
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular Csp3–H/C–C bond amination of alkyl azides for the selective synthesis of cyclic imines and tertiary amines†
Xiaojin
Wen‡
a,
Xinyao
Li‡
a,
Xiao
Luo
a,
Weijin
Wang
a,
Song
Song
 *ab and
Ning
Jiao
*a
*ab and
Ning
Jiao
*a
aState Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Xue Yuan Rd. 38, Beijing 100191, China. E-mail: ssong@bjmu.edu.cn; jiaoning@pku.edu.cn
bState Key Laboratory of Drug Research, Shanghai Institute of Materia Medical, Chinese Academy of Sciences, Shanghai 201203, China
First published on 7th April 2020
Abstract
The intramolecular Csp3–H and/or C–C bond amination is very important in modern organic synthesis due to its efficiency in the construction of diversified N-heterocycles. Herein, we report a novel intramolecular cyclization of alkyl azides for the synthesis of cyclic imines and tertiary amines through selective Csp3–H and/or C–C bond cleavage. Two C–N single bonds or a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond are efficiently constructed in these transformations. The carbocation mechanism differs from the reported metal nitrene intermediates and therefore enables metal-free and new transformation.
N double bond are efficiently constructed in these transformations. The carbocation mechanism differs from the reported metal nitrene intermediates and therefore enables metal-free and new transformation.
Introduction
N-Heterocycles are undoubtedly important chemicals in organic synthesis, and have been considered as key functionality regulators in pharmaceuticals.1 The intramolecular nitrogen insertion into Csp3–H and/or C–C bonds provides an efficient approach to N-heterocycles.2–5 The pioneering groups of Aubé4 and Pearson5 developed the intramolecular Schmidt reactions2 and made significant achievements for various N-heterocycle synthesis.3 The earliest intramolecular aliphatic C–N bond formation named the Hofmann–Loffler–Freytag reaction5 always started from unstable halogenated amines to construct N-heterocycles. Over the past two decades, the aliphatic C–H amination has achieved great progress via the C–H activation strategy.6 However, most of these reactions required electron withdrawing directing groups and delivered amide products (Scheme 1a). Beginning with Breslow's pioneering work,7 a metal-nitrene strategy was successfully applied in intramolecular Csp3–H bond N insertion, providing elegant approaches to amides bearing N–H bonds (Scheme 1a).8 Thus, the development of direct aliphatic C–H/C–C amination is still highly desirable.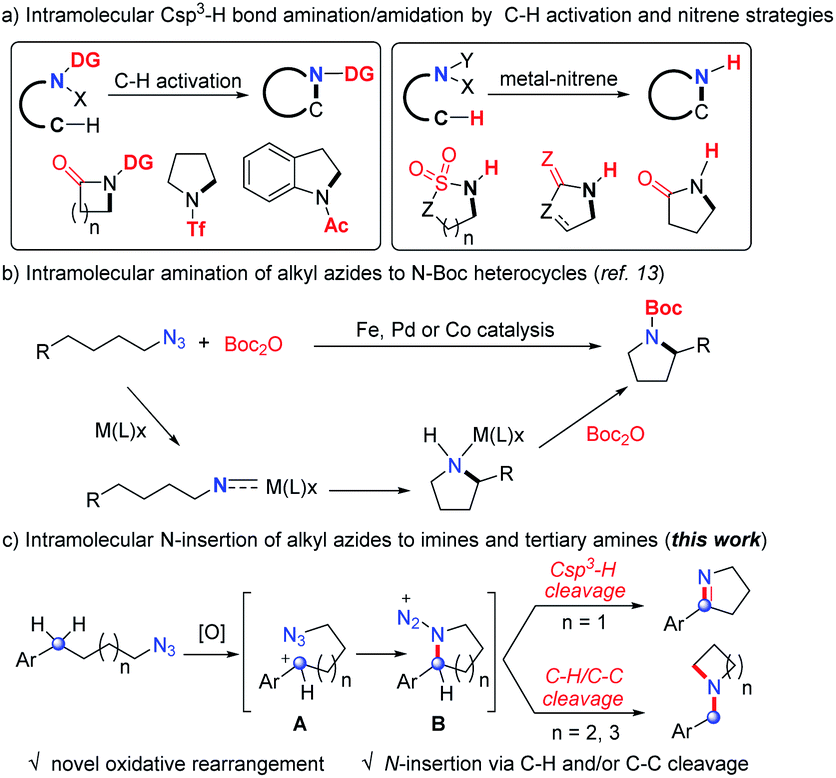 | ||
| Scheme 1 Intramolecular N-insertion of the Csp3–H bond. | ||
Organic azides are synthetically useful in drug discovery, bioconjugation and materials science.9 Although the intramolecular Csp3–H bond amination/amidation of aryl azides10 and sulfonyl azides11 has achieved great progress, the corresponding transformation of alkyl azides12 was rarely developed until recent results.13 In 2013, Betley and coworkers demonstrated the pioneering intramolecular aliphatic C–H amination of alkyl azides catalyzed by an iron catalyst (Scheme 1b).13a The groups of van der Vlugt,13c Lin,13d,e de Bruin,13c,f and Chi13g independently developed the same elegant intramolecular cyclization of alkyl azides by iron, palladium or cobalt catalysis to deliver N-Boc heterocycles (Scheme 1b), in which the involved nitrene type intermediates required an equivalent of Boc2O reagent to liberate the active catalyst to complete the catalytic cycle (Scheme 1b). Despite the advances of the above strategies (Scheme 1a and b), these intramolecular aliphatic amination/amidation processes always delivered N-carbonyl or sulfonyl heterocycles with the formation of one C–N single bond.
