosa. | |||||||||
| Reference 4 | H | H | NO 2 | 0.125 | 1 | 1 | >64 | 1 | 0.11 |
| 14 | H | H | NHAc | 0.06 | 16 | >64 | >64 | 64 | nd |
| 15 | H | H | H | 0.5 | >64 | 2 | >64 | >64 | 4.0 |
| 16 | H | H | F | 0.5 | 0.5 | 2 | >64 | 2 | nd |
| 17 | H | H | NH2 | 32 | 2 | 4 | >64 | >64 | 26.8 |
| 18 | H | NO2 | H | 0.25 | >64 | 0.5 | >64 | >64 | 2.2 |
| 19 | H | NH2 | H | 2 | 32 | 4 | >64 | >64 | nd |
| 20 | NO2 | H | H | 2 | 8 | 16 | >64 | >64 | nd |
| 21 | H | H | NHCONH2 | ≤0.03 | 0.5 | 4 | >64 | 64 | 8.7 |
| 22 | H | H | CN | 0.06 | 0.5 | 0.25 | 2 | 0.25 | 0.08 |
| 23 | H | H | CF3 | ≤0.03 | 0.5 | 64 | >64 | 16–32 | nd |
| 24 | H | H |
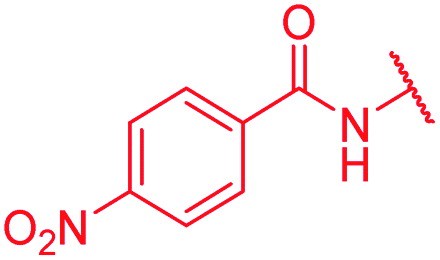
|
>64 | 0.5 | >64 | >64 | >64 | 2.5 |
| 25 | OH | i-PrO | NO2 | >64 | >64 | >64 | >64 | >64 | >50 |
| 26 | OH | i-PrO | NH2 | 0.01 | 2 | 2–4 | 32 | >64 | 50 |
| 27 | H | H | SO2CH3 | ≤0.03 | 2 | 4 | >64 | >64 | nd |
| 28 |
|
8 | 16 | >64 | >64 | >64 | 16 | ||
| 29 |
|
≤0.03 | ≤0.03 | 1 | >64 | 1 | 1.4 | ||
| 30 |

|
≤0.03 | ≤0.03 | 1 | >64 | 64 | 0.5 | ||
| 31 |

|
≤0.03 | ≤0.03 | 0.25 | 0.5 | ≤0.03 | 0.9 | ||
| 32 |
|
≤0.03 | ≤0.03 | 0.25 | 0.5 | 0.25 | 0.5 | ||
| 33 |
|
≤0.03 | 0.06 | 1 | >64 | 4 | nd | ||
| 34 |
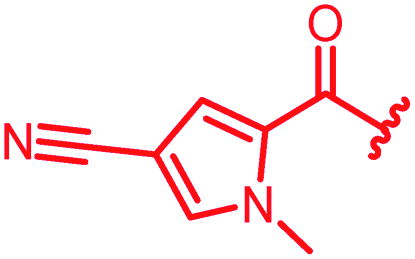
|
≤0.03 | ≤0.03 | 0.5 | >64 | 64 | 0.7 | ||
| 35 |
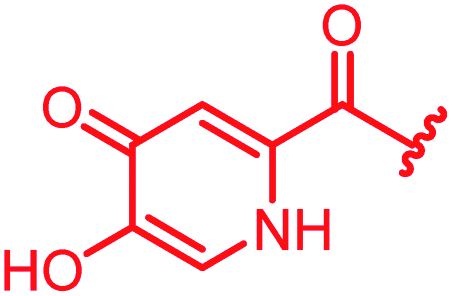
|
>64 | >64 | >64 | >64 | >64 | 3.6 | ||
| 36 |
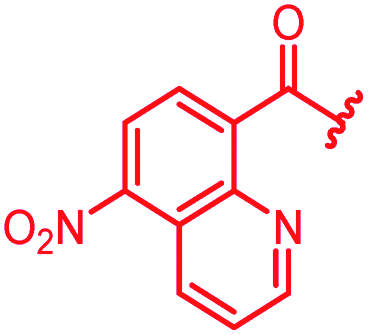
|
0.5 | 0.25 | >64 | >64 | >64 | 0.3 | ||
As cystobactamids are PABA oligomers, we wondered whether their elongation to a heptapeptide by the addition of another PNBA moiety to the N-terminus would further enhance potency. However, 24 showed a 20-fold reduced inhibitory activity in the gyrase assay and displayed worse antibacterial activity compared to 4. We then copied the substitution pattern of the oxidized D-ring of cystobactamids to the N-terminus. The obtained compound 25 neither showed acceptable gyrase inhibition nor any antibacterial activity. In contrast, the replacement of the nitro group with other electron-withdrawing substituents like trifluoromethyl (23) or fluorine (16) retained antibiotic activity or even improved them, as observed for a cyano (22) or a lactone moiety (29). The cyano group was identified as particularly favorable, because 22 also displayed a strongly increased activity against P. aeruginosa (2 μg mL−1vs. >64 μg mL−1 for 4); this substituent was therefore retained in most subsequent analogs. The introduction of a second substituent in ortho- or meta-position, as in 88–92, did not lead to improved properties (see Table S1 of the ESI†).
In attempts to replace the phenyl ring, heterocyclic moieties with different electronic properties and ring size were prepared, including the electron-poor pyridines 30 and 31, the thiophene 32 and the thiazole 33, and, due to its ability to interact with DNA bases,26 the electron-rich N-methyl pyrrole 34. A hydroxy-pyridinone heterocycle with iron-chelating properties was applied (35), as this moiety has been successfully attached to β-lactams as a siderophore mimic to enhance their permeation into Gram-negative bacteria through iron uptake pathways.27,28 Finally, the quinoline 36 should inform whether an extended π-system was favorable for gyrase binding.
The position of the nitrogen in pyridyl analogs mattered, because the 2-carboxyamide-5-cyano-substituted derivative 31 was highly potent against all tested strains, whereas the 3-carboxyamide-6-cyano derivative (30) was inactive against S. aureus and P. aeruginosa. A consistently high antimicrobial activity was also observed for the thiophene 32, whereas the pyrrole 34 and the quinoline 36 lost activity against S. aureus and P. aeruginosa. The introduction of an iron-chelating motif (35) led to a complete loss of antimicrobial activity and to significantly reduced anti-gyrase activity. After optimizing ring A, we decided to re-introduce the methoxy in the central asparagine, peculiar of the natural cystobactamids, to further assess its influence on the biological activity (ESI†). Remarkably, this molecule showed potent activity against all strains of the small panel of pathogens (analog 93, Table S1†).
In summary, the A-ring turned out to be a sensitive structural handle for the breadth of the antibacterial spectrum and the potency of cystobactamids. The most important findings are that a hydrogen bond acceptor and small substituents at the para-position are essential for a high antibiotic activity, and that the replacement of PABA with heterocyclic moieties is tolerated.
