
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective catalytic transformation of lignin with guaiacol as the only liquid product†
Xiaojun
Shen
 ab,
Qinglei
Meng
*a,
Qingqing
Mei
a,
Huizhen
Liu
abc,
Jiang
Yan
ab,
Jinliang
Song
a,
Dongxing
Tan
ab,
Bingfeng
Chen
a,
Zhanrong
Zhang
a,
Guanying
Yang
a and
Buxing
Han
*abcd
ab,
Qinglei
Meng
*a,
Qingqing
Mei
a,
Huizhen
Liu
abc,
Jiang
Yan
ab,
Jinliang
Song
a,
Dongxing
Tan
ab,
Bingfeng
Chen
a,
Zhanrong
Zhang
a,
Guanying
Yang
a and
Buxing
Han
*abcd
aBeijing National Laboratory for Molecular Science, CAS Key Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Centre for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: mengqinglei@iccas.ac.cn; hanbx@iccas.ac.cn
bSchool of Chemistry and Chemical Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cPhysical Science Laboratory, Huairou National Comprehensive Science Centre, Beijing 101407, P. R. China
dShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
First published on 16th December 2019
Abstract
Guaiacol is an important feedstock for producing various high-value chemicals. However, the current production route of guaiacol relies heavily on fossil resources. Using lignin as a cheap and renewable feedstock to selectively produce guaiacol has great potential, but it is a challenge because of its heterogeneity and inert reactivity. Herein, we discovered that La(OTf)3 could catalyze the transformation of lignin with guaiacol as the only liquid product. In the reaction, La(OTf)3 catalyzed the hydrolysis of lignin ether linkages to form alkyl-syringol and alkyl-guaiacol, which further underwent decarbonization and demethoxylation to produce guaiacol with a yield of up to 25.5 wt%, and the remaining residue was solid. In the scale-up experiment, the isolated yield of guaiacol reached up to 21.2 wt%. To our knowledge, this is the first work to produce pure guaiacol selectively from lignin. The bio-guaiacol may be considered as a platform to promote lignin utilization.
Introduction
In the chemical industry, guaiacol is considered as an important raw material for the syntheses of many value-added chemicals, such as spices (vanillin, veratraldehyde and eugenol),1,2 pesticides,3 drugs (eugenol, potassium guaiacolsulfonate, guaifenesin, berberine, and isoprenaline),4–8 a plant growth regulator (2-methoxy-5-nitrophenol sodium salt) and so on. Based on a global market survey, the demand for the above high-value chemicals increases year by year. Typically, annual demand for guaifenesin, vanillin, and eugenol is 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 16000, and 7300 t per year, respectively,5,9–12 all of which may be synthesized from guaiacol. Therefore, the demand for guaiacol is large. However, current production of guaiacol is mainly based on the methylation of catechol which is an expensive downstream chemical from fossil resources, using additional methylation reagents, for example, methanol and methyl chloride (Fig. 1A).13 Lignin is an aromatic abundant renewable source that is rich in guaiacyl (G) units, whether in the woody or herbaceous plants.14–16 Hence, using lignin as a cheap and renewable feedstock to produce guaiacol has considerable potential, and can decrease the dependence on fossil resources for producing this important chemical.15,17,18 Moreover, guaiacol would be used to produce more chemicals, both in category and quantity, if we could obtain cheap and renewable guaiacol from lignin.
000, 16000, and 7300 t per year, respectively,5,9–12 all of which may be synthesized from guaiacol. Therefore, the demand for guaiacol is large. However, current production of guaiacol is mainly based on the methylation of catechol which is an expensive downstream chemical from fossil resources, using additional methylation reagents, for example, methanol and methyl chloride (Fig. 1A).13 Lignin is an aromatic abundant renewable source that is rich in guaiacyl (G) units, whether in the woody or herbaceous plants.14–16 Hence, using lignin as a cheap and renewable feedstock to produce guaiacol has considerable potential, and can decrease the dependence on fossil resources for producing this important chemical.15,17,18 Moreover, guaiacol would be used to produce more chemicals, both in category and quantity, if we could obtain cheap and renewable guaiacol from lignin.