Inspired by these results, we speculated that the oxidative generation of carbocation A may trigger the formation of cyclic intermediate B (Scheme 1c), which may undergo other transformations in the absence of transition-metal catalysts and provide opportunities for new products. Herein, we described a novel intramolecular nitrogen insertion into a Csp3–H and/or C–C bond of alkyl azides to deliver cyclic imines and tertiary amines (Scheme 1c). The aliphatic C–H or C–C bond was selectively cleaved with the efficient formation of two C–N single bonds or a CN double bond.
Results and discussion
According to our previous element incorporation reactions through the carbocation intermediates generated in situ with the DDQ oxidant,14 we chose azide 1a as the model substrate to investigate our speculation. As expected, dihydropyrrole 2a was obtained in 75% yield in the presence of DDQ and TFA at 60 °C (Table 1, entry 1). Two C–H bonds were cleaved and a CN double bond was constructed along with the release of N2 in this case. TEMPO or CAN as the oxidant gave inferior yields (entries 2–3), while PIDA or NHPI could not execute the conversion of 1a to 2a (entries 4–5). The chlorinated solvent afforded better yields than that of other solvents such as DMSO, toluene, or MeCN (entries 6–9), and the reaction delivered the highest yield in TCE (entry 9). The pKa of acids influenced the reaction strongly (entries 10–12). 2a was obtained in only 10% yield in the presence of acetic acid (entry 10), while MsOH or TfOH failed to facilitate this transformation (entries 11–12). The treatment of 1a with 0.4 mL of TFA afforded 2a in a satisfactory 73% isolated yield (entry 13). Lowering the temperature hampered the reactivity (entry 14).
|
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Acid | Solvent | Yield of 2ab |
| a Reaction conditions: 1a (0.3 mmol), oxidant (0.36 mmol) and acid (0.2 mL) in a solvent (0.5 mL) at 60 °C for 12 h. b Yield determined by 1H NMR spectroscopy with dibromomethane as an internal standard. c Performed with TFA (0.4 mL). d Isolated yields. e Performed at room temperature. DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone, CAN = cerium ammonium nitrate, TEMPO = (2,2,6,6-tetramethylpiperidin-1-yl)oxyl, NHPI = N-hydroxyphthalimide, PIDA = phenyliodine diacetate, TFA = trifluoroacetic acid, MsOH = methanesulfonic acid, TfOH = trifluoromethanesulfonic acid, and TCE = 1,1,2,2-tetrachloroethane. | ||||
| 1 | DDQ | TFA | DCE | 75% |
| 2 | CAN | TFA | DCE | 18% |
| 3 | TEMPO | TFA | DCE | 8% |
| 4 | NHPI | TFA | DCE | 0 |
| 5 | PIDA | TFA | DCE | 0 |
| 6 | DDQ | TFA | DMSO | 0 |
| 7 | DDQ | TFA | PhMe | 64% |
| 8 | DDQ | TFA | MeCN | 46% |
| 9 | DDQ | TFA | TCE | 77% |
| 10 | DDQ | AcOH | TCE | 10% |
| 11 | DDQ | MsOH | TCE | 0 |
| 12 | DDQ | TfOH | TCE | 0 |
| 13c | DDQ | TFA | TCE | 84% (73%)d |
| 14e | DDQ | TFA | TCE | 76% |
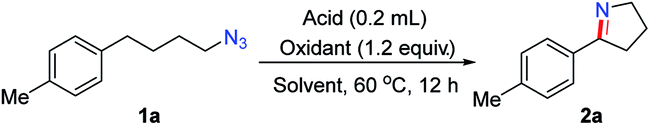
We explored the generality of this intramolecular Csp3–H nitrogen insertion for δ-aryl alkyl azides under standard reaction conditions (Table 2). Substrates bearing electron-donating substituents (MeO, tBu, PhO) at the aryl ring worked smoothly to afford the corresponding cyclic imines 2c–e in good yields. The electron-withdrawing substituents (F, Cl) caused low reactivity, resulting in pyrrolines 2f–g in diminished yields (26–31%). Substituents at the meta or ortho position of the arene rings 1h–j slightly affected the efficiency. Besides arenes, the heteroaryl azide 2-(4-azidobutyl)thiophene 1k was transformed to 2k in 32% yield. The substituents on the alkyl chain influenced this reaction slightly (2l–o). The cyclic imines 2 were easily converted to diversified heterocycles.15 Compared to the well-established approaches to cyclic imines, the present intramolecular N-insertion protocol features mild conditions and high atom economy.

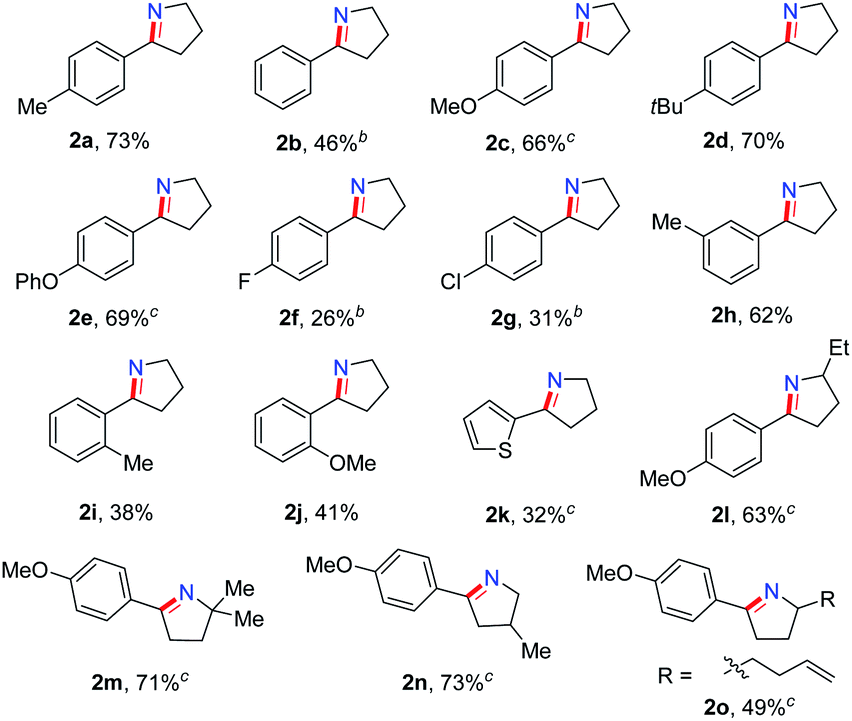
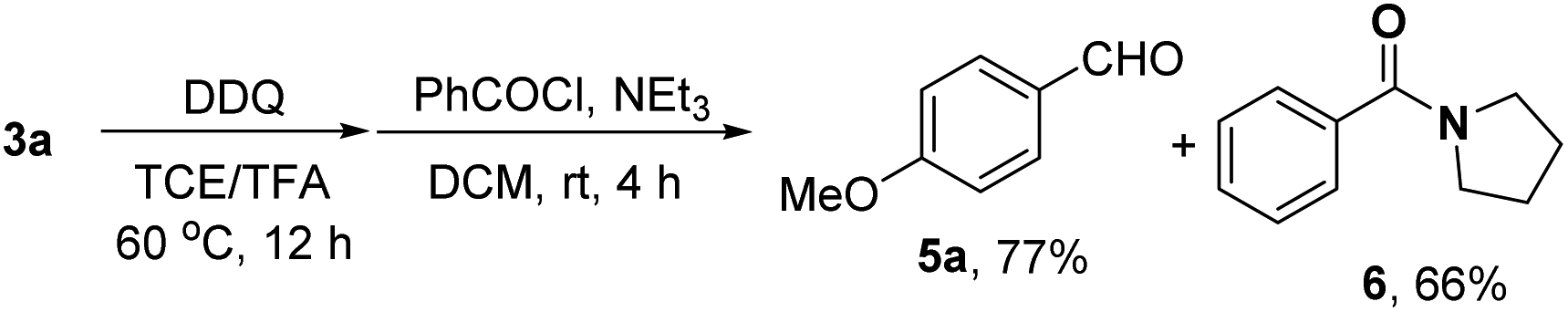
In order to synthesize a six-membered cyclic imine, we conducted the reaction of alkyl azide 3a under standard conditions. However, the target imine product 4a was not detected (eqn (1)). We conducted the capture experiment by the addition of benzoyl chloride to the reaction of 3a (eqn (2)). Aldehyde 5a and amide 6 were obtained in 77% and 66% yields, respectively (eqn (2)), which indicated that the azide 3a was converted to amine via an imine cation intermediate and a hydrolysis process (for the detailed mechanism, see Scheme 2 and 3).
 | (1) |
 | (2) |
 | ||
| Scheme 2 Proposed mechanism. | ||
 | ||
| Scheme 3 Energy profile for the DDQ-mediated amination of alkyl azides 1 and 3. | ||
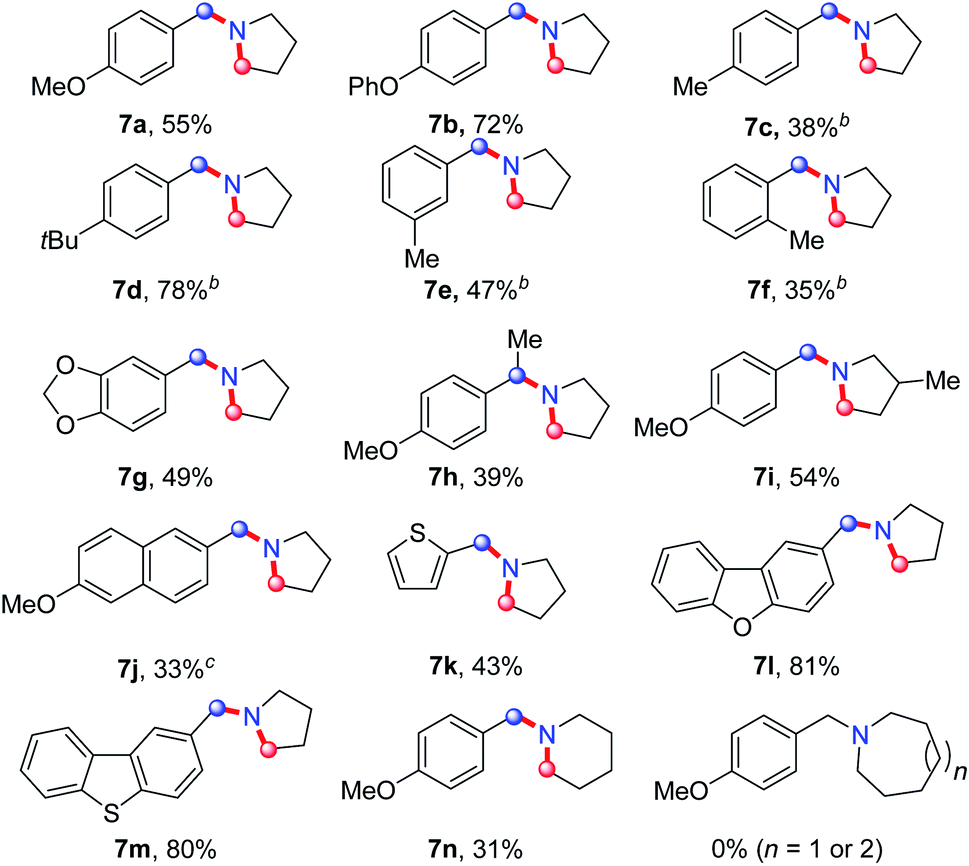
On the basis of this result, we investigated the one-pot reaction of alkyl azide 3 with DDQ and TFA followed by in situ reduction. We were delighted to find that the corresponding cyclic tertiary amine 7a was obtained in 55% yield (Table 3). The substituent on the arene slightly influenced the yield and a series of N-Bn pyrrolidines were synthesized in moderate yields. The azide substrates bearing alkyl substituents also smoothly delivered benzyl-substituted 7h or pyrrolidine 7i in moderate yield. In addition, naphthalene, thiophene, dibenzofuran and dibenzothiophene were all well tolerated to afford cyclic tertiary amines 7j–m in 33–81% yields. It is noteworthy that the transformation of 3 to 7 with the release of nitrogen as the only by-product, is thus highly atom-economic. Moreover, the present strategy cleaves the Csp3–Csp3 bond16 without strained rings or assisted functional groups. Besides pyrrolidine, piperidine derivative 7n also could be synthesized by the intramolecular N-insertion of alkyl azide 3n. Unfortunately, the present strategy could not be applied in the construction of seven- or eight-membered N-heterocycles.
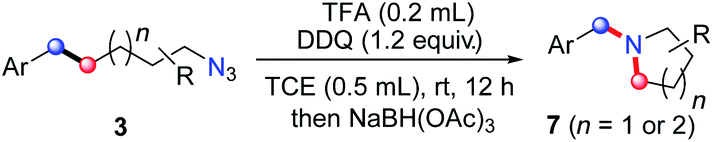
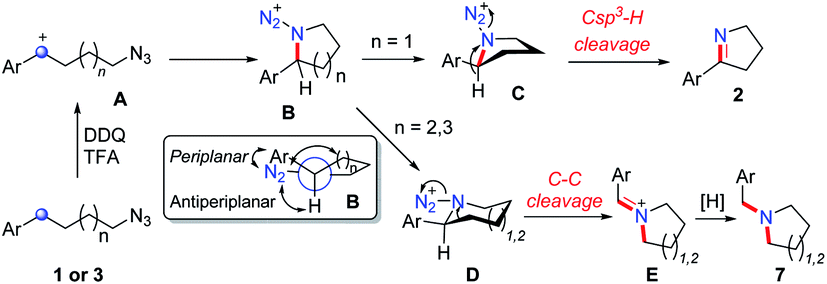
Based on the above experiments, we proposed the possible mechanism of the reaction (Scheme 2). The oxidation of alkyl azides 1 and 3 at the benzylic position by DDQ with TFA provides benzylic cation intermediate A, which is attacked by the azide group to generate cyclic intermediate B. In the most stable conformation of B, the aryl group should stand on the equatorial bond, which makes a small torsion angle with the azide moiety. As a result, the following Schmidt rearrangement of B with the concerted release of N2 and the aryl shift is unfavorable through periplanar migration, while the hydrogen or alkyl shift is potentially feasible through antiperiplanar migration. The five-membered ring species C undergoes deprotonation with the release of N2 to afford cyclic imine 2, while the six-membered ring intermediate D undergoes 1,2-alkyl migration to generate the imine cation E, which is sequentially reduced to deliver tertiary amine 7.
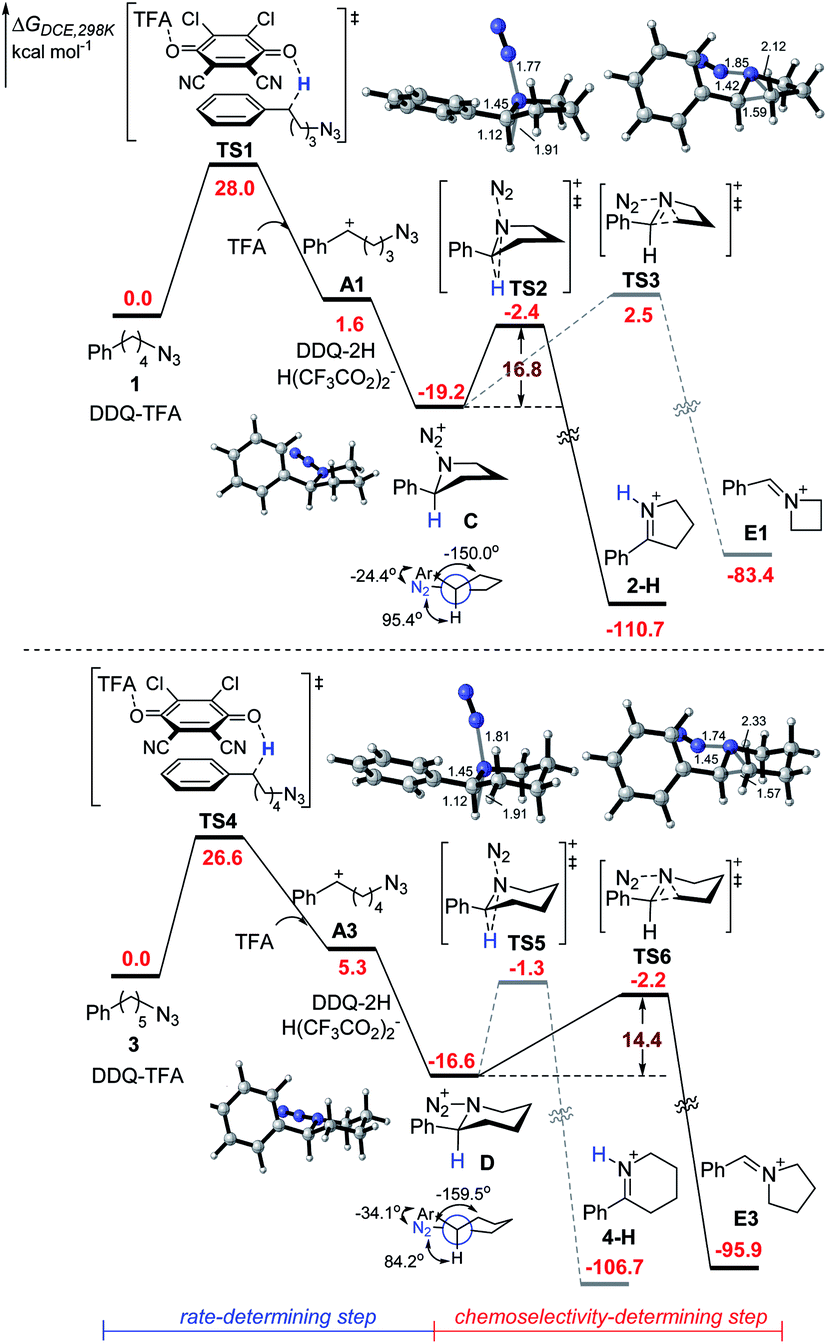
To further understand the mechanism, we performed preliminary DFT calculations on the model reaction of alkyl azides 1 and 3 with DDQ and TFA (Scheme 3).17 We first studied the oxidation of 1 at the benzylic position by DDQ with TFA through O-attack hydride transfer pathway, which is the most thermodynamically favorable pathway in some similar cases.18 The hydride transfer from 1 to the complex of DDQ and TFA through TS1 requires a Gibbs free energy barrier of 28.0 kcal mol−1 to form the benzylic carbocation intermediate A1 and DDQH-TFA− anion, which could be stabilized by another TFA molecule to afford DDQ-2H and H(CF3CO2)2− species. Subsequently, the azide moiety would attack the formed carbocation in A1 to generate five-membered ring C, which is exothermic by 19.2 kcal mol−1. In the most stable conformation of C, the phenyl group on the equatorial bond has a small torsion angle (−24.4°) with the azide moiety, while the benzylic hydrogen and alkyl group have big dihedral angles (95.4° and −150.0°, respectively) with the azide moiety. Therefore, the following Schmidt rearrangement2 of C with the concerted release of N2 and the hydrogen or alkyl shift is potentially feasible through antiperiplanar migration. The Schmidt rearrangement with the 1,2-H shift through the antiperiplanar transition state TS2 with a free energy barrier of 16.8 kcal mol−1 gives 2-H. The barrier of the 1,2-alkyl shift to imine cation E1 through TS3 (ΔG‡ = 21.7 kcal mol−1) is much higher than that of the 1,2-H shift pathway.
Alternatively, the hydride transfer from 3 to the complex of DDQ and TFA through TS4 requires a Gibbs free energy barrier of 26.6 kcal mol−1 to form the benzylic carbocation A3. The azide moiety is favorable to attack the intramolecular carbocation to generate six-membered ring D, which is exothermic by 16.6 kcal mol−1. In the most stable conformation of D, the dihedral angle of the azide moiety with the alkyl group increases to −159.5°, while the one with hydrogen decreases to 84.2°. This is likely to provide an advantage for the 1,2-alkyl shift. The following Schmidt rearrangement of D including the 1,2-H shift through TS5 requires a free energy barrier of 15.3 kcal mol−1 to give 4-H. In contrast with C, D undergoes a 1,2-alkyl shift through TS6 with a free energy barrier of 14.4 kcal mol−1, which is favorable compared to the 1,2-H shift pathway, indicating that the 1,2-alkyl shift pathway becomes predominant. Reviewing the whole energy profile, it is revealed that the oxidation with hydride transfer is the rate-determining step, while the chemoselectivity in the nitrogenation of alkyl azides is essentially controlled by the conformation of the cyclic intermediate and the ring-side in the Schmidt rearrangement process. The experimentally observed electronic effects on the Ar group are consistent with the first oxidation step with hydride transfer as the rate-determining step (see the ESI† for details).
Conclusions
In summary, we have demonstrated a novel metal-free intramolecular Csp3–H/C–C amination of alkyl azides for the synthesis of cyclic imines and tertiary amines. Two C–N single bonds or a CN double bond are efficiently constructed in these transformations through the highly selective benzyl Csp3–H or C–C bond cleavage. The mechanistic studies and DFT calculation indicate a carbocation pathway for this novel protocol. The present chemistry not only provides a new approach to N-heterocycles, but also expands the transformation and application of C–H/C–C amination in organic synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Key Research and Development Project (No. 2019YFC1708902), the National Natural Science Foundation of China (No. 21632001, 21602005, and 81821004), and the Drug Innovation Major Project (2018ZX09711-001), Peking University (No. PKU2020PKYZX004) is appreciated. Major changes in the central level support projects (2060302), and the Open Research Fund of Shanghai Key Laboratory of Green Chemistry and Chemical Processes are greatly appreciated. We thank Xiaoxue Yang in this group for reproducing the results of 2m and 7l.Notes and references
- (a) A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199–2238 CrossRef CAS PubMed; (b) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- (a) R. F. Schmidt, Ber., 1924, 57, 704–706 Search PubMed; (b) P. A. S. Smith, J. Am. Chem. Soc., 1948, 70, 320–323 CrossRef CAS; (c) L. H. Briggs, G. C. De Ath and S. R. Ellis, J. Chem. Soc., 1942, 61–63 RSC; (d) H. Wolfe, Org. React., 1946, 3, 307–336 Search PubMed; (e) P. A. S. Smith, Molecular Rearrangements, John Wiley & Sons, New York, 1963 Search PubMed; (f) R. A. Abramovich and E. P. Kyba, The Chemistry of the Azido Group, John Wiley & Sons, London, 1971 Search PubMed; (g) A. Wrobleski, T. C. Coombs, C. W. Huh, S.-W. Li and J. Aubé, Org. React., 2012, 78, 1–320 CAS; (h) M. Szostak and J. Aubé, Chem. Rev., 2013, 113, 5701–5765 CrossRef CAS PubMed.
- (a) J. Aubé and G. L. Milligan, J. Am. Chem. Soc., 1991, 113, 8965–8966 CrossRef; (b) J. Aubé, G. L. Milligan and C. J. Mossman, J. Org. Chem., 1992, 57, 1635–1637 CrossRef; (c) V. Gracias, G. L. Milligan and J. Aubé, J. Am. Chem. Soc., 1995, 117, 8047–8048 CrossRef CAS; (d) G. L. Milligan, C. J. Mossman and J. Aubé, J. Am. Chem. Soc., 1995, 117, 10449–10459 CrossRef CAS; (e) A. Wrobleski, K. Sahasrabudhe and J. Aubé, J. Am. Chem. Soc., 2004, 126, 5475–5481 CrossRef CAS PubMed.
- (a) W. H. Pearson and J. M. Schkeryantz, Tetrahedron Lett., 1992, 33, 5291–5294 CrossRef CAS; (b) W. H. Pearson, R. Walavalkar, J. M. Schkeryantz, W. Fang and J. D. Blickensdorf, J. Am. Chem. Soc., 1993, 115, 10183–10194 CrossRef CAS.
- For selected reviews of C-H amination, see: (a) H. M. L. Davies and M. S. Long, Angew. Chem., Int. Ed., 2005, 44, 3518–3520 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (c) J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092–4106 RSC; (d) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed; (e) J. J. Topczewski and M. S. Sanford, Chem. Sci., 2015, 6, 70–76 RSC; (f) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed; (g) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed; (h) F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 5061–5074 RSC.
- (a) A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1883, 16, 558–560 CrossRef; (b) K. Lçffler and C. Freytag, Chem. Ber., 1909, 42, 3427–3431 CrossRef; (c) S. Lang and J. A. Murphy, Chem. Soc. Rev., 2006, 35, 146–156 RSC; (d) P. S. Baran, K. Chen and J. M. Richter, J. Am. Chem. Soc., 2008, 130, 7247–7249 CrossRef PubMed; (e) C. Martínez and K. Muñiz, Angew. Chem., Int. Ed., 2015, 54, 8287–8291 CrossRef PubMed; (f) E. A. Wappes, S. C. Fosu, T. C. Chopko and D. A. Nagib, Angew. Chem., Int. Ed., 2016, 55, 9974–9978 CrossRef CAS PubMed; (g) P. Becker, T. Duhamel, C. J. Stein, M. Reiher and K. Muñiz, Angew. Chem., Int. Ed., 2017, 56, 8004–8008 CrossRef CAS PubMed; (h) S. Liu, J. Li, D. Wang, F. Liu, X. Liu, Y. Gao, J. Dai and X. Cheng, Chin. J. Chem., 2019, 37, 570–574 CrossRef CAS.
- (a) R. Breslow and S. H. Gellman, J. Chem. Soc., Chem. Commun., 1982, 1400–1401 RSC; (b) R. Breslow and S. H. Gellman, J. Am. Chem. Soc., 1983, 105, 6728–6729 CrossRef CAS.
- (a) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911–922 CrossRef CAS PubMed; (b) S.-M. Au, J.-S. Huang, W.-Y. Yu, W.-H. Fung and C.-M. Che, J. Am. Chem. Soc., 1999, 121, 9120–9132 CrossRef CAS; (c) E. Milczek, N. Boudet and S. Blakey, Angew. Chem., Int. Ed., 2008, 47, 6825–6828 CrossRef CAS PubMed; (d) M. E. Harvey, D. G. Musaev and J. Du Bois, J. Am. Chem. Soc., 2011, 133, 17207–17216 CrossRef CAS PubMed; (e) S. M. Paradine and M. C. White, J. Am. Chem. Soc., 2012, 134, 2036–2039 CrossRef CAS PubMed; (f) J. W. Rigoli, C. D. Weatherly, J. M. Alderson, B. T. Vo and J. M. Schomaker, J. Am. Chem. Soc., 2013, 135, 17238–17241 CrossRef CAS PubMed; (g) J. M. Alderson, A. M. Phelps, R. J. Scamp, N. S. Dolan and J. M. Schomaker, J. Am. Chem. Soc., 2014, 136, 16720–16723 CrossRef CAS PubMed; (h) S. M. Paradine, J. R. Griffin, J. Zhao, A. L. Petronico, S. M. Miller and M. C. White, Nat. Chem., 2015, 7, 987–994 CrossRef CAS PubMed; (i) S. Y. Hong, Y. Park, Y. Hwang, Y. B. Kim, M.-H. Baik and S. Chang, Science, 2018, 359, 1016–1021 CrossRef CAS PubMed; (j) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed; (k) K. J. Stowers, K. C. Fortner and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 6541–6544 CrossRef CAS PubMed; (l) C. G. Espino, P. M. Wehn, J. Chow and J. Du Bois, J. Am. Chem. Soc., 2001, 123, 6935–6936 CrossRef CAS; (m) C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378–15379 CrossRef CAS PubMed; (n) D. N. Zalatan and J. Du Bois, J. Am. Chem. Soc., 2008, 130, 9220–9221 CrossRef CAS PubMed.
- For selected reviews, see: (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040–1052 CrossRef CAS PubMed; (c) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed; (d) E. Leemans, M. D'hooghe and N. De Kimpe, Chem. Rev., 2011, 111, 3268–3333 CrossRef CAS PubMed.
- (a) S. Murata, R. Yoshidome, Y. Satoh, N. Kato and H. Tomioka, J. Org. Chem., 1995, 60, 1428–1434 CrossRef CAS; (b) K. Sun, R. Sachwani, K. J. Richert and T. G. Driver, Org. Lett., 2009, 11, 3598–3601 CrossRef CAS PubMed; (c) Q. Nguyen, K. Sun and T. G. Driver, J. Am. Chem. Soc., 2012, 134, 7262–7265 CrossRef CAS PubMed; (d) Q. Nguyen, T. Nguyen and T. G. Driver, J. Am. Chem. Soc., 2013, 135, 620–623 CrossRef CAS PubMed; (e) O. Villanueva, N. M. Weldy, S. B. Blakey and C. E. MacBeth, Chem. Sci., 2015, 6, 6672–6675 RSC; (f) I. T. Alt, C. Guttroff and B. Plietker, Angew. Chem., Int. Ed., 2017, 56, 10582–10586 CrossRef CAS PubMed; (g) T. G. Driver, Org. Biomol. Chem., 2010, 8, 3831–3846 RSC.
- (a) J. V. Ruppel, R. M. Kamble and X. P. Zhang, Org. Lett., 2007, 9, 4889–4892 CrossRef CAS PubMed; (b) M. Ichinose, H. Suematsu, Y. Yasutomi, Y. Nishioka, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2011, 50, 9884–9887 CrossRef CAS PubMed; (c) H. Lu, K. Lang, H. Jiang, L. Wojtas and X. P. Zhang, Chem. Sci., 2016, 7, 6934–6939 RSC; (d) H. Lu, H. Jiang, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2010, 49, 10192–10196 CrossRef CAS PubMed; (e) J. A. McIntosh, P. S. Coelho, C. C. Farwell, Z. J. Wang, J. C. Lewis, T. R. Brown and F. H. Arnold, Angew. Chem., Int. Ed., 2013, 52, 9309–9312 CrossRef CAS PubMed; (f) H. Lu, C. Li, H. Jiang, C. L. Lizardi and X. P. Zhang, Angew. Chem., Int. Ed., 2014, 53, 7028–7032 CrossRef CAS PubMed; (g) T. K. Hyster, C. C. Farwell, A. R. Buller, J. A. McIntosh and F. H. Arnold, J. Am. Chem. Soc., 2014, 136, 15505–15508 CrossRef CAS PubMed; (h) P. Dydio, H. M. Key, H. Hayashi, D. S. Clark and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 1750–1753 CrossRef CAS PubMed.
- (a) J. E. Forsee and J. Aubé, J. Org. Chem., 1999, 64, 4381–4385 CrossRef CAS; (b) B. T. Smith, V. Gracias and J. Aubé, J. Org. Chem., 2000, 65, 3771–3774 CrossRef CAS PubMed; (c) D. J. Gorin, N. R. Davis and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 11260–11261 CrossRef CAS PubMed; (d) A. Kapat, E. Nyfeler, G. T. Giuffredi and P. Renaud, J. Am. Chem. Soc., 2009, 131, 17746–17747 CrossRef CAS PubMed; (e) M. Szostak and J. Aubé, J. Am. Chem. Soc., 2010, 132, 2530–2531 CrossRef CAS PubMed; (f) R. Liu, O. Gutierrez, D. J. Tantillo and J. Aubé, J. Am. Chem. Soc., 2012, 134, 6528–6531 CrossRef CAS PubMed; (g) H. F. Motiwala, C. Fehl, S.-W. Li, E. Hirt, P. Porubsky and J. Aubé, J. Am. Chem. Soc., 2013, 135, 9000–9009 CrossRef CAS PubMed; (h) X. Sun, C. Gao, F. Zhang, Z. Song, L. Kong, X. Wen and H. Sun, Tetrahedron, 2014, 70, 643–649 CrossRef CAS; (i) X.-J. Wang, Y. Su, R. Li and P. Gu, J. Org. Chem., 2018, 83, 5816–5824 CrossRef CAS PubMed; (j) M. Charaschanya and J. Aubé, Nat. Commun., 2018, 9, 934–942 CrossRef PubMed.
- (a) E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591–595 CrossRef CAS PubMed; (b) D. A. Iovan, M. J. T. Wilding, Y. Baek, E. T. Hennessy and T. A. Betley, Angew. Chem., Int. Ed., 2017, 56, 15599–15602 CrossRef CAS PubMed; (c) B. Bagh, D. L. J. Broere, V. Sinha, P. F. Kuijpers, N. P. van Leest, B. de Bruin, S. Demeshko, M. A. Siegler and J. I. van der Vlugt, J. Am. Chem. Soc., 2017, 139, 5117–5124 CrossRef CAS PubMed; (d) N. C. Thacker, Z. Lin, T. Zhang, J. C. Gilhula, C. W. Abney and W. Lin, J. Am. Chem. Soc., 2016, 138, 3501–3509 CrossRef CAS PubMed; (e) Z. Lin, N. C. Thacker, T. Sawano, T. Drake, P. Ji, G. Lan, L. Cao, S. Liu, C. Wang and W. Lin, Chem. Sci., 2018, 9, 143–151 RSC; (f) P. F. Kuijpers, M. J. Tiekink, W. B. Breukelaar, D. L. J. Broere, N. P. van Leest, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Chem. –Eur. J., 2017, 23, 7945–7952 CrossRef CAS PubMed; (g) K.-P. Shing, Y. Liu, B. Cao, X.-Y. Chang, T. You and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 11947–11951 CrossRef CAS PubMed; (h) D. L. J. Broere, B. de Bruin, J. N. H. Reek, M. Lutz, S. Dechert and J. I. van der Vlugt, J. Am. Chem. Soc., 2014, 136, 11574–11577 CrossRef CAS PubMed; (i) D. L. J. Broere, N. P. van Leest, B. de Bruin, M. A. Siegler and J. I. van der Vlugt, Inorg. Chem., 2016, 55, 8603–8611 CrossRef CAS PubMed; (j) N. P. van Leest, L. Grooten, J. I. van der Vlugt and B. de Bruin, Chem. –Eur. J., 2019, 25, 5987–5993 CrossRef CAS PubMed.
- (a) C. Qin and N. Jiao, J. Am. Chem. Soc., 2010, 132, 15893–15895 CrossRef CAS PubMed; (b) F. Chen, C. Qin, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2011, 50, 11487–11491 CrossRef CAS PubMed; (c) C. Qin, T. Shen, C. Tang and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 6971–6975 CrossRef CAS PubMed; (d) J. Liu, X. Wen, C. Qin, X. Li, X. Luo, A. Sun, B. Zhu, S. Song and N. Jiao, Angew. Chem., Int. Ed., 2017, 56, 11940–11944 CrossRef CAS PubMed; (e) J. Liu, X. Qiu, X. Huang, X. Luo, C. Zhang, J. Wei, J. Pan, Y. Liang, Y. Zhu, Q. Qin, S. Song and N. Jiao, Nat. Chem., 2019, 11, 71–77 CrossRef CAS PubMed; (f) Y. Liang, Y.-F. Liang and N. Jiao, Org. Chem. Front., 2015, 2, 403–415 RSC.
- (a) M. Ringwald, R. Sturmer and H. H. Brintzinger, J. Am. Chem. Soc., 1999, 121, 1524–1527 CrossRef CAS; (b) C. A. Figueira and P. T. Gomes, Catal. Lett., 2015, 145, 762–768 CrossRef CAS; (c) H. Karoui, C. Nsanzumuhire, F. L. Le Moigne and P. Tordo, J. Org. Chem., 1999, 64, 1471–1477 CrossRef CAS PubMed.
- For selected reviews of C–C cleavage, see: (a) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610–618 RSC; (b) A. Dermenci, J. W. Coe and G. Dong, Org. Chem. Front., 2014, 1, 567–581 RSC; (c) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS PubMed; (d) M. Murakami and N. Ishida, J. Am. Chem. Soc., 2016, 138, 13759–13769 CrossRef CAS PubMed; (e) M. Tobisu and N. Chatani, Chem. Soc. Rev., 2008, 37, 300–307 RSC; (f) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed; (g) X. Wu and C. Zhu, Chin. J. Chem., 2019, 37, 171–182 CAS.
- All of the geometry optimizations and frequency calculations were performed with the M06-2X functional implemented in Gaussian 09. All of the energies discussed in the paper are Gibbs free energies at the def2-TZVP basis set based on the structures with the PCM solvation correction in DCE at the 6-31+G(d,p)basis set. Computational details and references are given in the ESI.†.
- (a) B. Chan and L. Radom, J. Phys. Chem. A, 2007, 111, 6456–6467 CrossRef CAS PubMed; (b) X. Guo, H. Zipse and H. Mayr, J. Am. Chem. Soc., 2014, 136, 13863–13873 CrossRef CAS PubMed; (c) S. Yamabe, S. Yamazaki and S. Sakaki, Int. J. Quantum Chem., 2015, 115, 1533–1542 CrossRef CAS; (d) A. S. K. Tsang, A. S. K. Hashmi, P. Comba, M. Kerscher, B. Chan and M. H. Todd, Chem. –Eur. J., 2017, 23, 9313–9318 CrossRef CAS PubMed; (e) C. A. Morales-Rivera, P. E. Floreancig and P. Liu, J. Am. Chem. Soc., 2017, 139, 17935–17944 CrossRef CAS PubMed; (f) A. Gouranourimi, A. Chipman, R. Babaahmadi, A. Olding, B. F. Yates and A. Ariafard, Org. Biomol. Chem., 2018, 16, 9021–9029 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization data and experimental procedures. See DOI: 10.1039/c9sc05522c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |