
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA water-based and metal-free dye solar cell exceeding 7% efficiency using a cationic poly(3,4-ethylenedioxythiophene) derivative†
Federico
Bella
 *a,
Luca
Porcarelli
*bc,
Daniele
Mantione
d,
Claudio
Gerbaldi
a,
Claudia
Barolo
e,
Michael
Grätzel
f and
David
Mecerreyes
cg
*a,
Luca
Porcarelli
*bc,
Daniele
Mantione
d,
Claudio
Gerbaldi
a,
Claudia
Barolo
e,
Michael
Grätzel
f and
David
Mecerreyes
cg
aGAME Lab, Department of Applied Science and Technology – DISAT, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 – Torino, Italy. E-mail: federico.bella@polito.it; Tel: +39 0110904643
bInstitute for Frontier Materials, Deakin University, Waurn Ponds, Geelong, VIC 3217, Australia
cPolymat, Institute for Polymer Materials, University of the Basque Country UPV/EHU, Joxe Mari Korta Center, Avda. Tolosa 72, 20018 – Donostia-San Sebastian, Spain. E-mail: luca_porcarelli001@ehu.es
dLaboratoire de Chimie des Polymères Organiques (LCPO – UMR 5629), Bordeaux INP, Université de Bordeaux, CNRS, 16 Av. Pey-Berland, 33607 – Pessac, France
eDepartment of Chemistry, NIS Interdepartmental Centre and INSTM Reference Centre, Università degli Studi di Torino, Via Pietro Giuria 7, 10125 – Torino, Italy
fLaboratory of Photonics and Interfaces, Institut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), Station 3, 1015 – Lausanne, Switzerland
gIkerbasque, Basque Foundation for Science, Maria Diaz de Haro 3, 48011 – Bilbao, Spain
First published on 14th January 2020
Abstract
A green, efficient and stable solar cell based only on water and safe and cheap elements of the periodic table is proposed in this work, finally consolidating (also from a sustainability viewpoint) the concept of “artificial photosynthesis” studied for decades by the scientific community. The concept of dye-sensitized solar cells is re-proposed here with a metal-free organic dye, an iodine-based electrolyte in a 100% aqueous environment and a new cathode (cationic PEDOT) synthesized for the first time with the aim of inhibiting the repulsion between the anions of redox couples and the PEDOT:PSS matrix commonly used as the counter-electrode. This elegant setup leads to a record efficiency of 7.02%, the highest value ever obtained for a water-based solar cell and, in general, for a photovoltaic device free of both organic solvents and expensive/heavy metals.
Introduction
Aqueous solar cells (ASCs) are an emerging technology in the field of hybrid photovoltaics (PV) due to their sustainability, stability, transparency and possible indoor use.1 Generally, ASCs are prepared by the complete replacement of the organic solvents present in the electrolyte of dye-sensitized solar cells (DSSCs) with water, thus making the device more environmentally friendly and less dangerous in terms of flammability and toxicity (in the case of breakage).2 After the pioneering work of O'Regan in 2010,3 research work in the field of ASCs became extremely active as the solvent change in the electrolyte involves a wide range of variations in these regenerative photoelectrochemical cells. The synthesis of novel dyes with engineered wettability,2b,4 the development of new redox pairs or the adoption of unconventional concentration levels of traditional redox shuttles,5 and the formulation of hydrogels to guarantee better long-term stability6 are just some of the research lines under thorough investigation.About 95% of the publications on ASCs report the use of platinum as the cathode,1a which accounted for the current record efficiency of 5.97% demonstrated by Lin et al. in 2015.7 However, the use of platinum counter-electrodes is in contrast with the concept of inexpensiveness that should characterize third generation hybrid solar cells. In this framework, electrically conductive polymers may represent valid alternative cathode materials for ASCs. Among different conducting polymers, poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) is the most promising due to its high conductivity, easy processing, low cost and commercial availability.8 Boschloo's and Hagfeldt's groups published efficiency values between 1.4% and 5.5% for ASCs using PEDOT:PSS cathodes and cobalt redox mediators, but the reference cell lost 45% of its initial performance after only 200 h under 1 sun at room temperature.9 Fayad et al. obtained a 4.50% efficiency by means of a thiolate/disulphide redox couple with the same PEDOT:PSS cathode.4a
Printing, blading and roll-to-roll technologies were proposed to fabricate standard DSSCs (not ASCs) starting from a PEDOT:PSS aqueous dispersion.10 Even if such a process is feasible and rapid, devices are often obtained with low current density, fill factor and efficiency values, which are typically attributed to both the worsening of the conductivity (with respect to that of platinum) and the quality of the cathode/electrolyte interface.11 The latter is reasonably due to the electrostatic repulsion between the negative PSS units of the cathode and triiodide anions (I3−) of the electrolyte, the latter having to be reduced through an electrocatalytic step.
To overcome the limitations of traditional cathodes based on PEDOT:PSS dispersions, here we propose a poly(3,4-ethylenedioxythiophene) derivative bearing a cationic ammonium moiety with an iodide counter-anion (cPEDOT). The ammonium PEDOT derivative is soluble in aqueous media and – given the absence of PSS units, replaced with a cationic moiety – avoids electrostatic repulsion issues with I3− anions in the aqueous electrolyte solution. Aqueous solutions of cPEDOT were easily spin-coated on conductive glass, and thermally induced crosslinking led to stable cathodes when used with a 100% aqueous electrolyte based on the I−/I3− redox couple. Unprecedented efficiencies of >7% under 1 sun were obtained, exceeding those provided by Pt-based counterparts, and ASCs were able to retain 96% of their initial efficiency after 1200 h under full sun irradiation. To the best of our knowledge, this is the first example of a dye-sensitized device able to avoid the use of organic solvent-based electrolytes, platinum, cobalt and ruthenium, while outperforming the devices assembled with one or more of these heavy/rare metals.
Results and discussion
PEDOT is commonly commercialized in the form of an aqueous dispersion with PSS, the latter playing the role of stabilizing the water-based liquid system. In this work, we prepared a water soluble PEDOT derivative that does not require the use of PSS. The 3,4-ethylenedioxythiophene (EDOT) derivative monomer was prepared by adding an ammonium iodide moiety to a heteroatom bi-cycle separated by a short ether linkage. The synthesis consisted of two steps as shown in Scheme 1A: a classical Williamson etherification and a quaternization via a strong methylating agent. In the first step, commercially available hydroxymethyl-EDOT (1) was treated with a strong inorganic base, such as sodium hydride and 2-chloro-N,N-dimethylethylamine (2) in N,N-dimethylformamide (DMF). An excess of the cheap amine and sodium hydride ensured high yield and a pure product (3). The second step was quaternization with methyl iodide. Also in this case, a large excess of the methylating agent and the fact that the quaternized salt precipitated in the reaction solvent guaranteed easy manipulation and a quantitative yield (4). The polymerization reaction was performed in chloroform solution using iron trichloride as the oxidant. A black precipitate was obtained (5), which was washed with the organic media, dispersed in water and dialyzed to eliminate any excess of the monomer or oxidant.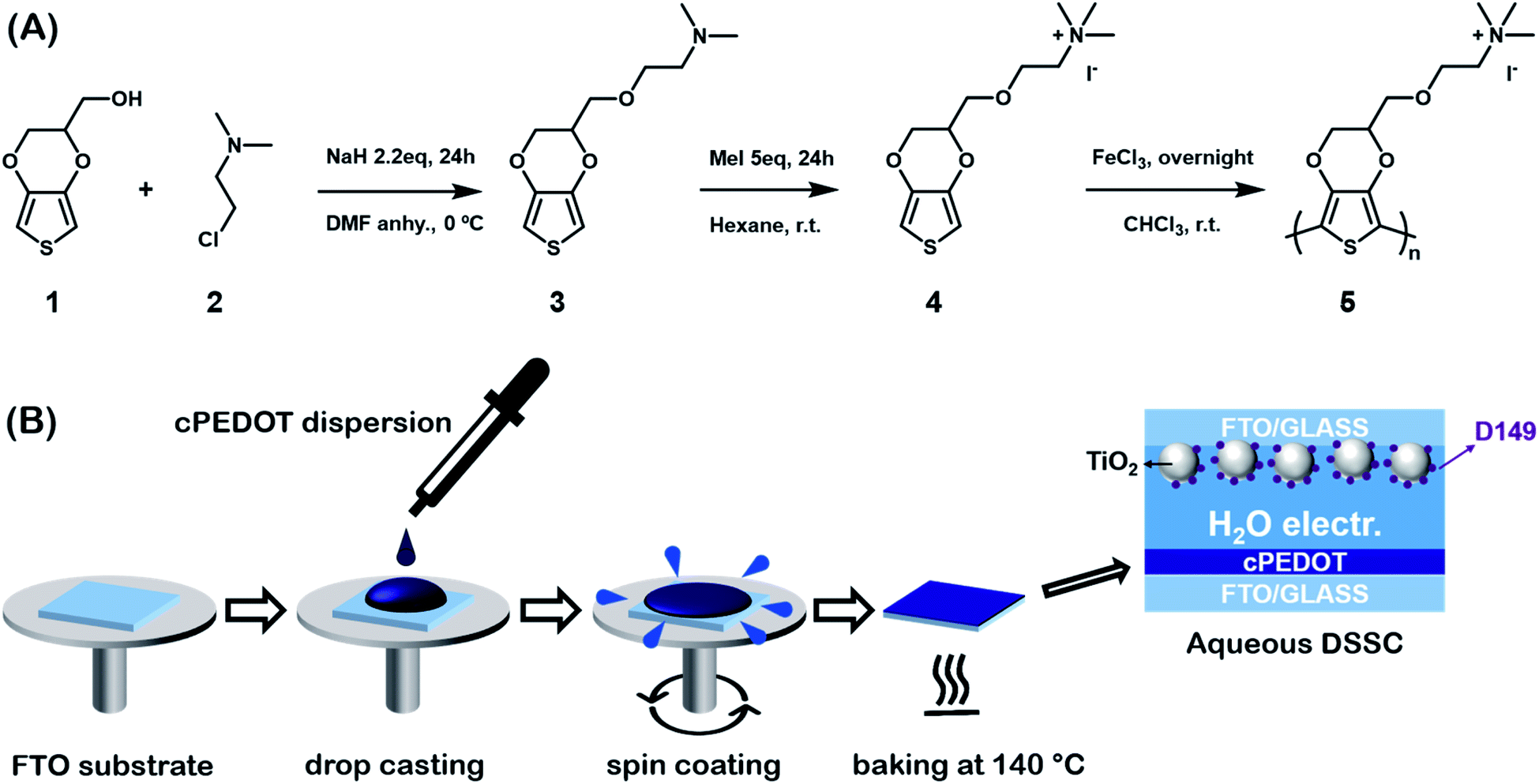 | ||
| Scheme 1 (A) Synthetic procedure to obtain the cPEDOT derivative; (B) schematic procedure for the fabrication of cPEDOT counter electrodes for ASCs. | ||
The resulting polymer was characterized via NMR, IR and UV-Vis-NIR (see Fig. S1–S7, ESI†); for this purpose, a cPEDOT film was prepared by drop-casting the dialyzed polymer solution and then it was completely dried before measurements and briefly exposed to an ambient atmosphere. The hygroscopicity of the film was attested by the broad band at around 3300 cm−1. The signal of amine stretching was visible at 1216 cm−1 and the absence of any signals at low wavenumbers, belonging to the wagging of amine protons, confirmed the absence of N–H bonds. C–O stretching was clearly visible at 1052 cm−1 and the bands of thiophene were more blue-shifted than those for the standard EDOT monomer, with the maximum around 1612 cm−1. The UV-Vis-NIR spectrum showed how ammonium moieties absorbed in the UV region; on the other hand, it was possible to observe the almost complete absence of polarons or bipolarons around 1100 nm. Also, the area around 800 nm, belonging to the conjugation across the thiophene backbone, highlighted a decrease of the absorbance correlated with reduced conjugation with respect to PEDOT:PSS.
cPEDOT films were prepared according to the very simple procedure shown in Scheme 1B. The cPEDOT dispersion was mixed with (3-glycidyloxypropyl)trimethoxysilane (GOPS) (the latter at a 2.5 wt% concentration), sonicated for 5 min and spin-coated over a ITO substrate. The films were baked at 140 °C for 3 h to allow formation of the siloxane network and complete the fabrication of polymer-based cathodes.12 This step, recently developed and under further investigation in the organic electronics field,13 led to a cPEDOT-based electrode which is extremely stable in an aqueous environment.
The electrocatalytic activity of the as-fabricated cPEDOT-based cathodes was compared to that of standard Pt-coated FTO. The cyclic voltammetry (CV) plots are shown in Fig. 1A, presenting the two typical pairs of oxidation and reduction peaks of the I−/I3− mediator. Since the electrocatalytic activity is directly related to the cathodic current density and inversely related to the separation (EPS) between anodic and cathodic peaks, it was found that cPEDOT showed smaller EPS and larger cathodic current density when compared to the Pt counterpart.
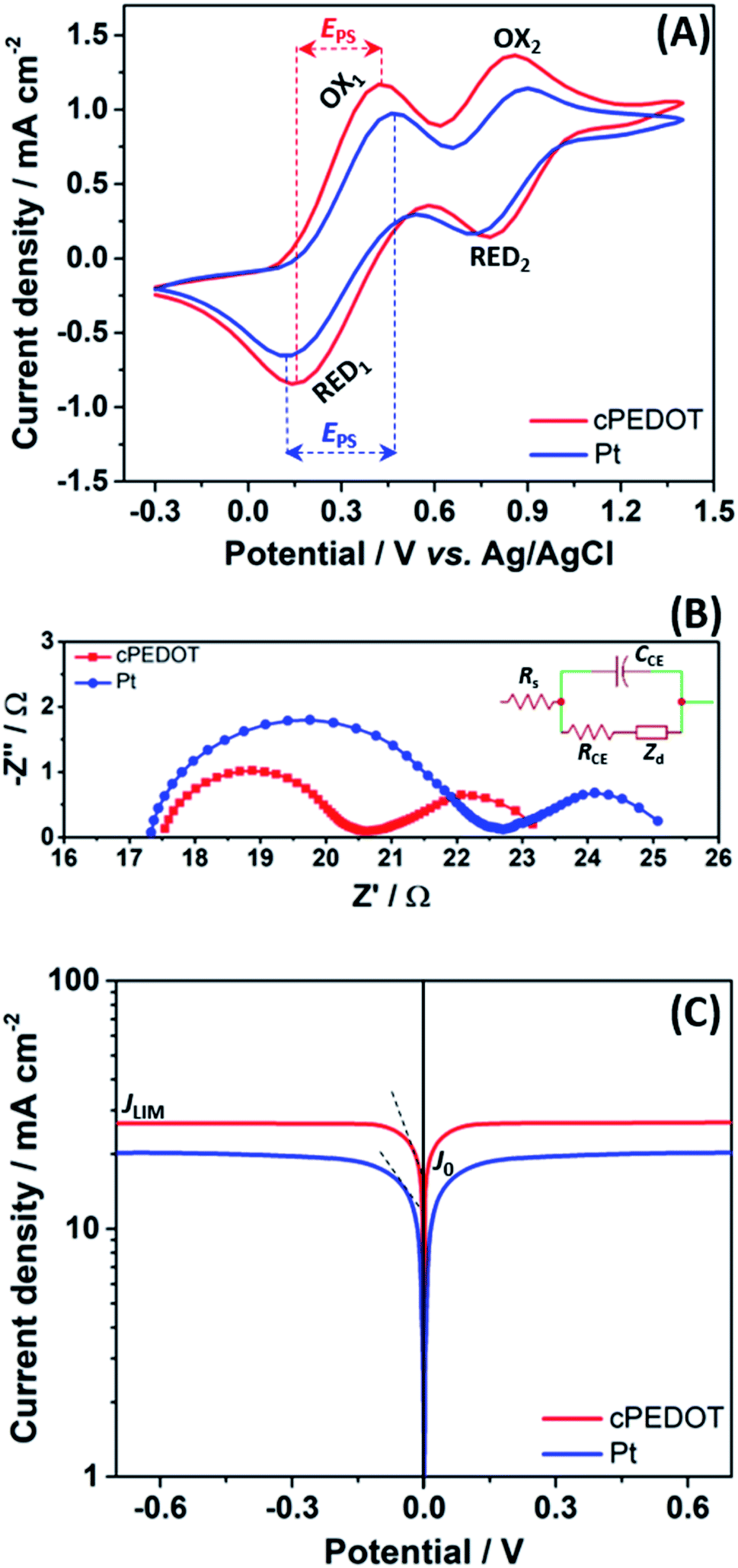 | ||
| Fig. 1 (A) Cyclic voltammograms of various cathodes combined with I−/I3− redox species, recorded at a scan rate of 5 mV s−1. Process #1 is responsible for the main function of the cathode towards the reduction/oxidation of I3−/I− (I3− + 2e− ↔ 3I−), whereas process #2 corresponds to the redox reaction of I3−/I2 (3I2 + 2e− ↔ 2I3−). (B) Nyquist plots for dummy cells fabricated with different electrodes (inset: the equivalent circuit used to fit experimental points). (C) Tafel polarization curves for dummy cells fabricated with different electrodes. | ||
The reaction kinetics in terms of the diffusion coefficient (D) was calculated using the Randles–Sevcik equation:
| IPC = (2.687 × 105)n3/2v1/2D1/2AC | (1) |
Such an excellent electrocatalytic activity for cPEDOT electrodes was further investigated by electrochemical impedance spectroscopy (EIS), carried out using symmetrical (dummy) cells with two electrodes. The Nyquist plots shown in Fig. 1B contain the two expected semicircles, the high-frequency one indicating the charge-transfer resistance (RCE) at the cathode/electrolyte interface and the low-frequency one indicating the Nernst diffusion impedance (Zd) for the iodine-based redox shuttle. The impedance parameters determined from the equivalent circuit modeling (inset of Fig. 1B) indicated a clearly reduced RCE value of 3.29 Ω for the cPEDOT electrode when compared to that of the Pt counterpart (5.33 Ω), while no marked differences were detected in the diffusion resistance values (2.57 and 2.61 Ω, respectively). RCE represents a pivotal parameter to evaluate the electrocatalytic activity of a solar cell cathode, and its influence on the ASC photovoltaic parameters (mainly ascribed to the reduced interface loss of charge transfer) will be discussed later in the manuscript. Furthermore, the same variation trend of RCE can be detected from the Tafel polarization experiment. As illustrated in Fig. 1C, the anodic and cathodic branches of cPEDOT show a larger slope than their Pt counterpart, and this is well consistent with the results derived from the CV and EIS experiments, again confirming the superior electrocatalytic activity of the cPEDOT cathode toward I3− reduction. The tangent line corresponding to the Tafel zone allows the extrapolation of the exchange current density (J0), which is directly related to the RCE using the following equation:
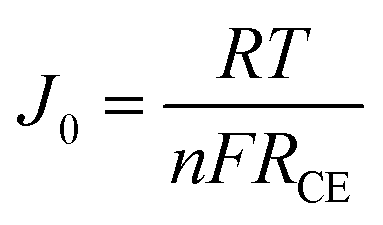 | (2) |
To quantify the effect of the enhanced electrocatalytic activity of cPEDOT cathodes compared to the standard Pt-based ones, ASC batches were assembled and tested under 1 sun AM1.5G irradiation intensity. The indole-based organic dye D149 was used for cell photoanodes, while the electrolyte consisted of 1.0 M NaI and 10 mM I2 in water; in other words, these solar cells do not contain heavy/rare metals in their redox shuttles, sensitizers and cathodes. Photocurrent density vs. photovoltage curves of the best ASCs are shown in Fig. 2A, while the average values of the photovoltaic parameters [i.e., short-circuit current density (Jsc), open-circuit potential (Voc), fill factor (FF) and power conversion efficiency (PCE)] are listed in Table 1. It is noteworthy that the best ASC fabricated with cPEDOT achieved a PCE of 7.02%, which is the highest value ever obtained for these devices, which is rather outstanding considering the green and cheap device components at the base of these cells. Tables S1 and S2 (ESI†) present a comparison between different experimental setups to fabricate ASCs, highlighting their effect on device performance: the ASC device is still in its infancy, and a broader knowledge of the engineering (and lab-related) aspects behind this technology should be acquired.
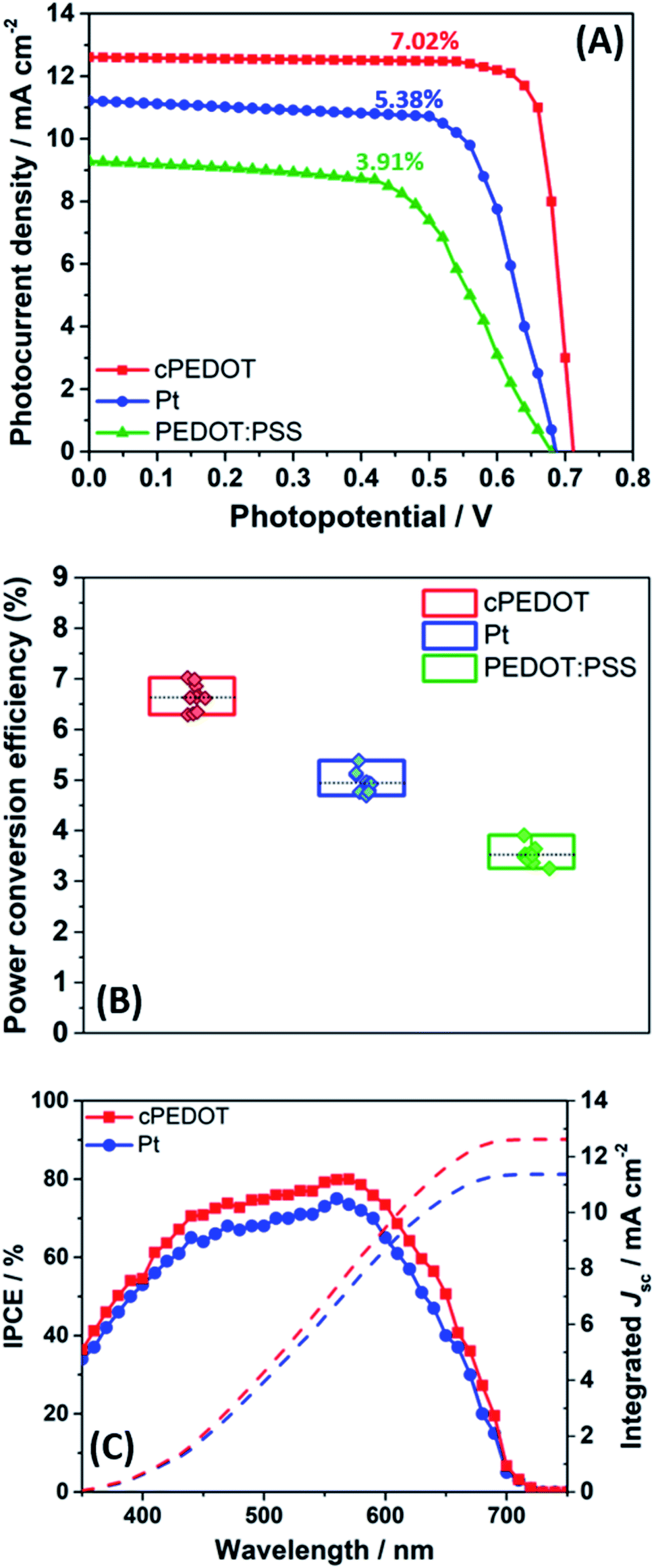 | ||
| Fig. 2 (A) Photocurrent density vs. photovoltage curves for the three best ASCs fabricated with different cathodes and measured under 1 sun irradiation (AM1.5G). (B) Statistics of 30 devices collected over 3 different batches: PCE values are shown for ASCs assembled with different cathodes. (C) IPCE and integrated Jsc for the same cPEDOT- and Pt-based devices shown in panel A. | ||
| Cathode | J sc (mA cm−2) | V oc (V) | FF | PCE (%) | ΔPCE |
|---|---|---|---|---|---|
| Pt | 11.05 ± 0.08 | 0.66 ± 0.01 | 0.68 ± 0.01 | 4.95 ± 0.12 | — |
| cPEDOT | 12.41 ± 0.09 | 0.69 ± 0.01 | 0.77 ± 0.01 | 6.64 ± 0.16 | +34% |
| PEDOT:PSS | 8.88 ± 0.16 | 0.65 ± 0.01 | 0.61 ± 0.01 | 3.53 ± 0.10 | −29% |
An overview of the statistics of 30 devices assembled with different cathodes is shown in Fig. 2B, highlighting the rather good reproducibility of cell fabrication. It is worth noting that all the photovoltaic parameters of cPEDOT devices are much better than those of the ASCs using Pt as the cathode, confirming the superior electrocatalytic activity of the former toward the reduction of I3−. The previously described features, in terms of cathodic current density and peak separation in CV, exchange current density and limiting current density in Tafel experiments, and charge-transfer resistance in EIS measurements, all reflected the obtained photovoltaic parameters, especially in terms of Jsc and FF values.
The integrated photocurrents calculated from the overlap integral of the IPCE spectra (Fig. 2C) with the standard AM1.5G solar emission spectrum are 12.62 and 11.37 mA cm−2 for the two best cells assembled with cPEDOT and Pt cathodes, respectively; these values are rather close to those measured by J–V experiments, which are shown in Fig. 2A (12.61 and 11.22 mA cm−2, respectively).
As mentioned in the Introduction, the possibility of easily depositing Pt-free cathodes is a fundamental aspect for the development of large-scale, cheap hybrid solar cells. In our work, we also compared the response of the newly developed material with the performance of a batch of cells fabricated using the classic commercial PEDOT:PSS, deposited by spin-coating starting from an aqueous dispersion having the same content of active material as in the case of cPEDOT. The data shown in Fig. 2A, B and Table 1 highlight that the results obtained with cPEDOT-based devices markedly exceed those measured in the presence of the classic anionic groups (e.g., PSS) characterizing the commercial product. Indeed, the ammonium moiety present in the cPEDOT active compound allows overcoming of the electrostatic repulsions occurring between PSS and I3−, boosting the regeneration of the redox couple and the solar cell efficiency.
J–V data were also collected at different irradiation intensities (in the 0.2–1.0 sun range), and the results are plotted in Fig. 3A. For both cathodes (Pt and cPEDOT), Jsc values were proportional to the incident light, without the presence of evident or slight deviations from linearity. This accounts for the absence of mass transport limitation occurring between the cell electrodes. This was further proved by monitoring photocurrent transients employing a modulation (on/off) of the incident light as shown in Fig. 3B. The plot shows a situation analogous to the ideal mass transport case, where the photocurrent rapidly reaches a maximum when the light is switched on, thereafter remaining constant. Such a condition was maintained at all the illumination levels tested, confirming not only the excellent quality of aqueous electrolytes (compared to the acetonitrile-based counterparts), but also the ability of cPEDOT cathodes to regenerate the huge amount of I3− ions produced at the photoanode side rapidly and efficiently.
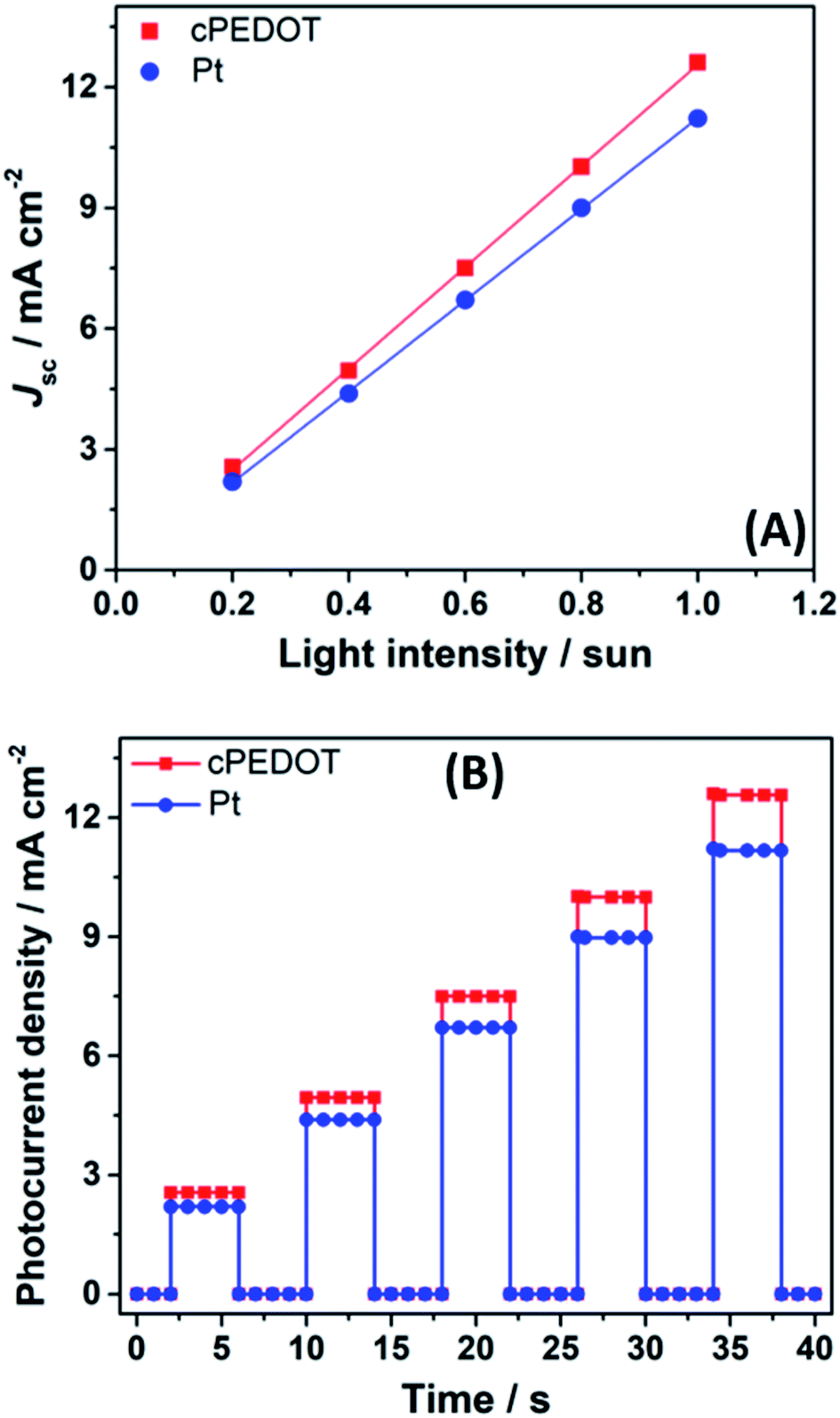 | ||
| Fig. 3 (A) Jsc values plotted as a function of different light intensities for ASCs assembled with different cathodes. (B) Transient photocurrent measurements under different light intensities for the two cells in panel A. | ||
To evaluate the stability of the ASCs assembled with different cathodes, we placed the three batches of cells (initial PCE listed in Fig. 2B) under a LED light beam with an irradiation intensity equal to 1 sun. The test was carried out at ambient temperature and lasted for 50 days (1200 h); photovoltaic performance was tested every 50 h by measuring the J–V curve. Fig. 4A shows that both cPEDOT- and Pt-based ASCs demonstrated a remarkable stability, retaining 96% and 94%, respectively, of their initial efficiency after 1200 h under simulated sunlight. This excellent result could be further improved by adopting sealing techniques closer to the industrial ones (e.g., glass-frits) and consolidates the promising prospects of this generation of water-based photovoltaic devices that do not require rare or heavy metals. In contrast, devices assembled with the traditional PEDOT:PSS showed efficiencies decreasing with time, with a loss of 17% of the initial performance at the end of the experiment. From a simple visual analysis, we noted the progressive discoloration of the cathodes due to the progressive solubilization of the PEDOT:PSS layer in the aqueous environment; this was not observed in the case of cPEDOT, the integrity of which was completely preserved by the GOPS treatment.
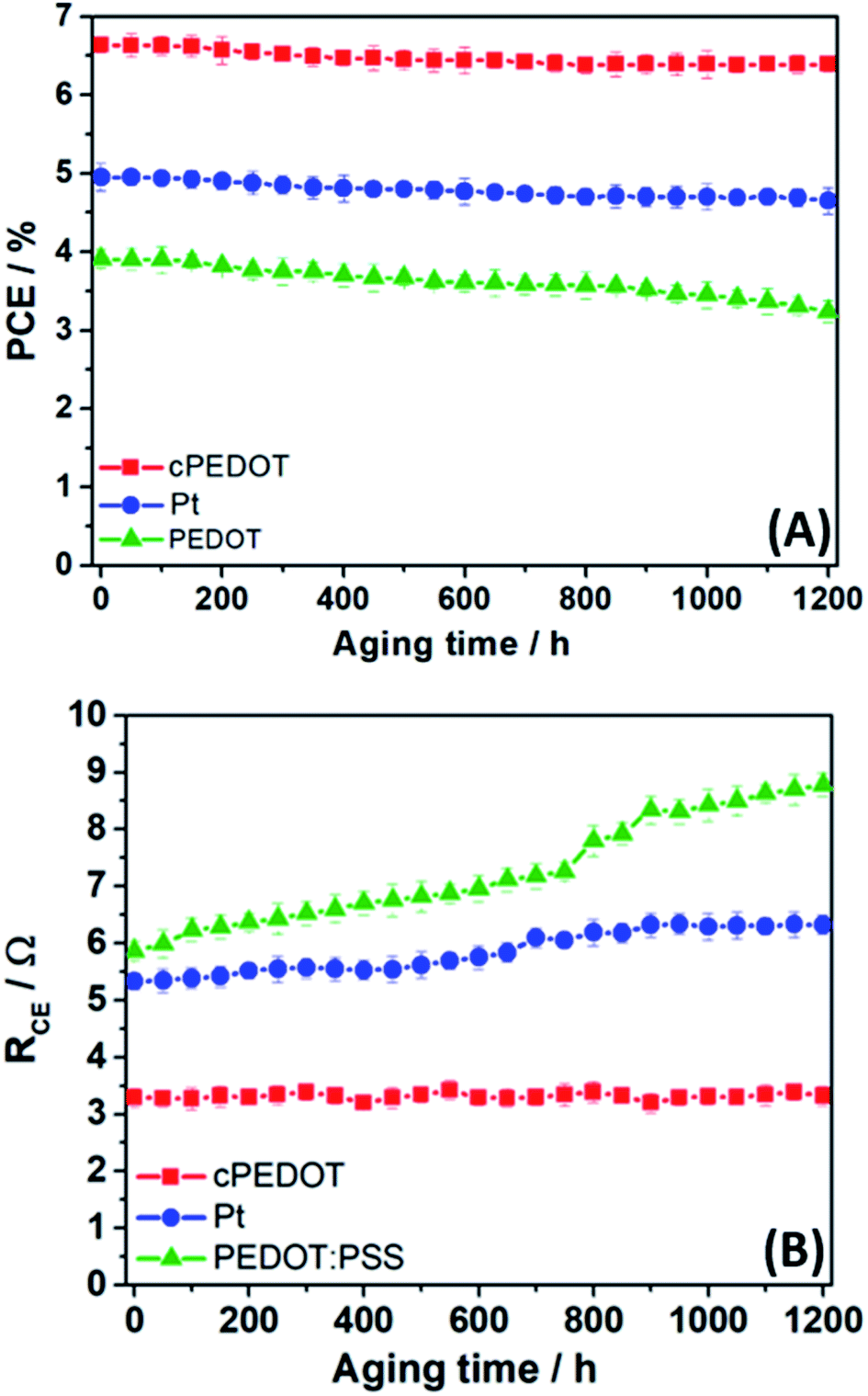 | ||
| Fig. 4 (A) Evolution of photovoltaic performances of ASCs assembled with different cathodes and kept at ambient temperature under 1 sun LED irradiation. Each point represents the average of a batch of 10 devices. (B) Evolution of RCE for ASCs assembled with different cathodes, where each point represents the average of a batch of 10 devices. RCE values were determined by EIS, after carrying out two CV scans (0 V → 1 V → −1 V → 0 V, scan rate: 50 mV s−1) of 50 h each. | ||
Aside from photovoltaic performance, the electrochemical stability of cathodes is another crucial factor that must be seriously considered from the viewpoint of practical applications of ASCs. To assess this point, we set up a protocol based on the following steps: (1) freshly assembled symmetrical dummy cells were tested with EIS at ambient temperature; (2) after 50 h, two CV scans (0 V → 1 V → −1 V → 0 V, scan rate: 50 mV s−1) were run; (3) after 30 s relaxation at 0 V, EIS measurement was repeated to determine RCE; (4) steps 2 and 3 were repeated up to a total time of 1200 h. The evolution of RCE values with time is shown in Fig. 4B. The dummy cells based on Pt and cPEDOT cathodes were stable over time. A slight increase in RCE (then stabilized) was observed for Pt systems, as already investigated by other researchers who attributed this behavior to an initial catalyst poisoning in the presence of the redox mediator I−/I3−.14 On the other hand, the standard PEDOT:PSS cathodes showed a continuous worsening of their performances, due to the increasing charge transfer resistance accompanied by the progressive solubilization of the polymer in the aqueous electrolyte.
The long-term stability of ASCs still needs in-depth studies to be compared to those of the corresponding organic solvent-based devices. In particular, future steps should cover two different approaches. The first relates to the stability of the semiconductor/dye/electrolyte interface: we never detected an appreciable desorption of D149 dye in the aqueous electrolyte, but analytical investigations should be set up to give a more precise quantification of this (eventual) phenomenon. This also presupposes the development of specific protocols for this purpose, since quantitatively desorbing D149 from TiO2 is not as simple as removing the traditional N719 dye with a basic aqueous solution. Secondly, the absence of H2 production must also be verified under conditions of asymmetrical irradiation (i.e., half-irradiated and half-dark surface) of modules assembled with aqueous electrolytes. Globally, a broad and synergistic effort is required from the scientific community in this emerging PV sector.
Conclusions
The evolution towards building-integrated photovoltaic architectures and indoor sun-powered technologies requires the development of solar cells that can operate under low/indirect illumination, with tunable transparency degrees, based on abundant, cheap and sustainable materials and fabricated with non-flammable and toxic components.In this work, we have demonstrated the first solar cell able to provide efficiencies higher than 7% (markedly stable under prolonged irradiation) making use of dyes, electrolytes and cathodes without metals, with particular emphasis on the absence of organic solvents in the electrolyte and on the synthesis of a new cationic PEDOT derivative able to match, and also exceed, the performances obtained using platinum.
These remarkable results pave the way for the novel generation of aqueous solar cells, as a leading technology for indoor environment applications and for smart integration into buildings and greenhouses, both emerging fields in the context of renewable energy.
Experimental section
Materials and characterization
The reactants were obtained from Sigma-Aldrich, and the solvents were purchased from Fischer Scientific and used without further purification. Hydroxymethyl EDOT (1) was obtained from Molekula s.r.l. 1D and 2D-NMR spectra were recorded at ambient temperature with a 400 MHz Bruker Avance III. The following abbreviations are used to describe peak patterns where appropriate: br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublets, dtd = doublet of triplet of doublets. Coupling constants (J) are reported in Hertz (Hz); DMSO-d6 was used as the solvent. Fourier transform infrared spectroscopy (FTIR) measurements were performed on a Brüker Alpha-p FTIR spectrometer. Spectra were collected from 4000 to 250 cm−1 with the following settings: 42 scans per sample and spectral resolution: 4 cm−1. High resolution mass spectrometry (HRMS) was carried out by direct injection in a Waters model SYNAPT G2 HDMS™, using a Q-TOF detector and positive electrospray ionization.Synthesis of 3,4-ethylenedioxythiophene N,N-dimethyl ethylamine (EDOT-DMEA) (3)
In a 100 mL round-bottom flask, 1.36 g (9.4 mmol, 1 eq.) of 1 were dissolved in 50 mL of dimethylformamide (DMF) together with 4.08 g (28 mmol, 3 eq.) of 2-chloro-N,N-dimethylethan-1-amine (2) and degassed with nitrogen. Then, 1.58 g of NaH (66 mmol, 7 eq.) were added portionwise, and a continuous flow of nitrogen was used to eliminate hydrogen. After 18 h of stirring at room temperature, DMF was evaporated and the residue was dissolved in EtOAc (250 mL) and washed with water (3 × 100 mL). The organic phase was dried over Na2SO4, the solid was separated by filtration and the solvent was evaporated under vacuum. The product was a viscous brownish oil that tended to crystallize. Yield was 82% (1.88 g).1H NMR (400 MHz, DMSO-d6, δ): 6.57 (s, 1H), 4.29 (dtd, J = 7.4, 5.1, 2.3 Hz, 1H), 4.24 (dd, J = 11.7, 2.3 Hz, 2H), 3.97 (dd, J = 11.7, 7.6 Hz, 2H), 3.64–3.57 (m, 3H), 3.52 (t, J = 5.9 Hz, 3H), 2.14 (s, 6H). 13C NMR (101 MHz, DMSO-d6, δ): 141.32, 141.28, 99.66, 99.59, 72.42, 69.22, 68.67, 65.44, 58.20, 45.55.
Synthesis of 3,4-ethylenedioxythiophene N,N,N-trimethyl-ethylammonium iodide (EDOT-TMEAI) (4)
A solution of 3, containing 1.42 g (5.8 mmol, 1 eq.) in 125 mL of hexane, was reacted with 2.175 mL (35 mmol, 6 eq.) of methyliodide; the system was stirred for 24 h at room temperature. A yellowish powder formed immediately after addition and its quantity increased with time.The yellow mixture was filtered and the solid was collected by Buchner filtration, washed with hexane (3 × 50 mL) and dried under vacuum to obtain 4 as a yellowish powder (2.31 g, quantitative). FTIR νmax/cm−1: 3112 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H), 2994, 2985 (C–H), 1551 (N–C) and 1474 (CC), 1158, 1008. 1H NMR (400 MHz, DMSO-d6, δ): 6.62–6.57 (m, 1H), 4.37 (dtd, J = 7.4, 5.0, 2.3 Hz, 1H), 4.27 (dd, J = 11.8, 2.4 Hz, 2H), 4.00 (dd, J = 11.8, 7.5 Hz, 1H), 3.93–3.83 (m, 3H), 3.71 (d, J = 4.9 Hz, 3H), 3.61–3.47 (m, 3H), 3.10 (s, 12H). 13C NMR (101 MHz, DMSO-d6, δ): 141.22, 99.85, 99.71, 72.15, 68.65, 65.30, 64.51, 53.11. HRMS (ESI) m/z: calculated 258.1158 Da, found 258.1167 Da.
C–H), 2994, 2985 (C–H), 1551 (N–C) and 1474 (CC), 1158, 1008. 1H NMR (400 MHz, DMSO-d6, δ): 6.62–6.57 (m, 1H), 4.37 (dtd, J = 7.4, 5.0, 2.3 Hz, 1H), 4.27 (dd, J = 11.8, 2.4 Hz, 2H), 4.00 (dd, J = 11.8, 7.5 Hz, 1H), 3.93–3.83 (m, 3H), 3.71 (d, J = 4.9 Hz, 3H), 3.61–3.47 (m, 3H), 3.10 (s, 12H). 13C NMR (101 MHz, DMSO-d6, δ): 141.22, 99.85, 99.71, 72.15, 68.65, 65.30, 64.51, 53.11. HRMS (ESI) m/z: calculated 258.1158 Da, found 258.1167 Da.
Synthesis of poly(3,4-ethylenedioxythiophene trimethylethylammonium iodide) (cPEDOT) (5)
0.1 g (0.253 mmol, 1 eq.) of EDOT-TMEAI (4) were dissolved in 5 mL of chloroform, together with 0.205 g (0.759 mmol, 3 eq.) of iron(III) trichloride hexahydrate. The resulting solution was stirred at room temperature overnight. Immediately the solution turned from yellow to a darker color, leading to a violet suspension with a black precipitate. The precipitate was separated from the solution and the solid product was extracted with water, forming a dark blue suspension that was dialyzed for 3 days, versus DI water, changing the water twice per day in order to eliminate all the iron and the unreacted monomer. At the end of dialysis, the concentration was set to 2 wt% by evaporating water under vacuum. Conversion was 92%.Fabrication of aqueous solar cells
Sodium iodide (NaI), iodine (I2), titanium tetrachloride (TiCl4), chenodeoxycholic acid (CDCA), chloroplatinic acid (H2PtCl6), tetrakis(dimethylamido)titanium(IV), ethanol, acetone, tert-butanol (t-BuOH) and acetonitrile (ACN) were purchased from Sigma-Aldrich. Deionized water (DI–H2O, 18 MΩ cm−1 at 25 °C) was obtained using a Direct-Q 3 UV Water Purification System (Millipore). Sensitizing dye 5-[[4-[4-(2,2-diphenylethenyl)phenyl]-1,2,3-3a,4,8b-hexahydrocyclopent[b]indol-7-yl]methylene]-2-(3-ethyl-4-oxo-2-thioxo-5-thiazolidinylidene)-4-oxo-3-thiazolidineacetic acid (D149) was purchased from Inabata Europe S.A. Fluorine-doped tin oxide (FTO) glass plates (sheet resistance 7 Ω sq−1, purchased from Solaronix) were cut into 2 cm × 1.5 cm sheets and used as substrates for the fabrication of both the photoanodes and the counter electrodes by an experimental procedure different from that adopted in our previous reports.2c,6cA glass pretreatment procedure was carried out to remove the contaminations that can affect the preparation of the compact underlayer and thus the cell performance. FTO glass was first cleaned with a detergent solution (Deconex®) in an ultrasonic bath for 45 min and then rinsed with water and ethanol. This initial step was followed by a 15 min UV/O3 treatment using cleaning apparatus (model no. 256–220, Jelight Company, Inc.) to remove any possible impurity residues.
As regards photoanodes, a TiO2 blocking layer was deposited at 120 °C from a tetrakis(dimethylamido)titanium(IV) precursor and deionized water using a Savannah 100 instrument (Cambridge NanoTech). The precursor vapors were pulsed into the reactor under a nitrogen atmosphere (99.999%); in detail, the titanium(IV) precursor was pulsed for 0.1 s and confined in the reactor for 30 s, followed by a 30 s purge to remove the excess precursor molecules and byproducts of the ALD process. Water pulsing followed and lasted for 0.015 s, and then the water was confined in the reactor for 30 s with a 30 s purge. 15 cycles of deposition contributed to 1 nm growth of TiO2; overall, 4 nm-thick layers were deposited and were not subjected to further thermal treatment.
In the second phase, two layers of mesoporous TiO2 were prepared on the top of the underlayer. The first layer was screen printed (with a 43T mesh frame and an AT-25PA model, Atma Champ Ent. Corp.) using a paste consisting of 30 nm diameter TiO2 particles (DSL 18NR-T, Dyesol) and the second one with 400 nm-sized particles (scattering layer with a porosity of 70%, obtained from the commercial product HPW-400NRD, CCIC). Each layer was heated to 110 °C for 5 min. The substrates were sintered on a hot plate with a ramped temperature profile, keeping the temperature at 125, 250, 325, 450 and 500 °C for 5, 5, 5, 15 and 15 min, respectively, with a 5 min ramp duration for each temperature step. The resulting TiO2 film thickness was 12.5 μm (7.5 μm + 5 μm), measured after the sintering process using a KLA Tencor Alpha-Step 500 surface profilometer. To increase the surface area of the TiO2 particles, a TiCl4 post-treatment with a 13 mM solution was performed for 30 min at 70 °C, which was followed by another sintering process at 500 °C for 30 min.
When required for DSSC fabrication, TiO2 electrodes were reactivated by heating with a hot gun for 30 min at 500 °C and subsequently soaked into a D149 dye solution (0.50 mM in t-BuOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ACN 1:1 with 0.90 mM CDCA as the coadsorbent); this dye was already considered for ASCs by Law et al.15 Dipping in the dye solution was carried out at ambient temperature for 5 h in the dark. After dye loading, the photoanodes were washed in acetone to remove residual dye not specifically adsorbed onto the TiO2 layer.
:ACN 1:1 with 0.90 mM CDCA as the coadsorbent); this dye was already considered for ASCs by Law et al.15 Dipping in the dye solution was carried out at ambient temperature for 5 h in the dark. After dye loading, the photoanodes were washed in acetone to remove residual dye not specifically adsorbed onto the TiO2 layer.
As regards Pt cathodes (fabricated for comparison purposes), cleaned FTO conductive glass was platinized by spreading a 5.0 mM H2PtCl6 solution onto the plate surface and heating up to 400 °C using a hot plate. For the cPEDOT cathodes, a PEDOT suspension (2 wt% in water) was doped with 2.5 wt% GOPS before cell fabrication and sonicated for 5 min. The resulting suspension was spin-coated (SPIN150 spin processor, SPS-Europe) on cleaned FTO glass and heated for 1 h at 140 °C. Solar cells were assembled using a 60 μm-thick thermoplastic Meltonix 1170-60 frame, and the electrolyte solution (1.0 M NaI and 10 mM I2 in H2O) was introduced through two holes pre-drilled in the cathode.
The cell was sealed with thermoplastic Meltonix covers and a glass coverslip. The sheet edges of FTO were coated by ultrasonic soldering (Cerasolzer alloy 246, MBR Electronics GmbH) to improve the electrical contact. An antireflection film (ARCTOP, Mihama Co.) was attached on the photoanode side.
Characterization of aqueous solar cell devices and components
Cyclic voltammetry (CV) was conducted on an electrochemical workstation (CS350, CorrTest, Wuhan) with a three-electrode electrochemical cell using the cathodes under study as the working electrode, Pt foil as the counter electrode and Ag/Ag+ as the reference electrode in a water solution containing 10 mM NaI, 1 mM I2 and 0.1 M LiClO4, fixing the scan rate at 5 mV s−1.A PARSTAT 2273 potentiostat/galvanostat equipped with a frequency response analyser (Princeton Applied Research, US) was used to perform electrochemical impedance spectroscopy (EIS) tests, applying a sinusoidal signal with an amplitude of 10 mV and a frequency varying in the range of 10−2 to 104 Hz. Measurements were carried out under dark conditions using symmetrical dummy cells (cathode/electrolyte/cathode) at zero bias potential. The dummy cells, with an active area of 0.20 cm2, consisted of the same aqueous redox electrolyte which is identical to that in ASCs.
Tafel-polarization measurements were performed using a symmetrical dummy cell as described above, and the scan rate was 5 mV s−1.
Current–voltage (I–V) characteristics of the solar cells were investigated using a Keithley 2400 source/meter and a Newport solar simulator (model 91160). The light power was regulated to the AM 1.5G solar standard at an intensity of 1000 W m−2 by using a reference Si photodiode equipped with a color-matched filter (KG-3, Schott) to reduce the mismatch between the simulated light and AM 1.5G to less than 4% in the wavelength region of 350–750 nm.16 When performing the I–V measurements, a black mask of 0.3 × 0.3 cm2 was used in order to avoid significant additional contribution from light impinging on the device outside the active area. I–V measurements were also performed at light intensities in the range of 0.2–1.0 sun, by using neutral density filters purchased from Newport. All the measurements were carried out on 10 different fresh cells per type of cathode, in order to verify the reproducibility of the obtained results.
The measurements of the incident photon-to-current conversion efficiency (IPCE) were performed by means of a computer-controlled setup consisting of a Xe light source (Spectral Products ASB-XE-175), a monochromator (Spectral Products CM 110), and a Keithley 2700 multimeter. The same certified reference solar cell previously mentioned was used for IPCE calibration. IPCE curves were recorded under bias light.17
A VeraSol-2 LED (class AAA) solar simulator with an irradiation intensity equal to 1 sun was used to perform long-term stability tests at ambient temperature and for 50 days (1200 h); the photovoltaic performance was tested every 50 h by measuring the J–V curves.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. P. received funding from the European Union's Horizon 2020 Research and Innovation Programme under Marie Skłodowska-Curie grant agreement no. 797295.References
- (a) F. Bella, C. Gerbaldi, C. Barolo and M. Grätzel, Chem. Soc. Rev., 2015, 44, 3431 RSC; (b) C. T. Li, R. Y. Y. Lin and J. T. Lin, Chem.–Asian J., 2017, 12, 486 CrossRef CAS PubMed.
- (a) C. Dong, W. Xiang, F. Huang, D. Fu, W. Huang, U. Bach, Y. B. Cheng, X. Li and L. Spiccia, Angew. Chem., Int. Ed., 2014, 53, 6933 CrossRef CAS PubMed; (b) R. Y. Y. Lin, T. M. Chuang, F. L. Wu, P. Y. Chen, T. C. Chu, J. S. Ni, M. S. Fan, Y. H. Lo, K. C. Ho and J. T. Lin, ChemSusChem, 2015, 8, 105 CrossRef CAS PubMed; (c) F. Bella, S. Galliano, G. Piana, G. Giacona, G. Viscardi, M. Grätzel, C. Barolo and C. Gerbaldi, Electrochim. Acta, 2019, 302, 31 CrossRef CAS.
- C. Law, S. C. Pathirana, X. Li, A. Y. Anderson, P. R. F. Barnes, A. Listorti, T. H. Ghaddar and B. C. ORegan, Adv. Mater., 2010, 22, 4505 CrossRef CAS PubMed.
- (a) R. Fayad, T. A. Shoker and T. H. Ghaddar, Dalton Trans., 2016, 45, 5622 RSC; (b) H. Tian, E. Gabrielsson, P. W. Lohse, N. Vlachopoulos, L. Kloo, A. Hagfeldt and L. Sun, Energy Environ. Sci., 2012, 5, 9752 RSC.
- (a) W. Yang, M. Söderberg, A. I. K. Eriksson and G. Boschloo, RSC Adv., 2015, 5, 26706 RSC; (b) W. Xiang, F. Huang, Y. B. Cheng, U. Bach and L. Spiccia, Energy Environ. Sci., 2013, 6, 121 RSC; (c) T. Daeneke, Y. Uemura, N. W. Duffy, A. J. Mozer, N. Koumura, U. Bach and L. Spiccia, Adv. Mater., 2012, 24, 1222 CrossRef CAS PubMed.
- (a) W. Xiang, D. Chen, R. A. Caruso, Y. B. Cheng, U. Bach and L. Spiccia, ChemSusChem, 2015, 8, 3704 CrossRef CAS PubMed; (b) S. Zhang, G. Y. Dong, B. Lin, J. Qu, N. Y. Yuan, J. N. Ding and Z. Gu, Sol. Energy, 2016, 127, 1927 CrossRef; (c) F. Bella, S. Galliano, M. Falco, G. Viscardi, C. Barolo, M. Grätzel and C. Gerbaldi, Green Chem., 2017, 19, 1043 RSC.
- R. Y. Y. Lin, F. L. Wu, C. T. Li, P. Y. Chen, K. C. Ho and J. T. Lin, ChemSusChem, 2015, 8, 2503 CrossRef CAS PubMed.
- (a) W. Song, X. Fan, B. Xu, F. Yan, H. Cui, Q. Wei, R. Peng, L. Hong, J. Huang and Z. Ge, Adv. Mater., 2018, 30, 1800075 CrossRef PubMed; (b) D. Mantione, I. del Agua, A. Sanchez-Sanchez and D. Mecerreyes, Polymers, 2017, 9, 354 CrossRef PubMed.
- (a) H. Ellis, R. Jiang, S. Ye, A. Hagfeldt and G. Boschloo, Phys. Chem. Chem. Phys., 2016, 18, 8419 RSC; (b) V. Leandri, H. Ellis, E. Gabrielsson, L. Sun, G. Boschloo and A. Hagfeldt, Phys. Chem. Chem. Phys., 2014, 16, 19964 RSC.
- (a) J. G. Chen, H. Y. Wei and K. C. Ho, Sol. Energy Mater. Sol. Cells, 2007, 91, 1472 CrossRef CAS; (b) W. Hong, Y. Xu, G. Lu, C. Li and G. Shi, Electrochem. Commun., 2008, 10, 1555 CrossRef CAS; (c) D. Yoo, J. Kim and J. H. Kim, Nano Res., 2014, 7, 717 CrossRef CAS.
- C. P. Lee, K. Y. Lai, C. A. Lin, C. T. Li, K. C. Ho, C. I. Wu, S. P. Lau and J. H. He, Nano Energy, 2017, 36, 260 CrossRef CAS.
- M. ElMahmoudy, S. Inal, A. Charrier, I. Uguz, G. G. Malliaras and S. Sanaur, Macromol. Mater. Eng., 2017, 302, 1600497 CrossRef.
- (a) A. Håkansson, S. Han, S. Wang, J. Lu, S. Braun, M. Fahlman, M. Berggren, X. Crispin and S. Fabiano, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 814 CrossRef; (b) D. Mantione, I. Del Agua, W. Schaafsma, M. Elmahmoudy, I. Uguz, A. Sanchez-Sanchez, H. Sardon, B. Castro, G. G. Malliaras and D. Mecerreyes, ACS Appl. Mater. Interfaces, 2017, 9, 18254 CrossRef CAS PubMed; (c) X. Strakosas, M. Sessolo, A. Hama, J. Rivnay, E. Stavrinidou, G. G. Malliaras and R. M. Owens, J. Mater. Chem. B, 2014, 2, 2537 RSC; (d) N. Chen, Y. Dai, Y. Xing, L. Wang, C. Guo, R. Chen, S. Guo and F. Wu, Energy Environ. Sci., 2017, 10, 1660 RSC.
- (a) W. Yang, Z. Li, X. Xu, L. Hou, Y. Tang, B. Deng, F. Yang, Y. Wang and Y. Li, Chem. Eng. J., 2018, 349, 782 CrossRef CAS; (b) L. Kavan, J. H. Yum and M. Grätzel, ACS Nano, 2011, 5, 165 CrossRef CAS PubMed.
- C. Law, O. Moudam, S. Villarroya-Lidon and B. C. O'Regan, J. Mater. Chem., 2012, 22, 23387 RSC.
- (a) C. C. P. Chiang, C. Y. Hung, S. W. Chou, J. J. Shyue, K. Y. Cheng, P. J. Chang, Y. Y. Yang, C. Y. Lin, T. K. Chang, Y. Chi, H. L. Chou and P. T. Chou, Adv. Funct. Mater., 2018, 28, 1703282 CrossRef; (b) L. Xu, C. Aumaitre, Y. Kervella, G. Lapertot, C. Rodríguez-Seco, E. Palomares, R. Demadrille and P. Reiss, Adv. Funct. Mater., 2018, 28, 1706291 CrossRef.
- (a) F. Bella, N. Vlachopoulos, K. Nonomura, S. M. Zakeeruddin, M. Grätzel, C. Gerbaldi and A. Hagfeldt, Chem. Commun., 2015, 51, 16308–16311 RSC; (b) A. Sacco, F. Bella, S. De La Pierre, M. Castellino, S. Bianco, R. Bongiovanni and C. F. Pirri, ChemPhysChem, 2015, 16, 960–969 CrossRef CAS PubMed; (c) R. Shanti, F. Bella, Y. S. Salim, S. Y. Chee, S. Ramesh and K. Ramesh, Mater. Des., 2016, 108, 560–569 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05596g |
| This journal is © The Royal Society of Chemistry 2020 |