Variations of the linker between A and B rings
A key structural difference between cystobactamids and the closely related albicidin 1 concerns the N-terminus, which is spanned by a para-hydroxy-2-methylcinnamoyl moiety in 1. To assess the impact of this substitution, 45, a hybrid of the two structures, was synthesized by replacing the PNBA unit of 4 with para-hydroxy-2-methylcinnamoyl. Compound 45 indeed possessed high antibiotic activity on all tested strains, including an MIC of 1 μg mL−1 against P. aeruginosa. As electron-donating substituents in the N-terminal ring of cystobactamids were unfavorable (see above), the para-hydroxy group of 45 was replaced by a cyano group to yield 46, but surprisingly, this change led to an overall unaltered antibacterial potency (Tables 3 and 7).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | R | MIC [μg mL−1] | E. coli gyrase IC50 [μM] | ||||
| E. coli ΔtolCa | E. coli WTb | P. ae. ΔmexABa | P. ae. WTc | S. aureus | |||
| a Strain provided by collaborators, see ESI. b DSM-1116. c PA14. d ATCC29213. e nd = not determined, P. ae. = P. aeruginosa. | |||||||
| Reference 22 |
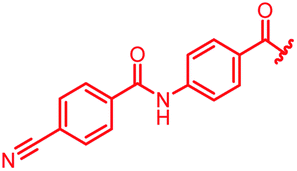
|
0.06 | 0.5 | 0.25 | 2 | 0.25 | 0.08 |
| 39 |
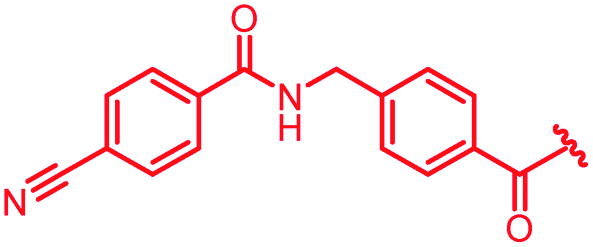
|
1 | >64 | >64 | >64 | >64 | nd |
| 40 |
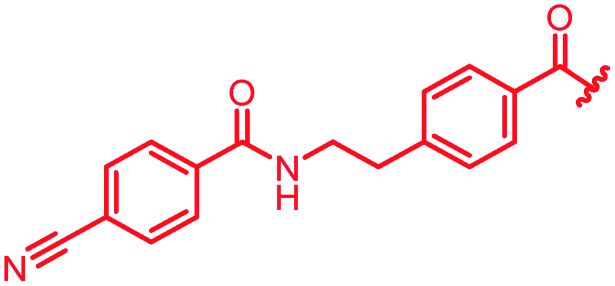
|
0.125 | 0.25 | 4 | >64 | >64 | nd |
| 41 |
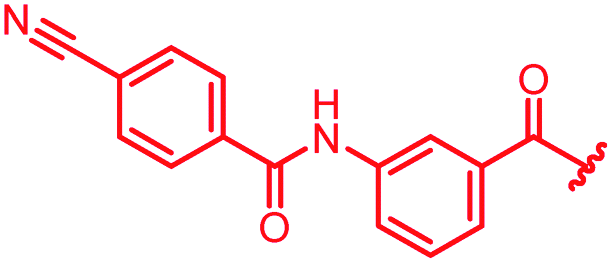
|
>64 | >64 | >64 | >64 | >64 | nd |
| 42 |
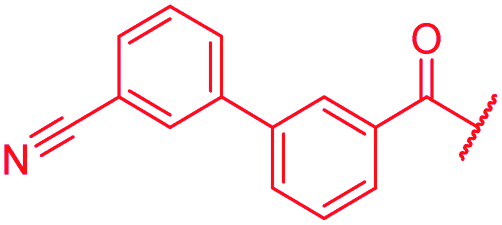
|
>64 | >64 | >64 | >64 | >64 | >50 |
| 43 |
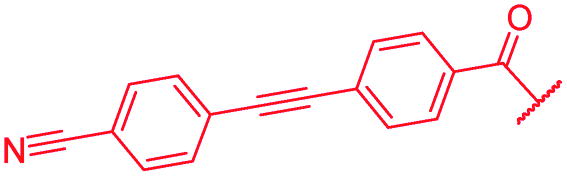
|
0.06 | 0.06 | 0.5 | >64 | ≤0.03 | 0.5 |
| 44 |
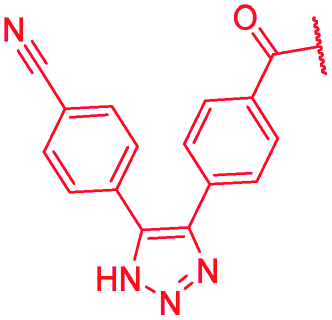
|
>64 | >64 | >64 | >64 | >64 | >50 |
| 45 |
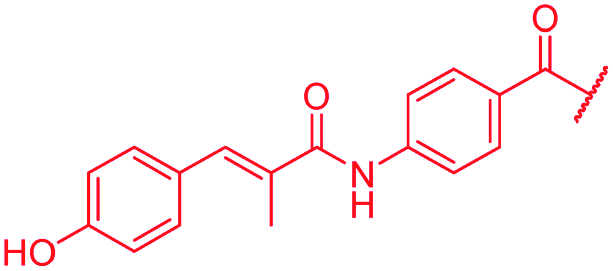
|
0.06 | 0.25 | 0.25 | 1 | 0.25 | nd |
| 46 |
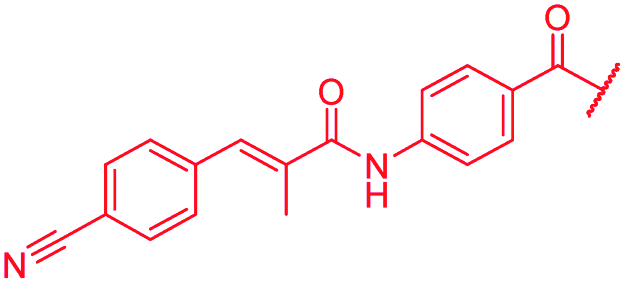
|
≤0.03 | 0.125 | 0.25 | 1 | 0.06 | 0.2 |
| 47 |
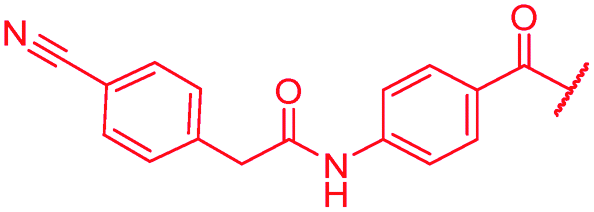
|
≤0.03 | 1 | >64 | >64 | >64 | 16.3 |
| 48 |
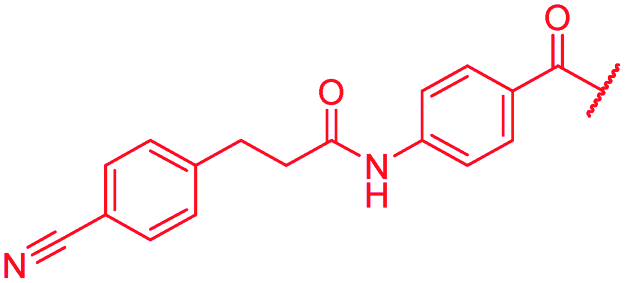
|
≤0.03 | 0.125 | 0.5 | >64 | >64 | 1.0 |
| 49 |
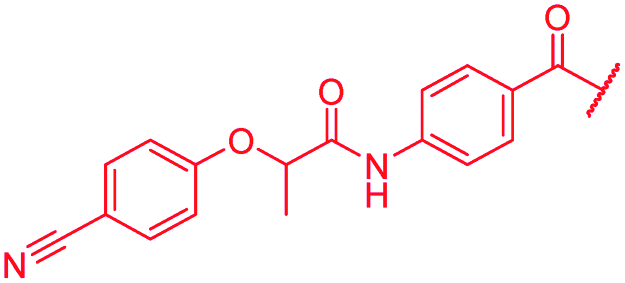
|
≤0.03 | 0.06 | >64 | >64 | >64 | 1.8 |
| 50 |

|
≤0.03 | 0.25 | >64 | >64 | >64 | 0.8 |
| 51 |
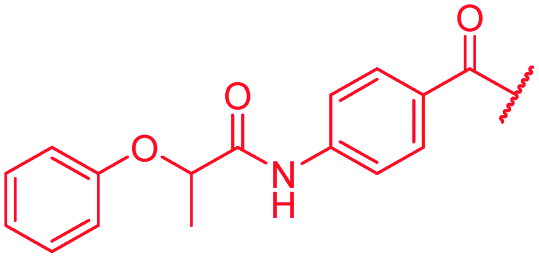
|
4 | >64 | >64 | >64 | >64 | nd |
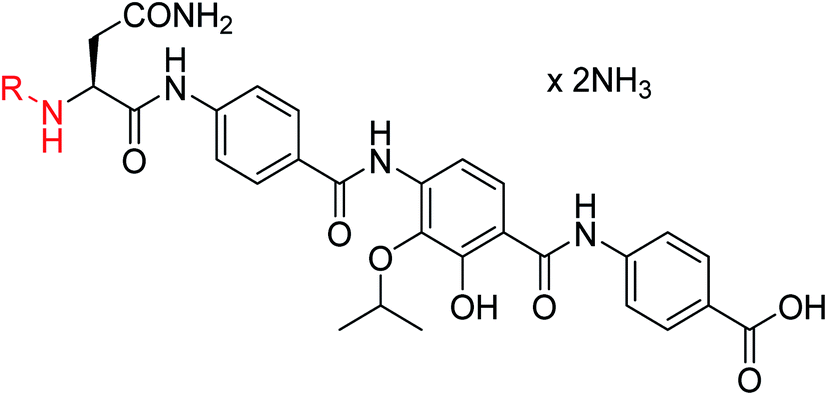
The fact that the aromatic A and B rings can be connected by a methacrylamide instead of an amide moiety inspired us to vary the length and chemical nature of this region. To access broader diversifications of the N-terminus, a modified synthetic strategy with a late introduction of pre-functionalized A and B rings was adopted (Scheme 4). First, the fragments 38 and 7 were coupled, followed by the cleavage of allyl- and Fmoc-protecting groups. The obtained deprotected tetrapeptide was then coupled to a diaryl unit that comprised rings A and B. The advantages of this approach over the one presented in Scheme 2 are the higher reactivity of the primary, aliphatic amine in the last coupling step and a shortened longest linear sequence (11 steps).
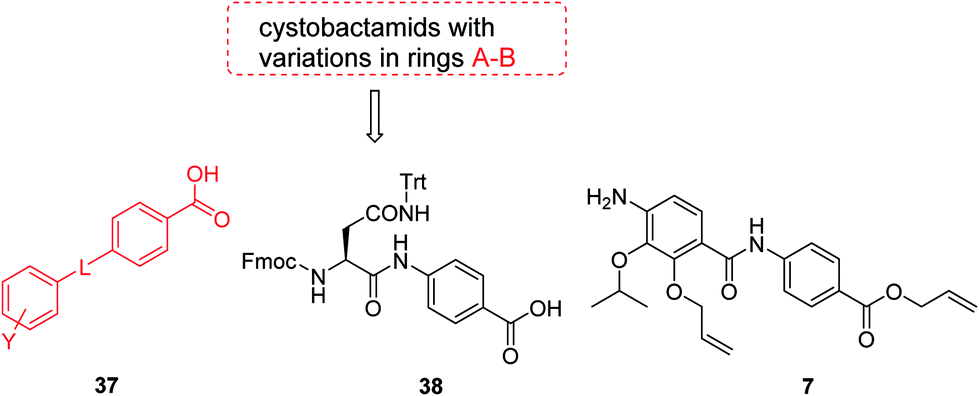 | ||
| Scheme 4 Retrosynthetic disconnection of cystobactamid with modified A–B rings. | ||
First, the CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CMe fragment of methacrylamide of 46 was replaced by saturated (CH2–CH2 as in 48), heteroatom-substituted (O–CHMe as in 49, S–CHMe as in 50) or truncated linkers (CH2 as in 47). In addition, one or two methylene units were introduced between the para-cyanobenzamide terminus of 22 and the B-ring to yield 39 and 40, respectively. The high cellular activity against E. coli was retained, but the fact that none of the compounds inhibited S. aureus or P. aeruginosa demonstrates that the structure–activity relationship (SAR) for a cellular activity is steep and organism-specific at this position.
CMe fragment of methacrylamide of 46 was replaced by saturated (CH2–CH2 as in 48), heteroatom-substituted (O–CHMe as in 49, S–CHMe as in 50) or truncated linkers (CH2 as in 47). In addition, one or two methylene units were introduced between the para-cyanobenzamide terminus of 22 and the B-ring to yield 39 and 40, respectively. The high cellular activity against E. coli was retained, but the fact that none of the compounds inhibited S. aureus or P. aeruginosa demonstrates that the structure–activity relationship (SAR) for a cellular activity is steep and organism-specific at this position.
The impact of the B-ring topology on activity was probed by 41 and 42, which possess a meta-connectivity on ring B; both compounds were completely inactive. In order to investigate whether the amide linking rings A and B in cystobactamid adopts a cis or trans conformation upon binding to gyrase, 43, linearized by an alkyne moiety, and the triazole 44, mimicking a cis-conformation, were synthesized. While 43 retained high gyrase inhibition and antibacterial activities, 44 was inactive in both assays. This led us to conclude that (i) a para-substitution at the B-ring and a stretched or trans-conformation between A and B is required for gyrase binding and (ii) the original amide linker of natural cystobactamids is not required, but can be replaced by a rod-like alkyne connector. However, the original linker between A and B rings had the best overall properties and was therefore retained in subsequent optimizations.
D-ring modifications
Two selected modifications of the D-ring were targeted. An analog of 4, devoid of the free hydroxy group, was assembled in 10 steps (ESI Schemes S4 and S5†). The analog 52 lost all activity against wild type pathogens, but prevented the growth of efflux-impaired strains, which underlines the crucial importance of the hydroxy group for antibiotic function. In line with this, also the masking of the hydroxy group by an allyl ether as in 13 led to a complete loss of antibacterial activity (Table 4). It is noteworthy that the amide bond between rings D and E is unusually acidic (pKa < 8), as manifested by the comparison of 1H-NMR data between the ammonium salt with the free acid analog of 4 (ESI Fig. S12†). Both the signals of the amide and the phenolic protons were absent in the spectra of the ammonium salt, which could be attributed to the presence of an anionic, hydrogen-bonded six-membered ring.|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | N-terminus | R | R1 | R2 | MIC [μg mL−1] | E. coli gyrase IC50 [μM] | ||||
|
|
E. coli ΔtolCa | E. coli WTb | P. ae. ΔmexABa | P. ae. WTc | S. aureus | |||||
| a Strain provided by collaborators, see ESI. b DSM-1116. c PA14. d ATCC29213. e nd = not determined, P. ae. = P. aeruginosa. | ||||||||||
| Reference 4 | NO 2 | i-Pr | OH | OH | 0.125 | 1 | 1 | >64 | 1 | 0.11 |
| 52 | NO2 | i-Pr | H | OH | 0.06 | >64 | 1 | >64 | >64 | nd |
| 129 | NO2 | i-Pr | H | OMe | >64 | >64 | >64 | >64 | >64 | nd |
| 13 | NO2 | i-Pr | OAllyl | OAllyl | >64 | >64 | >64 | >64 | >64 | nd |
| 53 | NO2 | i-Bu | OH | OH | 2 | >64 | >64 | >64 | 1 | 0.9 |
| 54 | CN | i-Bu | OH | OH | <0.03 | 0.25 | >64 | >64 | 0.25 | nd |
| 55 |
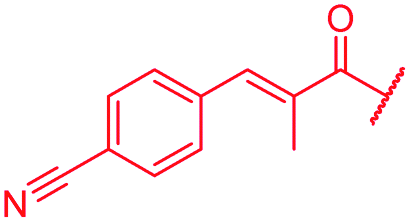
|
i-Bu | OH | OH | 0.06 | 0.5 | >64 | >64 | 0.06 | nd |
Next, the isopropoxy substituent of the D-ring was elongated by a single methylene unit to further increase the bulkiness of this structural feature, yielding 53 (Table 4). This rather subtle structural modification led to an eight-fold decrease of gyrase activity and a complete loss of activity against E. coli wild type and the P. aeruginosa ΔmexAB mutant. Two derivatives (54, 55) combining the isobutoxy residue and optimized N-terminal ring substitutions could restore the activity against E. coli WT, but not against P. aeruginosa. On the other hand, a natural cystobactamid with an ethoxy-substitution at the D ring also showed decreased antibacterial activity.11 Both results underline the importance of the D-ring substitution pattern and imply that the substituent pattern of natural cystobactamids is optimal.
Furthermore, a drastic re-scaffolding was investigated: cystobactamids target subunit A of bacterial DNA gyrase.8 As this subunit is responsible for interactions with DNA,29 we hypothesized that it could be beneficial to incorporate a motif into the antibiotic which itself has strong DNA binding properties. Among the high-affinity DNA minor groove recognition elements reported in the literature,30,31 we selected a minor groove binder developed by Renneberg and Dervan26 that contains a 4-hydroxybenzimidazole as well as an N-methylpyrrole moiety (a Hz/Py pair) and incorporated it into the cystobactamid scaffold. In an attempt to maintain the overall architecture of cystobactamids, the Hz/Py pair replaced the C and D rings of cystobactamids in 56 (Scheme 5). The synthesis of this compound was impaired by solubility and reactivity limitations, but succeeded with an approach that relied on the disconnection in the two fragments 58 and 59. The fragments were coupled following the activation of the carboxylic acid to an acyl fluoride to avoid potential side reactions of asparagine, such as intramolecular cyclization.32–34 In sum, 56 was assembled in 11 steps (longest linear sequence) in a total yield of 8.2% (Schemes S6 and S7†).
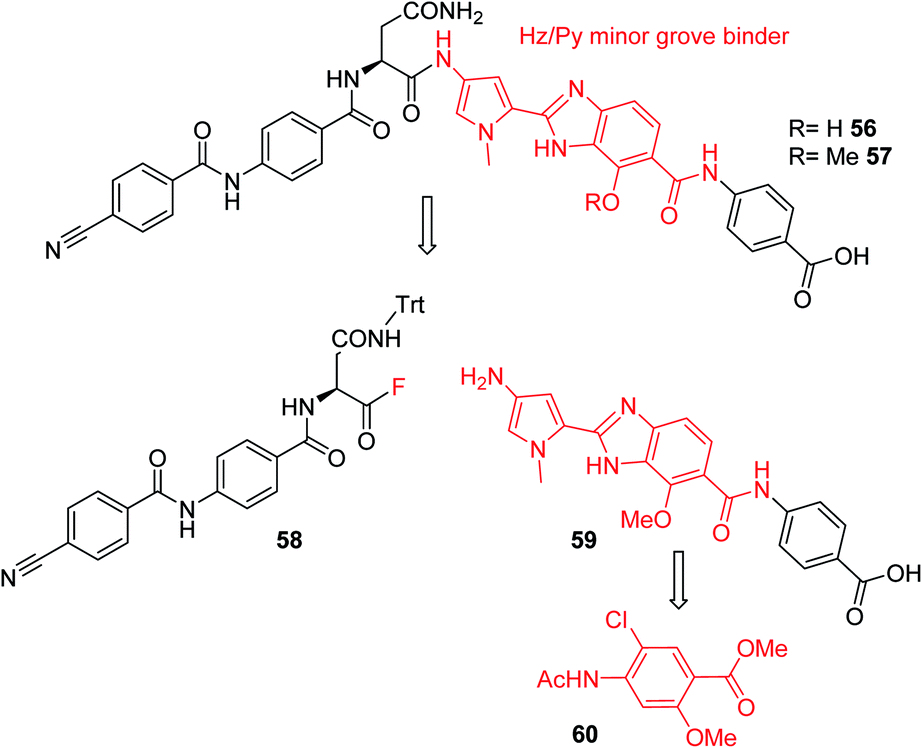 | ||
| Scheme 5 Retrosynthetic analysis followed for the synthesis of cystobactamid analog 56. The minor groove binding moiety is highlighted in red. | ||
Compound 56 inhibited bacterial gyrase with an IC50 of 10.9 μM, which represents an approximately 100-fold decrease in activity compared to 22. In consequence, 56 did not exert antibacterial activities at concentrations up to 64 μg mL−1 (ESI Table S2†). This result reflects that the re-scaffolding represents a high risk approach in the absence of information on cystobactamid-target interactions, and it also underlines the high importance of obtaining structural data on cystobactamid binding to inform a rational analog design.
A potential Achilles heel of oligo-PABA antibiotics was recently characterized by Süssmuth and coworkers: the endopeptidase AlbD, expressed by the Gram-negative Enterobacterium Pantoea dispersa, was shown to cleave the amide bond connecting C and D rings in 1 as well as in a tripeptide carrying a substitution pattern as in cystobactamids, thereby inactivating the antibiotics.35 Although the potential clinical relevance of this resistance mechanism is unclear, as albD has not been detected in human pathogens yet, we attempted to circumvent it by replacing the amide cleavage site with a 1,2,3-triazole as a bioisoster (Fig. 1).36 For this purpose, 61 was synthesized in 11 steps and an overall yield of 7% based on a retrosynthetic disconnection in two fragments (ESI Schemes S8 and S9†). The larger fragment 156 was synthesized by coupling 4-ethinylaniline to Fmoc-Asn(Trt)OH, followed by the installation of the diaryl unit 64. In the synthesis of fragment 154, the azide moiety was introduced into the advanced intermediate 158via Sandmeyer reaction using tBu-NO and TMS-azide in 70% yield. Finally, fragments 156 and 154 were coupled efficiently via copper-catalyzed 1,3-dipolar cycloaddition, and after cleavage of the acid-labile protecting groups, 61 was purified by preparative RP-HPLC. Remarkably, 61 displayed high potency against E. coli (MIC = 0.06 μg mL−1) and S. aureus (MIC ≤ 0.03 μg mL−1) and a good potency against P. aeruginosa (MICs of 2 and 8 μg mL−1 against mutant and wild type), in spite of a ca. 20-fold weaker inhibition of bacterial gyrase (ESI Table S3†). Thus, the “triazole cystobactamid” 61 has practically the same spectrum coverage as the corresponding analog bearing the original amide 22, but showed lower inhibitory properties at the molecular target. The stability of 61 was first tested in an in vitro assay with recombinant, purified AlbD. While albicidin and 22 were efficiently cleaved into their corresponding tetra- and dipeptides following a 6 h incubation with AlbD, 61 remained fully stable under these conditions (Fig. 1 and ESI Fig. S2–S4†). Further evidence that 61 indeed breaks AlbD-mediated resistance was obtained by MIC data against the AlbD producer P. dispersa: whereas P. dispersa was resistant against albicidin (MIC > 64 μg mL−1) and showed limited sensitivity towards the best natural cystobactamid 2 (MIC = 16 μg mL−1), 61 potently inhibited its growth with an MIC of 1 μg mL−1.
 | ||
| Fig. 1 Design of the AlbD-stable cystobactamid analog 61, with the amide-triazole replacement highlighted in red. The stabilities of 61 (retention time: 13.56 min) and 22 (retention time: 13.56 min) following a 6 h incubation with AlbD have been monitored by LC/UV/MS. Depicted is the UV absorption (190–400 nm) over time. The degradation products (retention times 10.29 min and 12.14 min) are depicted above the chromatograms. | ||
Thus, a bioisosteric substitution of the amide bond connecting rings C and D is tolerated, and the overall good spectrum coverage of 61 and its resistance-breaking properties make this molecule an interesting scaffold for further investigations.
E-ring modifications
For the investigation of the C-terminal region of the molecule, a retrosynthetic disconnection was chosen that featured a late-stage introduction of the E-ring (Scheme 6). In a first target compound, the E-ring was completely omitted to yield the pentapeptide 67. This compound was active only against mutant E. coli and possessed more than 100-fold reduced enzymatic activity compared to 22 (Table 5). This finding further underlines the importance of the full hexapeptide configuration for the Gram-negative antibacterial activity, that was demonstrated before by the inactive, N-terminally truncated albicidin16 or the tripeptide cystobactamid 507 harboring rings C–E.37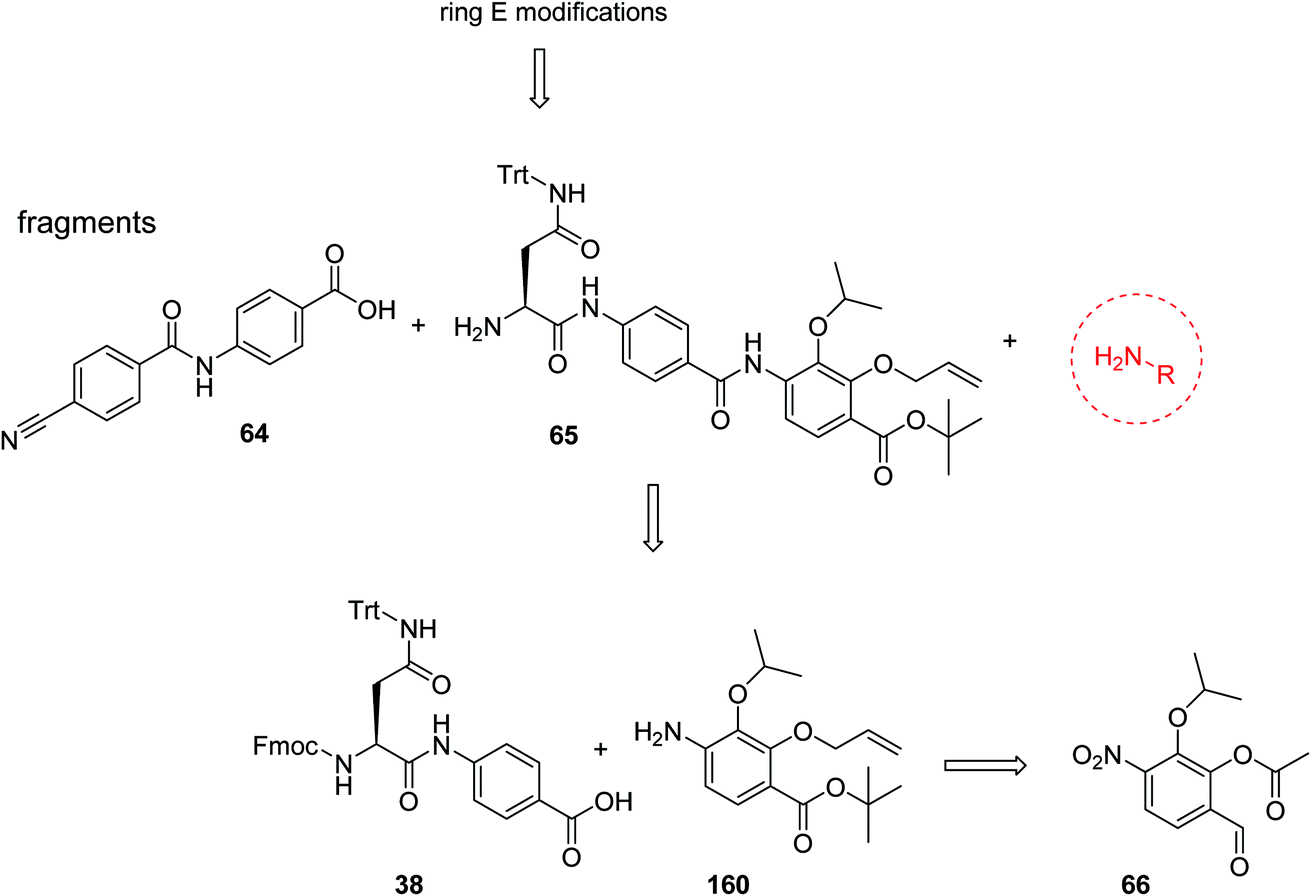 | ||
| Scheme 6 Retrosynthetic disconnection used for late stage variations of ring E. Fragments 65 and 64 are coupled first, followed by the final installation of ring E. | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Y | Ring E | MIC [μg mL−1] | EC gyrase IC50 [μM] | ||||
| E. coli ΔtolCa | E. coli WTb | P. ae. ΔmexABa | P. ae. WTc | S. aureus | ||||
| a Strain provided by collaborators, see ESI. b DSM-1116. c PA14. d ATCC29213. e nd = not determined, P. ae. = P. aeruginosa. | ||||||||
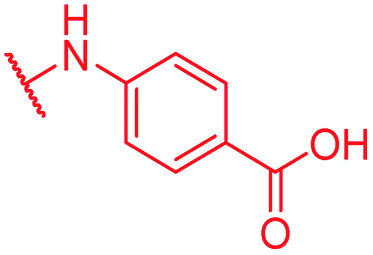
| Reference 22 | CN |
|
0.06 | 0.5 | 0.25 | 2 | 0.25 | 0.08 |
| 67 | CN | –OH | ≤0.03 | >64 | >64 | >64 | >64 | 9.4 |
| 68 | NO2 |
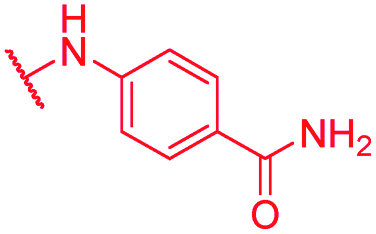
|
≤0.03 | 1 | >64 | >64 | 64 | 0.2 |
| 69 | NO2 |
|
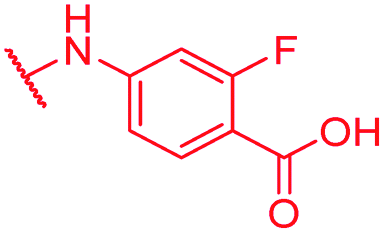
0.125 | 1 | 1 | >64 | 2 | nd |
| 70 | CN |
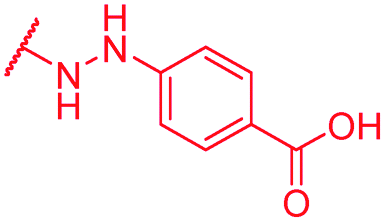
|
0.25 | >64 | >64 | >64 | >64 | nd |
| 71 | CN |
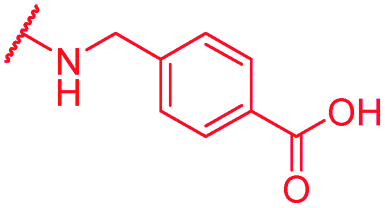
|
0.25 | >64 | >64 | >64 | >64 | nd |
| 72 | CN |
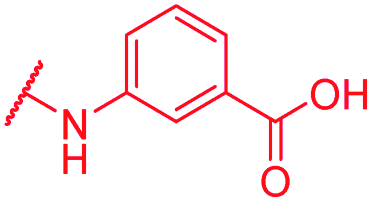
|
≤0.03 | 0.5 | >64 | >64 | 2 | nd |
| 73 | CN |
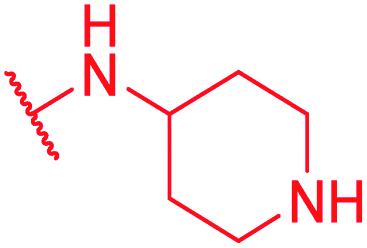
|
>64 | >64 | >64 | >64 | >64 | 0.5 |
| 74 | CN |
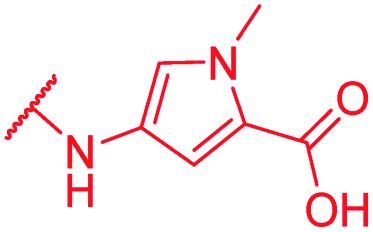
|
≤0.03 | 0.5 | 8 | >64 | 64 | nd |
| 75 | CN |
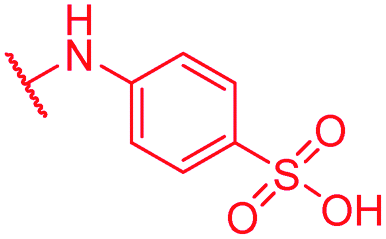
|
0.25 | >64 | >64 | >64 | >64 | nd |
| 76 | CN |
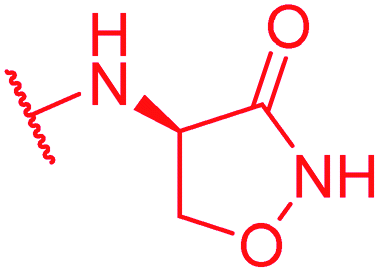
|
0.5 | >64 | >64 | >64 | >64 | nd |
| 77 | CN |
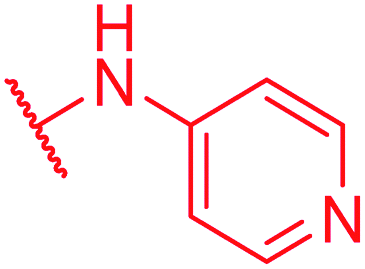
|
≤0.03 | >64 | >64 | >64 | >64 | 0.9 |
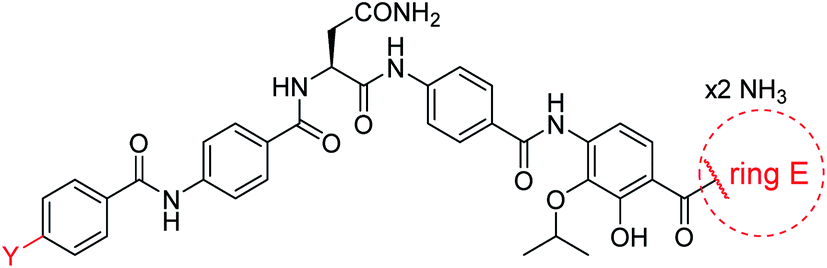
Secondly, the C-terminal carboxylic acid of 4 was replaced by an amide in 68. The compound was highly active in the gyrase assay, but only active against wild type E. coli. Introduction of an ortho-fluoro atom (69) retained the antibacterial spectrum of 4 with no activity against wild type P. aeruginosa. Moreover, 129, the methyl ester analog of 52, was completely inactive against efflux-impaired and wild type strains (Table 4), and similar effects were observed upon the replacement of the carboxylic acid by a sulfonic acid as in 75. More drastic modifications of the E-ring were realized by a replacement of the acidic PABA with basic 4-amino-pyridyl (77) and 4-amino-piperidyl (73) moieties, or the cycloserine 76. These switches in basicity of the C-terminus led to a drastic loss of antibacterial activity of these analogs, thereby illustrating the importance of the carboxylic acid. The insertion of a methylene group between the terminal arene and the amide linking it to the D-ring, which disrupts the conjugated π-system, also led to an almost complete loss of activity. It is noteworthy that a similar change between A and B-rings, as in 39, was not tolerated either (see above).
In contrast, the activity against E. coli strains was retained with a meta-substituted aminobenzoic acid terminus (as in 72) and with an N-methyl pyrrole moiety (as in 74) replacing the phenyl ring. However, both analogs clearly lost potency compared to a PABA-C-terminus.
Next, we investigated a conundrum of the E-ring substitution pattern: compared to the first-discovered cystobactamid 3, both the removal of a substituent (des-isopropoxy, to yield 2) and the addition of a substituent (hydroxy-919-2 = coralmycin A) were reported to enhance antibacterial activity.9 To assess whether an unsubstituted or a hydroxy/isopropoxy-substituted E-ring is superior, the latter pattern was combined with the five best N-terminal substitutions reported above (Table 6). The obtained analogs 78–82 showed comparable and high activity against E. coli and S. aureus, but two of them were less active against P. aeruginosa compared to their counterparts 31 and 32. This led us to conclude that the unsubstituted PABA unit was the superior E ring substitution for a broad spectrum coverage that includes P. aeruginosa.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | R | MIC [μg mL−1] | EC gyrase IC50 [μM] | ||||
| E. coli ΔtolCa | E. coli WTb | P. ae. ΔmexABa | P. ae. WTc | S. aureus | |||
| a Strain provided by collaborators, see ESI. b DSM-1116. c PA14. d ATCC29213. e nd = not determined, P. ae. = P. aeruginosa. | |||||||
| Reference 78 |
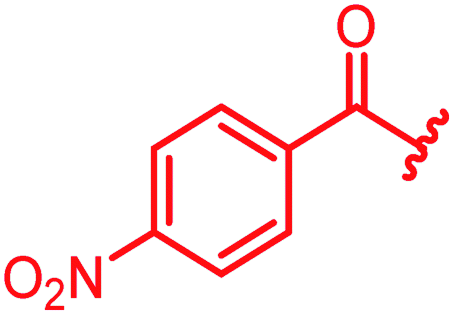
|
0.125 | 0.5 | 1 | >64 | 0.05 | 0.2 |
| 79 |
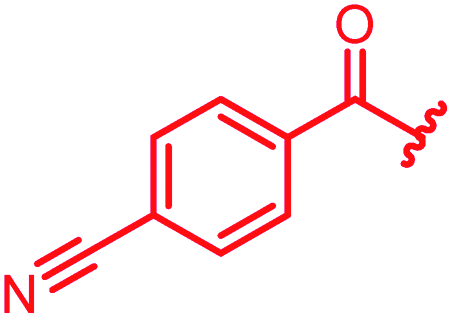
|
≤0.03 | 0.25 | 1 | 2 | ≤0.03 | 0.6 |
| 80 |

|
≤0.03 | 0.06 | 0.5 | >64 | ≤0.03 | nd |
| 81 |
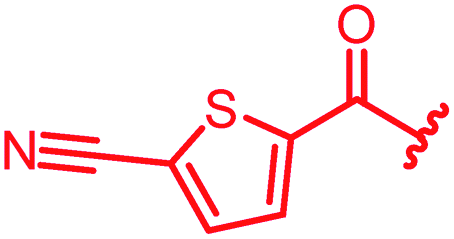
|
0.06 | 0.125 | 2 | >64 | 0.06 | nd |
| 82 |
|
≤0.03 | 0.125 | 0.25 | 1 | 0.06 | 0.2 |
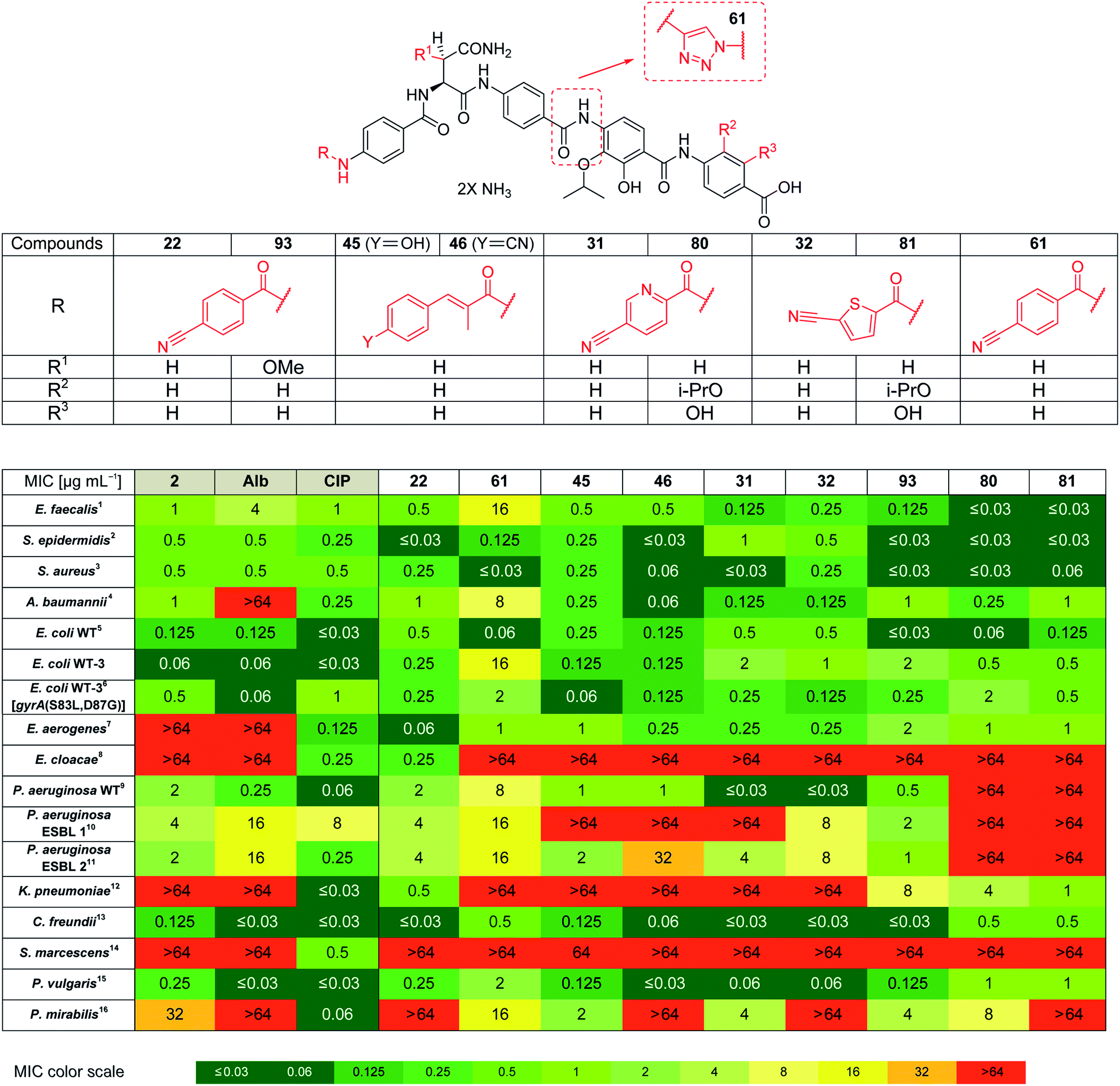
Based on the structural modifications in A, B, D and E rings as well as in selected linker regions described above, a small set of ten cystobactamid derivatives that were most potent on the small microbial strain panel and/or showed activity on the P. aeruginosa wild type strain was selected. This set was tested on a larger panel of Gram-negative and -positive pathogens of clinical interest, including A. baumannii, Klebsiella pneumoniae and other Enterobacteriaceae. The two natural products 1 (synthesized according to the reported experimental procedure),122 and ciprofloxacin served as reference compounds.
Remarkably, eight derivatives displayed potent (MIC ≤ 1 μg mL−1) activity against Enterobacter aerogenes, and three derivatives were active against K. pneumoniae, whereas both 1 and 2 were inactive (Table 7). Also spectrum gaps against E. aerogenes, Enterobacter cloacae and Proteus mirabilis could be closed by synthetic analogs.
| a Alb = albicidin, CIP = ciprofloxacin, ESBL = extended spectrum β-lactamases, 1ATCC-29212, 2DSM-28765, 3ATCC29213, 4DSM-30008, 5DSM-1116, 6E. coli based on the WT-3 wild type strain, carrying the mutations indicated above, 7DSM-30053, 8DSM-30054, 9PA14 10DSM-24600, 11DSM-46316, 12DSM-30104, 13DSM-30039, 14DSM-30121, 15DSM-2140, 16DSM-4479. Additional bacterial strain information is present in the experimental part. |
|---|
|
For A. baumannii, nine compounds reached MICs ≤ 1 μg mL−1, and the most potent congener 46 had a strongly improved activity (MIC = 0.06 μg mL−1) compared to ciprofloxacin (0.32 μg mL−1) or Cys 861-2 (1 μg mL−1).
The activity against E. coli WT-3, a strain that has a ca. 100-fold reduced fluoroquinolone sensitivity due to mutations at the active site of gyrase, was also assessed. The fact that the activity of most cystobactamids was not affected by the mutations supports the hypothesis that the binding site of the natural products is different from that of ciprofloxacin.
For all different ring substitutions, compounds that show a reduced gyrase affinity, but still potently kill E. coli were observed. This effect may either be due to a particularly high uptake, or due to the presence of a second, yet unidentified antibacterial target. To explore the latter option, a chemoproteomic study using accordingly modified chemical probes is regarded as an attractive future endeavor. To better understand the contributions of uptake to the observed SAR, mass spectrometry-based intracellular concentration measurements in a panel of strains with enhanced outer membrane permeability or impaired efflux are warranted in future studies.
For the cystobactamids 2 and 22 no signs of cytotoxicity, assessed by an MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide)-based assay, were visible within the tested concentration range of 0.1–10 μM. This is in line with previously published cytotoxicity data on cystobactamid analogs.8,11
Derivative 93 has the broadest antibacterial spectrum coverage. Remarkably, it possesses activity in the low μg mL−1 range against all tested bacteria except of E. cloacae and Serratia marcescens. These results indicate that the presence of the methoxy-group in the central Asn moiety is beneficial for the spectrum coverage. On the other hand, it is noteworthy that 93 does not exhibit the lowest MIC values against all the tested strains. For example, the lowest MIC values against K. pneumoniae, A. baumannii and Enterococcus faecalis were obtained with 81/22, 46 and 80/81, respectively. This suggests that strain-specific effects influence the activity of cystobactamid derivatives. Given that recent changes in the regulatory guidelines for the clinical testing of antibiotics encourage pathogen-specific antibiotic developments (in addition to classical paths focused on the body site of infection), it is well-conceivable that several different cystobactamids could be developed as narrow-spectrum antibiotics against single pathogens.
The current work allows deriving the following structure–activity relationships (Fig. 2): the full length of the molecule is essential for the activity, shortening it of a single aromatic unit either at the N- or C-terminus led to a complete loss of activity. For optimal inhibition of gyrase and good antibacterial properties, ring A should have a hydrogen bond acceptor, with a cyano group conferring the best antibacterial properties. The optimal position of the substituent on ring A is para. The replacement of the phenyl ring with heterocycles is possible and has a beneficial effect on the activity. Finally, the presence of a short rigid spacer, such as a substituted double bond, between ring A and carbonyl is well tolerated. The para-connectivity of the aromatic building blocks is also essential for the B-ring, while a relocation of the amino group to the meta-position has a drastic negative effect on the activity. The omission of the methoxy group in aliphatic linker region is well tolerated; this finding is in line with a recent study on natural albicidins.18 However, the enlarged panel of bacterial pathogens investigated in this study suggests that the presence of the methoxy group is overall beneficial for a broader spectrum coverage.
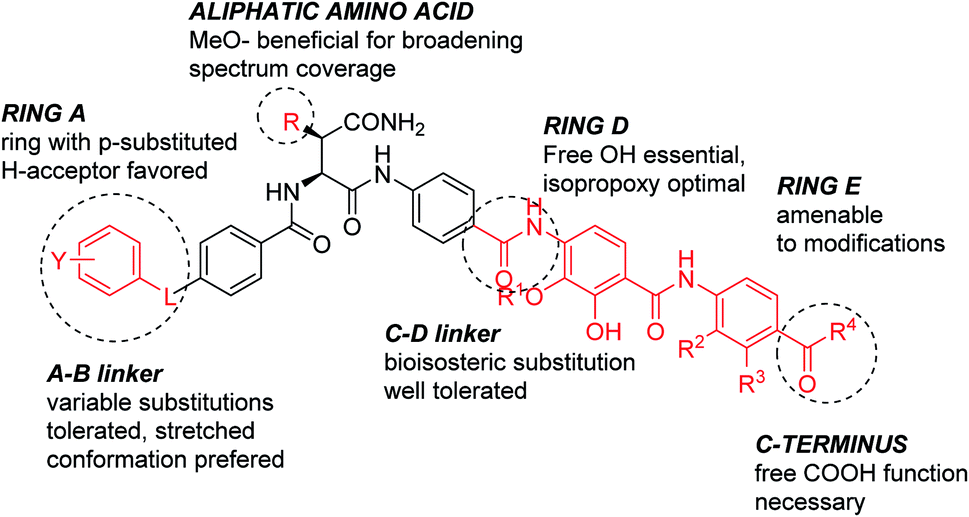 | ||
| Fig. 2 Structure–activity relationships of cystobactamids established in this work. | ||
The D-ring represents a critical part of the molecule; the enlargement of the isopropoxy group was associated with a drastic loss of activity; the same is true for a removal or etherification of the hydroxyl group. The latter finding stands in contrast to a recent report on a corresponding dehydroxy-albicidin that retained its activity (against a different panel of strains).18 Finally, ring E carrying an acidic function was found to be essential for antimicrobial activity. Among all variations tested, the E-ring substitution pattern of the natural product 2 was found to be optimal.
Cystobactamid-based targeted bacterial imaging
In addition to optimizing the cystobactamid scaffold to an antibiotic, we explored its potential for bacterial imaging. We speculated that structural elements of the cystobactamids could serve as a carrier of imaging moieties due to their capability of penetrating into a broad range of clinically relevant Gram-positive and -negative bacteria. A binding to gyrase is not required to fulfill this purpose.Indeed, there is a strong medical need to improve the diagnosis of infections at body sites that are hardly accessible for sampling. Therefore, noninvasive molecular probes that can selectively visualize bacteria based on bacteria-specific nutrient uptake pathways,38,39 enzymatic functions,40,41 or antibiotic scaffolds42–44 received considerable attention recently.45 Based on the structure–activity relationships established above, the tetrapeptidic N-terminus of 4 was selected as the carrier, and the aromatic rings D and E of the original scaffold were omitted and replaced by an imaging modality (Fig. 3a and b). Ring C was functionalized with an alkynyl moiety to enable a conjugation of the carrier with its payload by click chemistry. For this purpose, the Bodipy-based fluorophore BDP-FL-azide was coupled to the tetrapeptide to yield 85 (Scheme S11†). The free Bodipy–azide 84 was used as non-targeted control. Both 85 and 84 had no effect on bacterial growth (ESI Fig. S6†); however, pronounced differences in their labeling properties were observed: images taken after four hours of incubation clearly indicated a strong fluorescent signal for Gram-positive (S. aureus) as well as Gram-negative (E. coli, A. baumannii and K. pneumoniae) bacteria treated with 85, while that was not the case for free 84 (Fig. 3c, ESI Fig. S9†). This result was confirmed by a quantitative analysis by flow cytometry for E. coli (ESI Fig. S7†). Interestingly, an unlabeled subpopulation was observable after treatment with 85, but the molecular reasons of this phenomenon are unclear at this stage and deserve further attention in future studies.
 | ||
| Fig. 3 Labeling of bacterial cells with fluorescent or fluorogenic cystobactamid-conjugate. (a) Design of cystobactamid probe, its structure and the fluorescent dye used. (b) Schematic representation of the basic principle of the strategy and chemical structure of the Bodipy-cystobactamid conjugate. (c) Labeling of indicated bacterial species with Bodipy-conjugate or free Bodipy as monitored by confocal microscopy after 4 h of incubation with 10 μM compound in PBS. Scale bars: 11 μm. (d) Fluorescence intensity (Bodipy-FI) of free Bodipy or Bodipy-conjugate was quantified by flow cytometry. Histogram depicts Bodipy-FI vs. cell count. Exemplary result of one experiment after 5 h of incubation shown. (e) Bodipy-conjugate uptake quantified by flow cytometry. E. coli WT was incubated for 1 or 5 h in PBS with 85 or Bodipy (84). Quantification of two replicate experiments of Bodipy-conjugate uptake and propidium iodide (PI) staining as measured by flow cytometry. Plot depicts the percentage of cells classified as Bodipy- or PI-positive. (f) The bacterial uptake of cystobactamid conjugated to the fluorogenic dye malachite green (MG) or free MG was monitored over time with a fluorescence plate reader. 87 (free MG) or 86 (Cysto-MG) were added at a final concentration of 10 μM to E. coli_FAP6.2, and the fluorescence was recorded at indicated wavelengths for 16 h. (g) Chemical structures of Cysto-MG conjugate and free MG. | ||
In additional experiments, the fluorogen-activating-protein/malachite green (FAP/MG) system recently developed by us38 was applied to probe whether cyctobactamids conjugated with synthetic dyes are internalized by E. coli, or whether the compound binds to the outside of the cell wall. For this purpose, the cystobactamid-MG conjugate 86 was synthesized (see the ESI†) and uptake as indicated by fluorescence emission was monitored over time (Fig. 3f and g). The signal reached a maximum after 5 hours and clearly indicated an intracellular accumulation of the cystobactamid-based carrier. The translocation occurred at a rate comparable to that of free MG 87, which was used as a positive control and presumably enters the cell by passive diffusion.
The findings imply that the C-terminal truncation of cystobactamid leads to the loss of its antibiotic property (in accordance with the SAR findings above), but bacterial recognition and translocation are retained. However, the translocation mechanism of the imaging conjugates might differ from the (hitherto unkown) translocation mechanism of cystobactamid antibiotics. Apart from applications in bacterial imaging, such conjugates may also find use in a Trojan-horse strategy of antibiotic delivery.6 Future studies, such as competition assays with eukaryotic cells, will clarify whether the N-terminal motif could be utilized to specifically carry payloads into bacterial vs. mammalian cells.
