
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective/electro-oxidative intermolecular [3 + 2] annulation for the preparation of indolines†
Qingqing
Wang‡
a,
Pan
Wang‡
a,
Xinlong
Gao
a,
Dan
Wang
a,
Shengchun
Wang
a,
Xingan
Liang
a,
Liwei
Wang
a,
Heng
Zhang
 *a and
Aiwen
Lei
*ab
*a and
Aiwen
Lei
*ab
aCollege of Chemistry and Molecular Sciences, Institute for Advanced Studies (IAS), Wuhan University, Wuhan 430072, P. R. China. E-mail: hengzhang@whu.edu.cn; aiwenlei@whu.edu.cn
bNational Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang 330022, P. R. China
First published on 15th January 2020
Abstract
Compared with the reported intramolecular electro-oxidative cyclization of alkenyl amines or vinyl anilines for the preparation of pyrrolidines or indolines, the intermolecular version is less studied. Herein, this electrochemical intermolecular oxidative annulation of anilines and alkenes for the preparation of indolines proceeded under external oxidant-free conditions. The most noteworthy achievement of our work is the facile generation of indolines with quaternary centers at the 2-position. In addition, alkenes and anilines bearing various functional groups can be well tolerated. Remarkably, electrolyte-free conditions were used in an electrochemical flow cell, which shows the application potential of this method.
Introduction
Indoline derivatives are the core skeletons widely found in natural products, pharmaceuticals and functionalized materials, such as physovenine, vallesamidine, indapamide and cannabinoid receptor modulators.1 In recent years, substantial effort has been made to develop efficient methods for the synthesis of indolines. The dearomatization of indoles is the classical method to synthesize indolines.2 In addition, transition metal-catalyzed dehydrogenative C–H/N–H coupling reactions to synthesize indolines via inter- or intramolecular annulation have occupied a predominant position.3 Despite major progress in this field, stoichiometric amounts of oxidants such as Cu(II),3a–c Ag(I),3d benzoquinone (BQ)3e,f or selectfluor3g (with or without transition metal catalysts3h–l) are generally required. Under these reaction conditions, toxic or undesirable byproducts are not avoidable. It is more appealing to develop external oxidant-free reaction protocols to access indoline derivatives directly based on the rule of atom economy and sustainable chemistry.Electrochemical oxidation offers an efficient and mild alternative to the use of hazardous chemical oxidants and sometimes demonstrates unique reaction selectivity compared with chemical oxidation.4,5 Besides, electro-oxidation-induced direct C–H bond functionalization might be an efficient and environmentally friendly strategy to construct C–C and C–X bonds.5 Many methods have been developed to synthesize functionalized indolines. Among them, electrosynthesis methods can significantly reduce pollution. In the seminal studies on electrochemical cyclization of alkenyl amines,6 anodic oxidative coupling reactions have provided pyrrolidines or lactams under mild conditions either through direct electrolysis or using redox catalysts (Scheme 1a).6a–f,h However, these studies have focused on intramolecular C–N coupling. It is very difficult to achieve intermolecular annulation for the synthesis of indolines. Herein, we report a versatile regioselective/electrooxidative intermolecular [3 + 2] annulation method under oxidant-free conditions to synthesize substituted indolines (Scheme 1b).
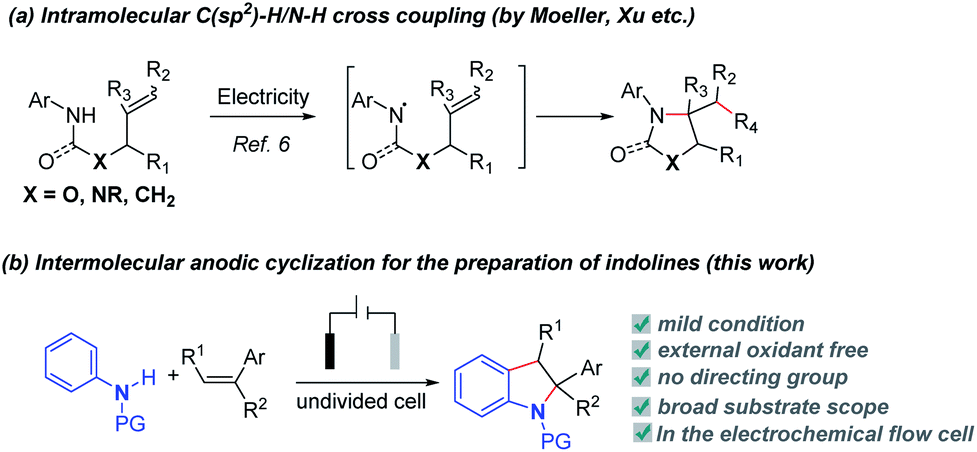 | ||
| Scheme 1 Electrochemical synthesis of N-containing heterocycles. | ||
Results and discussion
Our studies commenced with N-(4-methoxyphenyl)-4-methylbenzenesulfonamide (1a) and α-methylstyrenes (2a) (Table 1). The reaction was carried out in an undivided cell under constant current electrolysis (CCE) with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ, 10 mol%) and nBu4NBF4 as the supporting electrolyte. After considerable effort, 94% yield of the product 3aa could be obtained at 10 mA for 2 h with excellent regioselectivity (Table 1, entry 1). DDQ is well known for facilitating many transformations relying on electron and hydrogen abstraction.7 Notably, lower yields were obtained in this transformation in the absence of DDQ or HOAc (entries 2 and 3). Moreover, the yield was diminished (62%) by using Na2CO3 instead of HOAc (entry 5). Thereafter, the solvent effect was also explored. Inferior reactivity could be observed by using acetonitrile or 1,2-dichloroethane (entries 6 and 7). In addition, slightly lower yields were obtained by either decreasing or increasing the operating current (entries 8 and 9). In order to test the electrode effect, carbon cloth, a nickel plate, or an iron plate was applied as the cathode which furnished 3aa in 57–78% yields (entries 10–12). The reaction yield decreased dramatically when the reaction was opened to air (entry 13). No reaction took place without electric current under an air atmosphere (entries 14 and 15).|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yieldb (%) |
| a Reaction conditions: graphite rod anode (ϕ 6 mm), Pt plate cathode (15 mm × 15 mm × 0.3 mm), constant current = 10 mA, 1a (0.20 mmol), 2a (0.40 mmol), nBu4NBF4 (0.20 mmol), MeCN/DCE (4/2 mL), rt, 2 h (3.7 F mol−1), undivided cell, and nitrogen. b Isolated yields. c N.D. = not detected. | ||
| 1 | Standard conditions | 94 |
| 2 | No DDQ | 45 |
| 3 | No HOAc | 54 |
| 4 | BQ instead of DDQ | 61 |
| 5 | Na2CO3 instead of HOAC | 62 |
| 6 | MeCN only | 54 |
| 7 | DCE only | 37 |
| 8 | 6 mA, 4 h | 78 |
| 9 | 15 mA, 2 h | 85 |
| 10 | Carbon cloth as cathode | 75 |
| 11 | Ni plate as cathode | 57 |
| 12 | Fe plate as cathode | 78 |
| 13 | In air | 60 |
| 14 | No electric current | N.D.c |
| 15 | No electric current, in air | N.D.c |
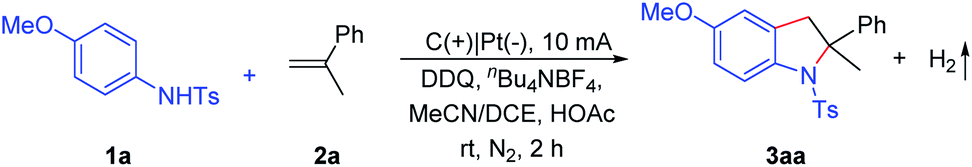
With the optimized reaction conditions in hand, we screened the substrate scope of alkenes (Scheme 2). Various α-methylstyrenes 2b–2i with meta- or para-substitution could be converted to the corresponding indolines 3ab–3ai in 61–95% yields. Notably, the electrophilic functional groups, such as fluoro, chloro, and bromo, were well tolerated. Gratifyingly, 6-methyl-1-methylene-2,3-dihydro-1H-indene (2j) also reacted efficiently to obtain the spiroindoline derivative (3aj, 62%). Remarkably, thiophene derivative 2k and 2-isopropenylnaphthalene (2l) reacted with anilines to furnish final products 3ak–3al in good to moderate yields. Importantly, diene 2m underwent selective electro-oxidative [3 + 2] annulation smoothly, giving the mono-cyclization product 3am in 53% yield. Subsequently, α-alkylstyrenes bearing linear or cyclic alkyl groups 2n–2p proved to be suitable substrates, and the desired indolines 3an–3ap were isolated in 