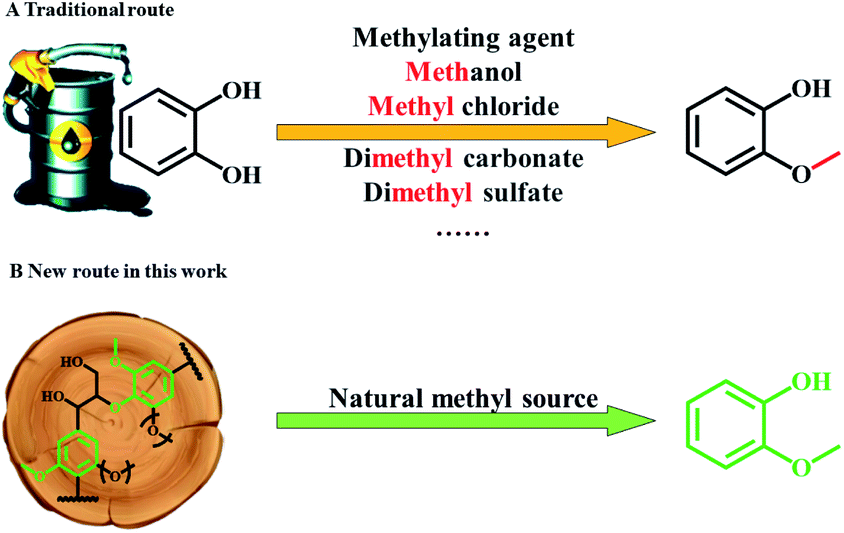 | ||
| Fig. 1 Production route to guaiacol: (A) traditional route, (B) new route in this work. | ||
However, lignin has high heterogeneity and low reactivity, because it is predominately bonded by a series of inert C–O and C–C linkages (accounting for approximately 70% and 30%, respectively).16 Although some technologies, such as hydrogenation,19–21 oxidation,22 hydrolysis,12,23,24 and multiple strategies,25 have been developed to depolymerize lignin into low-molecular-weight lignin monomers, the primary products from these processes are mixtures of compounds (phenols, arenes, aromatic acids, and so on) due to the complex structure of lignin,14,26 and guaiacol can be generated in low yield as one of the monomer products in the mixtures.27–30 The liquid mixtures usually make product separation and purification processes very complicated. It is therefore attractive to selectively transform lignin into a single value-added chemical, such as guaiacol, but this is particularly challenging to achieve efficiently.31
Herein, we discovered that Lewis acid La(OTf)3 could efficiently catalyze the selective transformation of lignin into guaiacol (Fig. 1B). As a catalytic system, La(OTf)3 efficiently catalyzed the hydrolysis of ether linkages in lignin to form alkyl-syringol and alkyl-guaiacol products. Alkyl-guaiacol products further underwent decarbonization to produce guaiacol, while alkyl-syringol was transformed into guaiacol via decarbonization and demethoxylation. The unique feature of this methodology is that guaiacol can be directly produced from the transformation of lignin with high yield, and the methylation process is not required in this route.
Results and discussion
In this work, we firstly screened catalytic systems and optimized the reaction conditions for the transformation of organosolv lignin from Eucalyptus. Initially, lignin transformation was studied with various catalysts in methanol (Table 1, entries 1–8). It was found that no guaiacol was generated without catalyst (Table 1, entry 1). As we know, ether linkages can be efficiently cleaved with Lewis acid catalysts.12,32–37 However, guaiacol could not be effectively obtained from lignin using weak Lewis acid catalysts, such as La(NO3)3, Al(OTf)3 and LaCl3 (Table 1, entries 2–4). Unlike the above acids, the triflate salts with stronger Lewis acidity could effectively catalyze the production of guaiacol from lignin with various yields (Table 1, entries 5–8 and Fig. S1A†). Methane was the only gaseous product (Fig. S1B†). We also analysed the reaction mixtures by HPLC, and the spectra clearly indicated that guaiacol was essentially the sole product (Fig. S1C†), establishing this transformation of lignin, delivering guaiacol as the only liquid product. The results above reveal that catalytic activity of La(OTf)3 was the highest among these catalysts, and the yield of guaiacol could reach 14.9%. As is well known, a larger water exchange rate constant (WERC) value is beneficial to the fast hydrolysis of the ether linkages.38 Among the metal cations, the La3+ ion has one of the largest WERC values.33,37,39 Furthermore, the higher temperature and stronger acidity cooperatively promoted the decarbonization of the side chain to form guaiacol.21 Therefore, La(OTf)3 could effectively hydrolyze the ether linkages and cleave the side chains in lignin.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalytic system | Temperature (°C) | Catalyst (mg) | Guaiacol yield (wt%) b | |
| Catalyst | Solvent/water (mL/mL) | ||||
| a Reaction conditions: 50 mg lignin, 20 mg catalyst, 4 mL methanol, 0.01 mL water if needed, 24 h, 0.1 MPa Ar, and 500 rpm. b Guaiacol yield is calculated based on the weight of lignin. c Without catalyst. d Ethanol/water instead of methanol/water. e Cyclohexane/water instead of methanol/water. f Toluene/water instead of methanol/water. g Substrate is conifer wood enzymatic mild acidolysis lignin from pine (EMAL-p). h Substrate is grass enzymatic mild acidolysis lignin from bamboo (EMAL-b). | |||||
| 1c | — | 4.0/0.00 | 270 | 20 | 0.0 |
| 2 | La(NO3)3 | 4.0/0.00 | 270 | 20 | 0.0 |
| 3 | Al(OTf)3 | 4.0/0.00 | 270 | 20 | 0.6 |
| 4 | LaCl3 | 4.0/0.00 | 270 | 20 | 1.2 |
| 5 | HOTf | 4.0/0.00 | 270 | 20 | 6.6 |
| 6 | Fe(OTf)3 | 4.0/0.00 | 270 | 20 | 7.0 |
| 7 | Yb(OTf)3 | 4.0/0.00 | 270 | 20 | 7.8 |
| 8 | La(OTf)3 | 4.0/0.00 | 270 | 20 | 14.9 |
| 9 | La(OTf)3 | 4.0/0.01 | 270 | 20 | 22.5 |
| 10 | La(OTf)3 | 4.0/0.01 | 250 | 20 | 8.6 |
| 11 | La(OTf)3 | 4.0/0.01 | 255 | 20 | 11.4 |
| 12 | La(OTf)3 | 4.0/0.01 | 260 | 20 | 18.9 |
| 13 | La(OTf)3 | 4.0/0.01 | 265 | 20 | 21.7 |
| 14 | La(OTf)3 | 4.0/0.01 | 275 | 20 | 22.5 |
| 15d | La(OTf)3 | 4.0/0.01 | 270 | 20 | 1.2 |
| 16e | La(OTf)3 | 4.0/0.01 | 270 | 20 | 0 |
| 17f | La(OTf)3 | 4.0/0.01 | 270 | 20 | 0 |
| 18g | La(OTf)3 | 4.0/0.01 | 270 | 20 | 25.5 |
| 19h | La(OTf)3 | 4.0/0.01 | 270 | 20 | 12.6 |
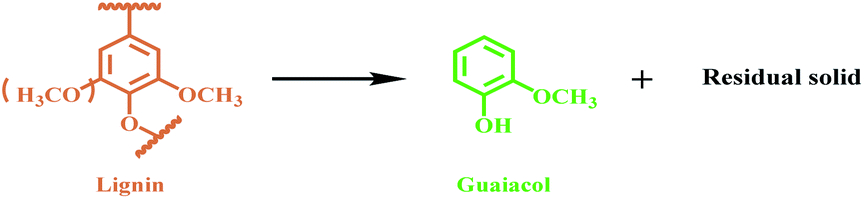
It is known that hydrolysis of ether linkage is much easier than alcoholysis.26,31 We found that water could improve the guaiacol yield (Table S1†), and a 22.5 wt% yield of guaiacol could be obtained (Table 1, entry 9). However, too much water in the catalytic system limited the production of guaiacol (Table S1,† entries 1–5), presumably due to the poor solubility of organosolv lignin in water.40 According to previous studies, metal triflates exhibit excellent stability at high temperature.33,37,39 In this strategy, the reaction temperature had a significant effect on the yield of guaiacol. As shown in Table 1, the yield of guaiacol increased from 8.6 to 22.5 wt% with the temperature increasing from 250 to 270 °C, and then became independent of temperature (Table 1, entries 9–14). In addition, the solvents also influence the yield of guaiacol (Table 1, entries 9 and 15–17). Guaiacol was hardly obtained in ethanol, cyclohexane, or toluene. This is understandable because methanol is generally an effective hydrogen donor.41 With methanol steam reforming, methanol could release hydrogen for catalytic transfer hydrogenation of the lignin intermediate products (for details see Table 2 and Fig. S7†), whereas ethanol, cyclohexane, and toluene are not good hydrogen donors. Furthermore, lignin has very low solubility in cyclohexane and toluene and is therefore not favorable for the transformation of lignin.42,43 Therefore, methanol/water was the optimum solvent. In addition, the yield of guaiacol from lignin increased gradually from 3 to 22.5 wt% with the increase of La(OTf)3 dosage (Table 1, entry 9 and Table S1† entries 6–9). Under the determined optimal conditions, we also investigated the transformation of other lignin samples from conifer wood (EMAL-p) and grass (EMAL-b) lignin using La(OTf)3 as the catalyst, and the yields of guaiacol were 25.5 and 12.6 wt%, respectively (Table 1, entries 18 and 19), further indicating the excellent catalytic performance of La(OTf)3.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Temperature (°C) | Conversion (%) | Yield (%) | |||
| 1 | 2 | 3 | 4 | |||
| a Reaction conditions: 50 mg lignin model compound a, 20 mg La(OTf)3, 0.1 MPa Ar, 500 rpm. 24 h, and methanol/water (4/0.01, v/v). | ||||||
| 1 | 220 | >99 | >99 | 79 | — | — |
| 2 | 230 | >99 | >99 | 95 | 4 | — |
| 3 | 240 | >99 | >99 | 86 | 12 | — |
| 4 | 250 | >99 | >99 | 71 | 17 | 11 |
| 5 | 260 | >99 | >99 | 49 | 18 | 31 |
| 6 | 270 | >99 | >99 | 31 | 11 | 49 |
To gain further information on the transformation of the lignin, lignin structures before and after the reaction were examined using 2D-HSQC NMR.14,44,45 As shown in Fig. 2, Aα (δC/δH 71.4/4.87, purple), Aβ (δC/δH for G units: 84.1/4.32, purple; δC/δH for S units: 87.1/4.11, purple) and Aγ (δC/δH 59.9/3.81, purple) corresponding to the side chain of β-O-4 were almost disappeared. Other units (β–β and β-5) were no longer observed after the catalytic reaction. These results confirmed that the lignin was dissociated in the reactions, and perhaps beyond just the β-ethers. The cross signals correlating with the syringyl (S) (δC/δH 104.0/6.72, red) and oxidized syringyl (S′) (δC/δH 106.3/7.30, red) units were no longer present in the HSQC spectrum after the reaction, and only guaiacyl (G) units (δC/δH 111/6.8, 115/6.7, 119/6.6, green) were observed, suggesting that demethoxylation of syringyl units had occurred to form guaiacyl units.14 This is consistent with the aforementioned analysis of the liquid products. To further confirm the uniqueness of guaiacol in the liquid product, we used deuterated methanol (methanol-d4) as the solvent in the lignin transformation. The 2D-HSQC NMR spectra of the obtained liquid product revealed that only guaiacol could be detected (Figs. S1D and 1E†).
 | ||
| Fig. 2 The 2D-HSQC NMR spectra of the organosolv lignin from Eucalyptus (top) and reaction residue (bottom) transformation (reaction conditions: Table 1, entry 9). | ||
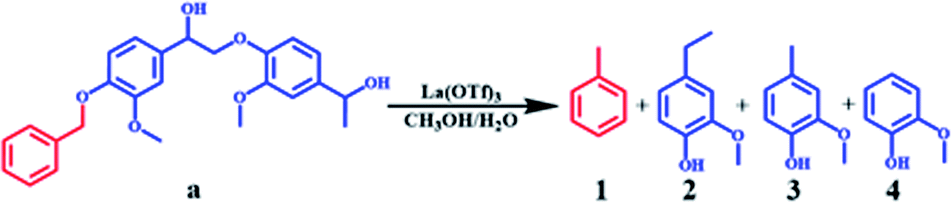
To further study the catalytic mechanism, the conversion of lignin model compound a was performed, and the results are shown in Table 2, Fig. 3 and S2.† As shown in Table 2, La(OTf)3 could efficiently catalyze the conversion of a into guaiacol. The results illustrated that the β-O-4 linkage was broken to generate 4-ethylguaiacol at 220 °C. Moreover, as the reaction temperature increased, the C–C bond in the ethyl group of 4-ethylguaiacol was gradually cleaved to form 4-methylguaiacol and then further transformed into guaiacol,21,25,46 which was futher established by the decarbonization of 4-ethylguaiacol (Fig. 3). As shown in Fig. 3A, 4-methylguaiacol was initially the major liquid product due to the cleavage of the C–C bond between Cα and Cβ in the side chain of 4-ethylguaiacol. With an extension of the reaction time, the remaining methyl chain in 4-methylguaiacol was cleaved, and then guaiacol was formed.21,47 Correspondingly, methane was generated as the main gaseous product (Fig. 3), which confirmed that guaiacol was indeed formed by successive cleavage of the C–C bond in the side chain, as depicted in Fig. 3B. This is different from various reported studies.12,33,34,36 Although Lewis acids have been used as catalysts12,36 or one of the active components of the catalytic system33,34 for transformation of lignin into aromatic chemicals, only a trace of guaiacol was usually generated together with various other compounds, mainly because the reaction was conducted at much lower reaction temperatures in the reported work. In addition, we also used ethanol as a solvent to study the reaction (Fig. S4†). The product distribution was different from that in methanol. 4-Vinylguaiacol was the major product in ethanol for the side-chain elimination reaction, which revealed again that methanol was an excellent organic solvent.40,48,49
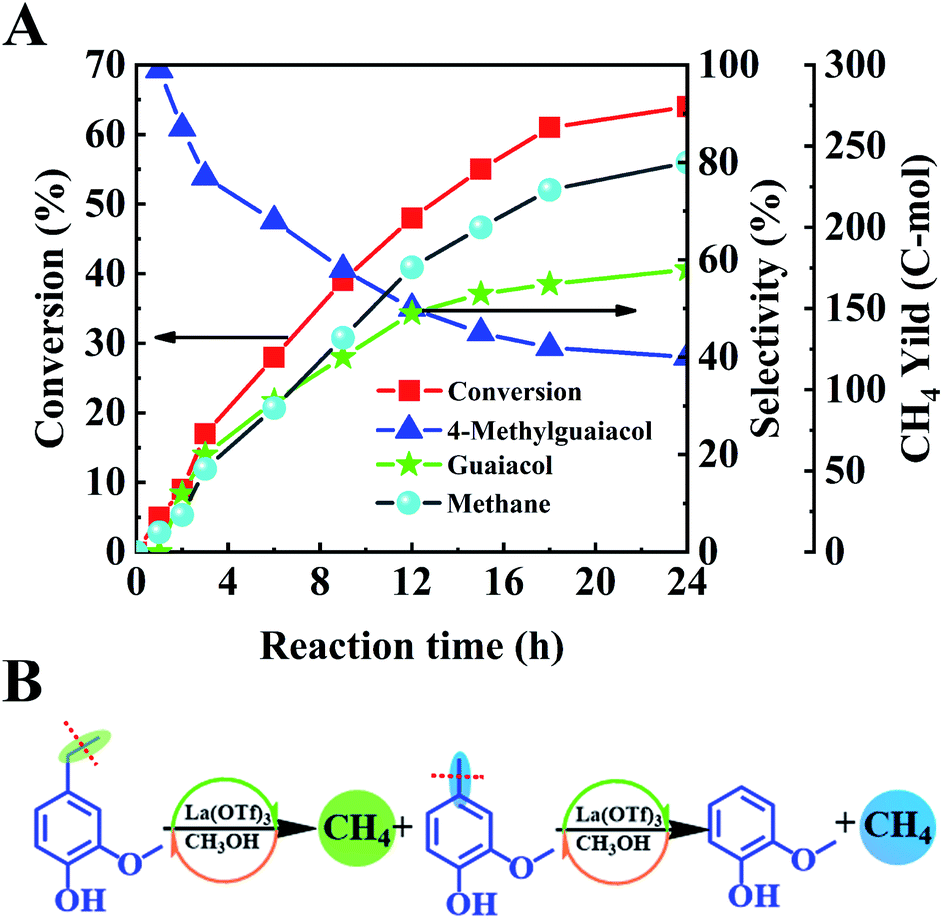 | ||
| Fig. 3 (A) The time-course of the product distributions for the C–C bond cleavage of the ethyl group in 4-ethyl guaiacol with the La(OTf)3 catalyst. Reaction conditions: 4-ethyl guaiacol (50 mg), La(OTf)3 (20 mg), methanol 4 mL, 10 μL water, 270 °C, 0.1 MPa Ar, and stirring at 800 rpm. (B) Reaction pathway for C–C bond cleavage of the ethyl group in 4-ethyl guaiacol. | ||
Although the amount of S units is higher than that of G units in the organosolv lignin, guaiacol was the only monomeric product observed and the corresponding residual solid contained only G units, as determined by NMR. Therefore, the reactions of S units in lignin model compound b (see ESI†) and 2, 6-dimethoxy-4-methylphenol were also investigated (Fig. S5 and S6†). The yield of alkyl-syringol was almost negligible, whereas guaiacol was generated in high yield. The results of these experiments using model compounds are therefore consistent with the lignin transformation results in revealing that even syringyl units really do produce guaiacol (via demethoxylation) under these conditions.
Under optimal conditions, we also performed a scale-up experiment using a larger amount of organosolv lignin. After the separation and purification, a 21.2 wt% yield of isolated guaiacol (0.32 g) could be obtained from 1.50 g lignin (Fig. 4 and S7†), indicating that pure guaiacol with appreciable yield could be produced from real lignin via this strategy.
 | ||
| Fig. 4 Scale-up production of guaiacol from lignin. Reaction conditions: 1.50 g organosolv lignin, 0.6 g La(OTf)3, 120 mL methanol, 0.3 mL water, 270 °C, 0.1 MPa Ar, and 24 h. | ||
Conclusions
In summary, guaiacol can be selectively produced from lignin using Lewis acid La(OTf)3 as a catalyst in methanol/water solvent. The yield of guaiacol reached up to 25.5 wt%, and the remaining residue is solid. As a catalytic system, La(OTf)3 efficiently catalyzed the hydrolysis of ether linkages in lignin to form alkyl-syringol and alkyl-guaiacol products. Alkyl-guaiacol products further underwent decarbonization to produce guaiacol, while alkyl-syringol was transformed into guaiacol via decarbonization and demethoxylation. In the scale-up experiment, 0.32 g of guaiacol can be obtained from 1.50 g lignin, and the residue is solid. This work opens the way to produce pure guaiacol from lignin selectively and bridges plant-based lignin and fossil-based products, affording a green and sustainable strategy that is of great importance for the valorization of lignin.Experimental
Catalytic performance
The reaction was carried out in a Teflon-lined stainless-steel reactor of 20 mL with a magnetic stirrer. In a typical experiment, desired amounts of reactant, catalyst (e.g. La(OTf)3), solvent (e.g. methanol) and H2O (if used) were loaded into the reactor. The reactor was sealed and purged with Ar to remove the air at room temperature. Then the reactor was placed in a furnace at desired temperature controlled by a PID temperature controller (model SX/A-1, Beijing Tianchen Electronic Company), and the stirrer was started. After the reaction, the reactor was placed in ice water and the gas released was collected in a gas bag. The gas sample was analyzed by using a GC (Agilent 4890D) equipped with a TCD detector and a packed column (carbon molecular sieve TDX-01, 1 m in length and 3 mm in diameter) using argon as the carrier gas. A known amount of n-dodecane as the internal standard was added into the reactor. After that, the liquid reaction mixture was centrifuged, and the supernatant liquid was collected for qualitative analysis using a GC-MS (Agilent 5977A, HP-5MS capillary column (30 m × 0.25 mm × 0.25 μm)) and quantitative analysis using a GC (Agilent 7890) equipped with a flame ionization detector (FID) and a HP-5MS capillary column. The yield of guaiacol was calculated from the GC data. The HPLC analysis of the supernatant liquid was conducted on an Agilent LC-20AT liquid chromatograph with a C18 chromatographic column and a UV-Vis detector (275 nm). The mobile phase was acetonitrile with a flow rate of 0.8 mL min−1, and the temperature was kept at 50 °C. The chromatography was run for 60 min. In the case with lignin as the reactant, the residual solid was washed with deionized water, then dried in a vacuum at 60 °C for 48 h over the P2O5 desiccant and characterized by 2D-HSQC NMR in DMSO-d6 (Bruker Advance III 600) according to the reported procedure.14,44 When deuterated methanol (methanol-d4) was used as the solvent, the procedure was the same as that described for the above reaction in methanol. After that, the liquid mixture was centrifuged and the methanol-d4 phase was directly characterized using a 2D-HSQC NMR (Bruker Advance III 400) without adding additional deuterated solvent.The scale-up reaction was performed in a batch reactor of Hastelloy C-276 (500 mL internal volume, Sen Long Instruments Company, Beijing, China). In the reaction, 1.50 g organosolv lignin, 0.6 g La(OTf)3, 120 mL methanol, and 0.3 mL water were added into the reactor. The reactor was sealed and purged with Ar to remove the air at room temperature. Then the reactor was heated to 270 °C under stirring at 800 rpm, and the reaction was maintained for 24 h at 270 °C. After the reaction, the reactor was immediately cooled to room temperature and a gas was released and collected in a gas bag. The liquid product was concentrated in vacuo. The product was purified on a silica-gel column with ethyl acetate–hexane as the solvent (1:9, v/v).
Preparation of organosolv lignin from hardwood
Preparation of organosolv lignin was conducted according to a literature method.27 In the experiment, Eucalyptus powder (50 g) was added to 70% isopropanol/water solution (500 mL). The batch reactor was heated to 220 °C under stirring at 1000 rpm, and the reaction was maintained for 1 h at 220 °C. After the reaction, the batch reactor was immediately cooled to room temperature in the ice water. The reaction mixture was filtered via a Buchner funnel. The filtrate was poured into ten volumes dilute HCl solution with a pH of 2.0 to precipitate the dissolved lignin. After centrifugation, the organosolv lignin was collected, and washed with dilute HCl solution several times. Finally, purified organosolv lignin was obtained by freeze-drying.Preparation of enzymatic mild acidolysis lignins (EMALs) from softwood and grass
The method was similar to that reported in the literature.50,51 The raw sawdust (pine and bamboo) was milled for 1 h at 450 rpm in a planetary ball mill (Fritsch GMBH, Idar-Oberstein, Germany). The ball-milled wood (5.0 g) was then suspended in acetate buffer (0.05 mol L−1, 100 mL, pH 4.8), with the loading of 100 FPU cellulase (Celluclast 1.5 L) and 100 FPU β-glucosidase (Novozyme 188). The reaction mixture was incubated at 50 °C in a rotary shaker (150 rpm) for 48 h. After enzymatic hydrolysis, the residue was collected by centrifugation and washed with acidified water (pH 2) followed by freeze-drying. The residues were then treated with 0.05 M HCl in acidic dioxane–water (85:15, v/v) at 86 °C under a nitrogen atmosphere for 2 h. The resulting mixture was filtered and the filtrate containing lignin was collected. The solid residue was washed with fresh dioxane–water (85:15, v/v) until the filtrate became clear. The filtrate and washings were combined and then neutralized with solid sodium bicarbonate. The neutralized solution was concentrated and finally precipitated in a large quantity of acidified water (10 volumes, pH 2). The precipitated lignin was collected by centrifugation and freeze-dried under high vacuum. To remove the carbohydrate remaining in the lignin, the lignin was dissolved in 90% acetic acid (20 mL), and then the lignin solution was dropped into 10 volumes of acidic water (pH 2) producing a lignin precipitate. The precipitated lignin was washed with acidified water several times and then freeze-dried. EMAL-p and EMAL-b stand for the lignin from pine (conifer wood lignin) and bamboo (grass), respectively.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported financially by National Key Research and Development Program of China (2017YFA0403103), National Natural Science Foundation of China (21703258, 21733011, 21890761, and 21533011), Beijing Municipal Science & Technology Commission (Z181100004218004), and Chinese Academy of Sciences (QYZDY-SSW-SLH013).References
- K. Li and J. W. Frost, J. Am. Chem. Soc., 1998, 120, 10545–10546 CrossRef CAS.
- L. J. Esposito, K. Formanek, G. Kientz, F. Mauger, V. Maureaux, G. Robert and F. Truchet, in Van Nostrand's Encyclopedia of Chemistry, ed. G.D. Considine, John Wiley & Sons, Ltd., 2005, p. 1 Search PubMed.
- L. Yang, W. Zhou, K. Seshan and Y. Li, J. Mol. Catal. A: Chem., 2013, 368–369, 61–65 CAS.
- N. Mahé, B. Nicolaï, M. Barrio, M.-A. Perrin, B. Do, J.-L. Tamarit, R. Céolin and I. B. Rietveld, Cryst. Growth Des., 2013, 13, 3028–3035 CrossRef.
- A. A. Khalil, U. u. Rahman, M. R. Khan, A. Sahar, T. Mehmood and M. Khan, RSC Adv., 2017, 7, 32669–32681 RSC.
- S. L. Bhanawase and G. D. Yadav, Catal. Today, 2017, 291, 213–222 CrossRef CAS.
- D. Liu and W. Zhang, in Pharmaceutical Process Chemistry, ed. T. Shioiri, K. Izawa and T. Konoike, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, p. 273 Search PubMed.
- C. S. P. Sastry, K. E. Rao and U. V. Prasad, Talanta, 1982, 29, 917–920 CrossRef CAS PubMed.
- P. C. Rodrigues Pinto, E. A. Borges da Silva and A. E. Rodrigues, in Biomass Conversion: The Interface of Biotechnology, Chemistry and Materials Science, ed. C. Baskar, S. Baskar and R. S. Dhillon, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, p. 381 Search PubMed.
- Eugenol Market Forecast, Trend Analysis & Competition Tracking – Global Market insights 2017 to 2026, 2018, available electronically at https://www.factmr.com/report/513/eugenol-market Search PubMed.
- Global Guaifenesin (API) Sales Market Report 2018, 2018, available electronically at https://www.marketresearchhub.com/report/global-guaifenesin-api-sales-market-report-2018-report.%20html Search PubMed.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS PubMed.
- R. Bal, M. Tada, T. Sasaki and Y. Iwasawa, Angew. Chem., Int. Ed., 2006, 45, 448–452 CrossRef CAS PubMed.
- S. D. Mansfield, H. Kim, F. Lu and J. Ralph, Nat. Protoc., 2012, 7, 1579 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna and M. Keller, Science, 2014, 344, 1246843 CrossRef PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS PubMed.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- Y. Li, L. Shuai, H. Kim, A. H. Motagamwala, J. K. Mobley, F. Yue, Y. Tobimatsu, D. Havkin-Frenkel, F. Chen, R. A. Dixon, J. S. Luterbacher, J. A. Dumesic and J. Ralph, Sci. Adv. Mater., 2018, 4, eaau2968 Search PubMed.
- L. Dong, L. Lin, X. Han, X. Si, X. Liu, Y. Guo, F. Lu, S. Rudić, S. F. Parker, S. Yang and Y. Wang, Chem, 2019, 5, 1521–1536 CAS.
- W. Lan, J. Behaghel de Bueren and J. Luterbacher, Angew. Chem., Int. Ed., 2019, 131, 2675 CrossRef.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249 CrossRef CAS PubMed.
- M. Wang, M. Liu, H. Li, Z. Zhao, X. Zhang and F. Wang, ACS Catal., 2018, 8, 6837–6843 CrossRef CAS.
- H. Zeng, D. Cao, Z. Qiu and C.-J. Li, Angew. Chem., Int. Ed., 2018, 57, 3752–3757 CrossRef CAS PubMed.
- X.-J. Shen, P.-L. Huang, J.-L. Wen and R.-C. Sun, Fuel Process. Technol., 2017, 167, 491–501 CrossRef CAS.
- J. Gong, A. Imbault and R. Farnood, Appl. Catal., B, 2017, 204, 296–303 CrossRef CAS.
- H. Pińkowska, P. Wolak and A. Złocińska, Chem. Eng. J., 2012, 187, 410–414 CrossRef.
- A. Toledano, L. Serrano and J. Labidi, J. Chem. Technol. Biotechnol., 2012, 87, 1593–1599 CrossRef CAS.
- Q. Mei, H. Liu, X. Shen, Q. Meng, H. Liu, J. Xiang and B. Han, Angew. Chem., Int. Ed., 2017, 56, 14868–14872 CrossRef CAS PubMed.
- A. C. Atesin, N. A. Ray, P. C. Stair and T. J. Marks, J. Am. Chem. Soc., 2012, 134, 14682–14685 CrossRef CAS PubMed.
- R. Jastrzebski, S. Constant, C. S. Lancefield, N. J. Westwood, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2016, 9, 2074–2079 CrossRef CAS PubMed.
- X. Huang, J. Zhu, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ChemSusChem, 2016, 9, 3262–3267 CrossRef CAS.
- P. J. Deuss, C. W. Lahive, C. S. Lancefield, N. J. Westwood, P. C. J. Kamer, K. Barta and J. G. de Vries, ChemSusChem, 2016, 9, 2974–2981 CrossRef CAS PubMed.
- P. J. Deuss, C. S. Lancefield, A. Narani, J. G. de Vries, N. J. Westwood and K. Barta, Green Chem., 2017, 19, 2774–2782 RSC.
- H. Wang, H. Wang, E. Kuhn, M. P. Tucker and B. Yang, ChemSusChem, 2018, 11, 285–291 CrossRef CAS PubMed.
- S. Kobayashi and K. Manabe, Acc. Chem. Res., 2002, 35, 209–217 CrossRef CAS PubMed.
- H.-J. Song, J. Deng, M.-S. Cui, X.-L. Li, X.-X. Liu, R. Zhu, W.-P. Wu and Y. Fu, ChemSusChem, 2015, 8, 4250–4255 CrossRef CAS PubMed.
- T. Renders, S. Van den Bosch, T. Vangeel, T. Ennaert, S.-F. Koelewijn, G. Van den Bossche, C. M. Courtin, W. Schutyser and B. F. Sels, ACS Sustainable Chem. Eng., 2016, 4, 6894–6904 CrossRef CAS.
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640–1647 RSC.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- H. Kim and J. Ralph, Org. Biomol. Chem., 2010, 8, 576–591 RSC.
- H. Kim, J. Ralph and T. Akiyama, BioEnergy Res., 2008, 1, 56–66 CrossRef.
- S. Song, J. Zhang, G. Gözaydın and N. Yan, Angew. Chem., Int. Ed., 2019, 58, 4934 CrossRef CAS PubMed.
- Y. Shao, Q. Xia, L. Dong, X. Liu, X. Han, S. F. Parker, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, S. Yang and Y. Wang, Nat. Commun., 2017, 8, 16104 CrossRef PubMed.
- X. Ouyang, X. Huang, J. Zhu, M. D. Boot and E. J. M. Hensen, ACS Sustainable Chem. Eng., 2019, 7, 13764–13773 CrossRef CAS.
- L.-P. Xiao, S. Wang, H. Li, Z. Li, Z.-J. Shi, L. Xiao, R.-C. Sun, Y. Fang and G. Song, ACS Catal., 2017, 7, 7535–7542 CrossRef CAS.
- J.-L. Wen, S.-N. Sun, T.-Q. Yuan, F. Xu and R.-C. Sun, Bioresour. Technol., 2013, 150, 278–286 CrossRef CAS PubMed.
- H. Li and G. Song, ACS Catal., 2019, 9, 4054 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Scheme S1, Table S1 and Fig. S1–S7. See DOI: 10.1039/c9sc05892c |
| This journal is © The Royal Society of Chemistry 2020 |