In vitro and in vivo profile of 22
The cystobactamid analog 22 showed excellent inhibitory properties against Gram-negative and Gram-positive bacteria, while exhibiting low toxicity against HepG2 cells. Stability studies conducted with 2 demonstrated a half-life t1/2 of 117 and >400 min in mouse and human hepatocytes, respectively, and stabilities of >90%, 66% and 78% after 4 h incubation in mouse, rat and human plasma, respectively.The ability of four bacterial strains to develop resistance against 22 was investigated next. We observed frequencies of resistance (FoR) between 3.8 × 10−8 (against E. coli) and 1.6 × 10−9 (against A. baumannii) at four times MIC, whereas the FoR's of ciprofloxacin were between 2.2 × 10−8 and 1.5 × 10−10 in the same panel. Clones of A. baumannii that were resistant to 22 exhibited a fitness loss in terms of impaired growth. However, a fitness loss was not observed for the other strains (ESI Fig. S10†). Serial passaging experiments demonstrated that a high (256 fold) level of resistance against 22 was generated within 4 and 8 passages against E. coli ATCC25922ΔtolC and P. aeruginosa PA14ΔmexAB. Compared to ciprofloxacin, this level of resistance formation was reached faster in case of E. coli, but more slowly in case of P. aeruginosa (ESI Fig. S11†). Although an optimal resistance profile (FoR's of <10−9 across all strains) is not reached by 22, the data encouraged us to conduct the first in vivo test with a cystobactamid. As the synthesis of 22 was scalable to a > 100 mg range, this analog was selected for subsequent studies. First, a dose of 5 mg kg−1 of 22 was administered intravenously (i.v.) into male CD-1 outbred mice to determine the pharmacokinetic (PK) parameters of the compound. A half-life around 1 h, an area under the curve (AUC) of around 2 μg mL−1 h−1 and a volume of distribution of 3.5 L kg−1 was observed in plasma (Table S8† and Fig. 4a). Thus, the compound was nearly cleared 8 hours after administration. Next, we determined plasma levels using a subcutaneous route of administration, as this frequently results in an initially slower accumulation and elimination. The maximum concentration Cmax after subcutaneous (s.c.) administration of 5 mg kg−1 was determined at 128 ng mL−1, and the tmax at 1.7 hours. The mean residence time was augmented from 1 to 3 hours compared to the intravenous route, and the bioavailability was calculated to be 25% (Table S6† and Fig. 4a). A multiple dosing PK study was conducted to assess plasma as well as urine levels after multiple administration of 22. Initially, 22 was administered at 10 mg kg−1 i.v. for a rapid accumulation, then 22 was dosed three times s.c. at 10 mg kg−1 after 2, 6 and 10 hours. After 24 hours plasma levels dropped again below 100 ng mL−1. In urine, high compound levels were observed without accumulation. Even after 24 hours urine levels of 22 were higher than 1 μg mL−1 (Fig. 4b).
 | ||
| Fig. 4 Pharmacokinetics and pharmacodynamics of 22 (CN-DM-861). (a) Compound levels in plasma and urine after 5 mg kg−1 i.v. and s.c. administration into CD-1 mice (n = 3). (b) Compound levels in plasma and urine after 10 mg kg−1 i.v. at t = 0 h and 10 mg kg−1 s.c. at t = 2, 6 and 10 h into CD-1 mice (n = 3). (c–e) Neutropenic thigh infection model in neutropenic CD-1 mice (n = 6). The E. coli strain ATCC25922 was used that was sensitive to 22 (MIC = 0.02 μg mL−1) and levofloxacin (MIC = 0.04 μg mL−1). Cfu were determined in muscle (c), kidney (d) and lung (e). 8.2 mg kg−1 levofloxacin was administered i.p. at t = 2, 6 and 10 h and served as a positive control. Vehicle-treated animals received vehicle used for 22 at t = 2 h (i.v.) and t = 4,8 and 12 h (s.c.). 22 was administered in two dosing groups: group 1 were administered 7.5 mg kg−1 i.v. at t = 2 h and 20 mg kg−1 s.c. at t = 4, 8 and 12 h. Group 2 were administered 7.5 mg kg−1 i.v. at t = 2 h and 10 mg kg−1 s.c. at t = 4, 8 and 12 h. ***p < 0.001; ****p < 0.0001. | ||
We then performed an in vivo tolerance study in male CD-1 mice. Compound 22 was administered four times intravenously (i.v.) every 6 hours in doses of 5, 10 and 20 mg kg−1, corresponding to total doses of 20, 40, and 80 mg kg−1 per day, respectively. While no adverse effect was observed at 5 and 10 mg kg−1, one animal in the group treated at 20 mg kg−1 showed adverse effects like a moderate loss of activity, appeared ungroomed and pale between the 3rd and 4th dose. These signs remained the same until 24 h. As a result, 22 was administered at maximal single i.v. doses of 12.5 mg kg−1 or less to avoid any toxicity issue.
With these encouraging results from PK and tolerance studies, we probed the antibacterial efficacy of 22 in a pharmacodynamics (PD) study using the neutropenic thigh infection model and the E. coli strain ATCC25922. Mice were treated with either levofloxacin, 22 (high or low dose) or vehicle (consisting of the same formulation as used for 22). Treatment of mice started two hours after infection, and bacterial loads in lung, muscle and kidney were assessed 24 hours after infection. In muscle, the bacterial load was reduced by 5 log-units for both 22 dosing groups as well as for the levofloxacin treated group (Fig. 4c). In kidney we observed a complete bacterial clearance in all dosing groups compared to vehicle-treated animals (Fig. 4d). Even lung tissue was reached by 22, demonstrated by a reduction of the bacterial load by 3 log units (Fig. 4e). Furthermore, we determined the compound levels in muscle, lung and kidney. Whereas we detected around 100 ng mL−1 in muscle for both cystobactamid-treated groups (low and high dosing), only low amounts of 22 were observed in kidney and lung tissue, with slightly higher levels for the high dosing group (Table S9†). The experiments show that 22 does not only reach the primary bacterial reservoir in muscle, but is also active in other organs like kidney or lung, which are affected due to secondary seeding of the bacteria.
As no dose dependency was observed in this first in vivo experiment, we investigated a lower daily dose in a second trial. In the second in vivo experiment, an optimized formulation was used to enable a more conventional dosing scheme with i.v. administrations of 22 at 1, 7, 13, and 19 h post infection in three dose groups (20 mg kg−1 per day, 40 mg kg−1 per day and 50 mg kg−1 per day). The reduction of colony-forming units (cfu) at the lower dose of 20 mg kg−1 per day (−3.97 log units) was smaller than those obtained at 40 and 50 mg kg−1 per day. In line with the first in vivo trial, no dose dependency was observed for the 40 and 50 mg kg−1 per day doses (−5.32 and −4.89 log units, respectively). Compared to the cfu count in untreated mice, 1 h post infection, 22 could not only maintain stasis, but reach a reduction of cfu counts at all total doses of 20, 40 and 50 mg kg−1 per day (Fig. S12†).
In comparison to fluoroquinolones used as comparators in both in vivo experiments, 22 had to be administered more frequently and at higher doses, and the cfu reductions were slightly lower, implying that the in vivo efficacy of 22 is slightly inferior to the gold standard in this model.
In summary, the first in vivo proof of concept for a cystobactamid (or any other PABA-based antibiotic) strongly encourages the further optimization and development of this novel antibiotic class.
Conclusions
Based on the recently established synthetic access to cystobactamids, we developed three different synthetic routes for a first, extensive SAR study at five different positions of the molecule. The antibacterial activity of cystobactamids was substantially improved and/or tuned against several, medically relevant Gram-negative pathogens. While the overall architecture of the cystobactamids could not be altered, substantial structural modifications (e.g. amide replacements, heterocycles) were tolerated or even favorable, illustrating the optimization potential of this antibiotic lead structure. The functional versatility of the scaffold was demonstrated by conjugates that were devoid of an antibiotic activity, but served as bacterial imaging probes. Finally, a first proof of concept in vivo was provided in a neutropenic thigh infection model in mice using the optimized cystobactamid analog 22. Overall, the current study leverages and highlights the great potential of this novel chemical scaffold to yield new antibiotics against resistant bacterial pathogens.Funding
The study was funded by an internal Pre4D grant of the HZI and by a scholarship of the HZI graduate school for G. Testolin. The studies were co-funded by the German Centre for Infection Research (DZIF; Grant no TTU09.710) and the BMBF Project “Wirkstoffentwicklung auf Basis von Naturstoffen zur Bekämpfung von Infektionskrankheiten” (no GGNATM27).Authors contribution
GT designed and synthesized cystobactamids, analyzed the data and wrote the manuscript. CG, TM, AB, AR, CL, MMH, WAME designed and synthesized cystobactamids. JvdH, CG and JK performed AlbD-related experiments. VF performed imaging experiments. KC, HP, JK, AV and JH profiled cystobactamids in antibacterial and gyrase and resistance assays. KR and SS performed in vivo experiments. RWH, RM, MB designed studies, analyzed data and wrote the manuscript.Conflicts of interest
GT, MB, CG, RM, JH, HP, MH, WAME, RWH and AR are co-inventors on patent applications on natural and synthetic cystobactamids.Acknowledgements
The animal studies were conducted in accordance with the recommendations of the European Community (Directive 86/609/EEC, 24 November 1986). All animal procedures for experiments depicted in Fig. 4 were performed in strict accordance with the German regulations of the Society for Laboratory Animal Science (GV-SOLAS) and the European Health Law of the Federation of Laboratory Animal Science Associations (FELASA). Animals were excluded from further analysis if sacrifice was necessary according to the human endpoints established by the ethical board. The experiments were approved by the ethical board of the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg, Germany (LAVES; permit No. and 33.19-42502-04-15/1857). We also thank Ulrike Beutling and Heike Overwin for HRMS and LCMS measurements, and Frank Surup and Christel Kakoschke for NMR measurements. We thank Carine Taillier, Nathalie Semenadisse, and Laurence Conraux for resistance studies. The in vivo tolerance studies and the animal studies depicted in Fig. S12† were performed in strict accordance under the UK Home office License and the European Health Law of the Federation of Laboratory Animal Science Associations (FELASA). The study was approved by the Agenda Resources (Alderley Park) Animal Welfare and Review Board (Project Licence No. PA67E0BAA E5). We thank Janine Schreiber and Jennifer Wolf for technical assistance.References
- M. S. Butler, M. A. Blaskovich and M. A. Cooper, J. Antibiot., 2017, 70, 3–24 CrossRef CAS PubMed.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher and N. Magrini, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef PubMed.
- L. L. Silver, Clin. Microbiol. Rev., 2011, 24, 71–109 CrossRef CAS PubMed.
- R. Tommasi, D. G. Brown, G. K. Walkup, J. I. Manchester and A. A. Miller, Nat. Rev. Drug Discovery, 2015, 14, 529–542 CrossRef CAS PubMed.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299–304 CrossRef CAS PubMed.
- P. Klahn and M. Brönstrup, Nat. Prod. Rep., 2017, 34, 832–885 RSC.
- B. S. Moore, G. T. Carter and M. Brönstrup, Nat. Prod. Rep., 2017, 34, 685–686 RSC.
- S. Baumann, J. Herrmann, R. Raju, H. Steinmetz, K. I. Mohr, S. Hüttel, K. Harmrolfs, M. Stadler and R. Müller, Angew. Chem., Int. Ed., 2014, 53, 14605–14609 CrossRef CAS PubMed.
- Y. J. Kim, H. J. Kim, G. W. Kim, K. Cho, S. Takahashi, H. Koshino and W. G. Kim, J. Nat. Prod., 2016, 79, 2223–2228 CrossRef CAS PubMed.
- S. Cociancich, A. Pesic, D. Petras, S. Uhlmann, J. Kretz, V. Schubert, L. Vieweg, S. Duplan, M. Marguerettaz, J. Noëll, I. Pieretti, M. Hügelland, S. Kemper, A. Mainz, P. Rott, M. Royer and R. D. Süssmuth, Nat. Chem. Biol., 2015, 11, 195–197 CrossRef CAS PubMed.
- S. Hüttel, G. Testolin, J. Herrmann, T. Planke, F. Gille, M. Moreno, M. Stadler, M. Brönstrup, A. Kirschning and R. Müller, Angew. Chem., Int. Ed., 2017, 56, 12760–12764 CrossRef PubMed.
- J. Kretz, D. Kerwat, V. Schubert, S. Grätz, A. Pesic, S. Semsary, S. Cociancich, M. Royer and R. D. Süssmuth, Angew. Chem., Int. Ed., 2015, 54, 1969–1973 CrossRef CAS PubMed.
- D. Trauner, B. Cheng and R. Müller, Angew. Chem., Int. Ed., 2017, 56, 12755–12759 CrossRef PubMed.
- T. Planke, M. Moreno, S. Hüttel, J. Fohrer, F. Gille, M. D. Norris, M. Siebke, L. Wang, R. Müller and A. Kirschning, Org. Lett., 2019, 21, 1359–1363 CrossRef CAS PubMed.
- M. Moeller, M. D. Norris, T. Planke, K. Cirnski, J. Herrmann, R. Müller and A. Kirschning, Org. Lett., 2019, 21, 8369–8372 CrossRef CAS PubMed.
- D. Kerwat, S. Grätz, J. Kretz, M. Seidel, M. Kunert, J. B. Weston and R. D. Süssmuth, ChemMedChem, 2016, 11, 1899–1903 CrossRef CAS PubMed.
- S. Grätz, D. Kerwat, J. Kretz, L. von Eckardstein, S. Semsary, M. Seidel, M. Kunert, J. B. Weston and R. D. Süssmuth, ChemMedChem, 2016, 11, 1499–1502 CrossRef PubMed.
- L. von Eckardstein, D. Petras, T. Dang, S. Cociancich, S. Sabri, S. Grätz, D. Kerwat, M. Seidel, A. Pesic, P. C. Dorrestein, M. Royer, J. B. Weston and R. D. Süssmuth, Chem.–Eur.J., 2017, 23, 15316–15321 CrossRef CAS PubMed.
- L. Rostock, R. Driller, S. Grätz, D. Kerwat, L. von Eckardstein, D. Petras, M. Kunert, C. Alings, F. J. Schmitt, T. Friedrich, M. C. Wahl, B. Loll, A. Mainz and R. D. Süssmuth, Nat. Commun., 2018, 9, 3095 CrossRef PubMed.
- R. O'Shea and H. E. Moser, J. Med. Chem., 2008, 51, 2871–2878 CrossRef PubMed.
- K. Sakamoto, K. Sato, A. Shigenaga, K. Tsuji, S. Tsuda, H. Hibino, Y. Nishiuchi and A. Otaka, J. Org. Chem., 2012, 77, 6948–6958 CrossRef CAS PubMed.
- X. Hu, S. J. Dawson, Y. Nagaoka, A. Tanatani and I. Huc, J. Org. Chem., 2016, 81, 1137–1150 CrossRef CAS PubMed.
- P. Marfey, Carlsberg Res. Commun., 1984, 49, 591–596 CrossRef CAS.
- K. Fujii, Y. Ikai, T. Mayumi, H. Oka, M. Suzuki and K.-I. Harada, Anal. Chem., 1997, 69, 3346–3352 CrossRef CAS.
- K. Fujii, Y. Ikai, H. Oka, M. Suzuki and K.-I. Harada, Anal. Chem., 1997, 69, 5146–5151 CrossRef CAS.
- D. Renneberg and P. B. Dervan, J. Am. Chem. Soc., 2003, 125, 5707–5716 CrossRef CAS PubMed.
- M. F. Brown, M. J. Mitton-Fry, J. T. Arcari, R. Barham, J. Casavant, B. S. Gerstenberger, S. Han, J. R. Hardink, T. M. Harris, T. Hoang, M. D. Huband, M. S. Lall, M. M. Lemmon, C. Li, J. Lin, S. P. McCurdy, E. McElroy, C. McPherson, E. S. Marr, J. P. Mueller, L. Mullins, A. A. Nikitenko, M. C. Noe, J. Penzien, M. S. Plummer, B. P. Schuff, V. Shanmugasundaram, J. T. Starr, J. Sun, A. Tomaras, J. A. Young and R. P. Zaniewski, J. Med. Chem., 2013, 56, 5541–5552 CrossRef CAS PubMed.
- C. J. McPherson, L. M. Aschenbrenner, B. M. Lacey, K. C. Fahnoe, M. M. Lemmon, S. M. Finegan, B. Tadakamalla, J. P. O'Donnell, J. P. Mueller and A. P. Tomaras, Antimicrob. Agents Chemother., 2012, 56, 6334–6342 CrossRef CAS PubMed.
- F. Collin, S. Karkare and A. Maxwell, Appl. Microbiol. Biotechnol., 2011, 92, 479–497 CrossRef CAS PubMed.
- J. W. Trauger, E. E. Baird and P. B. Dervan, Chem. Biol., 1996, 3, 369–377 CrossRef CAS PubMed.
- P. B. Dervan and B. S. Edelson, Curr. Opin. Struct. Biol., 2003, 13, 284–299 CrossRef CAS PubMed.
- C. Kaduk, H. Wenschuh, M. Beyermann, K. Forner, L. A. Carpino and M. Bienert, Lett. Pept. Sci., 1996, 2, 285–288 CrossRef CAS.
- L. A. Carpino, M. Beyermann, H. Wenschuh and M. Bienert, Acc. Chem. Res., 1996, 29, 268–274 CrossRef CAS.
- L. A. Carpino, D. Sadat-Aalaee, H. G. Chao and R. H. DeSelms, J. Am. Chem. Soc., 1990, 112, 9651–9652 CrossRef CAS.
- L. Vieweg, J. Kretz, A. Pesic, D. Kerwat, S. Grätz, M. Royer, S. Cociancich, A. Mainz and R. D. Süssmuth, J. Am. Chem. Soc., 2015, 137, 7608–7611 CrossRef CAS PubMed.
- E. Bonandi, M. S. Christodoulou, G. Fumagalli, D. Perdicchia, G. Rastelli and D. Passarella, Drug Discovery Today, 2017, 22, 1572–1581 CrossRef CAS PubMed.
- M. Moreno, W. A. M. Elgaher, J. Herrmann, N. Schläger, M. M. Hamed, S. Baumann, R. Müller, R. W. Hartmann and A. Kirschning, Synlett, 2015, 26, 1175–1178 CrossRef CAS.
- K. Ferreira, H. Y. Hu, V. Fetz, H. Prochnow, B. Rais, P. P. Müller and M. Brönstrup, Angew. Chem., Int. Ed., 2017, 56, 8272–8276 CrossRef CAS PubMed.
- X. Ning, S. Lee, Z. Wang, D. Kim, B. Stubblefield, E. Gilbert and N. Murthy, Nat. Mater., 2011, 10, 602–607 CrossRef CAS PubMed.
- Y. Kong, H. Yao, H. Ren, S. Subbian, S. L. Cirillo, J. C. Sacchettini, J. Rao and J. D. Cirillo, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12239–12244 CrossRef CAS PubMed.
- F. J. Hernandez, L. Huang, M. E. Olson, K. M. Powers, L. I. Hernandez, D. K. Meyerholz, D. R. Thedens, M. A. Behlke, A. R. Horswill and J. O. McNamara, Nat. Med., 2014, 20, 301–306 CrossRef CAS PubMed.
- M. van Oosten, T. Schäfer, J. A. Gazendam, K. Ohlsen, E. Tsompanidou, M. C. de Goffau, H. J. Harmsen, L. M. Crane, E. Lim, K. P. Francis, L. Cheung, M. Olive, V. Ntziachristos, J. M. van Dijl and G. M. van Dam, Nat. Commun., 2013, 4, 2584 CrossRef PubMed.
- Q. Zhang, Q. Wang, S. Xu, L. Zuo, X. You and H. Y. Hu, Chem. Commun., 2017, 53, 1366–1369 RSC.
- L. Zhang, Y. Liu, Q. Zhang, T. Li, M. Yang, Q. Yao, X. Xie and H. Y. Hu, Anal. Chem., 2018, 90, 1934–1940 CrossRef CAS PubMed.
- X. Wang and N. Murthy, Sci. Transl. Med., 2014, 6, 259fs243 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04769g |
| This journal is © The Royal Society of Chemistry 2020 |