81–93% yields. Additionally, 1,1-diphenylethylene derivatives 2q–2u were converted to the corresponding 2,2-diarylindolines which were difficult to access because of significant steric issues. To our delight, the desired products 3aq–3au were formed in high yields. Moreover, the reaction could be extended to styrenes 2v–2x and afforded the indoline products 3av–3ax in 59–78% yields. Notably, this method could also be efficiently extended to (E)-1,2-diphenylethene to afford the 2,3-fused indolines (3ay). Moreover, other olefins (e.g. alkyl olefins) have been tried, but only a trace amount of the desired products could be obtained (Scheme S4†).
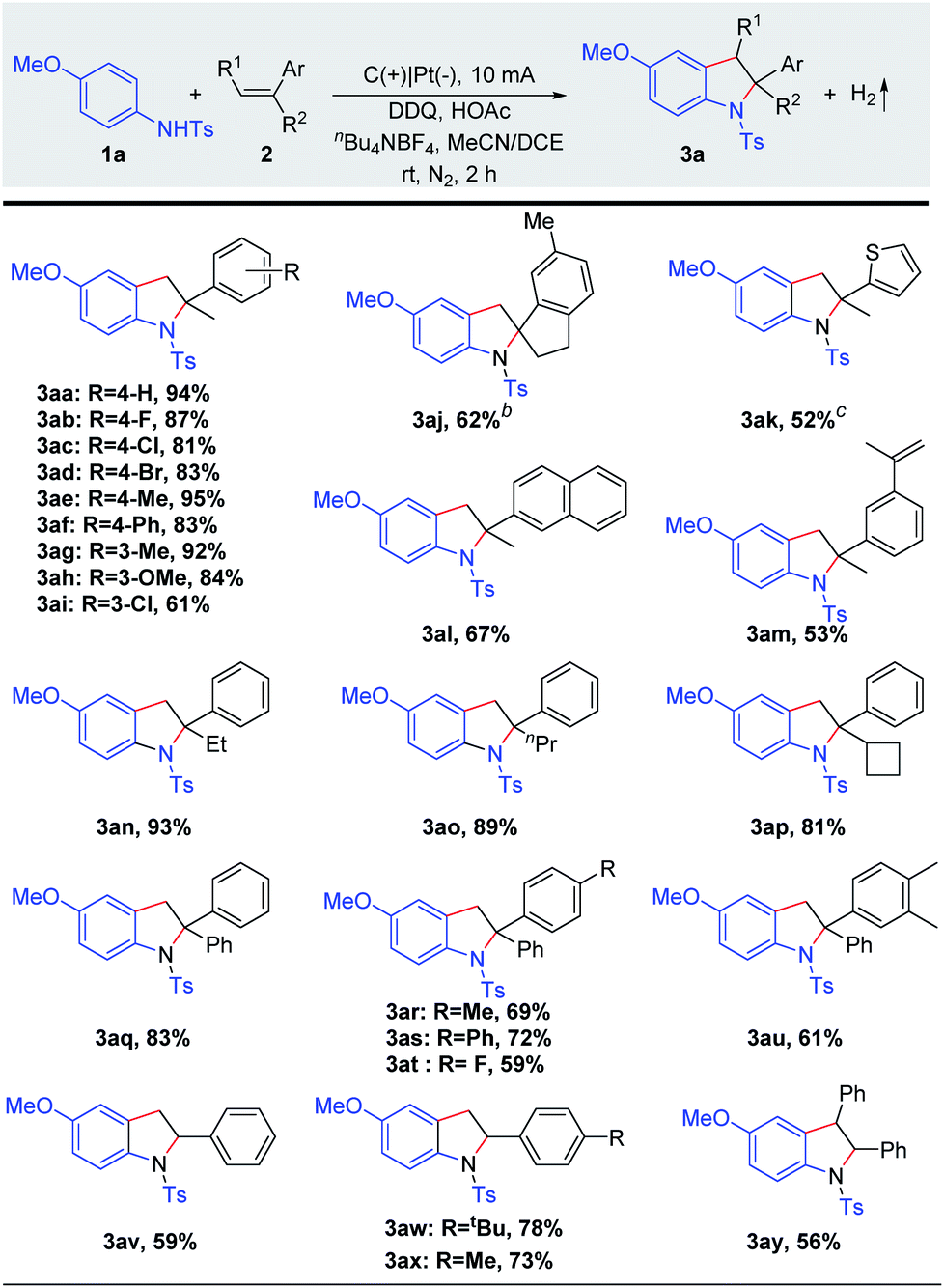 | ||
| Scheme 2 Substrate scope of alkenes.a a Reaction conditions: carbon rod anode (ϕ 6 mm), Pt plate cathode (15 mm × 15 mm × 0.3 mm), constant current = 10 mA, 1a (0.20 mmol), 2 (0.40 mmol), HOAc (0.20 mmol), 10 mol% DDQ, solvent (MeCN/DCE = 4/2 mL), undivided cell, nitrogen, and 2 h (3.7 F mol−1). Isolated yields are shown. b 4 h. c 8 mA, and 3 h. | ||
Subsequently, the scope of anilines was explored (Scheme 3). The strong electron-rich substituted amines 4-methyl-N-(4-(methylthio)phenyl)benzenesulfonamide 1b and N-(benzo[d][1,3]dioxol-5-yl)-4-methylbenzenesulfonamide 1c could be tolerated to obtain the products 3ba and 3ca in high yield under constant current electrolysis. As for the reaction of N-(4-(tert-butyl) phenyl)-4-methylbenzenesulfonamide (3d), low yield was obtained. At the same time, considering the electronic effect, we have also made efforts to try other substituted amines in an undivided cell under constant current or constant voltage electrolysis. We found that a trace amount of products could be monitored in these reactions (Scheme S2†). When substituted N-tosylanilines 1e–1i were used, indolines 3ea–3ia were obtained in good yields (58–82%). Subsequently, we speculated that applying a sulfonyl group as the protecting group might be helpful in manipulating reactivity. Therefore, different N-sulfonylanilines 1j–1p were prepared and well tolerated under the standard reaction conditions, giving indolines 3ja–3pa in high yields (66–84%). Except for N-Ts, anilines with other protecting groups (1q and 1r) have been tried, which could give the desired products 3qa and 3ra in 53% and 56% yields.
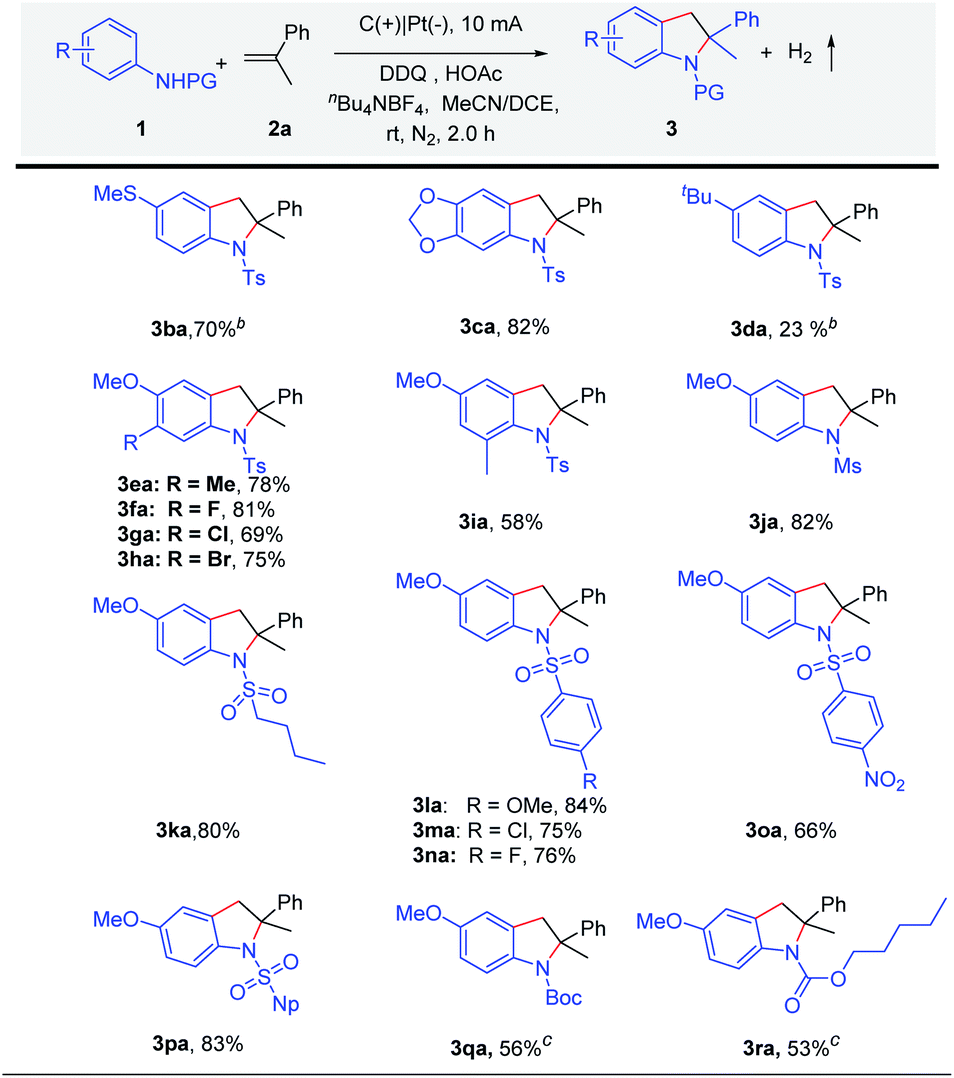 | ||
| Scheme 3 Substrate scope of amines.a aReaction conditions: carbon rod anode (ϕ 6 mm), Pt plate cathode constant current = 10 mA, 1 (0.2 mmol), 2a (0.4 mmol), HOAc (0.2 mmol), 10 mol% DDQ, solvent (MeCN/DCE = 4/2 mL), undivided cell, N2, and 2 h (3.7 F mol−1). Isolated yields are shown. nBu4NBF4 (0.1 M) was added as the supporting electrolyte. b Na2CO3 (0.2 mmol), I = 8 mA, 5 h. c Na2CO3 (0.2 mmol), I = 10 mA, and 4 h. | ||
Recently, electrochemical flow cells have been successfully used in a variety of organic transformations.9–16 Considering the difficulty of electrolyte post-treatment and the price of electrolytes, we tried to achieve this transformation in flow cells in the absence of the electrolyte. Gratifyingly, indoline derivatives could be obtained under electrolyte-free conditions in the presence of only 5 mol% DDQ, which shows the application potential of this method (Scheme 4). Various alkenes (e.g. α-methylstyrenes, styrene, 1,1-diphenylethylene derivatives, and heterocyclic olefin) could be well tolerated.
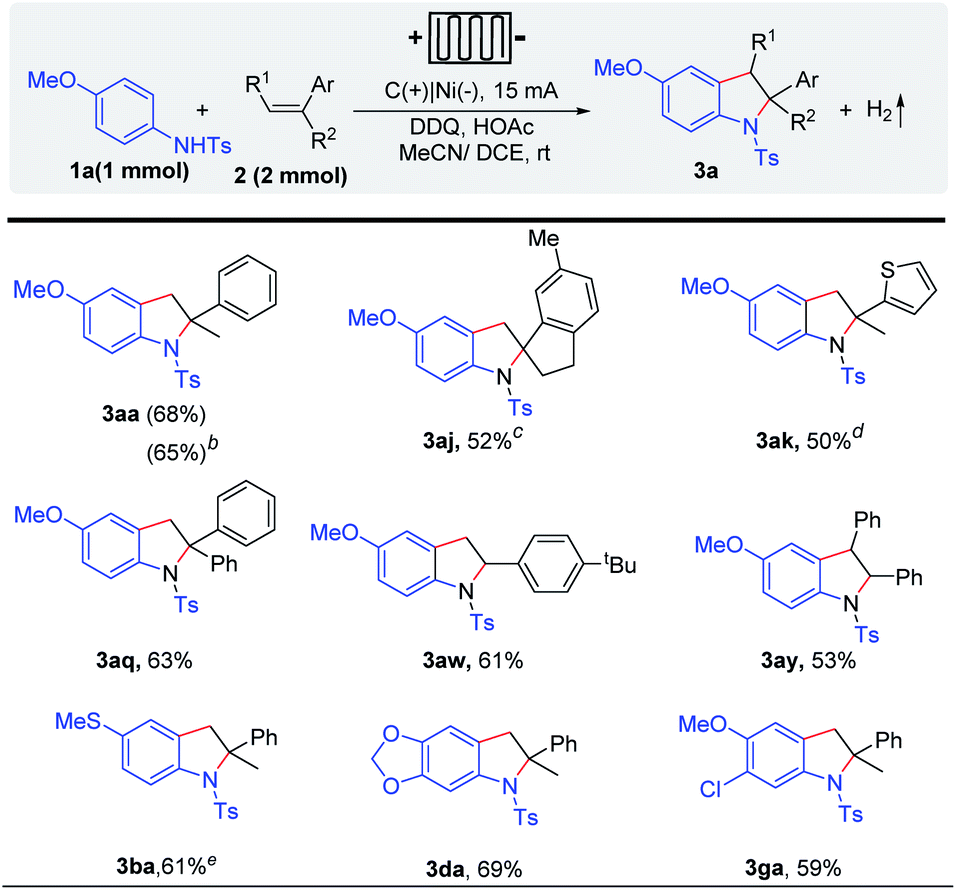 | ||
| Scheme 4 Substrate scope in the electrochemical flow cell.a aReaction conditions: I = 10 mA, 1a (1.0 mmol), 2 (2.0 mmol), HOAc (0.2 mmol), 5 mol% DDQ, solvent (MeCN/DCE = 10/5 mL), in the electrochemical flow cell, nitrogen, and 8 h. Isolated yields are shown. b 4 mmol scale, MeCN/DCE (50 mL/25 mL). c 10 h. d 8 mA, 10 h. e 8 mA, and 5 h. | ||
To further demonstrate the utility of the electrochemical method, the scale-up reaction was carried out in the 5 mmol scale. Considering the price of platinum electrodes, a nickel plate was employed as the cathode. In a continuous-flow reactor, indoline 3aa could be obtained in 65% yield with a good selectivity and efficiency in a gram scale synthesis (Scheme 5a). Furthermore, the optimized conditions were applied to electro-oxidative [3 + 2] annulation in an undivided cell in the 5.0 mmol scale. Thus, indoline 3aa could be obtained in 80% yield (Scheme 5b).
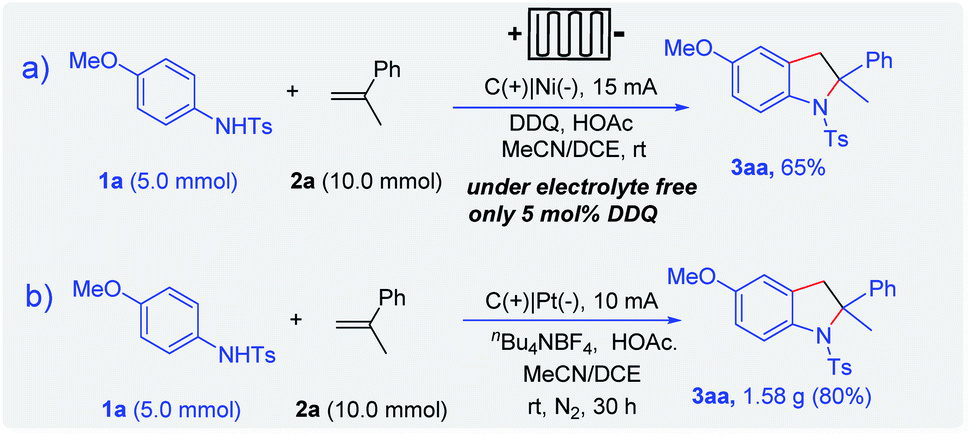 | ||
| Scheme 5 Gram scale experiment. | ||
Moreover, as illustrated in Scheme 6, deprotection of the N-Ts or N-Boc group proceeded smoothly to separately give 2-methyl-2-phenylindoline (4a) in 99% and 95% yields on a larger scale (up to 5.5 mmol). Disubstituted indolines possess a significant synthetic value in medicinal chemistry.8 Interestingly, we envisioned that the oxidative dehydrogenation of indoline (3aw) may achieve the 2-position indole derivative (4b) in 69% yield by a one pot two-step method in the same undivided cell under constant current electrolysis without separation of 3aa (Scheme 7).
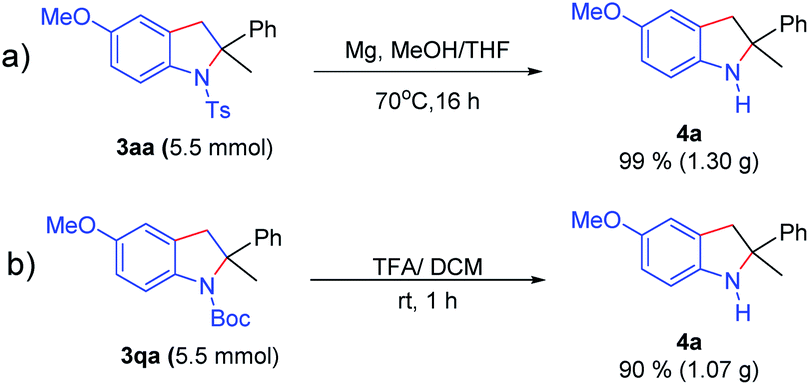 | ||
| Scheme 6 The deprotection of the N-Ts and N-Boc group. | ||
 | ||
| Scheme 7 One pot two-step process for 2-position indole synthesis. | ||
In order to clarify the reaction pathway of this reaction, some control experiments were carried out as shown in Scheme 5. When (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO, 1 equiv.) was added into the reaction between 1a and 2a under the standard conditions, no desired product was observed (Scheme 8a). Thus, this transformation was proposed to proceed via a radical pathway. Then 5.0 equiv. of triethyl phosphite was added to the reaction mixture to trap the radical intermediates.4o,17 Interestingly, the phosphorylation product 4c was obtained in 50% yield, which suggested the generation of a carbon radical during the reaction (Scheme 8b). Moreover, in the absence of 2a and DDQ, the homocoupling product 4d could be obtained with the formation of 4e in 53% yield at the same time, which suggested that nitrogen radicals were generated and then transformed into carbon radicals subsequently (Scheme 8c). Furthermore, electron paramagnetic resonance (EPR) experiments were carried out when electrolysis was performed for 30 minutes in the absence of styrene 2a and DDQ. The EPR spectra show that an organic conjugated radical is formed (see Fig. S1†). Therefore, the above control experiments and EPR results might reveal the existence of a carbon radical intermediate under the reaction conditions.
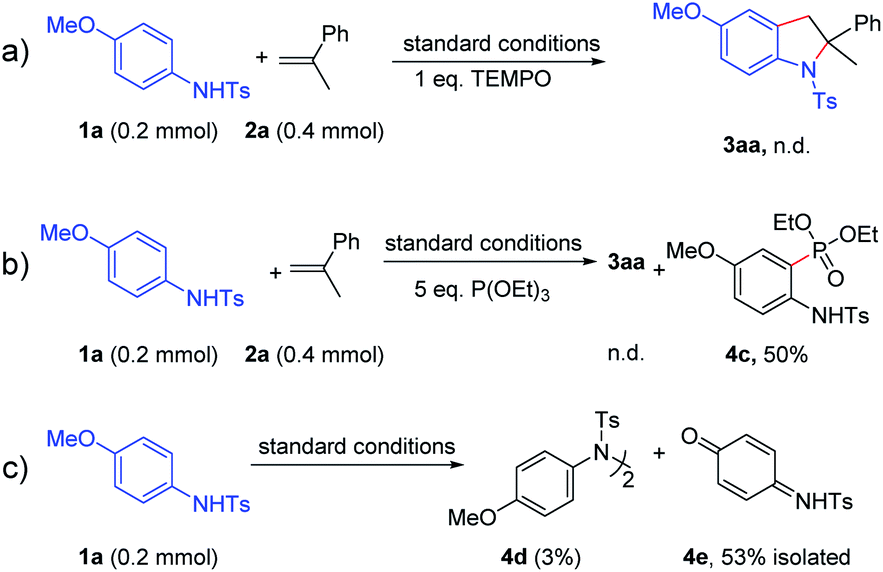 | ||
| Scheme 8 Mechanistic studies. | ||
To gain a deeper insight into the mechanism of this transformation, cyclic voltammetry (CV) experiments were conducted. First, as shown in Fig. 1a, no obvious oxidation peak was observed for α-methylstyrenes 2a in the region of 0.0–2.0 V vs. Ag/AgCl. However, N-(4-methoxyphenyl)-4-methylbenzenesulfonamide 1a gave an oxidation wave at 1.49 V vs. Ag/AgCl. The results suggested that 1a was easier to oxidize than 2a. However, the electrochemical behavior of 1a did not change in the presence of HOAc, indicating that HOAc may mainly serve as a proton source for hydrogen evolution. However, when α-methylstyrene 2a was added, a catalytic current was observed, which showed radical addition between 1a and alkene 2a (Fig. 1a, the red line). Moreover, when DDQ was added, a slight catalytic current was observed; the peak currents of Ox1 and Ox2 increased slightly from 16.8 to 17.9 μA, which indicated that aniline 1a was not mainly oxidized by DDQ. In other words, it may only mean that the reaction was slow and did not occur at the electrode surface (see Fig. S3†). Furthermore, UV experiments also demonstrated that there was no interaction between DDQ and 1a (Fig. 1b).
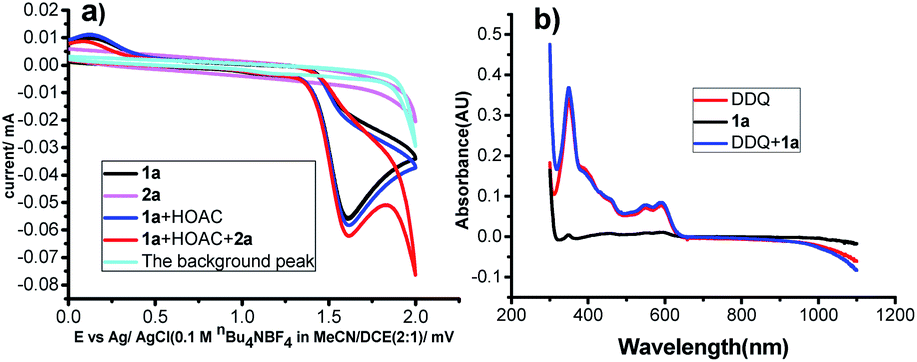 | ||
| Fig. 1 (a) Cyclic voltammograms. (b) UV experiments. | ||
In addition, kinetic studies of this reaction were carried out by detecting the initial rate with different concentrations of 1a and 2b by HPLC analysis. The reaction demonstrated a first order dependence on 1a and was independent of the concentration of 2b, which indicated that the anodic oxidation of 1a may be the rate determining step (for details see Fig. S5†).
Based on the experimental results and literature reports,10 a plausible mechanism is outlined in Scheme 9. The reaction is initiated by the anodic oxidation of aniline 1a. The subsequent deprotonation can produce N-radical species I, which can resonate to liberate the C-radical species II. Intermediate III can be formed through the radical addition between II and alkene 2a. Subsequently, intermediate III is oxidized either through anodic oxidation or by DDQ. Finally, the target molecule 3aa is generated through the intermolecular cyclization. Concomitant cathodic reduction of the proton leads to the formation of dihydrogen.
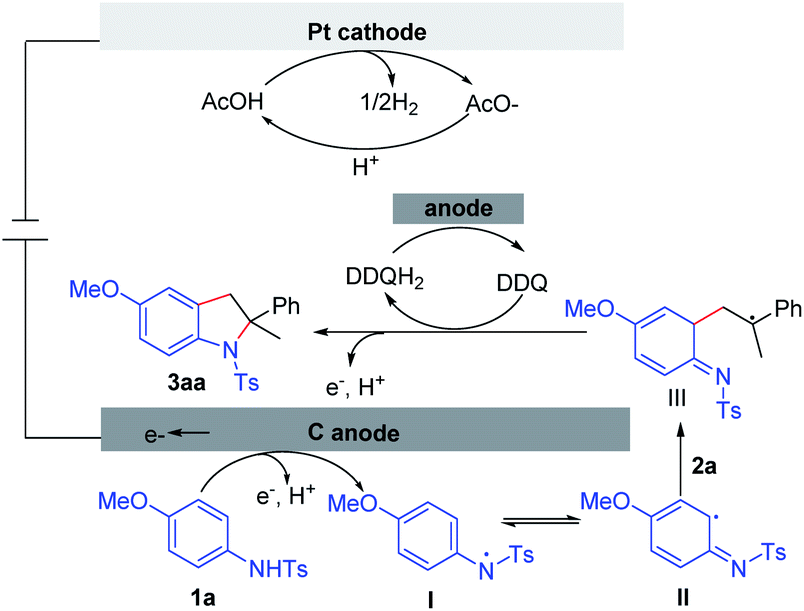 | ||
| Scheme 9 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed a novel method for the electrochemical intermolecular [3 + 2] annulation of anilines and alkenes. This method was external oxidant-free, which provided a simple and atom-economic way to synthesize functionalized indolines. A wide range of functional groups proved to be compatible under our optimized conditions. Besides, in the absence of electrolyte, indolines could be obtained in the electrochemical flow cell, which shows the great application potential of this method. Control experiments and mechanistic studies suggested that a carbon radical was involved in the reaction pathway.Experimental
General procedure for regioselective/electro-oxidative intermolecular [3 + 2] annulation for the preparation of indolines
An undivided cell was equipped with a carbon anode and a platinum cathode and connected to a DC regulated power supply. N-(4-Methoxyphenyl)-4-methylbenzenesulfonamide (0.20 mmol), prop-1-en-2-ylbenzene (0.40 mmol), nBu4NBF4 (0.1 M), 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (0.02 mmol), AcOH (0.2 mmol) and CH3CN/DCE (4/2 mL) were combined and added. The bottle was equipped with a graphite electrode as the anode and a platinum electrode (1.5 × 1.5 × 0.3 cm3) as the cathode and was then charged with nitrogen. The reaction mixture was stirred and electrolyzed at a constant current of 10 mA for 2.0 h. When the reaction was finished, the solution was extracted with EtOAc (3 × 10 mL) and H2O (3 × 10 mL). The combined organic layer was dried with Na2SO4 and filtered. The solvent was removed with a rotary evaporator. The pure product was obtained by flash chromatography on silica gel using petroleum ether and ethyl acetate as the eluent (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21701127, 21520102003, and 21390402), the Hubei Province Natural Science Foundation of China (2017CFB152 and 2017CFA010), the Fundamental Research Funds for the Central Universities (213413000050), and the Scientific Research Foundation of Wuhan University (413100043 and 413100021). The Program of Introducing Talents of Discipline to Universities of China (111 Program) is also appreciated. Qingqing Wang is grateful to the guidance and advice from Dr Yi-Hung Chen in this work.Notes and references
- For selected examples, see: (a) E. J. Glamkowski and P. A. Eieitano, J. Med. Chem., 1979, 22, 106–109 CrossRef CAS PubMed; (b) K. Shishido, E. Shitara, H. Komatsu, K. Hiroya, K. Fukumoto and T. Kametani, J. Org. Chem., 1986, 51, 3007–3011 CrossRef CAS; (c) M. Zhang, X.-M. Huang, L.-Q. Shen and Y. Qin, J. Am. Chem. Soc., 2009, 131, 6013–6020 CrossRef CAS PubMed; (d) J. D. Trzupek, D. J. Lee, B. M. Crowley, V. M. Marathias and J. S. Danishefsky, J. Am. Chem. Soc., 2010, 132, 8506–8512 CrossRef CAS PubMed; (e) S. Thakrar, N. Pandya, H. Vala, A. Bavishi, A. Radadiya, C. Pannecouque and A. K. Shah, Chem. Biol. Interface, 2012, 2, 107–113 CAS; (f) Z.-D. Pan, S. M. Pound, N. R. Rondla and C. J. Douglas, Angew. Chem., Int. Ed., 2014, 53, 5170–5174 CAS; (g) A. C. S. Reddy, V. S. K. Choutipalli, J. Ghorai, V. Subramanian and P. Anbarasan, ACS Catal., 2017, 7, 6283–6288 CrossRef CAS.
- (a) R. Kuwano, K. Kaneda, T. Ito, K. Sato, T. Kurokawa and Y. Ito, Org. Lett., 2004, 6, 2213–2215 CrossRef CAS PubMed; (b) R. Kuwano and M. Kashiwabara, Org. Lett., 2006, 8, 2653–2655 CrossRef CAS PubMed; (c) D. Liu, G. Zhao and L. Xiang, Eur. J. Org. Chem., 2010, 21, 3975–3984 CrossRef.
- (a) J.-J. Li, T.-S. Mei and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452–6455 CrossRef CAS PubMed; (b) S. W. Youn, T. Y. Ko and Y. H. Jang, Angew. Chem., Int. Ed., 2017, 56, 6636–6640 CrossRef CAS PubMed; (c) E. S. Sherman, S. R. Chemler, T. B. Tan and O. Gerlits, Org. Lett., 2004, 6, 1573–1575 CrossRef CAS PubMed; (d) E. S. Sherman, P. H. Fuller, D. Kasi and S. R. Chemler, J. Org. Chem., 2007, 72, 3896–3905 CrossRef CAS PubMed; (e) C. E. Houlden, C. D. Bailey, J. G. Ford, M. R. Gagne, G. C. Lloyd-Jones and K. I. Booker-Milburn, J. Am. Chem. Soc., 2008, 130, 10066–10067 CrossRef CAS PubMed; (f) M. K. Manna, A. Hossian and R. J. Jana, Org. Lett., 2015, 17, 672–675 CrossRef CAS PubMed; (g) G.-Z. Zhang, Y.-D. Luo, Y.-Z. Wang and L.-M. Zhang, Angew. Chem., Int. Ed., 2011, 50, 4450–4454 CrossRef CAS PubMed; (h) Y. Ni, Q.-L. Yu, Q.-H. Liu, H.-H. Zuo, H.-B. Yu, W.-J. Wei, R.-Z. Liao and F.-R. Zhong, Org. Lett., 2018, 20, 1404–1408 CrossRef CAS PubMed; (i) U. Sharma, R. Kancherla, T. Naveen, S. Agasti and D. Maiti, Angew. Chem., Int. Ed., 2014, 53, 11895–11899 CrossRef CAS PubMed; (j) D.-B. Zhao, S. Vsquez-Cspedes and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 1657–1661 CrossRef CAS PubMed; (k) Y. Gao, Y.-B. Huang, W.-Q. Wu, K.-F. Huang and H.-F. Jiang, Chem. Commun., 2014, 50, 8370–8373 RSC; (l) L. Ye, K.-Y. Lo, Q.-S. Gu and D. Yang, Org. Lett., 2017, 19, 308–311 CrossRef CAS PubMed.
- (a) C. B. Kazuhiro and T. A. Masahiro, Chem. Commun., 1994, 21, 2485–2486 Search PubMed; (b) T. K. Nokami, R. J. Soma, Y. M. Yamamoto, T. Y. K. C. Kamei and J. I. Yoshida, Beilstein J. Org. Chem., 2012, 8, 456–460 CrossRef CAS PubMed; (c) A. Jutand, Contribution of Itami, Chem. Rev., 2008, 108, 2300–2347 CrossRef CAS PubMed; (d) E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 25, 302–308 CrossRef PubMed; (e) J. I. Yoshida, A. Shimizu and R. Hayash, Chem. Rev., 2018, 118, 4702–4730 CrossRef CAS PubMed; (f) Y.-Y. Jiang, K. Xu and C.-C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed; (g) C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS; (h) N. K. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS PubMed; (i) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC; (j) S. Tang, Y.-C. Liu and A.-W. Lei, Chem, 2018, 4, 27–45 CrossRef CAS; (k) N. Sauermann, H. T. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS; (l) H. Yi, G.-T. Zhang, H.-M. Wang, Z.-Y. Huang, J. Wang, A. K. Singh and A.-W. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed; (m) J.-W. Wu, Y. Zhou, Y.-C. Zhou, C. W. Chiang and A.-W. Lei, ACS Catal., 2017, 7, 8320–8323 CrossRef CAS; (n) P. Wang, S. Tang, P. Huang and A. W. Lei, Angew. Chem., Int. Ed., 2017, 56, 3009–3013 CrossRef CAS PubMed; (o) Q.-Q. Wang, K. Xu, Y.-Y. Jiang, Y.-G. Liu, B.-G. Sun and C.-C. Zeng, Org. Lett., 2017, 81, 5517–5520 CrossRef PubMed; (p) P.-F. Huang, P. Wang, S.-C. Wang, S. Tang and A.-W. Lei, Green Chem., 2018, 20, 4870–4874 RSC; (q) F. Xu, Y.-J. Li, C. Huang and H.-C. Xu, ACS Catal., 2018, 8, 3820–3824 CrossRef CAS; (r) P. Qian, Z.-C. Yan, Z.-H. Zhou, K.-F. Hu, J.-W. Wang, Z.-B. Li, Z. -G. Zha and Z.-Y. Wang, Org. Lett., 2018, 20, 6359–6363 CrossRef PubMed; (s) Q.-Q. Wang, Y.-Y. Jiang, C.-C. Zeng and B.-G. Sun, Chin. J. Chem., 2019, 37, 352–358 CrossRef CAS; (t) J.-B. Chen, S.-D. Lv and S.-Y. Tian, ChemSusChem, 2019, 12, 115–132 CrossRef CAS PubMed.
- (a) B. R. Rosen, E. W. Werner, A. G. O'Brien and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5571–5574 CrossRef CAS PubMed; (b) T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, J. Am. Chem. Soc., 2017, 139, 12317–12324 CrossRef CAS PubMed; (c) T. Gieshoff, D. Schollmeyer and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 9437–9440 CrossRef CAS PubMed; (d) P. Qian, J.-H. Su, Y.-K. Wang, M.-X. Bi, Z.-G. Zha and Z.-Y. Wang, J. Org. Chem., 2017, 8212, 6434–6440 CrossRef PubMed; (e) M.-Y. Lin, K. Xu, Y.-Y. Jiang, Y.-G. Liu, B.-G. Sun and C.-C. Zeng, Adv. Synth. Catal., 2018, 360, 1665–1672 CrossRef CAS; (f) S. Tang, S.-Y. Wang, Y.-C. Liu, H.-J. Cong and A.-W. Lei, Angew. Chem., Int. Ed., 2018, 130, 4827–4831 CrossRef; (g) H. Zhang and A. W. Lei, Synthesis, 2018, 50, 83–96 CrossRef; (h) X.-L. Gao, P. Wang, L. Zeng, S. Tang and A.-W. Lei, J. Am. Chem. Soc., 2018, 140, 4195–4199 CrossRef CAS PubMed; (i) Z.-W. Hou, Z.-Y. Mao, H.-B. Zhao, Y. Y. Melcamu, X. Lu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 9168–9172 CrossRef CAS PubMed; (j) N. Sauermann, R. Mei and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5090–5094 CrossRef CAS PubMed; (k) S. Tang, D. Wang, Y.-C. Liu, L. Zeng and A.-W. Lei, Nat. Commun., 2018, 9, 798 CrossRef PubMed; (l) Q. L. Yang, X. Y. Wang, J. Y. Lu, L. P. Zhang, P. Fang and T. S. Mei, J. Am. Chem. Soc., 2018, 140, 11487–11494 CrossRef CAS PubMed; (m) Y.-T. Zhao and W.-J. Xia, Chem. Soc. Rev., 2018, 47, 2591–2608 RSC; (n) X. Hu, G. T. Zhang, F. X. Bu, L. Nie and A. W. Lei, ACS Catal., 2018, 8, 9370–9375 CrossRef CAS; (o) S. Zhang, L. J. Li, M.-Y. Xue, R.-K. Zhang, K. Xu and C.-C. Zeng, Org. Lett., 2018, 20, 3443–3446 CrossRef CAS PubMed; (p) K. Liu, S. Tang, T. Wu, S.-C. Wang, M.-Z. Zou, H.-J. Cong and A. W. Lei, Nat. Commun., 2019, 10, 1–10 CrossRef; (q) J.-W. Wang, P. Qian, K.-F. Hu, Z.-G. Zha and Z.-Y. Wang, ChemElectroChem, 2019, 6, 1–6 CrossRef; (r) P. Qian, Z.-C. Yan, Z.-H. Zhou, K. F. Hu, J. W. Wang, Z. B. Li, Z. G. Zha and Z. Y. Wang, J. Org. Chem., 2019, 84, 3148–3157 CrossRef CAS PubMed; (s) J. C. Siu, J. B. Parry and S. Lin, J. Am. Chem. Soc., 2019, 141, 2825–2831 CrossRef CAS PubMed; (t) W.-J. Kong, L. H. Finger, A. M. Messinis, R. Kuniyil, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2019, 141, 17198–17206 CrossRef CAS PubMed.
- (a) H.-C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2008, 130, 13542–13543 CrossRef CAS PubMed; (b) H.-C. Xu, J. M. Campbell and K. D. Moeller, J. Org. Chem., 2014, 79, 379–391 CrossRef CAS; (c) L. Zhu, P. Xiong, Z.-Y. Mao, Y. H. Wang, X.-M. Yan, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 2226–2229 CrossRef CAS PubMed; (d) F. Xu, L. Zhu, S. B. Zhu, X. Yan and H.-C. Xu, Chem.–Eur. J., 2014, 20, 12740–12744 CrossRef CAS PubMed; (e) H.-B. Zhao, Z.-W. Hou, Z.-J. Liu, Z.-F. Zhou, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 587–590 CrossRef CAS PubMed; (f) Z.-W. Hou, H. Yan, J.-S. Song and H.-C. Xu, Chin. J. Chem., 2018, 36, 909–915 CrossRef CAS; (g) S. Liang, C.-C. Zeng, X.-G. Luo, F.-Z. Ren, H.-Y. Tian, B.-G. Sun and D. Little, Green Chem., 2016, 18, 2222–2230 RSC; (h) X.-L. Yi and X.-L. Hu, Angew. Chem., Int. Ed., 2019, 58, 4700–4704 CrossRef CAS PubMed.
- A. E. Wendlandt and S. S. Stahl, Angew. Chem., Int. Ed., 2015, 54, 14638–14658 CrossRef CAS PubMed.
- (a) D. Robinson, T. Bertrand and W. Sherman, J. Chem. Inf. Model., 2016, 56, 886–894 CrossRef CAS PubMed; (b) X.-Y. Jiao and W. G. Bentrude, J. Am. Chem. Soc., 1999, 121, 6088–6089 CrossRef CAS.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- H. Chong, X. Y. Qian and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 6650–6653 CrossRef PubMed.
- C. Gütz, A. Stenglein and S. R. Waldvogel, Org. Process Res. Dev., 2017, 21, 771–778 CrossRef.
- G. Laudadio, W. de Smet, L. Struik, Y. Cao and T. J. Noël, J. Flow Chem., 2018, 8, 157–165 CrossRef PubMed.
- D. Wang, P. Wang, S. C. Wang, Y.-H. Chen, H. Zhang and A. W. Lei, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- A. A. Folgueiras-Amador and T. Wirth, J. Flow Chem., 2017, 7, 94–95 CrossRef CAS.
- A. A. Folgueiras-Amador, K. Philipps, S. Guilbaud, J. Poelakker and T. Wirth, Angew. Chem., Int. Ed., 2017, 56, 15446–15450 CrossRef CAS PubMed.
- G. Laudadio, N. J. W. Straathof, M. D. Lanting, B. Knoops, V. Hessel and T. Noël, Green Chem., 2017, 19, 4061–4066 RSC.
- L. Schulz, M. Enders, B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2017, 56, 4877–4881 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05729c |
| ‡ Qingqing Wang and Pan Wang contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |