
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilica-supported, narrowly distributed, subnanometric Pt–Zn particles from single sites with high propane dehydrogenation performance†
Lukas
Rochlitz
a,
Keith
Searles
a,
Jan
Alfke
ac,
Dmitry
Zemlyanov
 b,
Olga V.
Safonova
c and
Christophe
Copéret
*a
b,
Olga V.
Safonova
c and
Christophe
Copéret
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1-5, CH-8093 Zürich, Switzerland. E-mail: ccoperet@ethz.ch
bBirck Nanotechnology Center, Purdue University, 1205 West State Street, West Lafayette, Indiana 47907, USA
cPaul Scherrer Institut, CH-5232 Villigen, Switzerland
First published on 23rd December 2019
Abstract
The development of highly productive, selective and stable propane dehydrogenation catalysts for propene production is strategic due to the increasing need for propene and the availability of shale gas, an abundant source of light alkanes. In that context, the combination of surface organometallic chemistry (SOMC) and a thermolytic molecular precursor (TMP) approach is used to prepare bimetallic subnanometric and narrowly distributed Pt–Zn alloyed particles supported on silica via grafting of a Pt precursor on surface OH groups present in a Zn single-site containing material followed by a H2 reduction treatment. This material, that exhibits a Zn to Pt molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 in the form of alloyed Pt–Zn particles with a 0.2 to 0.4 fraction of the overall Zn amount remaining as ZnII sites on the silica surface, catalyzes propane dehydrogenation (PDH) with high productivity (703 gC3H6 gPt−1 h−1 to 375 gC3H6 gPt−1 h−1) and very low deactivation rates (kd = 0.027 h−1) over 30 h at high WHSV (75 h−1). This study demonstrates how SOMC can provide access to highly efficient and tailored catalysts through the stepwise introduction of specific elements via grafting to generate small, homogeneously and narrowly distributed supported alloyed nanoparticles at controlled interfaces.
:2 in the form of alloyed Pt–Zn particles with a 0.2 to 0.4 fraction of the overall Zn amount remaining as ZnII sites on the silica surface, catalyzes propane dehydrogenation (PDH) with high productivity (703 gC3H6 gPt−1 h−1 to 375 gC3H6 gPt−1 h−1) and very low deactivation rates (kd = 0.027 h−1) over 30 h at high WHSV (75 h−1). This study demonstrates how SOMC can provide access to highly efficient and tailored catalysts through the stepwise introduction of specific elements via grafting to generate small, homogeneously and narrowly distributed supported alloyed nanoparticles at controlled interfaces.
Introduction
Propene is the second most utilized building block of the petrochemical industry besides ethene, and its production has been dramatically influenced by the emergence of shale gas resources. Indeed, crackers have been converted from naphtha to ethane units, with ethane feedstocks predominantly producing ethene, thereby reducing propene production. To compensate for the resulting propene production gap, the development of on-purpose propene production technologies has thus been on the rise.1,2 The most used on-purpose technology is selective propane dehydrogenation (PDH), a highly energy intensive process (ΔH0298 = 124.3 kJ mol−1), industrially implemented mainly via two processes using a bimetallic Pt–Sn/Al2O3 (UOP Oleflex process) or a metal-oxide based Cr2O3/Al2O3 (Lummus Catofin process) catalyst.3 Due to the requirement of high reaction temperatures (500–700 °C) to reach reasonable conversion levels, one of the biggest issues for the existing systems is catalyst stability due to coke formation and sintering requiring constant and rapid regeneration.3 The development and understanding of catalytic systems with increased stability while keeping a high selectivity and productivity is thus an intense field of research. In particular, major research endeavors focused on bi- and multi-metallic systems related to the industrial Pt–Sn/Al2O3 catalyst. In all cases, a combination of Pt with a second metal (In, Sn, Ga, Cu, Zn, etc.) beneficially influenced the catalyst performance as compared to monometallic Pt catalysts.4–9 While the selectivity for most of the systems reaches high levels, all of them suffer from deactivation. Zn, a highly abundant and non-toxic metal, is of special interest as the second component in bimetallic systems for PDH but has been studied less extensively when compared to more prominent post-transition-metals Sn and Ga.3,10Most synthetic strategies for bimetallic Pt–Zn dehydrogenation catalysts rely on well-established impregnation techniques.9,11,12 While being simple catalyst preparation methods, impregnation techniques typically yield poorly defined systems with inhomogeneous distribution of the components due to complex dissolution/precipitation events that occur in aqueous conditions. In order to develop more controlled preparation methods, surface organometallic chemistry (SOMC)13–15 in combination with the thermolytic molecular precursor approach (TMP)16,17 has emerged as a powerful preparation technique. In particular, it has been shown that supported single-sites can be used as building blocks to generate supported nanoparticles with controlled interfaces, allowing the introduction of dopants at the interface between silica and the metal particles or alternatively yielding a bimetallic alloy supported on SiO2.17–20 A Pt–Ga alloy prepared through this approach displays high productivity and stability in the PDH reaction that has been attributed to surface dilution of Pt upon the introduction of gallium.21
In view of the known activity of Pt–Zn in PDH and improved performance of catalysts prepared via SOMC/TMP approach, we reason that SOMC could constitute an ideal way to generate small and narrowly dispersed Pt–Zn alloys by first installing ZnII single sites on a SiO2 support followed by anchoring of a Pt precursor on the ZnII/SiO2 material and a subsequent treatment under H2 to generate the desired particles. These silica-supported Pt–Zn nanoparticles were characterized by a multi-technique approach (CO and pyridine adsorption FTIR as well as X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) studies); they show high productivity (703 gC3H6/(gPt−1 h−1)), high selectivity (≥95%) and an outstanding stability (kd = 0.027) at high WHSV compared to other Pt–Zn based systems for PDH at 550 °C. This improved performance is attributed to the formation of subnanometric and narrowly distributed alloyed particles supported on ZnII modified silica that likely plays a role in stabilizing these particles under PDH reaction conditions.
Results and discussion
The bimetallic Pt–Zn system was prepared via SOMC in a three-step process involving first the generation of Zn single-sites, then grafting of Pt(II) molecular precursor onto regenerated OH groups followed by a hydrogen treatment. Grafting of [Zn(OSi(OtBu)3)2]2 on SiO2–700 followed by a thermal treatment at 600 °C yielded ZnII/SiO2 as a white solid that contains ZnII single sites (1.73 wt% Zn to 0.80 Zn per nm2), free of organic ligands along with isolated OH sites (0.66 OH per nm2) (Fig. 1(a)).22,23 In the next step, [Pt(OSi(OtBu)3)2(COD)] (1.2 equiv./OH)24,25 (COD = 1,5-cyclooctadiene) was grafted onto the surface OH groups of ZnII/SiO2; the resulting supernatant contained [Pt(OSi(OtBu)3)2(COD)] (0.35 equiv. Ptadded−1) and HOSi(OtBu)3 (0.35 equiv. Ptadded−1). The obtained white material – Pt(OSi(OtBu)3)(COD)ZnII/SiO2 – dried under high vacuum (10−5 mbar) contained 2.90 wt% Pt, 1.62 wt% Zn (1.66 equiv. Pt−1), 4.44 wt% C (24.9 equiv. Pt−1) and 0.79 wt% H (52.7 equiv. Pt−1). The too high C and H values (20 C and 39 H expected) can be explained by a chemisorption (coordination) of some of the released HOSi(OtBu)3 – formed during grafting of the Pt precursor – on the silica-support containing ZnII Lewis acid sites. This is evidenced by the low amount of observed HOSi(OtBu)3 released in solution compared to amount of remaining [Pt(OSi(OtBu)3)2(COD)], revealing that 45% of the expected HOSi(OtBu)3 is missing and remains chemisorbed at the surface of the support consistent with the elemental analysis. The IR spectrum revealed only partial consumption of the surface OH groups compared to ZnII/SiO2. This process is accompanied by the appearance of ν(C–H) at 3000–2800 cm−1 and δ(C–H) at 1500–1340 cm−1 (Fig. 1(b) and ESI Fig. S3†). All these data are consistent with grafting of the PtII precursor through surface OH groups and the retention of some silanol on the surface. Furthermore, solid-state NMR supports the formation of![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiOPt(OSi(OtBu)3)(COD) with signals at 1.5 ppm and 5.0 ppm in 1H SSNMR and signals at 30 ppm and 91 ppm in the 13C SSNMR spectrum, respectively (ESI Fig. S4 and S5†). The data is consistent with the formation of a grafted PtII complex together with Zn single-sites in Pt(OSi(OtBu)3)(COD)ZnII/SiO2. The material was subsequently treated under H2 at 600 °C for 8 h yielding a black material – Pt0Znδ+/SiO2 – suggesting the formation of particles (3.05 wt% Pt, 1.54 wt% Zn). HAADF-STEM images of Pt0Znδ+/SiO2 confirmed the formation of well-dispersed and narrowly distributed sub-nanometric particles (0.8 ± 0.2 nm) (Fig. 1(c)).
SiOPt(OSi(OtBu)3)(COD) with signals at 1.5 ppm and 5.0 ppm in 1H SSNMR and signals at 30 ppm and 91 ppm in the 13C SSNMR spectrum, respectively (ESI Fig. S4 and S5†). The data is consistent with the formation of a grafted PtII complex together with Zn single-sites in Pt(OSi(OtBu)3)(COD)ZnII/SiO2. The material was subsequently treated under H2 at 600 °C for 8 h yielding a black material – Pt0Znδ+/SiO2 – suggesting the formation of particles (3.05 wt% Pt, 1.54 wt% Zn). HAADF-STEM images of Pt0Znδ+/SiO2 confirmed the formation of well-dispersed and narrowly distributed sub-nanometric particles (0.8 ± 0.2 nm) (Fig. 1(c)).
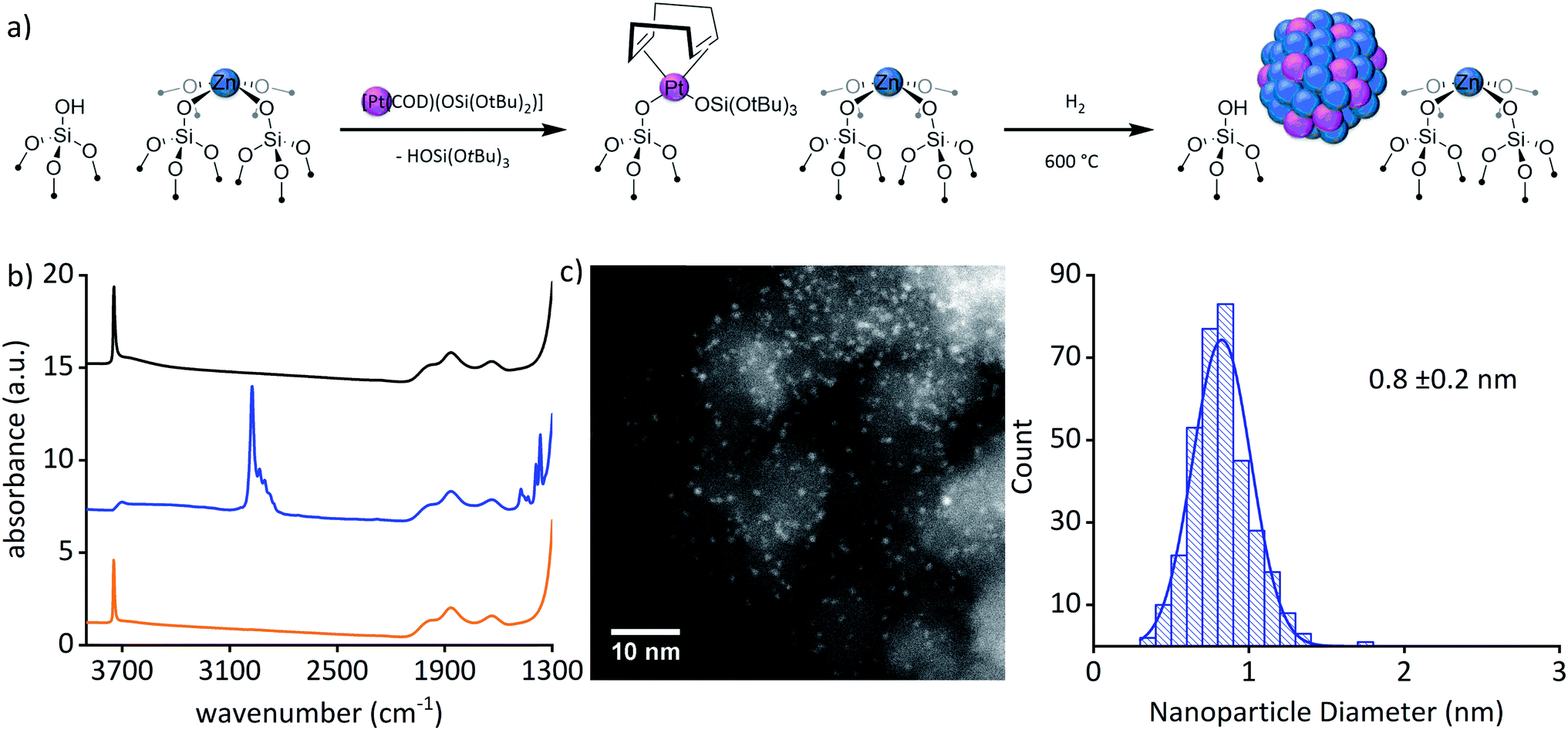 | ||
| Fig. 1 (a) Schematic representation of the synthesis of the material Pt0Znδ+/SiO2. (b) FTIR spectra of ZnII/SiO2 (orange), Pt(OSi(OtBu)3)(COD)ZnII/SiO2 (blue) and Pt0Znδ+/SiO2 (black). (c) Representative HAADF-STEM image and particle size distribution of Pt0Znδ+/SiO2. | ||
The IR spectrum of a Pt0Znδ+/SiO2 self-supporting pellet exposed to CO (3 mg, 12 mbar) shows an intense CO vibrational band centered at 2046 cm−1 that is red shifted by 38 cm−1 compared to what is observed for Pt particles (2084 cm−1) supported on silica – Pt0/SiO2 (2.2 ± 0.8 nm) – prepared by a similar approach (grafting of [Pt(OSi(OtBu)3)2(COD)] onto SiO2–700 followed by a treatment under H2 at 500 °C). The shift is ascribed to an inherent difference in the particle surfaces of the Pt0/SiO2 and Pt0Znδ+/SiO2 materials attributed to alloy formation of the two components in Pt0Znδ+/SiO2 (Fig. 2(a)).26 Additionally, H2 and CO chemisorption studies also support the inherent difference of the particle surface in Pt0Znδ+/SiO2 compared to Pt0/SiO2 (see ESI† for details). A background subtracted spectrum of Pt0Znδ+/SiO2 (12 mg, 120 mbar CO; Fig. 2(b)) shows weak bands at 2202 cm−1 and 2179 cm−1 consistent with CO adsorbed on ZnII sites, similar to those found in ZnII/SiO2 (13 mg, 123 mbar CO; 2206 cm−1) and ZnII sites that are likely to be in close proximity to the alloyed particles. Another band at 1906 cm−1 – not observable for Pt0/SiO2 – is consistent with a μ2-binding mode of CO on the Pt0Znδ+/SiO2 material. In addition to the CO IR studies, pyridine desorption was also used to probe the surface property of the material. Vibrational bands at 1538 cm−1, 1438 cm−1 and 1408 cm−1 are associated with the interaction of pyridine with Pt species on SiO2 and α-pyridyl species on Pt{111}.27,28 Bands at 1610 cm−1 and 1452 cm−1 – similar to ZnII/SiO2 – as well as 1595 cm−1 and 1445 cm−1 for Pt0Znδ+/SiO2 indicate two different types of Lewis acidic Zn sites – a stronger and a weaker one – on the surface of the material.22,29 No bands above 1610 cm−1 indicate a low Brønsted acidity of the support (see ESI† for details).30 The combined results of the CO and pyridine IR studies suggest the formation of an alloyed Pt–Zn material along with residual surface ZnII sites.
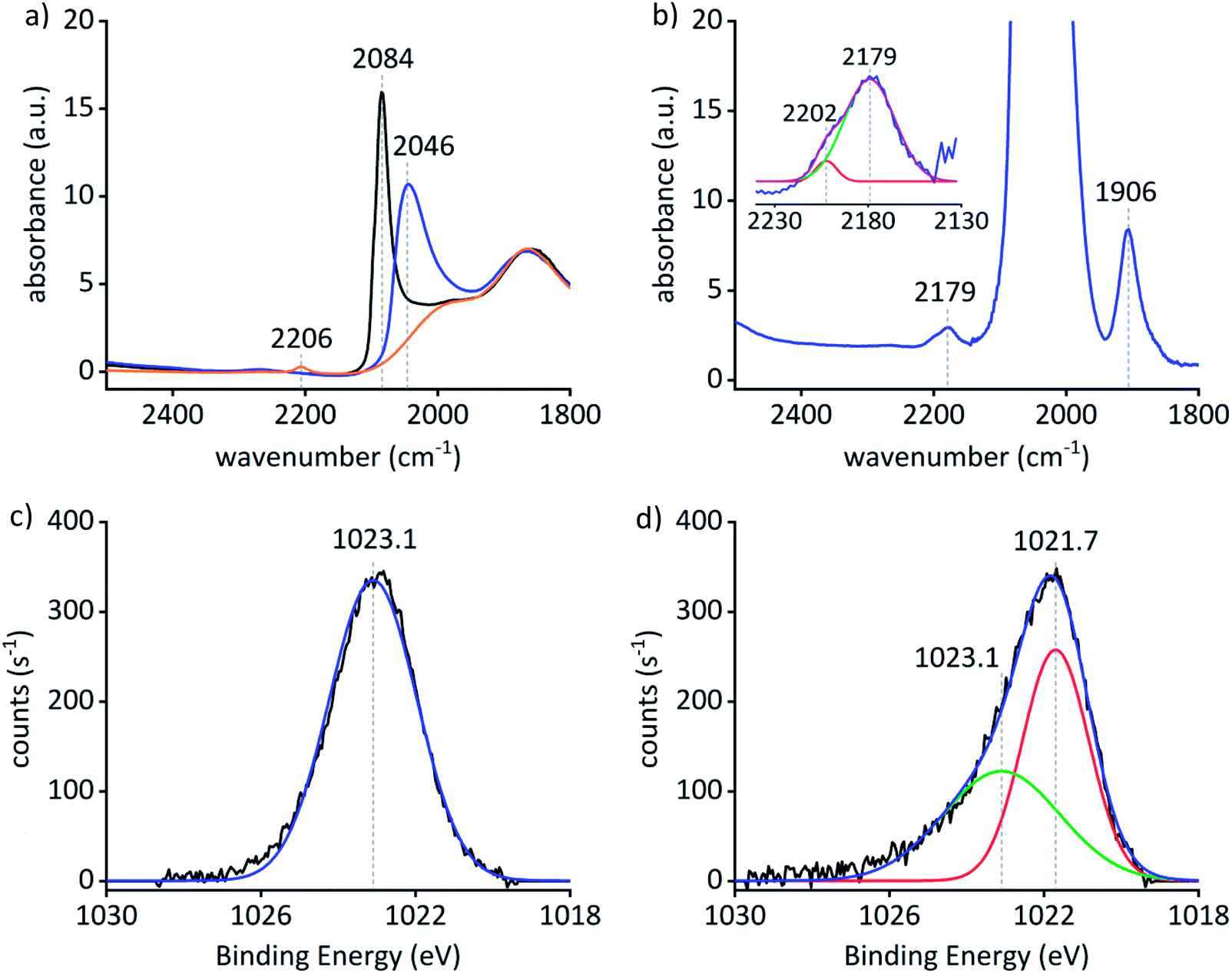 | ||
| Fig. 2 (a) CO adsorption on Pt0/SiO2 (black), ZnII/SiO2 (orange) and Pt0Znδ+/SiO2 (blue). The spectra are normalized to the υSiO vibrational frequency at 1865 cm−1. (b) Background subtracted spectrum of CO adsorption on Pt0Znδ+/SiO2 (blue) and two component Gaussian fit (red, green; cumulative fit (purple)) of the CO adsorption region around 2179 cm−1 of Pt0Znδ+/SiO2. (c) High resolution Zn 2p3/2 XPS spectrum of ZnII/SiO2 (black) and Gaussian–Lorentzian fit to the data (blue). (d) High resolution Zn 2p3/2 XPS spectrum for Pt0Znδ+/SiO2 (black), two component Gaussian–Lorentzian fit to the data (green: 45%; red: 55%) and cumulative fit (blue). | ||
XPS was used to gather further insight in the chemical state of Pt and Zn as well as the composition of the material. The results are summed up in Table 1. The Pt 4f7/2 binding energy in Pt0Znδ+/SiO2 is 71.6 eV which is shifted by +0.3 eV compared to Pt0/SiO2, likely due to the restructuring of the Pt 4f states upon alloying with Zn.31 The Zn 2p3/2 peak for ZnII/SiO2 consists of a symmetric component centered at 1023.1 eV (Fig. 2(c)), while the Zn 2p3/2 peak for Pt0Znδ+/SiO2 (Fig. 2(d)) is asymmetric and was therefore fitted with two components at 1021.7 eV (55% of area) and 1023.1 eV (45% of area). The lower binding energy component can be attributed to Zn0 (through ZnII reduction). This binding energy is close to the reported value for Zn metal (1021.8(2) eV),32 while the other value parallels what is found for ZnII/SiO2, in accordance with a minority of Zn remaining on the surface as ZnII.
X-ray absorption studies of the materials have also been performed at the Zn K-edge and Pt L3-edge in order to obtain a better understanding of the nature of Pt and Zn in the catalyst precursors and Pt0Znδ+/SiO2. Fig. 3(a) shows the Zn K-edge X-ray absorption near-edge structure (XANES) spectra of Pt(OSi(OtBu)3)(COD)ZnII/SiO2, Pt0Znδ+/SiO2 and Zn foil. An edge shift of −3.8 eV from Pt(OSi(OtBu)3)(COD)ZnII/SiO2 to Pt0Znδ+/SiO2 clearly indicates the reduction of ZnII to Zn0. The differences in shape and edge position of Pt0Znδ+/SiO2 compared to Zn foil are attributed to alloy formation of Zn with Pt along with ZnII species as supported by comparison of the derivative spectra of Pt(OSi(OtBu)3)(COD)ZnII/SiO2 and Pt0Znδ+/SiO2 (Fig. 3(b)) revealing the existence of a remaining oxidized Zn species in Pt0Znδ+/SiO2 – in accordance with the observations of adsorption IR studies and XPS – attributed to ZnII sites on the support surface. A linear combination fit of Pt(OSi(OtBu)3)(COD)ZnII/SiO2 and Zn foil spectra suggests 22% of Zn remaining as ZnII sites on the surface of Pt0Znδ+/SiO2 (ESI Fig. S21†). Analysis of the extended X-ray absorption fine structure (EXAFS) of the Zn K-edge of the material precursors Pt(OSi(OtBu)3)(COD)ZnII/SiO2 and ZnII/SiO2 (see Table 2 and ESI Fig. S24–S27 and Tables S4, S5†) reveals a significant elongation of the Zn–O bond distance in the former material compared to the latter, an indication for some changes in the local environment of Zn upon grafting of Pt(OSi(OtBu)3)2(COD), consistent with the interaction of the ZnII sites with the Pt precursor. In both materials the inclusion of a Zn–Zn instead of a Zn–Si path decreased the fit quality significantly – indicating the high dispersion of ZnII single sites in the precatalyst before the H2 treatment – also confirmed by a wavelet analysis of [Zn(OSi(OtBu)3)2]2 and Pt(OSi(OtBu)3)(COD)ZnII/SiO2 (ESI Fig. S28–S33†), clearly showing the disappearance of the Zn–Zn scattering pathway in the latter material. Detailed EXAFS analysis of Pt0Znδ+/SiO2 was not possible due to the presence of overlapping scattering paths, resulting in fits without physical meaning.
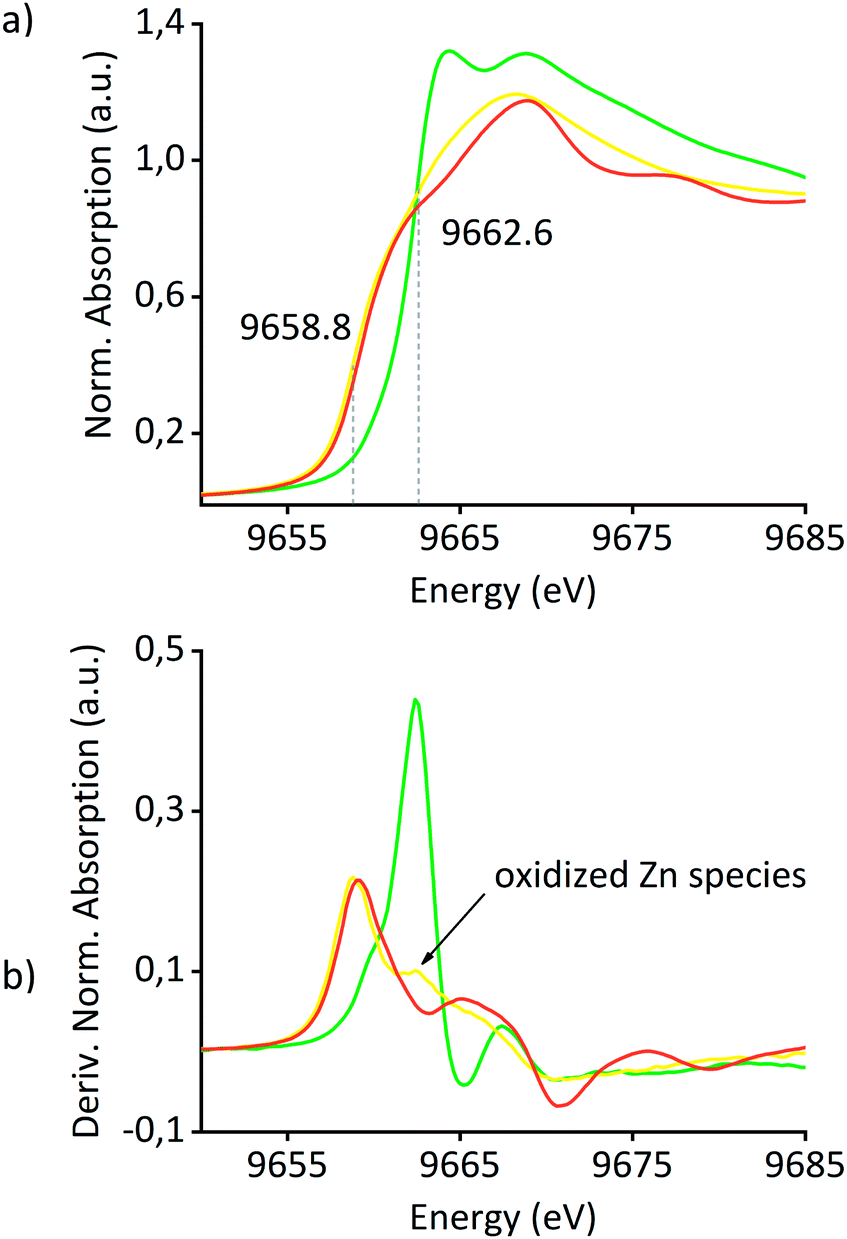 | ||
| Fig. 3 (a) XANES spectra of Pt(OSi(OtBu)3)(COD)ZnII/SiO2 (green), Pt0Znδ+/SiO2 (yellow) and Zn foil (red). (b) First derivative spectra of the same materials. | ||
| Sample | Neighbor, Nb | r [Å] | σ 2 [Å2] |
|---|---|---|---|
| a Samples recorded in transmission mode. b Number of specified neighbors. c Distance to neighbor. d Debye–Waller factor. | |||
| Pt L 3 -edge | |||
| Pt0Znδ+/SiO2 | Pt, 3.1 ± 1.4 | 2.62 ± 0.01 | 0.008 ± 0.002 |
| Zn, 6.7 ± 2.4 | 2.48 ± 0.03 | 0.022 ± 0.004 | |
| Pt0/SiO2 | Pt, 9.1 ± 0.4 | 2.747 ± 0.002 | 0.0058 ± 0.0002 |
|
|||
| Zn K-edge | |||
| ZnII/SiO2 | O, 3.5 ± 0.9 | 1.88 ± 0.02 | 0.016 ± 0.004 |
| Si, 0.7 ± 0.6 | 3.07 ± 0.04 | 0.005 ± 0.008 | |
| Pt(OSi(OtBu)3)(COD)ZnII/SiO2 | O, 3.8 ± 0.5 | 1.94 ± 0.01 | 0.011 ± 0.002 |
| Si, 0.7 ± 0.4 | 3.09 ± 0.02 | 0.002 ± 0.004 | |
XANES analysis of the Pt L3 edge of Pt(OSi(OtBu)3)(COD)ZnII/SiO2 and Pt0Znδ+/SiO2 follows similar trends as the Zn K edge (ESI Fig. S33 and S34†) with a strong decrease in white line intensity upon H2 treatment – indicating reduction of the corresponding metal – and a shift to lower edge energy, supporting what was observed for the Zn K edge. EXAFS analysis of the Pt L3 edge of Pt0/SiO2 and Pt0Znδ+/SiO2 (see Table 2) reveals a considerably shortened Pt–Pt bond distance in the bimetallic material consistent with structural changes and alloy formation. Furthermore, an approximately 2:1 (Zn:Pt) ratio of nearest neighbours for Pt0Znδ+/SiO2 suggests a 1:1 metal ratio in the nanoparticles (see ESI† for details). However, large errors on the coordination numbers and the fact that XAS only provides average data do not allow the precise determination of particle composition and homogeneity of the alloying. Based on the EA, XPS and XAS results, it can be concluded that Pt0Znδ+/SiO2 consists of alloyed, Pt–Zn nanoparticles supported on SiO2 with a fraction of 0.2 to 0.4 of the total Zn remaining as ZnII sites on the surface of the material.
The materials ZnII/SiO2, Pt0/SiO2 and Pt0Znδ+/SiO2 were then tested in the PDH reaction at 550 °C under flow conditions (50 ml min−1; 20% C3H8 in Ar) in a stainless-steel tubular reactor where negligible mass and heat transfer limitations occur (see calculations in ESI†). The results are summarized in Table 3. A very high initial productivity 703 gC3H6 gPt−1 h−1 with a conversion of 30.2% and selectivity of 98.1% to C3H6 could be achieved for Pt0Znδ+/SiO2 at a WHSV of 75 h−1. The high selectivity could be maintained over the course of 30 h time on stream with a final selectivity of 95.0% while the conversion dropped to a final 16.1% conversion (kd = 0.027 h−1 – see eqn (1)) and a productivity of 375 gC3H6 gPt−1 h−1.
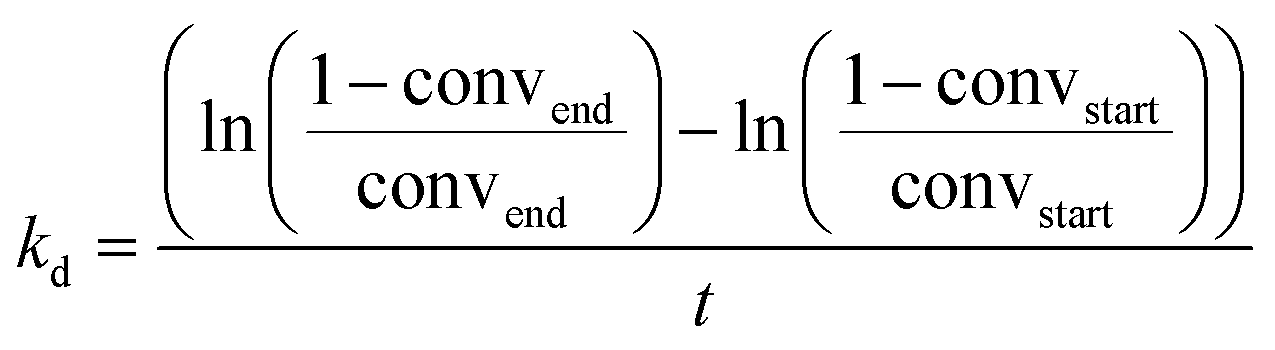 | (1) |
| Sample | Time [h] | Conversion [%] | Selectivityb [%] | Carbon balance [%] | Productivity [gC3H6/gPt−1 h−1] | WHSV [h−1] | k d [h−1] |
|---|---|---|---|---|---|---|---|
| a 50 ml min−1, 20% C3H8 in Ar. b Selectivity for C3H6, only volatile compounds taken into account. c kd = (ln((1 − convend)/convend) − ln((1 − convstart)/convstart))/t. | |||||||
| Pt0/SiO2 | 0.1 | 2.5 | 74.9 | >99 | 14.5 | 32 | 0.26 |
| 2 | 1.5 | 47.1 | 8.7 | ||||
| ZnII/SiO2 | 0.1 | 0.9 | 43.3 | >99 | — | 32 | 0 |
| 10 | 0.9 | 39.6 | — | ||||
| Pt0Znδ+/SiO2 | 0.1 | 35.3 | 97.6 | 97 | 350 | 32 | 0.014 |
| 30 | 26.6 | 96.3 | 264 | ||||
| Pt0Znδ+/SiO2 | 0.1 | 30.2 | 98.1 | 97 | 703 | 75 | 0.027 |
| 30 | 16.1 | 95.0 | 375 | ||||
Catalytic tests at lower WHSV of 32 h−1 showed a significant increase in the initial conversion up to 35.3%, while maintaining high selectivity (>96%) and an almost closed carbon balance (97%). In contrast, Pt0/SiO2 shows a very low initial productivity (14.5 gC3H6 gPt−1 h−1) and a high deactivation rate (kd = 0.26) over the course of 2 h, while ZnII/SiO2 shows comparable catalytic performance to SiO2–700 (1.2% conversion, 39–38% selectivity over 10 h) revealing the absence of catalytic activity of ZnII/SiO2 under these conditions.
Comparison of Pt0Znδ+/SiO2 to the monometallic Pt0/SiO2 shows a dramatic improvement in the productivity of the bimetallic system by more than one order of magnitude. Comparison of Pt0Znδ+/SiO2 to other Pt–Zn systems reveals superior stability of the reported system (ESI Table S9†). While most systems show high selectivity and conversion levels, significantly larger deactivation factors are observed in comparison to Pt0Znδ+/SiO2 or the studies include H2 co-feeding33 and significantly lower weight hourly space velocities to decrease deactivation rates. Furthermore, the metal based productivity and the stability of the reported system also surpass these of the recently published Pt–Ga based system (661 (357 after 20 h) gC3H6 gPt−1 h−1; kd = 0.041 h−1) prepared via the same SOMC/TMP approach.21
To further investigate the structural stability of Pt0Znδ+/SiO2 under PDH conditions in situ XAS studies were performed under the same conditions as the catalytic tests. The spectra of Pt0Znδ+/SiO2 and Pt0/SiO2 show no significant change at the Pt L3 edge over 8 h and 2 h, respectively. The Zn K edge spectra of Pt0Znδ+/SiO2 show a slight and consistent shift over 8 h, indicating a slight structural change for Zn. Preliminary analysis indicates the further – but not complete – reduction of ZnII sites to Zn0 (ESI Fig. S48 and S49†).
The very high productivity of the reported Pt0Znδ+/SiO2 system compared to most other Pt–Zn systems is attributed to the formation of subnanometric alloyed particles, probably a result of the high metal dispersion in the precatalyst before reduction. These particles show minor growth during catalysis over 30 h as shown by post-catalysis TEM analysis (ESI Fig. S8†). The narrow particle size distribution and the presence of remaining Lewis acidic ZnII sites on the materials surface could both play a significant role in stabilizing this catalyst. Post-catalysis characterization shows the formation of coke as well as minor particle growth (ESI Fig. S8 and S54†), which both are most likely contributing to catalyst deactivation.
Conclusions
This work shows that utilizing SOMC/TMP as a synthetic methodology enables the formation of narrowly distributed bimetallic Pt–Zn subnanometric particles supported on SiO2. The resulting high productivity and stability in the PDH reaction for the tested range of WHSV compared to other Pt–Zn systems are attributed to the combination of alloying and high metal dispersion. Furthermore, the remaining ZnII surface sites likely play a role to prevent sintering. SOMC/TMP is a unique synthetic tool to address the origin of catalytic performances in complex multi-metallic systems, where composition, size and support effects can play a crucial role; we are currently further investigating this approach as a general tool for the synthesis of model catalysts to establish detailed structure–activity relationships.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. S. R. thanks the Swiss National Science Foundation (SNSF fond number: 200021_169134) for funding. J. A. thanks the Sinergia project of the Swiss National Science Foundation (SNSF fond number: CRSII5_183495) for funding. We thank Dr Dmitry Lebedev and ScopeM for help with TEM measurements. Fabian Müller and Debora Thöny are acknowledged for help with TGA measurements. Ka Wing Chan is thanked for help with XAS interpretation. Scott Docherty is acknowledged for help with chemisorption measurements. Dr Gina Noh, David Trummer, Petr Šot and Scott Docherty are acknowledged for discussions. The whole Copéret group is acknowledged for help with XAS measurements.References
-
J. S. Plotkin, The Propylene Gap: How Can It Be Filled? – American Chemical Society, https://www.acs.org/content/acs/en/pressroom/cutting-edge-chemistry/the-propylene-gap-how-can-it-be-filled.html, accessed 1 November 2019 Search PubMed
.
-
J. S. Plotkin, The Propylene Quandary – American Chemical Society, https://www.acs.org/content/acs/en/pressroom/cutting-edge-chemistry/the-propylene-quandary.html, accessed 1 November 2019 Search PubMed
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS
- J. Liu, W. Zhou, D. Jiang, W. Wu, C. Miao, Y. Wang and X. Ma, Ind. Eng. Chem. Res., 2018, 57, 11265–11270 CrossRef CAS
- Z. Han, S. Li, F. Jiang, T. Wang, X. Ma and J. Gong, Nanoscale, 2014, 6, 10000–10008 RSC
- E. C. Wegener, Z. Wu, H.-T. Tseng, J. R. Gallagher, Y. Ren, R. E. Diaz, F. H. Ribeiro and J. T. Miller, Catal. Today, 2018, 299, 146–153 CrossRef CAS
- J. J. H. B. Sattler, I. D. Gonzalez-Jimenez, L. Luo, B. A. Stears, A. Malek, D. G. Barton, B. A. Kilos, M. P. Kaminsky, T. W. G. M. Verhoeven, E. J. Koers, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2014, 53, 9251–9256 CrossRef CAS PubMed
- Q. Zhang, K. Zhang, S. Zhang, Q. Liu, L. Chen, X. Li, C. Wang and L. Ma, J. Catal., 2018, 368, 79–88 CrossRef CAS
- J. T. Miller, V. J. Cybulskis, B. C. Bukowski, H.-T. Tseng, J. R. Gallagher, Z. Wu, E. Wegener, A. J. Kropf, B. Ravel, F. H. Ribeiro and J. Greeley, ACS Catal., 2017, 7, 4173–4181 CrossRef
- Z. Nawaz, Rev. Chem. Eng., 2015, 31, 413–436 CAS
- Y. Zhang, Y. Zhou, L. Huang, S. Zhou, X. Sheng, Q. Wang and C. Zhang, Chem. Eng. J., 2015, 270, 352–361 CrossRef CAS
- G. Liu, L. Zeng, Z.-J. Zhao, H. Tian, T. Wu and J. Gong, ACS Catal., 2016, 6, 2158–2162 CrossRef CAS
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed
- M. K. Samantaray, E. Pump, A. Bendjeriou-Sedjerari, V. D'Elia, J. D. A. Pelletier, M. Guidotti, R. Psaro and J.-M. Basset, Chem. Soc. Rev., 2018, 47, 8403–8437 RSC
- M. M. Stalzer, M. Delferro and T. J. Marks, Catal. Lett., 2015, 145, 3–14 CrossRef CAS
- K. L. Fujdala and T. D. Tilley, J. Catal., 2003, 216, 265–275 CrossRef CAS
- C. Copéret, Acc. Chem. Res., 2019, 52, 1697–1708 CrossRef PubMed
- K. Larmier, W.-C. Liao, S. Tada, E. Lam, R. Verel, A. Bansode, A. Urakawa, A. Comas-Vives and C. Copéret, Angew. Chem., Int. Ed., 2017, 56, 2318–2323 CrossRef CAS PubMed
- E. Lam, K. Larmier, P. Wolf, S. Tada, O. V. Safonova and C. Copéret, J. Am. Chem. Soc., 2018, 140, 10530–10535 CrossRef CAS PubMed
- G. Noh, E. Lam, J. L. Alfke, K. Larmier, K. Searles, P. Wolf and C. Copéret, ChemSusChem, 2019, 12, 968–972 CrossRef CAS PubMed
- K. Searles, K. W. Chan, J. A. Mendes Burak, D. Zemlyanov, O. Safonova and C. Copéret, J. Am. Chem. Soc., 2018, 140, 11674–11679 CrossRef CAS PubMed
- A. K. Cook and C. Copéret, Organometallics, 2018, 37, 1342–1345 CrossRef CAS
- N. Rendón, A. Bourdolle, P. L. Baldeck, H. Le Bozec, C. Andraud, S. Brasselet, C. Coperet and O. Maury, Chem. Mater., 2011, 23, 3228–3236 CrossRef
- P. Laurent, L. Veyre, C. Thieuleux, S. Donet and C. Copéret, Dalton Trans., 2013, 42, 238–248 RSC
- D. A. Ruddy, J. Jarupatrakorn, R. M. Rioux, J. T. Miller, M. J. McMurdo, J. L. McBee, K. A. Tupper and T. D. Tilley, Chem. Mater., 2008, 20, 6517–6527 CrossRef CAS
- A. G. T. M. Bastein, F. J. C. M. Toolenaar and V. Ponec, J. Catal., 1984, 90, 88–95 CrossRef CAS
- B. A. Morrow, I. A. Cody, L. E. Moran and R. Palepu, J. Catal., 1976, 44, 467–476 CrossRef CAS
- S. Haq and D. A. King, J. Phys. Chem., 1996, 100, 16957–16965 CrossRef CAS
- C. Morterra and G. Cerrato, Catal. Lett., 1991, 10, 357–363 CrossRef CAS
- R. Ferwerda, J. H. van der Maas and F. B. van Duijneveldt, J. Mol. Catal. A: Chem., 1996, 104, 319–328 CrossRef CAS
- J. A. Rodriguez and M. Kuhn, J. Chem. Phys., 1995, 102, 4279–4289 CrossRef CAS
- S. P. Kowalczyk, R. A. Pollak, F. R. McFeely, L. Ley and D. A. Shirley, Phys. Rev. B: Solid State, 1973, 8, 2387–2391 CrossRef CAS
- V. Galvita, G. Siddiqi, P. Sun and A. T. Bell, J. Catal., 2010, 271, 209–219 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, material characterization data, catalytic measurement details. See DOI: 10.1039/c9sc05599a |
| This journal is © The Royal Society of Chemistry 2020 |