
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-induced intramolecular charge transfer in the radical spirocyclisation of indole-tethered ynones†
Hon Eong
Ho
 a,
Angela
Pagano
b,
James A.
Rossi-Ashton
a,
James R.
Donald
a,
Ryan G.
Epton
a,
Jonathan C.
Churchill
a,
Michael J.
James
a,
Peter
O'Brien
a,
Richard J. K.
Taylor
*a and
William P.
Unsworth
*a
a,
Angela
Pagano
b,
James A.
Rossi-Ashton
a,
James R.
Donald
a,
Ryan G.
Epton
a,
Jonathan C.
Churchill
a,
Michael J.
James
a,
Peter
O'Brien
a,
Richard J. K.
Taylor
*a and
William P.
Unsworth
*a
aDepartment of Chemistry, University of York, York, YO10 5DD, UK. E-mail: richard.taylor@york.ac.uk; william.unsworth@york.ac.uk
bDepartment of Chemistry and Industrial Chemistry, University of Genova, via Dodecaneso, 31, 16146 Genova, Italy
First published on 13th December 2019
Abstract
Indole-tethered ynones form an intramolecular electron donor–acceptor complex that can undergo visible-light-induced charge transfer to promote thiyl radical generation from thiols. This initiates a novel radical chain sequence, based on dearomatising spirocyclisation with concomitant C–S bond formation. Sulfur-containing spirocycles are formed in high yields using this simple and mild synthetic protocol, in which neither transition metal catalysts nor photocatalysts are required. The proposed mechanism is supported by various mechanistic studies, and the unusual radical initiation mode represents only the second report of the use of an intramolecular electron donor–acceptor complex in synthesis.
Introduction
The use of visible-light-mediated photochemistry in synthesis has grown enormously in recent years,1,2 triggered by dramatic progress in the fields of photoredox catalysis,3 energy transfer4 and atom transfer processes.5 This has led to a renaissance in the use of single electron transfer (SET) processes in chemical synthesis, including renewed interest in the study of charge transfer in electron donor–acceptor (EDA) complexes.The phenomenon of charge transfer was first postulated in the 1950s,6,7 but received relatively little attention in synthetic chemistry8 until a surge in interest in the last decade,9 with an illustrative example recently reported by Glorius and co-workers summarised in Scheme 1a.10 In this study, it was shown that the coupling of indoles 1 and pyridinium salt acceptors 2 can be performed in the absence of a transition metal catalyst or photocatalyst. Key to this process is the formation of an intermolecular EDA complex A between an indole (the donor) and a pyridinium salt (the acceptor); the EDA complex can then absorb visible light to promote charge transfer to form radical 4, that can then fragment to give electrophilic radical 5 and couple with indole 1 to afford C-2 functionalised indole 3via a radical chain process.
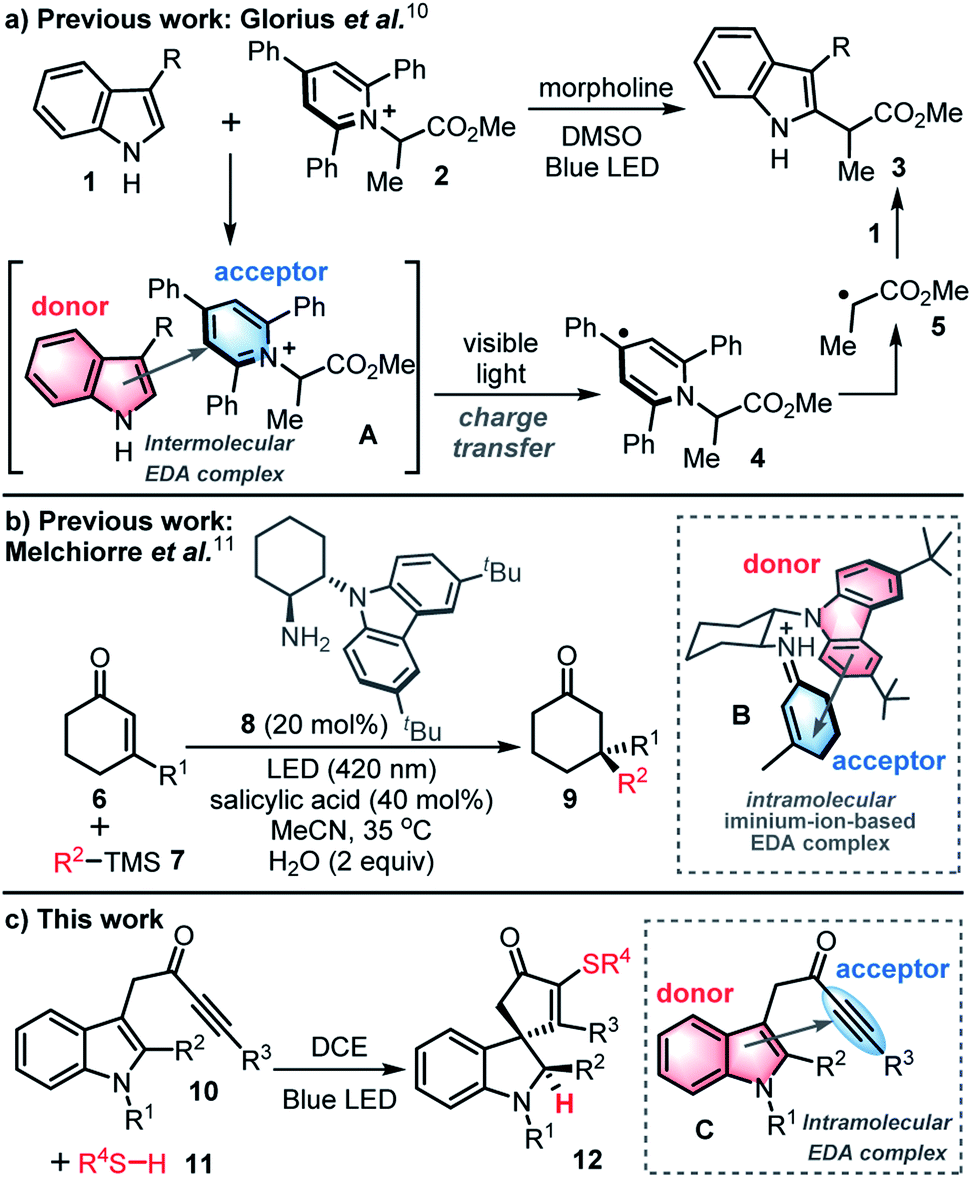 | ||
| Scheme 1 EDA complexes in synthetic chemistry (a, b = previous work, c = this work). DCE = 1,2-dichloroethane. LED = light emitting diode. | ||
This report and others,9,10 highlight the value of intermolecular EDA-mediated charge transfer for the construction of challenging chemical bonds. However, until a 2018 report by Melchiorre and co-workers,11,12 there were no reports of the use of photon-absorbing intramolecular EDA complexes being used in synthesis. In this seminal study, an enantioselective radical conjugate addition reaction is described that relies on visible-light-excitation of a chiral iminium-ion based intramolecular EDA complex B, itself formed in situ from a ketone 6 and organocatalyst 8 (Scheme 1b).11
Herein, a new synthetic method based on intramolecular EDA complexation and charge transfer is reported for only the second time. Thus, a novel radical dearomatising spirocyclisation13 of indole-tethered ynones 10 (ref. 14) and thiols 11 is described for the formation of spirocyclic indolines 12 with concomitant C–S bond formation (Scheme 1c).15 The reactions are catalyst-free and proceed via a thiyl radical-based chain process, that is thought to be self-initiated by the ynone starting material 10, through visible-light-induced charge transfer of an intramolecular EDA complex C. A wide range of sulfur-containing spirocycles 12 have been prepared using this mild, high yielding synthetic procedure. This unusual photocatalyst-free method to initiate radical chemistry was discovered by serendipity, and could easily have gone unnoticed, if not for the recent surge in interest in the study of charge transfer in electron donor–acceptor (EDA) complexes.
Results and discussion
The original aim of this project was to develop a radical spirocyclisation protocol based on more well-established methods for radical generation. We postulated that indole-tethered ynones 13 would react with electrophilic radicals via regioselective addition to the alkyne group (13 → 14), before cyclising on to the indole at its 3-position to form a spirocyclic radical intermediate (14 → 15). Radical 15 could then go on to form either spirocyclic indoline 16via hydrogen atom transfer (HAT), or spirocyclic indolenine 18via single electron oxidation followed by proton loss (Scheme 2). Oxidation of 16 to 18 in the presence of molecular oxygen, or other oxidants, was also predicted to be viable. Confidence in this general idea was raised by reports of radical-based dearomative processes involving phenol- and anisole-tethered alkynes.16,17 These studies confirm that electrophilic radicals can react with ynones with the desired regioselectivity, and that the so-formed radical species can go on to react with tethered aromatics; however, to the best of our knowledge, such a strategy had not been applied to indole-tethered alkynes prior to this report.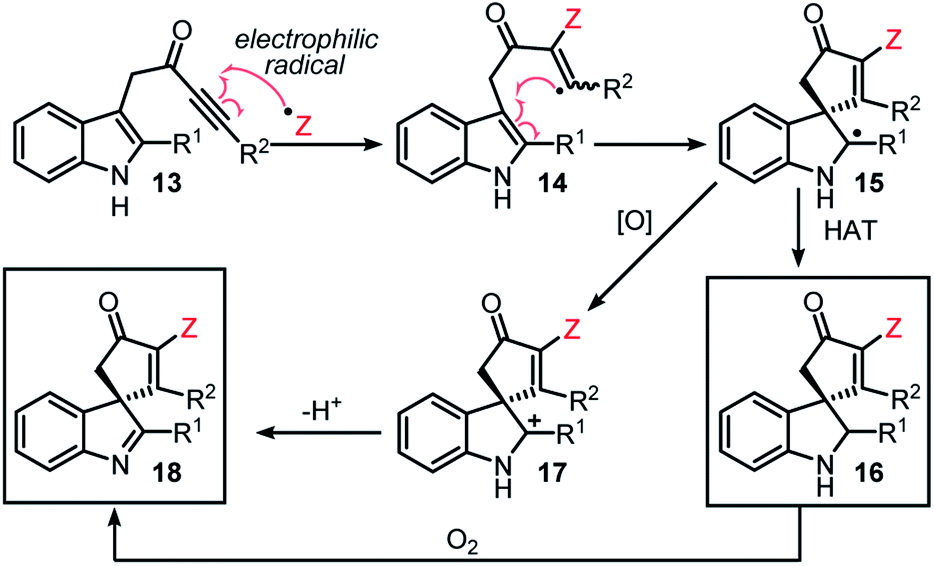 | ||
| Scheme 2 Planned reaction courses. | ||
This study began by examining the reaction of ynone 13a with a thiyl radical generated from thiophenol in situ. Thiyl radicals are versatile reactive intermediates in synthetic radical chemistry18 and we reasoned that the derived products could be relevant in drug discovery, in view of the prevalence of vinyl sulfides in bioactive compounds.19 A photoredox catalysis approach originally was chosen to generate the thiyl radical,20 which led to the development of the conditions summarised in Scheme 3 (see ESI† for full optimisation). Thus, the combination of thiophenol, catalytic Ru(bpy)3(PF6)2 and irradiation with a blue LED lamp (λmax = 455 nm, 60 W) at rt under air, enabled the conversion of indole-tethered ynone 13a into spirocyclic indolenine2118a in reasonable yield via an overall oxidative dearomative process (Scheme 3, a). Product 18a was synthesised along with indoline 16a as minor side-product, with 16a believed to be an intermediate on the route to indolenine 18a.22 Control experiments revealed that oxygen is required for the efficient formation of 18a (indoline 16a is the major product in the absence of oxygen, Scheme 3, d) and more intriguingly, a mixture of both products 18a and 16a is formed even in the absence of any photocatalyst (Scheme 3, e). In the absence of light, no reaction occurs (Scheme 3, f). This indolenine-forming reaction was also found to work on other ynone starting materials (5 examples, see ESI† page S8 for details).
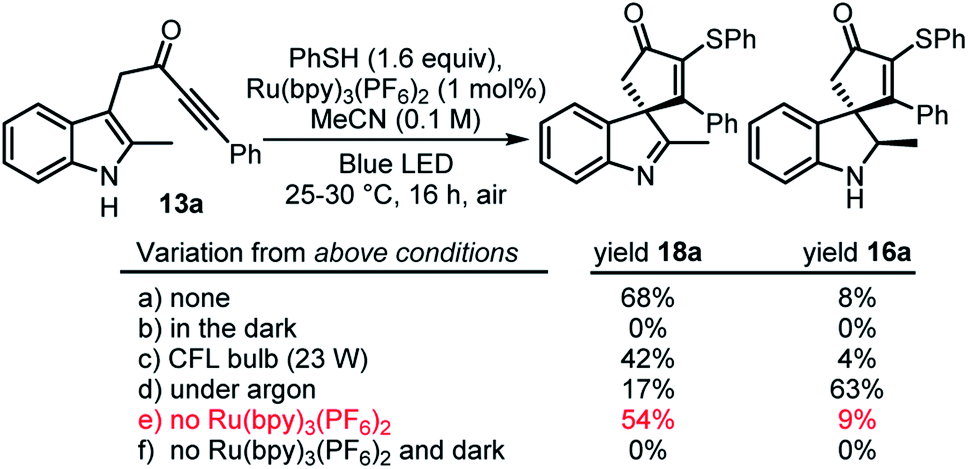 | ||
| Scheme 3 Control reactions; all yields are based on comparison to an internal standard in the 1H NMR spectrum of the unpurified reaction mixture except for ‘a’ which is an isolated yield. CFL = compact fluorescent lamp. | ||
The unexpected discovery that spirocyclisation can be achieved without Ru(bpy)3(PF6)2 was intriguing, both from a mechanistic standpoint, and in view of the clear practical and environmental benefits of avoiding metal-based photocatalysts. We therefore decided to explore this process in more detail and started by establishing whether the photocatalyst-free reaction conditions could be applied to other substrates. Additional optimisation was performed at this point (see ESI†), and based on this, the decision was made to prioritise the synthesis of indoline products 16/19 (via overall redox neutral reactions, c.f.Scheme 3, d) rather than indolenines 18 (overall oxidative processes, c.f.Scheme 3, a).23 Thus, the reactions were performed under argon rather than air; another change to the conditions was to switch the solvent from acetonitrile to 1,2-dichloroethane (see ESI† for further optimisation details).
The optimised conditions were then tested across a range of indole-tethered ynones and thiols, and the results summarised in Scheme 4 confirm that the reaction is broad in scope. Changes to the substituent on the indole nitrogen (NH and NMe), the indole 2-position and the ynone terminus (R3) were all tolerated (16a–n, 15–99%), with bulky TIPS-substituted ynone 13o (to form 16o) the only unsuccessful example. In cases where the indole C-2 position is substituted (16a–f) the products were isolated as the single diastereoisomer shown, with the assignment of relative stereochemistry based on X-ray crystallographic data for 16a.24,25
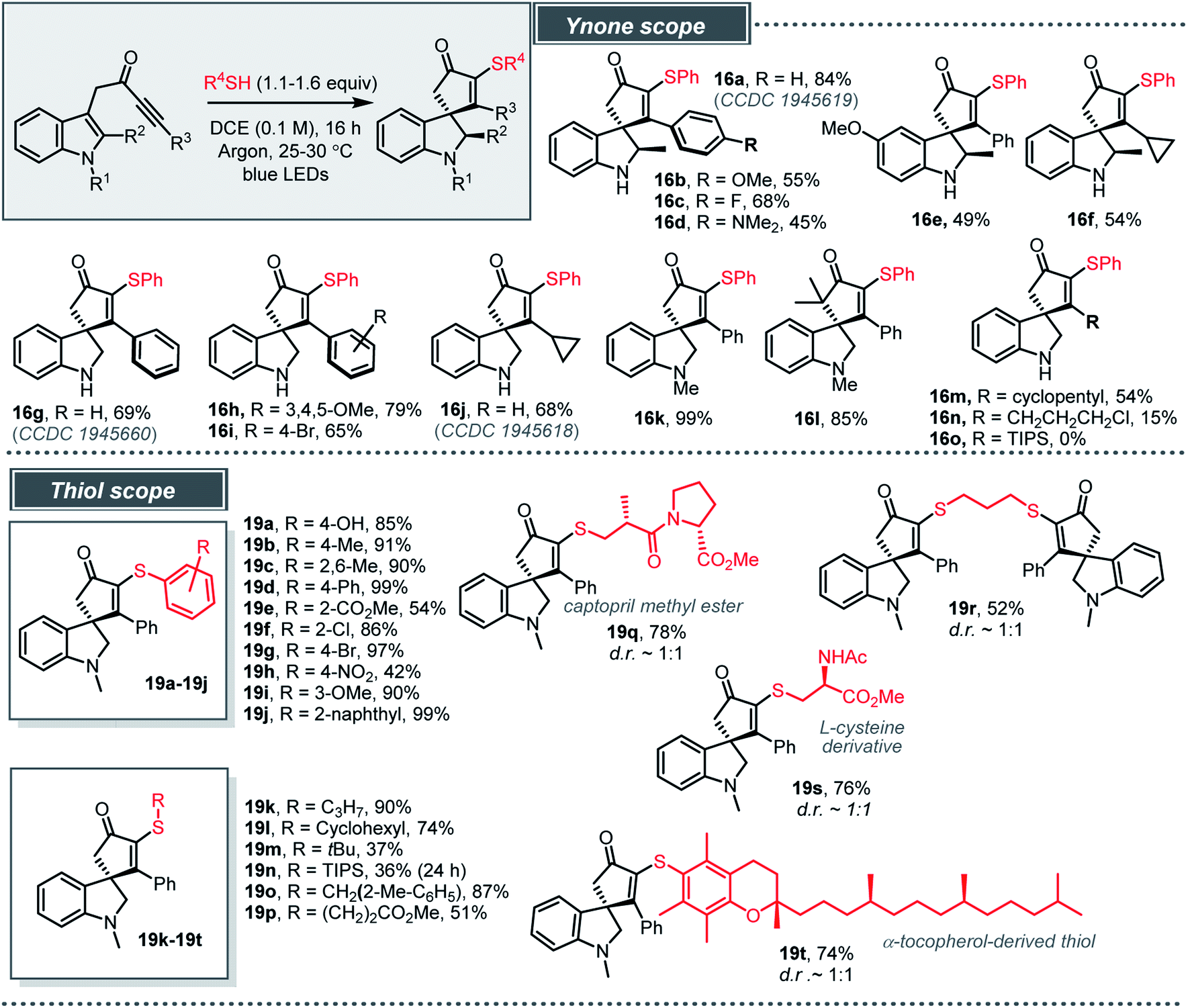 | ||
| Scheme 4 Light induced charge-transfer radical spirocyclisation. Isolated yields following column chromatography are given. | ||
The reactions also work well with a range of thiols (20 examples, 19a–19t, 36–99%). Various electronically diverse, substituted arylthiols were well-tolerated (19a–19j), with the more electron-rich examples typically the highest yielding. We were also pleased to observe that aliphatic thiols are compatible with the standard procedure (19k–19p); the S–H bond dissociation energy (BDE) for alkyl thiols (∼87 kcal mol−1) is usually higher than related aryl thiols (72–82 kcal mol−1), which can adversely affect their reactivity in radical reactions.18 More complex thiols have also been shown to work well using the standard procedure, including 1,3-propanedithiol (to make dimeric product 19r), a protected cysteine derivative and a peptide-like thiol (to make 19q and 19s) and an α-tocopherol-derived thiol (to make 19t).
Having established the synthetic method, attention then turned to understanding how the reactions operate in the absence of photocatalyst. We reasoned that visible-light activation of one of the starting materials must play a key role. Based on precedent, several possibilities could be envisaged:26,27 (1) light-induced homolytic cleavage of the thiol S–H bond to generate a thiyl radical would account for the observed reactivity, as this could start a radical chain reaction, as outlined earlier in Scheme 2; (2) similar reactivity could also originate from homolytic S–S bond cleavage of trace disulfides present in the thiol reagent; (3) photoexcitation of the alkyne to its triplet state could form an open-shell species theoretically capable of initiating radical chemistry.
Although we considered all of these scenarios to be unlikely under the influence of blue light (based on the energy typically needed to homolyse S–H/S–S bonds or to electronically excite alkynes),18,27 control experiments were nonetheless designed to examine these possibilities. The experiments were based on the hydrothiolation of simple alkynes; thus, each of alkynes 20 and 22 were reacted with n-propane thiol under blue LED irradiation at RT, but no conversion into vinyl sulfides 21 and 23 was observed, with the alkynes fully recovered in each case (Schemes 5a and 5b). The lack of hydrothiolation in these experiments supports the idea that the initiation mechanisms suggested above are invalid; thiyl radicals seem not to be formed directly from the thiol reagent (or trace amounts of disulfides) in the presence of either of these alkynes, and the lack of reactivity observed with ynone starting material 20, appears to rule out simple triplet excitation of the ynone functionality under blue light radiation. Instead, these results suggest that the indole-tethered ynone moiety 13 itself is critical to the observed reactivity. Indeed, compelling evidence that supports the involvement of the ynone in initiating thiyl radical chemistry was obtained when alkynes 20 and 22 were again reacted with n-propane thiol, but this time with the addition of sub-stoichiometric (20 mol%) ynone 13k; under these conditions, hydrothiolation products 21 and 23 were obtained in 54% and 80% yields, respectively.
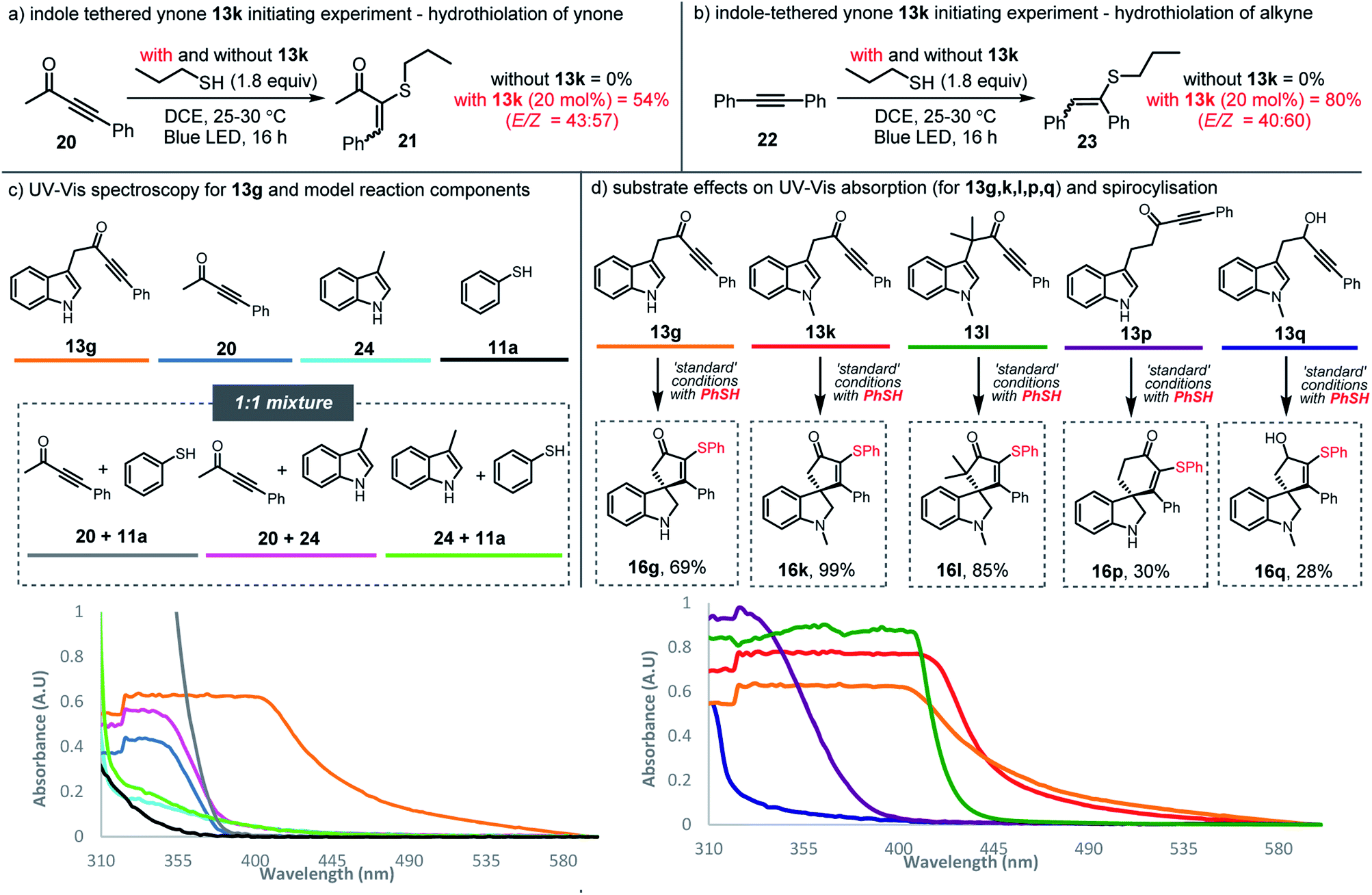 | ||
| Scheme 5 (a) Hydrothiolation of ynone 20; (b) Hydrothiolation of alkyne 22; (c) UV-Vis spectroscopy studies of indole-tethered ynone 13g compared with model reaction components; (d) UV-Vis spectroscopy of indole-tethered ynones (13g, 13k, 13l, 13p) and propargylic alcohol 13q. All UV-Vis experiments were performed in anhydrous 1,2-dichloroethane (0.02 M). | ||
Normally, neither ynones nor 3-substituted indoles would be expected to absorb visible light wavelength photons. However, the ynones 13 used in this study tend to be yellow in colour, and indeed, ynone 13g was found to absorb relatively strongly at around 455 nm (the λmax of the light source) when analysed using UV-Vis spectroscopy (orange line, Scheme 5c; for the emission spectra of 13g see the ESI†). In contrast, model ynone 20 (blue line), 3-substituted indole 24 (cyan line), and thiophenol 11a (black line) displayed little/no absorption in the same region. Equimolar mixtures of these compounds (20 + 11a in grey, 20 + 24 in pink and 24 + 11a in green) did absord in the visible region when measured at 0.02 M, although interestingly, when mixed at much higher concentration (>0.5 M) a significant bathochromic shift was observed for a mixture of ynone 20 and indole 24, indicating that the intermolecular interaction of these components can influence their absorption properties, albeit at concentrations well above those used in the synthetic reactions (see ESI, Scheme S3†). The addition of thiol 11a does not appear to influence the absorbance, with near-identical UV-Vis spectra obtained for both ynone 13k and a mixture of 20 and 24, with and without the inclusion of 11a (see ESI Scheme S4†).
The ability of the indole ynone starting materials 13 to absorb visible light around 455 nm appears to correlate well with the success of the radical cascade processes, as illustrated by the data presented in Scheme 5d. For example, alkynes 13p and 13q, showed low absorption at 455 nm (purple and blue lines respectively) and both the conversion and yields for these reactions were much lower than those for the standard substrates (c.f.Scheme 4).28 In contrast, ynones 13g, 13k and 13l, which all reacted efficiently to form 16g, 16k and 16l with full conversion and in good yields, show a clear red-shift into the visible region (orange, red and green lines respectively).29
We postulate that through-space interactions explain the enhanced visible light absorption of the reactive ynone systems, via formation of an intramolecular EDA complex of the form C (Scheme 6a) that enables the ynone to absorb relatively long wavelength visible light and initiate thiyl radical chemistry. A through bond process (i.e. involving conjugation via enolate formation)30 was also considered as a possible explanation for the red shift of typical ynone substrates, but was ruled out on the basis of the UV-Vis data and successful reaction of non-enolisable ynone 13l. The formation of an intramolecular EDA complex is also supported by time-dependent density functional theory (TDDFT) calculations performed on ynone 13k, which predicted a peak at 441 nm with an oscillator strength of 0.050 (Scheme 6b). This peak is composed entirely of a charge transfer excitation between the π orbitals of the indole HOMO and ynone LUMO (Scheme 6b).
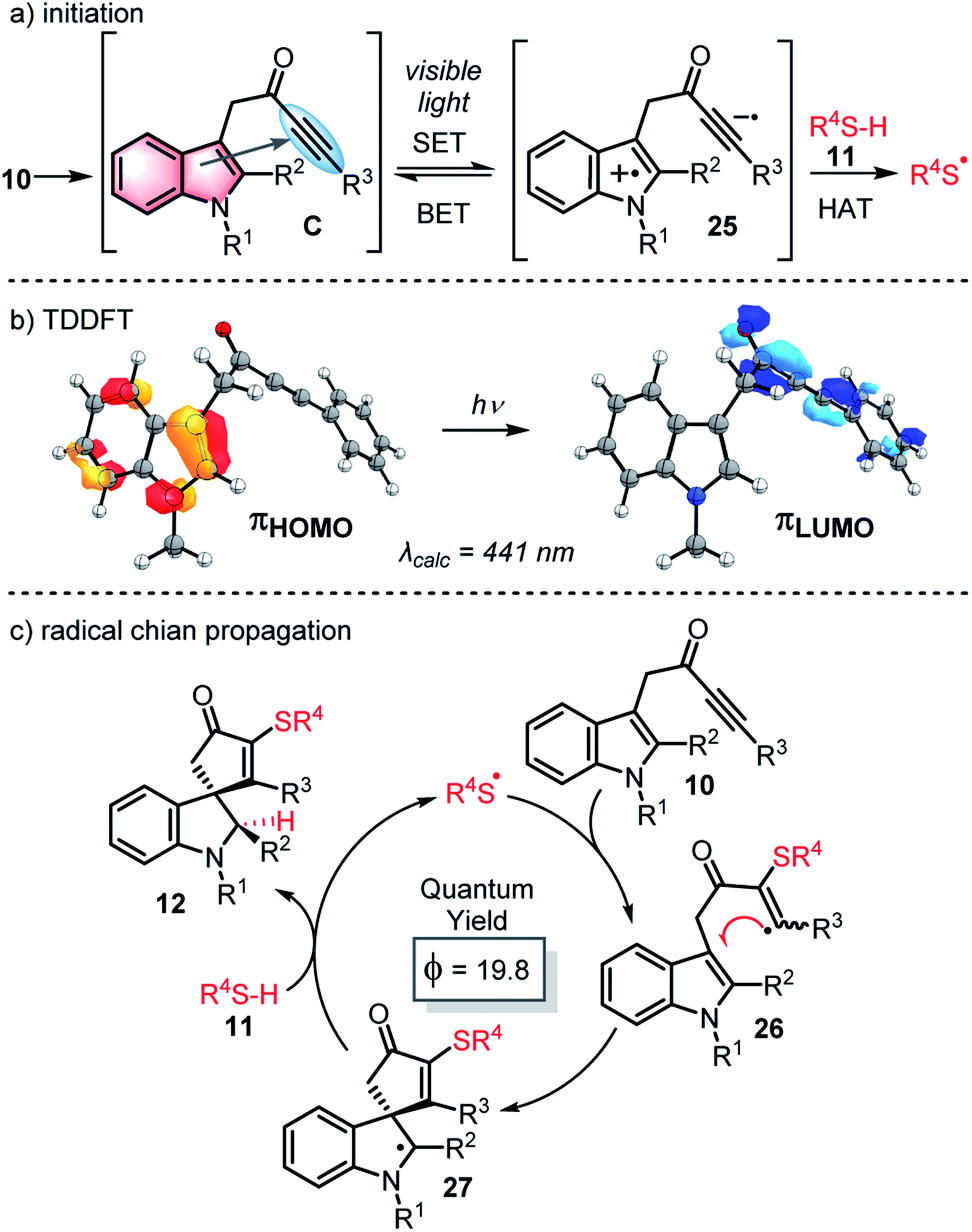 | ||
| Scheme 6 (a) Proposed mechanism for initiation; (b) TDDFT calculations; (c) proposed propagation cycle. | ||
Thus, a mechanism is proposed in which the formation of an EDA complex C is followed by visible light absorption to form a photoexcited state, loosely represented as charge transfer complex 25. This species may simply relax to reform EDA complex Cvia back electron transfer, or alternatively, the open shell excited state 25 could abstract a hydrogen atom from the thiol 11, thus generating the thiyl radical needed to start a radical cascade (Scheme 6a).31 At this point, a more typical radical chain process can operate (Scheme 6c), which likely proceeds by the addition of thiyl radical to the ynone (10 → 26), spirocyclisation (26 → 27) and hydrogen atom abstraction from thiol 11 (27 → 12), thus enabling chain propagation. Quantum yield measurements (ϕ = 19.8)32 support operation through a chain process.33,34
Conclusions
In summary, a new dearomative method for the synthesis of sulfur-containing spirocyclic indolines is described, based on the reaction of indole-tethered ynones with thiyl radicals generated in situ from thiols. The reactions are promoted by visible light, operate at RT under mild reaction conditions and need neither a transition metal catalyst nor added photocatalyst to proceed efficiently across a wide range of substrates. The reaction is thought to be self-initiated35 with visible-light-mediated photoexcitation of an intramolecular EDA complex formed between the indole and ynone moieties in the starting material leading to the formation an open shell excited charge-transfer complex, capable of abstracting a hydrogen atom from the thiol and initiating radical chain propagation.To the best of our knowledge, this is only the second report that details the use of intramolecular EDA complexes in synthesis. This rare radical activation mode was uncovered entirely by serendipity,36 and this is a feature of the discovery that we are keen to highlight, as we believe that intramolecular EDA complexes likely play key roles in other synthetic methods, but can go (or have gone) unnoticed.36d As awareness of the value of charge transfer processes in synthetic chemistry grows, we believe that many important new methods of this type will be discovered, both through design and by serendipity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the EPSRC (H. E. H. EP/N035119/1), the University of York, the Leverhulme Trust (for Early Career Fellowships, ECF-2015-013, W. P. U. and ECF-2019-135, M. J. J.) and Erasmus+ (for Angela Pagano) for financial support. We are also grateful for the provision of an Eleanor Dodson Fellowship (to W. P. U.) by the Department of Chemistry, University of York. Dr Adrian C. Whitwood and Rachel Parker are thanked for X-ray crystallography. We would also like to thank Prof. Giovanni Petrillo (University of Genova) Dr Alison Parkin and Prof. Neil Hunt (both University of York) for helpful discussions.Notes and references
- (a) H. Trommsdorff, Ann. Pharm., 1834, 11, 190 CrossRef; (b) H. D. Roth, Angew. Chem., Int. Ed., 1989, 28, 1193 CrossRef.
- For selected reviews of visible light photocatalysis, see: (a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (b) D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734 CrossRef CAS PubMed; (c) M. D. Kärkäs, J. A. Porco Jr and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683 CrossRef PubMed; (d) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850 CrossRef CAS PubMed; (e) T. Hering, A. U. Meyer and B. König, J. Org. Chem., 2016, 81, 6927 CrossRef CAS PubMed; (f) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034 CrossRef CAS PubMed.
- For selected reviews of photoredox catalysis, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) J. D. Nguyen, E. M. D'Amato, J. M. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed; (c) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed; (d) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed.
- For information on visible-light-induced organic photochemical reactions that proceed via energy-transfer pathways, see: Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586 CrossRef CAS PubMed and references cited therein.
- T. Courant and G. Masson, J. Org. Chem., 2016, 81, 6945 CrossRef CAS PubMed.
- R. S. Mulliken, J. Am. Chem. Soc., 1952, 74, 811 CrossRef CAS.
- For photophysical studies see: S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641 CrossRef CAS PubMed and references cited therein.
- For early synthetic examples, see: (a) M. A. Fox, J. Younathan and G. E. Fryxell, J. Org. Chem., 1983, 48, 3109 CrossRef CAS; (b) S. Sankararaman, W. A. Haney and J. K. Kochi, J. Am. Chem. Soc., 1987, 109, 7824 CrossRef CAS; (c) T. Gotoh, A. B. Padias and J. H. K. Hall, J. Am. Chem. Soc., 1991, 113, 1308 CrossRef CAS; (d) R. Rathore and J. K. Kochi, Adv. Phys. Org. Chem., 2000, 35, 193 CrossRef.
- For a useful review of this area, see: (a) C. G. S. Lima, T. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389 CrossRef CAS. For selected recent research articles, see: (b) M. Tobisu, T. Furukawa and N. Chatani, Chem. Lett., 2013, 42, 1203 CrossRef CAS; (c) J. Davies, S. G. Booth, S. Essafi, R. A. W. Dryfe and D. Leonori, Angew. Chem., Int. Ed., 2015, 54, 14017 CrossRef CAS PubMed; (d) Y. Cheng, X. Yuan, J. Ma and S. Yu, Chem.–Eur. J., 2015, 21, 8355 CrossRef CAS PubMed; (e) M. L. Spell, K. Deveaux, C. G. Bresnahan, B. L. Bernard, W. Sheffield, R. Kumar and J. R. Ragains, Angew. Chem., Int. Ed., 2016, 55, 6515 CrossRef CAS PubMed; (f) V. Quint, F. Morlet-Savary, J.-F. Lohier, J. Lalevée, A.-C. Gaumont and S. Lakhdar, J. Am. Chem. Soc., 2016, 138, 7436 CrossRef CAS PubMed; (g) A. Böhm, T. Bach and T. Chem, Chem.–Eur. J., 2016, 22, 15921 CrossRef PubMed; (h) L. Marzo, S. Wang and B. König, Org. Lett., 2017, 19, 5976 CrossRef CAS PubMed; (i) J. Zhang, Y. Li, R. Xu and Y. Chen, Angew. Chem., Int. Ed., 2017, 56, 12619 CrossRef CAS PubMed; (j) B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616 CrossRef CAS PubMed; (k) H.-H. Zhang and S. Yu, Org. Lett., 2019, 21, 3711 CrossRef CAS PubMed; (l) Q.-Q. Ge, J.-S. Qian and J. Xu, J. Org. Chem., 2019, 84, 8691 CrossRef CAS PubMed; (m) K. Liang, N. Li, Y. Zhang, T. Li and C. Xia, Chem. Sci., 2019, 10, 3049 RSC; (n) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429 CrossRef CAS PubMed; (o) Y. Liu, X.-L. Chen, K. Sun, X.-Y. Li, F.-L. Zeng, X.-C. Liu, L.-B. Qu, Y.-F. Zhao and B. Yu, Org. Lett., 2019, 21, 4019 CrossRef CAS PubMed; (p) J. J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700 CrossRef CAS PubMed.
- M. J. James, F. Strieth-Kalthoff, F. Sandfort, F. J. R. Klauck, F. Wagener and F. Glorius, Chem.–Eur. J., 2019, 25, 8240 CrossRef CAS PubMed.
- Z.-Y. Cao, T. Ghosh and P. Melchiorre, Nat. Comm., 2018, 9, 3274 CrossRef PubMed.
- For important contributions from the same group that involve intermolecular EDA complexes, see: (a) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750 CrossRef CAS PubMed; (b) E. Arceo, A. Bahamonde, G. Bergonzini and P. Melchiorre, Chem. Sci., 2014, 5, 2438 RSC; (c) M. Nappi, G. Bergonzini and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 4921 CrossRef CAS PubMed; (d) S. R. Kandukuri, A. Bahamonde, I. Chatterjee, I. D. Jurberg, E. C. Escudero-Adán and P. Melchiorre, Angew. Chem., Int. Ed., 2015, 54, 1485 CrossRef CAS PubMed; (e) Ł. Woźniak, J. J. Murphy and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 5678 CrossRef PubMed; (f) A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019 CrossRef CAS PubMed.
- For recent reports of radical dearomatising reactions of indoles, see: (a) J. Wu, Y. Dou, R. Guillot, C. Kouklovsky and G. Vincent, J. Am. Chem. Soc., 2019, 141, 2832 CrossRef CAS PubMed; (b) J. Wu, R. K. Nandi, R. Guillot, C. Kouklovsky and G. Vincent, Org. Lett., 2018, 20, 1845 CrossRef CAS PubMed; (c) J.-H. Ye, L. Zhu, S.-S. Yan, M. Miao, X.-C. Zhang, W.-J. Zhou, J. Li, Y. Lan and D.-G. Yu, ACS Catal., 2017, 7, 8324 CrossRef CAS.
- For transition metal (non-radical) transformations of indole-tethered ynones of this type, see: (a) M. J. James, J. Cuthbertson, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Angew. Chem., Int. Ed., 2015, 54, 7640 CrossRef CAS PubMed; (b) M. J. James, R. E. Clubley, K. Y. Palate, T. J. Procter, A. C. Wyton, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Org. Lett., 2015, 17, 4372 CrossRef CAS PubMed; (c) J. T. R. Liddon, M. J. James, A. K. Clarke, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Chem.–Eur. J., 2016, 22, 8777 CrossRef CAS PubMed; (d) A. K. Clarke, M. J. James, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Angew. Chem., Int. Ed., 2016, 55, 13798 CrossRef CAS PubMed; (e) J. T. R. Liddon, A. K. Clarke, R. J. K. Taylor and W. P. Unsworth, Org. Lett., 2016, 18, 6328 CrossRef CAS PubMed; (f) A. K. Clarke, J. M. Lynam, R. J. K. Taylor and W. P. Unsworth, ACS Catal., 2018, 8, 6844 CrossRef CAS; (g) H. E. Ho, T. C. Stephens, T. J. Payne, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, ACS Catal., 2019, 9, 504 CrossRef CAS.
- For selected indole dearomatisation methods, see: (a) K.-J. Wu, L. X. Dai and S.-L. You, Org. Lett., 2012, 14, 3772 CrossRef CAS PubMed; (b) S. J. Chambers, G. Coulthard, W. P. Unsworth, P. O'Brien and R. J. K. Taylor, Chem.–Eur. J., 2016, 22, 6496 CrossRef CAS PubMed; (c) V. Magné, F. Blanchard, A. Marinetti, A. Voituriez and X. Guinchard, Adv. Synth. Catal., 2016, 358, 3355 CrossRef; (d) M. J. James, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Angew. Chem., Int. Ed., 2016, 55, 9671 CrossRef CAS PubMed; (e) Y. Wang, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2017, 56, 15093 CrossRef CAS PubMed; (f) D. Ryzhakov, M. Jarret, R. Guillot, C. Kouklovsky and G. Vincent, Org. Lett., 2017, 19, 6336 CrossRef CAS PubMed; (g) J. T. R. Liddon, J. A. Rossi-Ashton, R. J. K. Taylor and W. P. Unsworth, Org. Lett., 2018, 20, 3349 CrossRef CAS PubMed.
- For examples of radical-based dearomative processes involving phenol- and anisole-tethered alkynes, see: (a) W. Wei, H. Cui, D. Yang, H. Yue, C. He, Y. Zhang and H. Wang, Green Chem., 2017, 19, 5608 RSC; (b) Y. Zhang, J. Zhang, B. Hu, M. Ji, S. Ye and G. Zhu, Org. Lett., 2018, 20, 2988 CrossRef CAS PubMed; (c) C. R. Reddy, S. Yarlagadda, B. Ramesh, M. R. Reddy, B. Sridhar and B. V. S. Reddy, Eur. J. Org. Chem., 2017, 2332 CrossRef CAS; (d) Y. Liu, Q.-L. Wang, C.-S. Zhou, B.-Q. Xiong, P.-L. Zhang, C. Yang and K.-W. Tang, J. Org. Chem., 2018, 83, 2210 CrossRef CAS PubMed; (e) L.-J. Wang, A.-Q. Wang, Y. Xia, X.-X. Wu, X.-Y. Liu and Y.-M. Liang, Chem. Commun., 2014, 50, 13998 RSC; (f) X.-H. Ouyang, R.-J. Song, B. Liu and J.-H. Li, Chem. Commun., 2016, 52, 2573 RSC; (g) D.-P. Jin, P. Gao, D.-Q. Chen, S. Chen, J. Wang, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2016, 18, 3486 CrossRef CAS PubMed; (h) H. Cui, W. Wei, D. Yang, J. Zhang, Z. Xu, J. Wen and H. Wang, RSC Adv., 2015, 5, 84657 RSC; (i) H.-L. Hua, Y.-T. He, Y.-F. Qiu, Y.-X. Li, B. Song, P. Gao, X.-R. Song, D.-H. Guo, X.-Y. Liu and Y.-M. Liang, Chem.–Eur. J., 2015, 21, 1468 CrossRef CAS PubMed. For selected examples of metal-free radical-cyclisation of alkyne: (j) J. Wen, W. Wei, S. Xue, D. Yang, Y. Lou, C. Gao and H. Wang, J. Org. Chem., 2015, 80, 4966 CrossRef CAS PubMed; (k) X.-H. Ouyang, R.-J. Song, Y. Li, B. Liu and J.-H. Li, J. Org. Chem., 2014, 79, 4582 CrossRef CAS PubMed; (l) H. Sahoo, A. Mandal, S. Dana and M. Baidya, Adv. Synth. Catal., 2018, 260, 1099 CrossRef; (m) X.-H. Yang, X.-H. Ouyang, W.-T. Wei, R.-J. Song and J.-H. Li, Adv. Synth. Catal., 2015, 357, 1161 CrossRef CAS; (n) C. Pan, B. Fang, W. Hu, X. Feng and J.-T. Yu, J. Org. Chem., 2016, 81, 2087 CrossRef CAS PubMed; (o) W. Wei, L. Wang, H. Yue, Y.-Y. Jiang and D. Yang, Org. Biomol. Chem., 2018, 16, 8379 RSC.
- For a relevant process based on a visible light mediated thiyl radical annulation of alkyne-tethered quinolinones, see: K. Kim, H. Choi, D. Kang and S. Hong, Org. Lett., 2019, 21, 3417 CrossRef CAS PubMed . EDA complex activation in the ground state was ruled out in this study.
- For a review on thiyl radicals in organic synthesis, see (a) F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587 CrossRef PubMed. For the bond dissociation energies of thiols and disulfides, see (b) N. A. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024 CrossRef CAS PubMed.
- (a) M. Feng, B. Tang, S. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef CAS PubMed; (b) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832 CrossRef CAS PubMed; (c) A. Gangjee, Y. Zeng, T. Talreja, J. J. McGuire, R. L. Kisliuk and S. F. Queener, J. Med. Chem., 2007, 50, 3046 CrossRef CAS PubMed; (d) B. Le Grand, C. Pignier, R. Létienne, F. Cuisiat, F. Rolland, A. Mas and B. Vacher, J. Med. Chem., 2008, 51, 3856 CrossRef CAS PubMed.
- For a recent example of thiyl radicals being used in photoredox mediated cross coupling, see: M. S. Oderinde, M. Frenette, D. W. Robbins, B. Aquila and J. W. Johannes, J. Am. Chem. Soc., 2016, 138, 1760 CrossRef CAS PubMed.
- For a review on spirocyclic indolenines, see: M. J. James, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Chem.–Eur. J., 2016, 22, 2856 CrossRef CAS PubMed.
- Re-subjecting an isolated sample of 16a to the reaction conditions led to full conversion into indolenine 18a over time, with atmospheric oxygen the likely oxidant (see ESI† for further details).
- We reasoned that running the reactions under argon rather than air would lead to a more reproducible procedure, and we also know that spiroindolenines can be easily prepared by oxidation of the corresponding spiroindolines if required (see ESI†).
- CCDC 1945619 (16a), 1945618 (16j), 1945660 (16g), 1645620 (13g), and 1645621 (13k) contain the crystallographic data, see ESI.†.
- When an analogous ynone substrate (13r) bearing a phenyl group at the indole C-2 position was tested, an inseparable mixture of indolenine product 18r and indoline 16r was obtained, as well as other minor impurities (the yields quoted below are based on NMR against an internal standard). The increased stability of the doubly benzylic radical that this reaction proceeds by, as well as the fact that this radical would be expected to undergo oxidation more easily, both account for the contrasting reactivity of this substrate compared with its alkyl substituted analogues. Although this reaction is not useful synthetically, the small amount of 16r produced did appear to be formed as a single diastereoisomer, most likely with the same relative stereochemistry as that obtained for products 16b–f
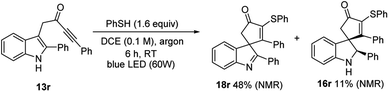 .
. - See ref. 2d and R. K. Mohamed, P. W. Peterson and I. V. Alabugin, Chem. Rev., 2013, 113, 7089. CrossRef PubMed.
- For radical hydrothiolation, see ref. 18a, and: (a) J. Healy, T. Rasmussen, S. Miller, I. R. Booth and S. J. Conway, Org. Chem. Front., 2016, 3, 439 RSC; (b) l. H. Rosa, M. L. B. Peixoto, G. R. Rosa, B. Godoi, F. Z. Galetto, M. G. M. D'Oca and M. Godoi, Tetrahedron Lett., 2017, 58, 3777 CrossRef; (c) J. József, L. L. Juhász and L. Somsák, New J. Chem., 2019, 43, 5670 RSC; (d) V. V. Levin, N. D. Zelinsky and A. D. Dilman, J. Org. Chem., 2019, 84, 8337 CrossRef CAS PubMed.
- The structural features of 13p and 13q (the elongated tether in 13p and reduction of the carbonyl group in 13q) were designed to disrupt formation of the EDA complex that is proposed to enable charge transfer and the resulting reaction.
- The blue LED spectral output is a bell curve centred at 455 nm, with sufficient irradiance in the 400–420 nm range to trigger slow radical initiation from substrates 13p and 13q. We believe these substrates to be competent thiyl radical chain carriers, as exemplified by the fact that an increase in yield for the conversion of 13q → 16q, from 28 to 65%, was observed upon the addition of 20 mol% of ynone 13k, a substrate with a significantly higher absorbance at 455 nm.
- L. Dell'Amico, A. Vega-Peñaloza, S. Cuadros and P. Melchiorre, Angew. Chem., Int. Ed., 2016, 55, 3313 CrossRef PubMed.
- Unfortunately, we have been unable to obtain clear structural information on the fate of the initiator species 25 following HAT. Analysis of the unpurified reaction a typical reaction between ynone 13l and thiophenol using ESI mass spectrometry led to the detection of peak representing a reduced version of the starting material (ynone 13l + H2 + H+), but no identifiable products could be isolated from the reaction.
- For more details on the method used to calculate quantum yield, see ESI page S9,† and for important background on such methods, see: (a) M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC; (b) K. Liang, N. Li, Y. Zhang, T. Li and C. Xia, Chem. Sci., 2019, 10, 3049 RSC.
- The proposed mechanism is further supported by deuterium labelling studies in which the reaction of ynone 13l with PhSD was performed, with the expected deuterated product (16l′, see ESI, pages S12 and S13†) being obtained. No significant difference in rate of reaction was observed when comparing this reaction with the analogous reaction with PhSH, suggesting that the HAT step is not rate determining (also see ESI, pages S12 and S13†).
- Attempts to provide additional evidence for the existence of the EDA complex using cyclic voltammetry were inconclusive (see ESI pages S15 and S16†).
- For useful perspective on catalysis of radical reactions, including especially relevant discussion of the concept of ‘smart initiation’, see: A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed.
- For examples and discussion of the importance of serendipity for discovery in synthetic chemistry, see: (a) S. Z. Zard, Chem. Commun., 2002, 1555 RSC; (b) R. N. Grimes, J. Organomet. Chem., 2013, 747, 4 CrossRef CAS; (c) M. Kazim, M. A. Siegler and T. Lectka, Org. Lett., 2019, 21, 2326 CrossRef CAS PubMed . For useful discussion on the discovery of unforeseen energy-transfer-based transformations, see:; (d) J. Ma, F. Strieth-Kalthoff, C. Henkel, M. Teders, A. Kahnt, W. Knolle, A. Gómez-Suárez, K. Dirian, W. Alex, K. Bergander, C. G. Daniliuc, B. Abel, D. M. Guldi and F. Glorius, Chem, 2019, 5, 2183 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1945620, 1945621, 1945618, 1945619 and 1945660. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05311e |
| This journal is © The Royal Society of Chemistry 2020 |