
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licencen → π* interactions as a versatile tool for controlling dynamic imine chemistry in both organic and aqueous media†
Hang
Chen‡
ab,
Hebo
Ye‡
a,
Yu
Hai
ab,
Ling
Zhang
a and
Lei
You
 *ab
*ab
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China. E-mail: lyou@fjirsm.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 31st January 2020
Abstract
The imine bond holds a prominent place in supramolecular chemistry and materials science, and one issue is the stability of imines due to their electrophilic nature. Here we introduced ortho-carboxylate groups into a series of aromatic aldehydes/imines for dictating imine dynamic covalent chemistry (DCC) through n → π* interactions, one class of widespread and yet underused non-covalent interactions. The thermodynamically stabilizing role of carboxylate-aldehyde/imine n → π* interactions in acetonitrile was elucidated by the movement of the imine exchange equilibrium and further supported by crystal analysis. Computational studies provided mechanistic insights for n → π* interactions, the strength of which can surpass that of CH hydrogen bonding and is dependent on the orientation of interacting sites based on natural bond orbital analysis. Moreover, the substituent effect and the combination of recognition sites allowed additional means for modulation. Finally, to show the relevance of our findings ortho-carboxylate containing aldehydes were used to regulate imine formation/exchange in water, and modification of the N-terminus of amino acids and peptides was achieved in a neutral buffer. This work represents the latest example of weak interactions governing DCC and sets the stage for assembly and application studies.
Introduction
The imine bond plays a notable role in supramolecular and dynamic covalent chemistry (DCC) as well as materials science.1–3 By rendering classical covalent bonds reversible, the field of DCC has been flourishing over the past decade.4–6 Among the most explored dynamic covalent reactions (DCRs) imine formation and exchange have been widely used for the assembly of discrete structures, such as macrocycles, cages, and knots.7–10 The scrambling of components between imine assemblies also offers many possibilities toward complex systems, including molecular machinery, self-healing systems, signaling networks, and biomimic interfaces.11–16 In addition, the imine linkage holds a paramount position in the rapid development of covalent organic frameworks, which have found utility in storage, separation, sensing, and catalysis.17–20 Furthermore, as an inherent reactive site in peptides/proteins the amine group provides a handle for the labeling of biomolecules and ensuing regulation.21–23Although many advances have been made in imine chemistry, one issue is the stability and thus robustness of the imine bond due to the electrophilic nature.24,25 This problem is further exacerbated in aqueous solution, the working medium of nature.21–23 The post-structural modification, such as the reduction of the imine bond, helps assuage the stability issue at the cost of reversibility.26 Despite the enhanced stability of hydrazone/oxime, their formation and exchange generally requires acid or nucleophilic catalysis.27–30 By taking a page from “neighboring group participation” in organic chemistry, adjacent recognition sites were used to control the imine. For example, salicylaldehyde derived imine (i.e., ortho-hydroxyl) is stabilized through enamine/imine tautomerization and resonance-assisted hydrogen bonding (Fig. 1a).31–34 For another important building block, pyridine-2-carboxaldehyde and its derivatives, their imine assemblies were often constructed with metal coordination (Fig. 1b).35–37 Moreover, an ortho-boronic acid unit was employed to modulate imine and hydrazone through B–N dative bonds (Fig. 1c).38–41 Such interplay between reversible covalent and supramolecular reactivity has been opening up ample opportunities ranging from dynamic assemblies and complex systems to functionalization.
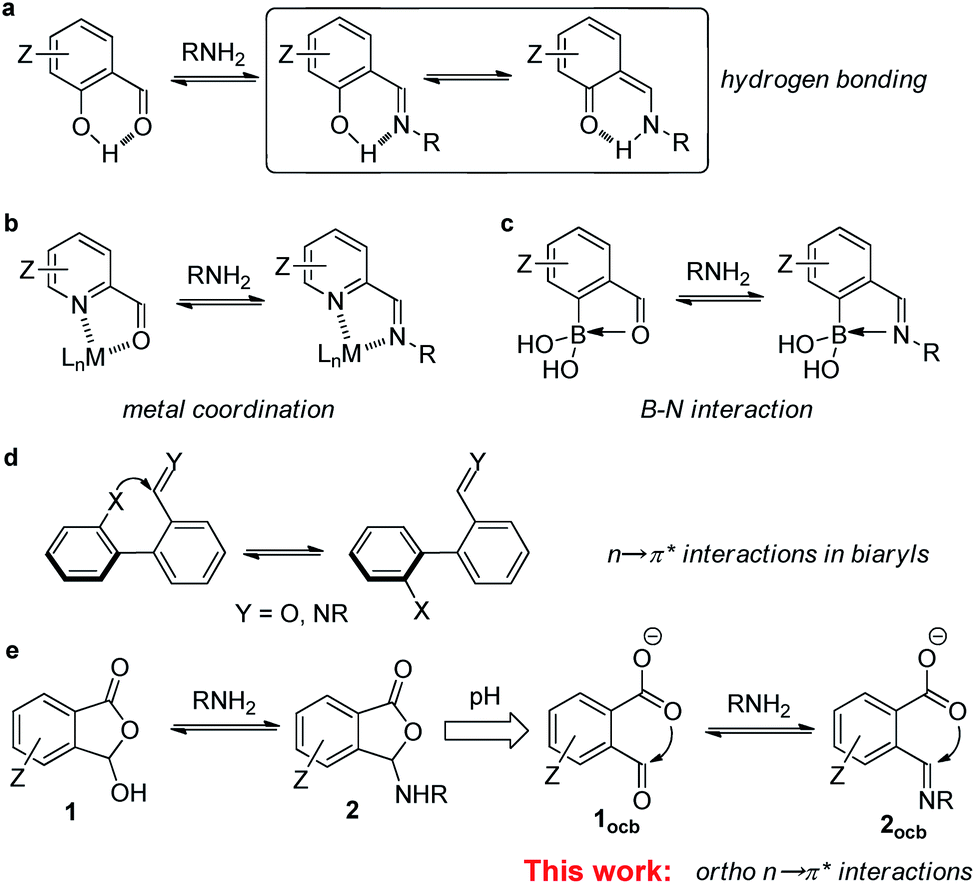 | ||
| Fig. 1 Neighboring supramolecular recognition sites for dictating imine chemistry. (a) Hydrogen bond with salicylaldehyde. (b) Metal coordination with pyridine-2-carboxaldehyde. (c) B–N dative bond with 2-formylphenylboronic acid. (d) n → π* interaction at the 2,2′ positions of biaryl. (e) The current work of ortho n → π* interaction with 2-formylbenzoic acid derivatives. | ||
As one class of underrated non-covalent interactions, orbital donor–acceptor interactions, such as n → π* interactions, have generated significant interest recently, as they are closely associated with molecular recognition, catalysis, and organic materials.42–49 The overlapping of the lone pair (n) and the π* orbital, along the Bürgi–Dunitz trajectory of a nucleophile approaching an electrophile, can result in the delocalization of electron density and thus stabilization.50,51 A large amount of effort has been devoted to the elucidation of the effect of carbonyl–carbonyl n → π* interactions on the structure, stability, and reactivity of peptides/proteins and their analogs.42,52–56 In addition to orbital interaction it was revealed that electrostatic interaction in terms of π-hole based carbon bonding contributes to lone pair-carbonyl interactions.57,58 As compared to the increasing popularity of halogen/chalcogen bonding59–63 and anion–π interactions,64–69 the use of n → π* interactions for regulating synthetic systems still remains scarce. It is thus imperative to diversify the molecules manifesting n → π* interactions and to further uncover new building units and regulating mechanisms.
Stimulated by the aforementioned neighboring effect, we wondered about the possible role of ortho n → π* interaction in imine DCC (Fig. 1e). In contrast to the supramolecular interactions in Fig. 1a–c, wherein aldehyde oxygen/imine nitrogen servers as an electron donor, we sought an interacting mode with aldehyde/imine carbon as an electron acceptor. The ortho-recognition sites also have the advantage of inherent adjacency, different from n → π* interactions at the 2,2′ position of biphenyls wherein conformational equilibrium can switch the interaction on and off (Fig. 1d).70–72 In the current work a suite of ortho-carboxyl aromatic aldehydes (1, Fig. 1e) and associated imines (2) allowed an in-depth evaluation of weak interactions dictating their reactivity and stability. The carboxylate-aldehyde/imine n → π* interactions provided stabilizing forces for anionic open aldehyde/imine in acetonitrile and resulted in the shift of the imine exchange equilibrium. Moreover, the critical role of n → π* interactions, including the interplay with CH hydrogen bonding, the regulation through the substituent effect, and the combination with different recognition motifs, was confirmed through a collection of experimental and computational evidence. Finally, to demonstrate the utility, aldehydes with ortho-carboxylate were employed for imine formation/exchange in water, and labeling of the N-terminus of amino acids and peptides was further achieved in a neutral buffer. These results established spatial orbital interaction as a valuable tool for controlling DCC and set the scene for assembly and functionalization endeavors.
Results and discussion
Hypothesis and design
2-Formylbenzoic acid exists in the ring form (1, Fig. 1e),73–76 with ring/chain tautomerization further dependent on imine formation (2, Fig. 1e). To our surprise, non-covalent interactions within 2-formylbenzoic acid and its analogs have escaped from attention. Open aldehyde/imine was thus mainly focused on to elucidate participating weak interactions, as base induced ring opening would afford the conjugate base form (1ocb and 2ocb). More importantly, we postulated that the incorporation of a donating carboxylate group at the ortho position of an arylaldehyde and its imine would stabilize the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Y (Y = O, NR) bond through carboxylate/CY n → π* interaction (Fig. 1e). In addition, DCRs of 1 with amines to create 2 would perturb the system in response to the change in electrophilicity from the aldehyde to its imine. Furthermore, the ionization of the carboxyl group to give carboxylate would offer us the opportunity to enhance the solubility of aldehyde 1/imine 2 and also to exert control on n → π* interactions and imine DCC in water. It is also worthwhile to mention that hydrogen bonding between aldehyde/imine CH and carboxyl oxygen (CH⋯O) could affect n → π* interactions (C⋯O) as they share the oxygen donor.
Y (Y = O, NR) bond through carboxylate/CY n → π* interaction (Fig. 1e). In addition, DCRs of 1 with amines to create 2 would perturb the system in response to the change in electrophilicity from the aldehyde to its imine. Furthermore, the ionization of the carboxyl group to give carboxylate would offer us the opportunity to enhance the solubility of aldehyde 1/imine 2 and also to exert control on n → π* interactions and imine DCC in water. It is also worthwhile to mention that hydrogen bonding between aldehyde/imine CH and carboxyl oxygen (CH⋯O) could affect n → π* interactions (C⋯O) as they share the oxygen donor.
To systematically examine the role of the ortho-carboxyl unit, a series of aromatic aldehydes with varied substituents was prepared (1a–1e, Fig. 2a). The derivatives of pyridine-2-carboxaldehyde (1f) and salicylaldehyde (1g) were also studied to probe potential cooperative effects from additional recognition sites. The parent aldehydes (3a–3g) and their imines (4a–4g) were employed as controls (Fig. 2a). By taking advantage of the reversibility of DCC systems, the thermodynamic effect of adjacent recognition sites, such as carboxyl, could be measured with in situ competing target and reference DCRs in one vessel (Fig. 2b). The free energy difference (ΔGex) of the imine exchange equilibrium would be directly reflective of the relative affinity of aldehydes toward amines (ΔG1 – ΔG2), thus providing a facile method for quantification.77–79 In essence, the movement of the imine exchange equilibrium would provide an estimation of participating weak interactions.
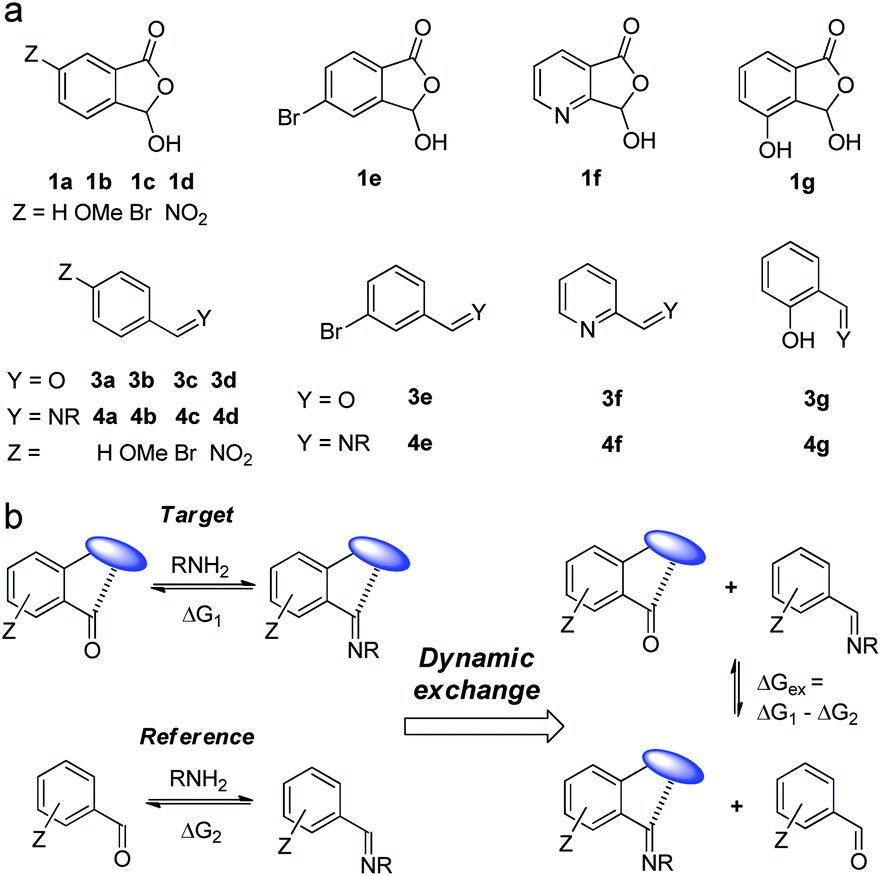 | ||
| Fig. 2 (a) Compounds (1) and controls (3 and 4) studied in this work. (b) Dynamic component exchange for probing the thermodynamic effect of nearby recognition sites (represented by the blue oval) on imine DCC. | ||
Imine formation and exchange in acetonitrile
The reaction of 1d (a of Fig. 2A) with 1-butylamine in CD3CN afforded the desired product quickly in a nearly quantitative yield (Fig. S3†). A resonance around 6.6 ppm emerged, indicative of the creation of the cyclic hemiaminal ether 2d (b of Fig. 2A). The domination of ring tautomers was also found in 1a–1f (Fig. S1†) as well as their corresponding amine adducts 2a–2f (Fig. S4–S15†). The anionic open forms were created by simply titrating the base DBU into 1/2 (Fig. S18–S26†) or performing DCRs with amines with DBU present (Fig. S27–S33†). Taking 1d as an example, the emergence of a peak at 10.8 pm with 1.2 equiv. of DBU (a of Fig. 3B) suggested the formation of the conjugate base of the aldehyde form (1docb). Base promoted ring opening was also observed with 2d, giving anionic imine 2docb (b of Fig. 3B). The open imine was detected for 2g even without the base, as evidenced by the appearance of imine CH at 8.9 ppm (Fig. S16†). The unique behavior of 2g was explained with the stabilizing role of enamine/imine tautomerization and hydrogen bonding for salicylaldehyde derived imine (Fig. 1a).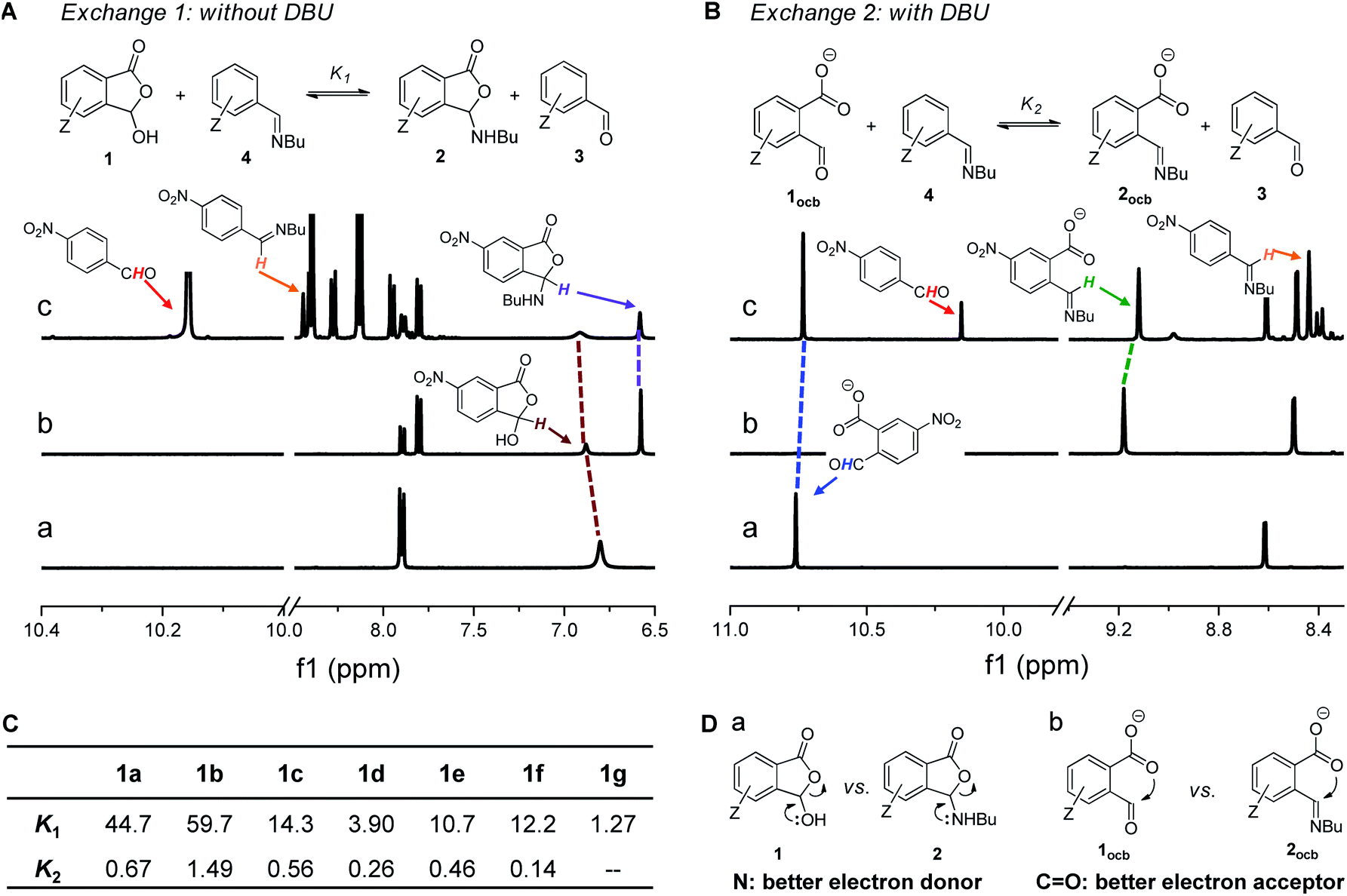 | ||
| Fig. 3 (A) Imine exchange 1 between 1 and 4, with 1H NMR spectra of 1d (a), 2d (b), and exchange (c) shown. (B) Imine exchange 2 between 1ocb and 4 in the presence of DBU, with 1H NMR spectra of 1docb (a), 2docb (b), and exchange (c) shown. (C) Summary of equilibrium constants for imine exchanges 1 and 2. (D) Illustration of the stabilizing effect of ortho-carboxyl (a) and carboxylate (b) on cyclic and open aldehyde/imine, respectively. Solvent: CD3CN. | ||
In order to probe the existence of n → π* interaction, aldehyde exchange was next conducted in CD3CN (Fig. 3). The competition between arylaldehyde (3) and its ortho-carboxyl derivative (1) for the reaction with 1-butylamine was examined (exchange 1 and 2). In the absence of DBU (Fig. 3A) amine assemblies incorporating a carboxyl (2) were favored in all cases (K1 > 1, Fig. 3C and S34–S41†). For example, the largest K1 value of 59.7 was obtained, overwhelmingly preferring 2b over 4b (p-OMe). A K1 value of 44.7, 14.3, and 3.90 was found for H (1a), p-Br (1c), and p-NO2 (1d), respectively (c of Fig. 3A). The smallest K1 (1.27) for 1g is consistent with the aforementioned effect of the salicylaldehyde motif. We interpreted the higher affinity of 1 toward amines with the stabilization of the lactone form of 2 (a of Fig. 3D) through the anomeric effect80,81 on hemiaminal ether carbon arising from overlapping between the lone pair of the nitrogen atom and σ* orbital of the C–O bond (n → σ*(C–O) interaction). Due to nitrogen being a better electron donor than oxygen, such an effect would be more dominant in 2 than 1.
Interestingly, there was a sharp decrease in the equilibrium constant of imine exchange (Fig. 3B) when DBU (1.2 equiv.) was added into the mixture of 1, 3, and 1-butylamine to form anionic open aldehyde 1ocb and imine 2ocb (K2, Fig. 3C and S42–S48†). For example, the base enabled the reversal of the position of the equilibrium for p-NO2, with aldehyde 1docb favored (K2 = 0.26, c of Fig. 3B). 1a (H), 1b (p-OMe), and 1c (p-Br) gave a K2 value of 0.67, 1.49, and 0.56, respectively, all of which are significantly smaller than K1. The smallest K2 (0.14) was found with 1f. These findings were rationalized as follows: since open aldehydes are more electron-deficient and thus better electron acceptors than their imines, through-space carboxylate-aldehyde n → π* interaction would become stronger than that of imine, resulting in enhanced stabilization and accumulation of aldehyde 1ocb (b of Fig. 3D). As a result, the thermodynamic effect of neighboring carboxyl can be readily modulated via changing the pH.
To further isolate the role of n → π* interaction from the electronic effect of carboxylate, analogous exchange reactions were run by using meta- and para-carboxylate-benzaldehyde (1aocb (meta) and 1aocb (para)), respectively (Fig. S49 and S50†). The carboxylate at the meta or para position is unlikely to interact directly with aldehyde and its imine but could influence them through the electronic effect. 1aocb (meta) and 1aocb (para) afforded K2 values of 2.01 and 1.65, different from the data of 1aocb (0.67). These results support the shift of the exchange equilibrium through ortho n → π* interaction, while meta/para carboxylate mainly serves as an electron-withdrawing group.
Crystal structures
The next goal was to reveal structural features of associated n → π* interactions. For example, in 1aocb the aldehyde and carboxylate adopt a torsion angle of 17 and 32° relative to the benzene plane, respectively (Fig. 4a).82 Such an orientation would be able to accommodate the orbital interaction. A distance of 2.88 Å was revealed between carboxylate oxygen and aldehyde carbon (O⋯C), further suggesting an attractive interaction. In the X-ray structure of imine 2aocb a torsion angle of 37 and 11° was found for imine and carboxylate, respectively, which also have close contact (2.79 Å for O⋯C, Fig. 4b). In addition, hydrogen bonds were found in the crystal packing, such as the one between imine nitrogen and ammonium NH. For pyridine derived imine 2focb, the co-planarity of CN with the pyridine plane was apparent, likely facilitated by a hydrogen bonding-bridged five-membered ring (Fig. 4c). Moreover, the carboxylate is significantly twisted (53°) in order to align with the imine carbon (3.00 Å for O⋯C). The structural parameters, in particular the torsion angles of and the distance between aldehyde/imine and carboxylate, provided further evidence for n → π* interactions.
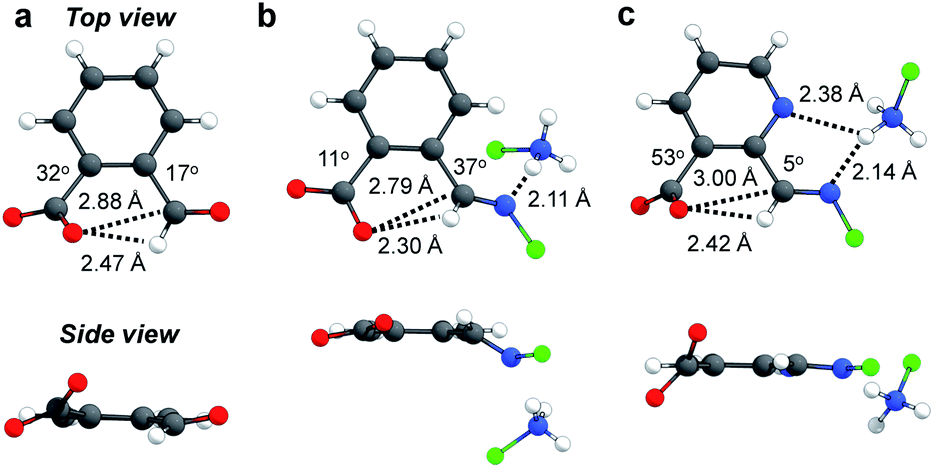 | ||
| Fig. 4 Crystal structures of 1aocb (a), 2aocb (b), and 2focb (c). For clarity the cyclohexyl group on the amine nitrogen is shown with a green sphere. The absolute value of the acute angle was used to reflect the torsion angle of carboxylate, aldehyde, or imine relative to the phenyl plane. | ||
Calculations of orbital interactions
DFT calculations were then conducted to offer more insights. The functional/basis set of M06-2X-D3/def2-TZVP was employed, and the PCM solvent model was applied for acetonitrile. The conformer of open conjugate base 1aocb with the n → π* interaction is dominant (99%, Fig. 5a and S51†), consistent with the X-ray structure. An analogous structure was also obtained for 2aocb (99%) as for 1aocb (Fig. 5b and S51†). The alignment of orbitals was further visualized through natural bond orbital (NBO) analysis, which offers an estimation of the interaction energy according to second-order perturbation theory (ΔE(2)).83 For example, the overlap of two oxygen lone pairs and the antibonding orbital of aldehyde was apparent within 1aocb, with NBO energies of 1.90 and 0.39 kcal mol−1, respectively (Fig. 5a). The sum (2.29 kcal mol−1) was used for analysis here, and a NBO energy of 1.32 kcal mol−1 (sum of 1.04 and 0.28 kcal mol−1) was found for carboxylate-imine n → π* interaction in 2aocb (Fig. 5b). In addition, NBO analysis revealed an n → σ*(CH) stabilization energy of 0.03 kcal mol−1 for the CH⋯O hydrogen bond in 1aocb (Table S2†), significantly smaller than the corresponding value for n → π* interaction (2.29 kcal mol−1). A similar trend was found with open imine 2aocb, preferring n → π* interaction over the CH hydrogen bond. Moreover, the molecular electrostatic potential (MEP) maps of 1aocb and 2aocb were calculated, and the attractive interaction between the electron-rich region of carboxylate oxygen and electron-deficient region of aldehyde/imine carbon was apparent as compared to the donor (carboxylate) and acceptor (aldehyde/imine) fragments (Fig. S52†). These results are consistent with the contribution of both orbital and electrostatic interactions for lone pair-carbonyl contacts.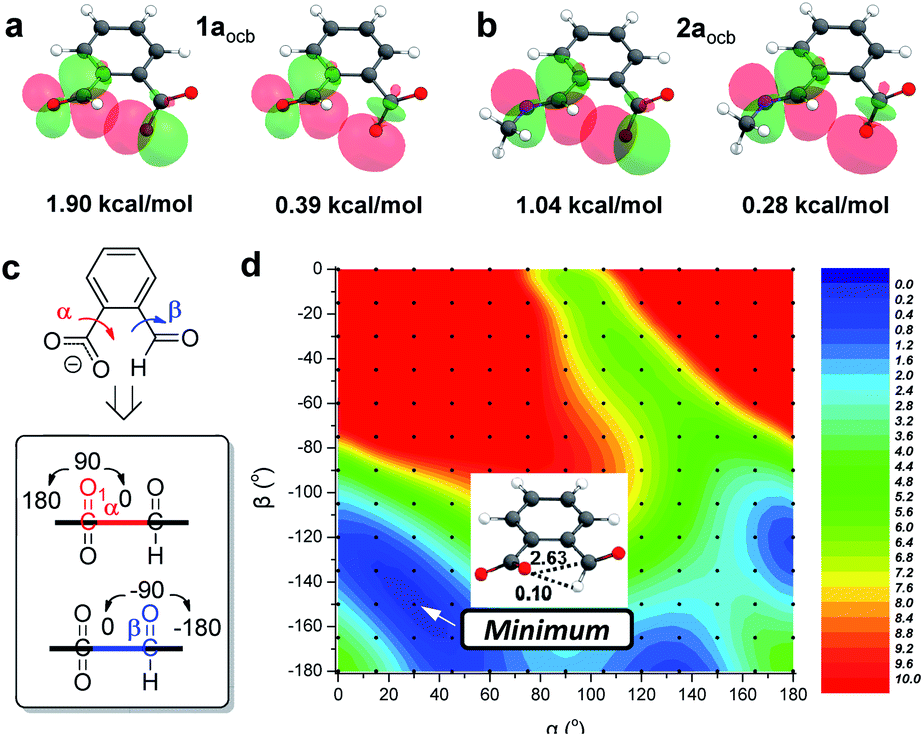 | ||
| Fig. 5 Optimized structures of open conjugate base (a) 1aocb and (b) 2aocb, with NBOs and energies listed. (c) Illustration of the torsion scan and definition of α and β. (d) A heat map of the energies of 1aocb (in kcal mol−1) versus (α, β) coordinates. The structure at the minimum and associated NBO energies (n → π* interaction and CH hydrogen bond, in kcal mol−1) are shown. | ||
To further elaborate the extent of weak interactions, a rigid potential energy surface scan of 1aocb was performed. The orientation of carboxylate (α) and aldehyde (β) relative to the phenyl plane was varied (Fig. 5c), and the minimized energies of ensuing rotamers are shown in a heat plot (Fig. 5d). The most stable isomer was found at (30°, −150°) in the blue region, echoing the X-ray (α = 32°, β = −163°) as well as geometry-optimized (α = 29°, β = −156°) structures of 1aocb. The n → π* interaction (2.63 kcal mol−1) was dominant compared to CH⋯O hydrogen bonding (0.10 kcal mol−1). Moreover, low-energy blue regions along the α and β axes were identified. In addition, there are two red regions (upper left and upper right) with overwhelmingly unfavorable energies due to strong electronic repulsion between carboxylate oxygen and aldehyde oxygen. The loss of conjugation of aldehyde and carboxylate with the aromatic ring at (90°, −90°) seriously undermines the stability (9.7 kcal mol−1). Nevertheless, a planar isomer at (0°, −180°) or (180°, −180°) is also unfavorable (4.5 kcal mol−1), as n → π* interaction can't contribute since the carboxylate resides on the nodal plane of the π* orbital of the carbonyl. Torsion scan analysis of 2aocb also verified its optimized structure (Fig. S53†). Overall, we interpreted these results as follows: to accommodate the alignment of orbitals and thus directionality of n → π* interaction and concomitantly to compensate the energy change as a result of varying conjugation, the torsion angle of aldehyde (α) and carboxylate (β) would be adaptive.
Substituent effects on n → π* interactions
With weak interactions governing aldehyde 1aocb/imine 2aocb elucidated, substituent effects were further examined as a means of regulating n → π* interactions (Fig. 6). The major structures of the open conjugate base forms for a suite of ortho-carboxylate aldehydes and imines (1ocb and 2ocb) were calculated (Table S2†), and NBO stabilization energies of n → π* interactions were summarized (Fig. 6a). In general a larger NBO value was found for aldehyde than for its associated imine, supporting the explanation in Fig. 3. Among para or meta-substituted aldehydes 1docb (p-NO2) gave the strongest n → π* interaction (4.49 kcal mol−1), while 1bocb (p-OMe) had the weakest (0.72 kcal mol−1). The data for 1aocb (H), 1cocb (p-Br) and 1eocb (m-Br) fell in the middle. A similar trend was found for imine 2, though the discrimination between substituents is modest. The sequence of NBO data largely falls in line with the electron-withdrawing ability of the para-substituent. Electron-donating p-OMe can stabilize the aldehyde/imine via the resonance effect, hence decreasing the electrophilicity and in turn weakening the n → π* interactions.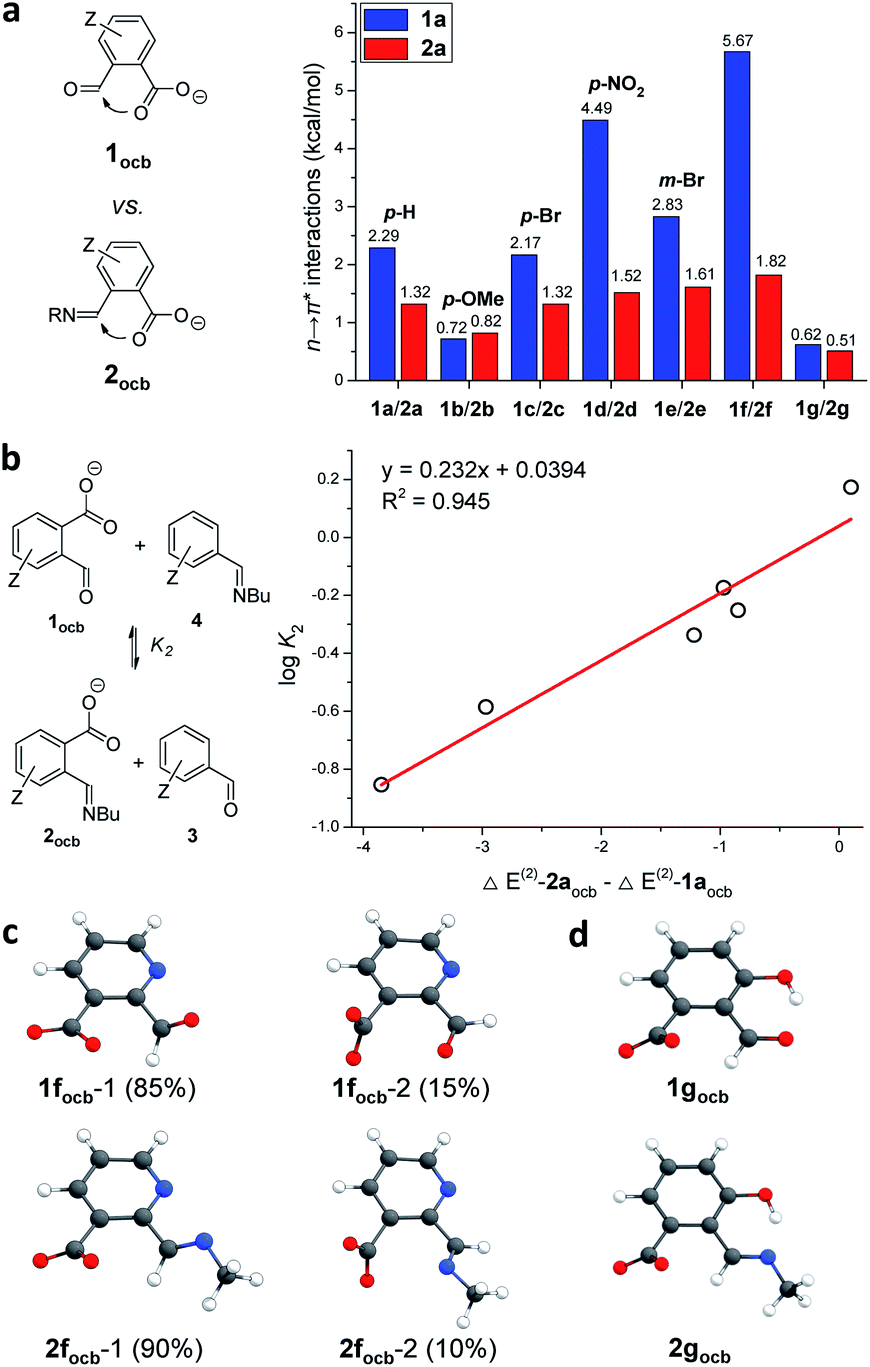 | ||
Fig. 6 (a) Comparison of NBO energies of n → π* interactions in 1ocb and 2ocb. (b) Correlation of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K2 with the difference in n → π* NBO energies of 1aocb and 2aocb. Optimized structures and populations of isomers of 1focb and 2focb (c), as well as 1gocb and 2gocb (d). K2 with the difference in n → π* NBO energies of 1aocb and 2aocb. Optimized structures and populations of isomers of 1focb and 2focb (c), as well as 1gocb and 2gocb (d). | ||
The interplay of n → π* interactions with other recognition motifs was then probed. For 1focb besides isomer 1focb-1 (85%) 1focb-2 accounted for 15% of the population (Fig. 6c). The decreased percentage of 1focb-1 over 1aocb (99%) is likely due to the electronic repulsion between pyridine nitrogen and carbonyl oxygen in 1focb-1. Similar results were also revealed with 2focb (Fig. 6c). The NBO energy of n → π* interactions is significant for 1focb-1 (5.67 kcal mol−1) and 2focb-1 (1.82 kcal mol−1, Fig. 6a). The enhancement of NBO energies of n → π* interactions in 1focb-1 and 2focb-1 as compared to 1aocb and 2aocb (2.29 and 1.32 kcal mol−1) can be attributed to the electron-withdrawing ability of pyridine. One conformer was dominant for 1gocb and 2gocb (Fig. 6d), as in the case of 1aocb and 2aocb. Moreover, NBO analysis revealed an n → π* interaction energy of 0.62 and 0.51 kcal mol−1 for 1gocb and 2gocb, respectively (Fig. 6a). These values are significantly smaller than the data for 1aocb and 2aocb. Hydrogen-bonding facilitated aldehyde/enol and imine/enamine tautomerization23 in the open forms of 1g and 2g would reduce the electrophilicity of aldehyde and imine, accordingly attenuating their corresponding orbital interactions with nearby carboxyl groups. Nevertheless, the contribution arising from neighboring carboxyl to the overall reaction is still significant, as revealed by imine exchanges with 1f/1g in Fig. 4. More importantly, a linear correlation of logK of exchange 2 with the difference in n → π* interaction energies of aldehydes 1ocb and imines 2ocb was obtained, further validating their contribution (Fig. 6b and Table S3†).
Imine DCC in aqueous solutions
Having gathered both experimental and theoretical evidence for n → π* interactions, the control of imine DCC in water was further explored. There is intensive interest in supramolecular and dynamic covalent chemistry in water, the environment of nature.84–86 Gratifyingly, the creation of open imines was found for the reactions of 1a–1g with ethanolamine (2.0 equiv.) in D2O (Fig. S54–S60†), as an excess amount of amine deprotonated the carboxyl to solubilize aldehyde 1 and imine 2 in the form of their anionic conjugate bases (1ocb and 2ocb). To gauge the thermodynamic effect of ortho carboxylate, the competition between 1 and 3 for reactions with ethanolamine (2.0 equiv.) was then examined in D2O (with 10% DMSO-d6, exchange 3 in Fig. 7a). Imine 2 was preferred in all cases (K3 > 1), supporting the shift of the imine exchange equilibrium toward the right (Fig. 7a and S61–S67†). A K value of 3.30 and 3.10 was revealed for 1f and 1g, respectively, while 1a–1e gave a K value around 1.30. Relative to acetonitrile different equilibrium constants were obtained in aqueous solution. To further probe solvent effects, exchange reactions between 1a and 4a were run in different solvent mixtures (Fig. S70–S77, Table S4†). Upon increasing the percentage of water there was an enhancement in the K value, indicating gradual movement of the equilibrium toward the right. One explanation comes from the stabilization of imine 2via hydrogen bonding with water in conjunction with n → π* interaction, and the different capability of forming hydrogen bonds between aldehyde oxygen and imine nitrogen with water would modulate corresponding n → π* interactions. Hydrogen bond and n → π* interaction working collectively to modulate the system have been reported.42,52,70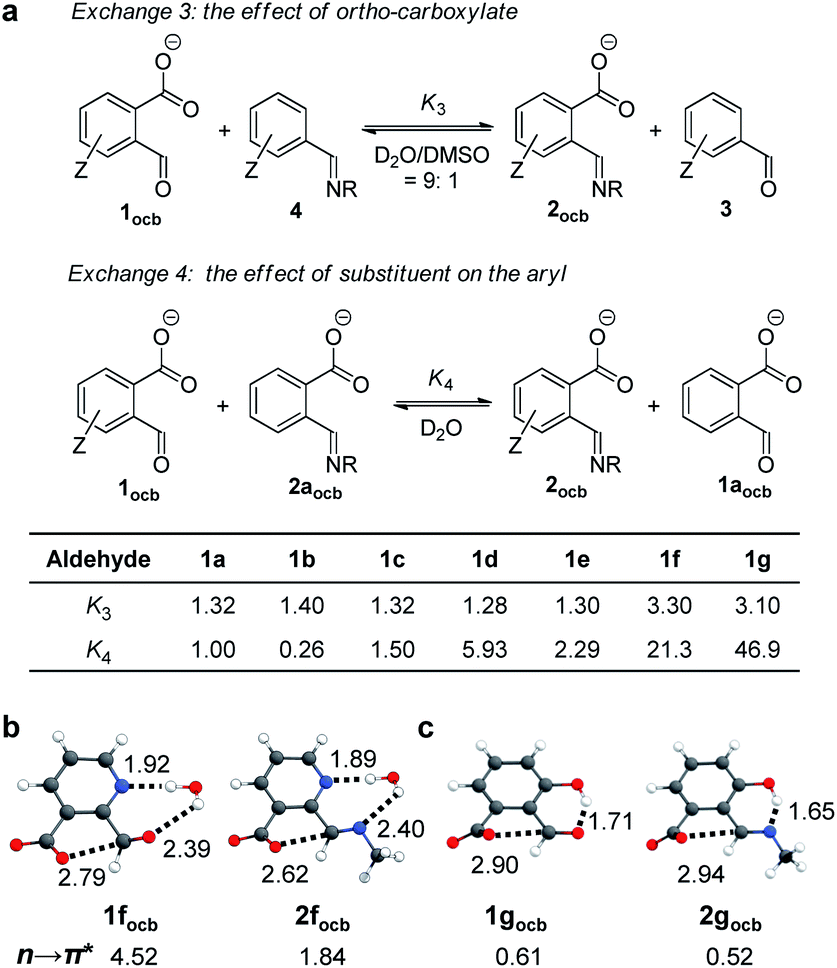 | ||
| Fig. 7 (a) Imine exchange 3 between between 1 and 4 in D2O:DMSO-d6 (9:1) as well as imine exchange 4 between 1 and 2a in D2O, with equilibrium constants listed. Optimized structures of 1focb and 2focb (b), as well as 1gocb and 2gocb (c), with distances (Å) and NBO energies (kcal mol−1) of n → π* interactions shown. | ||
Aldehydes 1f and 1g were further studied as they gave larger K3 values than others. A series of aldehyde exchanges with 1a as a reference was also conducted to identify the aldehyde exhibiting the best affinity for ethanolamine (3.0 equiv., exchange 4 in Fig. 7a and S78–S83†). Salicylaldehyde derived 1g gave the largest K value (46.9), strongly preferring its imine 2g (Fig. 7a). A K value of 21.3 was obtained with pyridine derived 1f. The enhanced affinity of 1f and 1g toward the amine is rationalized with stabilizing forces arising from nearby recognition sites of pyridine nitrogen or phenol along with n → π* interaction. DFT calculation of model compounds corroborated the hypothesis. Water bridged concerted hydrogen bonds were apparent, with n → π* NBO energies of 4.52 and 1.84 kcal mol−1 for 1f and 2f, respectively (Fig. 7b). With 1f there was a decrease in NBO data (4.52 kcal mol−1) of orbital interaction in water as compared to acetonitrile (5.67 kcal mol−1, Fig. 6a), suggesting that concerted hydrogen bonding with water attenuates n → π* interaction in 1f. However, the n → π* NBO value for 2f remained nearly the same between water (1.84 kcal mol−1) and acetonitrile (1.82 kcal mol−1). These trends explain the reversal in the equilibrium constant from acetonitrile (0.14) to water (3.30) for 1f. Phenol mediated intramolecular hydrogen bonds were found for 1g and 2g, which afforded n → π* NBO energies of 0.61 and 0.52 kcal mol−1, respectively (Fig. 7c). These values are similar to the data for acetonitrile, and n → π* interaction is weakened due to intramolecular hydrogen bonding. Although hydrogen bonding would play a dominant role in stabilizing imines 2f and 2g, ortho carboxylate still contributes, as reflected by a K3 value of 3.30 and 3.10 for 2f and 2g, respectively. These results indicate the feasibility of the combination of different recognition sites for modulating imine chemistry.
Imine DCC at physiological pH
Encouraged by the findings in water, imine formation and exchange were then studied at physiological pH. Aldehyde 1g was chosen as it gave the largest K for exchange 4. A K value of 2.90 was afforded for the competition of 1g and salicylaldehyde for ethanolamine at pH 7.4 (50 mM PBS buffer), again preferring 2g (Fig. S84†). The reaction of 1g (10 mM) with ethanolamine (1.2 equiv.) readily gave imine 2g, albeit with a modest yield (17%) at pH 7.4 (Fig. S85 and S86†). A slightly smaller yield (12%) was apparent at pH 7.0, while there was an increase in the yield of imine (30%) when the pH was switched to 7.8 (Fig. S87†). Moreover, the yields remained constant after a week (Table S5†). In order to optimize the extent of imine formation in a pH 7.4 buffer, a series of DCRs were performed while maintaining the concentration of ethanolamine at a constant (2.0 mM) and varying that of 1g. With an aldehyde/amine ratio around 20, a yield larger than 60% was obtained (Fig. 8a and S88†).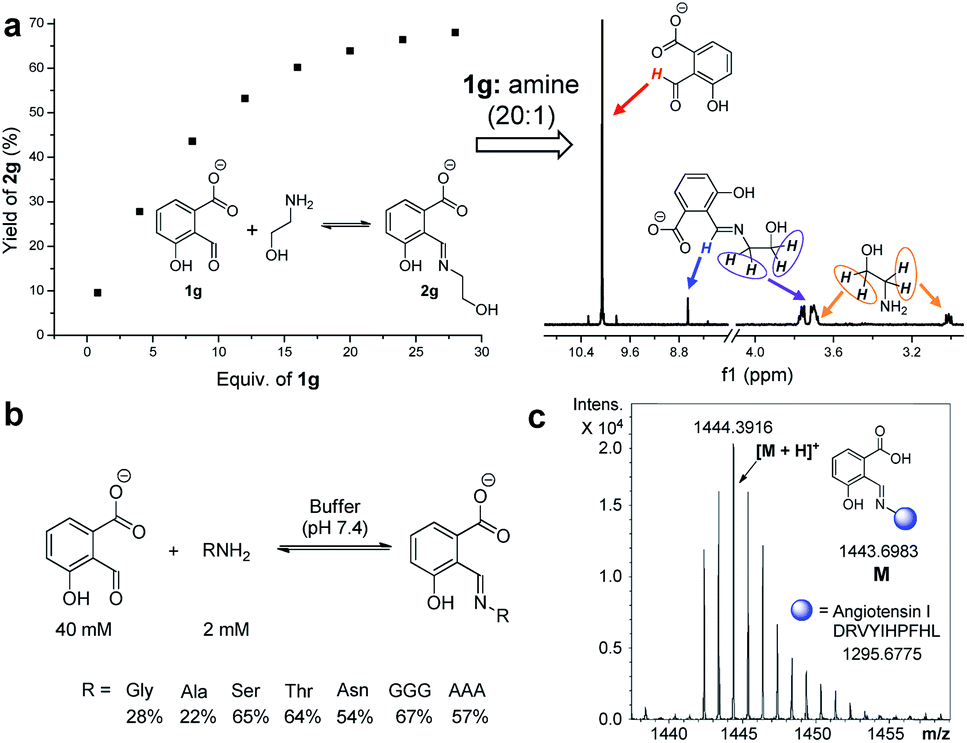 | ||
| Fig. 8 (a) The yield of the reaction of varied concentrations of 1g and ethanolamine (2.0 mM) in buffer (pH 7.4), with one 1H NMR spectrum shown. (b) Representative structures and yields of 2g incorporating amino acids/peptides in the buffer (pH 7.4). (c) ESI-mass spectrum of 2g incorporating the peptide angiotensin I in the buffer (pH 7.4). | ||
Finally, attention was turned to the modification of biomolecules. As an inherent reactive site in peptides/proteins the amine group provides a handle for bioconjugation.21,87,88 For example, the reaction of 1g (40 mM) with amino acids, such as glycine and alanine (2 mM each), afforded the corresponding imine with a yield of 28% and 22% at pH 7.4, respectively (Fig. 8b and S91–S100†). To further check the utility, the modification of several model peptides was examined, such as triglycine, trialanine, and angiotensin I (Fig. 8c). With an aldehyde/peptide ratio of 20:1, the desired product was found by ESI mass spectral analysis (Fig. S101–S108†). These results demonstrate potential applications in biological labelling.
Conclusions
In summary, we developed a facile platform for the modulation of dynamic imine chemistry with n → π* interactions. A detailed investigation of imine exchanges with a library of ortho-carboxyl aromatic aldehydes was conducted, and n → π* interactions played a stabilizing role in the anionic open aldehyde/imine, leading to the shift of imine exchange equilibrium. Moreover, theoretical calculations, including NBO analysis and torsion scan, provided deep insights for n → π* interactions, which can compete with CH hydrogen bonding and can be modulated by the substituent effect. In addition, the incorporation of ortho-carboxyl into salicylaldehyde and pyridine-2-carboxaldehyde demonstrated the feasibility of the use of different recognition sites. Aldehydes with ortho-carboxylate were further employed for the regulation of imine formation/exchange in water, and the modification of the N-terminus of amino acids and peptides was realized at neutral pH. The strategy of ortho-carboxyl containing aldehydes showcases the capability of even weak interactions, such as orbital interactions, for controlling the DCC, and will open new possibilities in dynamic assemblies. Considering the popularity of carbonyls and their derivatives in organic compounds, we anticipate that the use of n → π* interactions in supramolecular chemistry, catalysis, and functional materials has significant growth prospects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NSFC (21672214), the Recruitment Program of Global Youth Experts, and the Strategic Priority Research Program (XDB20000000) and the Key Research Program of Frontier Sciences (QYZDB-SSW-SLH030) of the CAS for funding.Notes and references
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- Y. Jia and J. Li, Chem. Rev., 2015, 115, 1597–1621 CrossRef CAS PubMed.
- H. Vardhan, A. Mehta, I. Nath and F. Verpoort, RSC Adv., 2015, 5, 67011–67030 RSC.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC.
- T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS.
- D. Zhang, T. K. Ronson and J. R. Nitschke, Acc. Chem. Res., 2018, 51, 2423–2436 CrossRef CAS PubMed.
- Y. Liu and Z.-T. Li, Aust. J. Chem., 2013, 66, 9–22 CrossRef CAS.
- G. Zhang and M. Mastalerz, Chem. Soc. Rev., 2014, 43, 1934–1947 RSC.
- A. Wilson, G. Gasparini and S. Matile, Chem. Soc. Rev., 2014, 43, 1948–1962 RSC.
- A. J. McConnel, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef PubMed.
- Q. Ji, R. C. Lirag and O. S. Miljanić, Chem. Soc. Rev., 2014, 43, 1873–1884 RSC.
- J. M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed.
- J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222–9239 CrossRef CAS PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- J. L. Segura, M. J. Mancheno and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
- J. Jiang, Y. Zhao and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 3255–3265 CrossRef CAS PubMed.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- C. Qian, Q. Y. Qi, G. F. Jiang, F. Z. Cui, Y. Tian and X. Zhao, J. Am. Chem. Soc., 2017, 139, 6736–6743 CrossRef CAS PubMed.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- J. M. Gilmore, R. A. Scheck, A. P. Esser-Kahn, N. S. Joshi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 5307–5311 CrossRef CAS PubMed.
- A. Bandyopadhyay, S. Cambray and J. Gao, Chem. Sci., 2016, 7, 4589–4593 RSC.
- M. Ciaccia and S. Di Stefano, Org. Biomol. Chem., 2015, 13, 646–654 RSC.
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC.
- M. Liu, M. A. Little, K. E. Jelfs, J. T. A. Jones, M. Schmidtmann, S. Y. Chong, T. Hasell and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 7583–7586 CrossRef CAS PubMed.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- P. Schmidt, C. Stress and D. Gillingham, Chem. Sci., 2015, 6, 3329–3333 RSC.
- L. Shen, N. Cao, L. Tong, X. Zhang, G. Wu, T. Jiao, Q. Yin, J. Zhu, Y. Pan and H. Li, Angew. Chem., Int. Ed., 2018, 57, 16486–16490 CrossRef CAS PubMed.
- S. R. Beeren and J. K. M. Sanders, Chem. Sci., 2011, 2, 1560–1567 RSC.
- C. M. Metzler, A. Cahill and D. E. Metzler, J. Am. Chem. Soc., 1980, 102, 6075–6082 CrossRef CAS.
- J. Crugeiras, A. Rios, E. Riveiros and J. P. Richard, J. Am. Chem. Soc., 2009, 131, 15815–15824 CrossRef CAS.
- K. W. Bentley and C. Wolf, J. Am. Chem. Soc., 2013, 135, 12200–12203 CrossRef CAS.
- M. Zhang, L. Li, Q. Lin, M. Tang, Y. Wu and C. Ke, J. Am. Chem. Soc., 2019, 141, 5154–5158 CrossRef CAS PubMed.
- J. Holub, G. Vantomme and J. M. Lehn, J. Am. Chem. Soc., 2016, 138, 11783–11791 CrossRef CAS PubMed.
- R. Lavendomme, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 12147–12158 CrossRef CAS PubMed.
- C. He, Z. Lin, Z. He, C. Duan, C. Xu, Z. Wang and C. Yan, Angew. Chem., Int. Ed., 2008, 47, 877–881 CrossRef CAS PubMed.
- P. M. S. D. Cal, J. B. Vicente, E. Pires, A. V. Coelho, L. F. Veiros, C. Cordeiro and M. P. Gois, J. Am. Chem. Soc., 2012, 134, 10299–10305 CrossRef CAS PubMed.
- S. Cambray and J. Gao, Acc. Chem. Res., 2018, 51, 2198–2206 CrossRef CAS PubMed.
- B. M. Chapin, P. Metola, V. M. Lynch, J. F. Stanton, T. D. James and E. V. Anslyn, J. Org. Chem., 2016, 81, 8319–8330 CrossRef CAS PubMed.
- B. Akgun and D. G. Hall, Angew. Chem., Int. Ed., 2018, 57, 2–19 CrossRef PubMed.
- R. W. Newberry and R. T. Raines, Acc. Chem. Res., 2017, 50, 1838–1846 CrossRef CAS PubMed.
- S. K. Singh and A. Das, Phys. Chem. Chem. Phys., 2015, 17, 9596–9612 RSC.
- I. W. Windsor, B. Gold and R. T. Raines, ACS Catal., 2019, 9, 1464–1471 CrossRef CAS PubMed.
- D. J. Schmucker, S. R. Dunbar, T. D. Shepherd and M. A. Bertucci, J. Phys. Chem. A, 2019, 123, 2537–2543 CrossRef CAS PubMed.
- A. Choudhary, K. J. Kamer and R. T. Raines, J. Org. Chem., 2011, 76, 7933–7937 CrossRef CAS PubMed.
- B. Sahariah and B. K. Sarma, Chem. Sci., 2019, 10, 909–917 RSC.
- A. Rahim, P. Saha, K. K. Jha, N. Sukumar and B. K. Sarma, Nat. Commun., 2017, 8, 1–13 CAS.
- J. Echeverría, Chem. Commun., 2018, 54, 3061–3064 RSC.
- H. B. Bürgi, J. D. Dunitz and E. Shelter, J. Am. Chem. Soc., 1973, 95, 5065–5067 CrossRef.
- R. G. Brennan, G. G. Prive, W. J. P. Blonski, F. E. Hruska and M. Sundaralingam, J. Am. Chem. Soc., 1983, 105, 7737–7742 CrossRef CAS.
- C. Siebler, R. S. Erdmann and H. Wennemers, Angew. Chem., Int. Ed., 2014, 53, 10340–10344 CrossRef CAS PubMed.
- C. Fufezan, Proteins: Struct., Funct., Genet., 2010, 78, 2831–2838 CrossRef CAS.
- B. C. Gorske, R. C. Nelson, Z. S. Bowden, T. A. Kufe and A. M. Childs, J. Org. Chem., 2013, 78, 11172–11183 CrossRef CAS PubMed.
- R. W. Newberry, B. VanVeller, I. A. Guzei and R. T. Raines, J. Am. Chem. Soc., 2013, 135, 7843–7846 CrossRef CAS PubMed.
- J. S. Laursen, J. Engel-Andreasen, P. Fristrup, P. Harris and C. A. Olsen, J. Am. Chem. Soc., 2013, 135, 2835–2844 CrossRef CAS.
- J. Echeverría, Cryst. Growth Des., 2018, 18, 506–512 CrossRef.
- R. Shukla and D. Chopra, CrystEngComm, 2018, 20, 3308–3312 RSC.
- H. Wang, W. Wang and W. Jin, Chem. Rev., 2016, 1169, 5072–5104 CrossRef PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, Chem. Rev., 2018, 118, 2010–2041 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- K. Selvakumar and H. B. Singh, Chem. Sci., 2018, 9, 7027–7042 RSC.
- M. Giese, M. Albrecht and K. Rissanen, Chem. Rev., 2015, 1151, 68867–68895 Search PubMed.
- Y. Zhao, Y. CotelleLe, L. Liu, J. López-Andarias, A. Bornhof, M. Akamatsu, N. Sakai and S. Matile, Acc. Chem. Res., 2018, 51, 2255–2263 CrossRef CAS PubMed.
- M. X. Wang, Acc. Chem. Res., 2011, 45, 182–195 CrossRef PubMed.
- D. Tuo, W. Liu, X. Wang, X. Wang, Y. Ao, Q. Wang, Z. Li and D. X. Wang, J. Am. Chem. Soc., 2019, 141, 1118–1125 CrossRef CAS.
- H. Zeng, P. Liu, G. Feng and F. Huang, J. Am. Chem. Soc., 2019, 141, 16501–16511 CrossRef.
- B. Jiang, W. Wang, Y. Zhang, Y. Lu, C. W. Zhang, G. Q. Yin, X. L. Zhao, L. Xu, H. Tan, X. Li, G. X. Jin and H. B. Yang, Angew. Chem., Int. Ed., 2017, 56, 14438–14442 CrossRef CAS PubMed.
- H. Zheng, H. Ye, X. Yu and L. You, J. Am. Chem. Soc., 2019, 141, 8825–8833 CrossRef CAS PubMed.
- E. C. Vik, P. Li, P. J. Pellechia and K. D. Shimizu, J. Am. Chem. Soc., 2019, 141, 16579–16583 CrossRef CAS PubMed.
- G. W. Breton, L. O. Davis, K. L. Martin and T. A. Chambers, Cryst. Growth Des., 2019, 19, 3895–3904 CrossRef CAS.
- P. Kovařícek and J. M. Lehn, Chem.–Eur. J., 2015, 21, 9380–9384 CrossRef PubMed.
- P. Kovaříček, A. C. Meister, K. Flídrová, R. Cabot, K. Kovaříčková and J. M. Lehn, Chem. Sci., 2016, 7, 3215–3226 RSC.
- E. T. Kool, D. H. Park and P. Crisalli, J. Am. Chem. Soc., 2013, 135, 17663–17666 CrossRef CAS PubMed.
- J. Liu, X. Wang, C. Zhao, J. Hao, G. Fang and S. Wang, J. Hazard. Mater., 2018, 344, 220–229 CrossRef CAS PubMed.
- A. G. Santana, E. Jiménez-Moreno, A. M. Gómez, F. Corzana, C. González, G. Jiménez-Oses, J. Jiménez-Barbero and J. L. Asensio, J. Am. Chem. Soc., 2013, 135, 3347–3350 CrossRef CAS PubMed.
- M. K. Chung, P. S. White, S. J. Lee, M. L. Waters and M. R. Gagné, J. Am. Chem. Soc., 2012, 134, 11415–11429 CrossRef CAS PubMed.
- M. K. Chung, S. J. Lee, M. L. Waters and M. R. Gagné, J. Am. Chem. Soc., 2012, 134, 11430–11443 CrossRef CAS PubMed.
- L. Somsák, Chem. Rev., 2001, 101, 81–135 CrossRef PubMed.
- K. B. Wiberg, W. F. Bailey, K. M. Lambert and Z. D. Stempel, J. Org. Chem., 2018, 83, 5242–5255 CrossRef CAS PubMed.
- O. Büyükgüngör and M. Odabaşoǧlu, Acta Crystallogr., 2006, 62, 2749–2750 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- S. Ulrich, Acc. Chem. Res., 2019, 52, 510–519 CrossRef CAS PubMed.
- X. Sun and T. D. James, Chem. Rev., 2015, 115, 8001–8037 CrossRef CAS PubMed.
- M. Mondal and A. K. H. Hirsch, Chem. Soc. Rev., 2015, 44, 2455–2488 RSC.
- J. I. MacDonald, H. K. Munch, T. Moore and M. B. Francis, Nat. Chem. Biol., 2015, 11, 326–331 CrossRef CAS PubMed.
- K. A. McCarthy, M. A. Kelly, K. Li, S. Cambray, A. S. Hosseini, T. van Opijnen and J. Gao, J. Am. Chem. Soc., 2018, 140, 6137–6145 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, X-ray data, NMR and mass spectra, and DFT calculations. CCDC 1964753 and 1964754. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05698j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |