
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A H2O2 self-sufficient nanoplatform with domino effects for thermal-responsive enhanced chemodynamic therapy†
Shichao
Zhang
a,
Changyu
Cao
a,
Xinyi
Lv
a,
Hanming
Dai
a,
Zhihao
Zhong
a,
Chen
Liang
a,
Wenjun
Wang
b,
Wei
Huang
d,
Xuejiao
Song
*a and
Xiaochen
Dong
 *ac
*ac
aKey Laboratory of Flexible Electronics (KLOFE), Institute of Advanced Materials (IAM), School of Physical and Mathematical Sciences, Nanjing Tech University (NanjingTech), Nanjing 211800, China. E-mail: iamxcdong@njtech.edu.cn; xjsong@njtech.edu.cn
bSchool of Physical Science and Information Technology, Liaocheng University, Liaocheng 252059, China
cSchool of Chemistry and Materials Science, Nanjing University of Information Science & Technology, Nanjing, 210044, China
dShaanxi Institute of Flexible Electronics (SIFE), Northwestern Polytechnical University (NPU), Xi'an 710072, China
First published on 8th January 2020
Abstract
Chemodynamic therapy (CDT), employing Fenton or Fenton-like catalysts to convert hydrogen peroxide (H2O2) into toxic hydroxyl radicals (˙OH) to kill cancer cells, holds high promise in tumor therapy due to its high selectivity. However, the anticancer efficacy is unsatisfactory owing to the limited concentration of endogenous H2O2. Herein, thermal responsive nanoparticles with H2O2 self-sufficiency are fabricated by utilizing organic phase change materials (PCMs) to encapsulate iron–gallic acid nanoparticles (Fe–GA) and ultra-small CaO2. PCMs, acting as the gatekeeper, could be melted down by the hyperthermia effect of Fe–GA under laser irradiation with a burst release of Fe–GA and CaO2. The acidic tumor microenvironment would further trigger CaO2 to generate a large amount of H2O2 and Ca2+. The self-supplied H2O2 would be converted into ˙OH by participating in the Fenton reaction with Fe–GA. Meanwhile, in situ generation of Ca2+ could cause mitochondrial damage and lead to apoptosis of tumor cells. With efficient tumor accumulation illustrated in in vivo photoacoustic imaging, Fe–GA/CaO2@PCM demonstrated a superior in vivo tumor-suppressive effect without inducing systemic toxicity. The study presents a unique domino effect approach of PCM based nanoparticles with thermal responsiveness, H2O2 self-supply, and greatly enhanced CDT effects, showing bright prospects for highly efficient tumor treatment.
Introduction
Chemodynamic therapy (CDT), a new type of reactive oxygen species (ROS)-based cancer treatment, depends on the in situ Fenton (Fenton-like) reaction to convert hydrogen peroxide (H2O2) into hydroxyl radicals (˙OH) by catalysis, and has high toxicity to cancer cells.1–4 Recently, various kinds of catalysts have been developed for CDT, such as Fe2+, Fe3+, Mn2+, Cu+, V2+ and Cr4+.5–16 Among these ions, Fe2+ and Fe3+ are typical Fenton ions and show great advantages in biocompatibility since iron is essential for cell growth, proliferation and oxygen delivery, as well as many other life processes. The typical Fenton reaction equation of iron is as follows:| Fe2+ + H2O2 → Fe3+ + OH− + ˙OH | (1) |
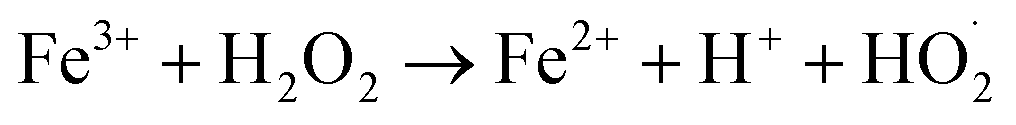 | (2) |
During such a cyclic process, no exogenous stimulation or oxygen is required. Compared with photodynamic therapy (PDT), the other kind of important ROS dependent therapy involving external laser irradiation and sufficient oxygen, CDT exhibits remarkable advantages in overcoming the obstacles of limited penetration depth of laser and hypoxic tumor microenvironments (TMEs).17–20 However, the chemodynamic efficacy is still restricted because of the limited endogenously produced H2O2, even though the concentration of H2O2 in many types of solid tumors has been reported to be higher than that in normal tissues.8,20–22 Under this circumstance, increasing the concentration of H2O2 or improving the catalytic capability of Fenton agents shows great promise in enhancing anticancer efficiency.
In response to the lack of H2O2 in tumors, various methods have been developed to increase the intratumoral H2O2 concentration.8,15,20–22 On the one hand, glucose oxidase loaded nanoparticles are utilized to oxidize intratumoral glucose to H2O2 and gluconic acid in the presence of O2via a glucose-metabolic reaction, increasing the H2O2 concentration. However, due to its oxygen dependence, the efficiency of H2O2 generation in this strategy is severely restricted by tumor hypoxia. On the other hand, metal peroxides (MPs), such as CuO2, have been utilized as H2O2 sources to produce H2O2 in acidic TMEs.8,21 Very recently, copper peroxide (CP) nanodots were successfully fabricated by Chen's group and used as an nanoagent to enhance CDT with self-supplying H2O2 in the acidic environment of endo/lysosomes.8 Although MPs show high efficiency in H2O2 generation, the poor stability in aqueous solution restricts their further application in cancer treatment, especially via intravenous injection. Therefore, developing a safer and more efficient nanocarrier, in which H2O2 production can be strictly controlled, is highly desirable for improving CDT efficiency.23–26
Organic phase-change materials (PCMs), which usually refer to materials with huge latent heats of fusion and exhibiting reversible solid–liquid transition at a nearly constant temperature, have attracted considerable interest in serving as thermo-responsive materials for drug release.27–32 Through adjusting the ratio of fatty acids or fatty alcohols, PCMs with different melting points (MPs) can be obtained. When the temperature is lower than the MP, the PCM plays the part of ‘gatekeeper’ to prevent the encapsulated drugs from premature release. Once the temperature is higher than the MP, a burst release of drugs can be achieved. In addition to small drugs, such as DOX, other kinds of materials, such as hydrophilic or hydrophobic nanoparticles, can also be encapsulated within PCMs, making them a promising candidate in the stimuli-sensitive drug delivery system.33
Herein, organic PCMs with a melting point of 46 °C were utilized to co-encapsulate hydrophilic iron–gallic acid (Fe–GA) nanoparticles (NPs) and ultra-small hydrophobic CaO2 nanoparticles to obtain Fe–GA/CaO2@PCM NPs for thermal responsive enhanced CDT (Scheme 1). Fe–GA nanoparticles in Fe–GA/CaO2@PCM were used not only as a photothermal therapy (PTT) agent to generate the hyperthermia effect, but also an ideal CDT agent.34–37 The PCM layer could be the gatekeeper to isolate CaO2 from the outside environment, thus enhancing the stability and reducing the premature release of CaO2. When the Fe–GA/CaO2@PCM NPs were irradiated with an 808 nm laser, the hyperthermia induced by Fe–GA caused the melting of the PCM, causing the burst release of CaO2 NPs. The CaO2 NPs could further be triggered by acidic TMEs to produce a large amount of H2O2 and Ca2+, achieving high efficacy in Fe–GA based CDT and Ca2+ induced mitochondrial damage, respectively.21,38 Meanwhile, the PTT effect of Fe–GA could not only kill cancer cells, but also accelerate the generation of ˙OH.3 Under the guidance of photoacoustic imaging and fluorescence imaging, Fe–GA/CaO2@PCM NPs exhibited high accumulation in the tumor site via enhanced permeability and retention (EPR) effects. With the H2O2 self-sufficient CaO2 NPs, great performance in inhibiting tumor growth was shown in PTT/CDT combined treatment. Benefiting from their good biocompatibility and thermal responsiveness, the Fe–GA/CaO2@PCM NPs could be a potential multifunctional nanoplatform with on-demand H2O2 self-supply for enhanced PTT/CDT treatments.
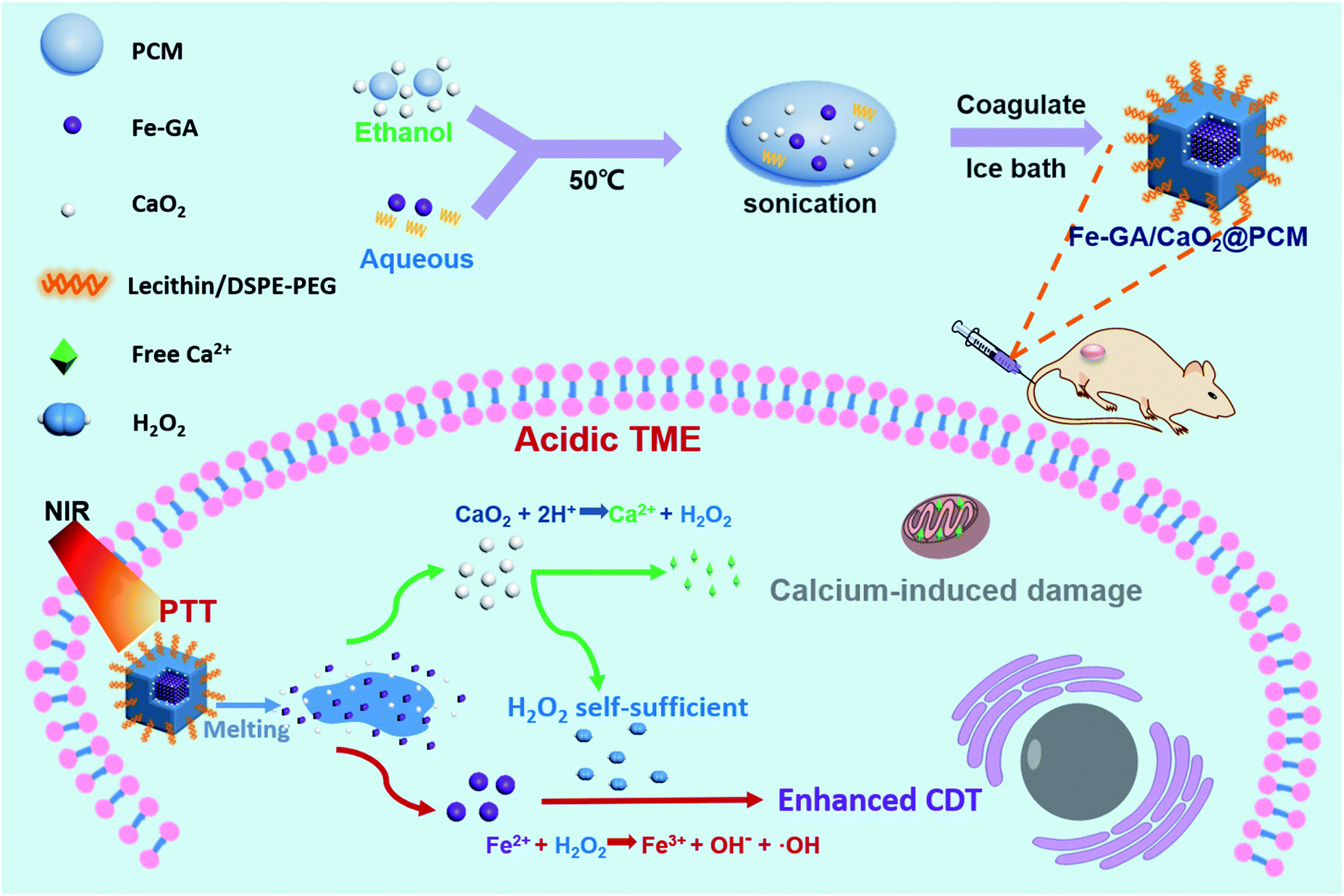 | ||
| Scheme 1 A scheme showing the fabrication of Fe–GA/CaO2@PCM and its application in H2O2 self-sufficient chemodynamic therapy. | ||
Results and discussion
The thermal responsive Fe–GA/CaO2@PCM NPs were prepared as shown in Scheme 1. First, organic PCMs with a melting point of 46 °C were fabricated by mixing 1-hexadecanol with oleic acid at a ratio of 3.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in ethanol. The solid PCM can be obtained for the following experiment via evaporating the ethanol. Next, Fe–GA NPs with an average size of 11 nm (Fig. S1a and b†) were successfully synthesized through a coordination method and ultra-small CaO2 NPs were synthesized according to the literature, respectively.35,39 The scanning electron microscopy (SEM) image and dynamic light scattering (DLS) revealed that the CaO2 NPs have a uniform size of about 18 nm (Fig. 1a and S1c†). X-ray diffraction (XRD) has confirmed the successful synthesis of CaO2 (Fig. S2a†). To obtain the Fe–GA/CaO2@PCM NPs, PCMs were utilized to co-encapsulate Fe–GA NPs and CaO2 NPs via a resolidification method. With the modification of lecithin and DSPE-mPEG, the Fe–GA/CaO2@PCM NPs exhibited excellent dispersibility and stability. The final capacity of CaO2 and Fe in Fe–GA/CaO2@PCM NPs was determined to be 8.1% and 12.7% (w/w) characterized by inductively coupled plasma mass spectrometry (ICP-MS), respectively. As revealed by the SEM and TEM images in Fig. 1b, c and S2b,† Fe–GA/CaO2@PCM NPs exhibit a cubic-like morphology with uniform distribution. Interestingly, upon laser irradiation, the NPs were melted down and the morphology turned out to be chaotic. Meanwhile, the diameters of the Fe–GA/CaO2@PCM NPs were decreased from 130 nm to 55 nm with laser irradiation (Fig. 1d). The sharply decreased diameter further confirms the thermal responsive profile of Fe–GA/CaO2@PCM NPs.
:1 in ethanol. The solid PCM can be obtained for the following experiment via evaporating the ethanol. Next, Fe–GA NPs with an average size of 11 nm (Fig. S1a and b†) were successfully synthesized through a coordination method and ultra-small CaO2 NPs were synthesized according to the literature, respectively.35,39 The scanning electron microscopy (SEM) image and dynamic light scattering (DLS) revealed that the CaO2 NPs have a uniform size of about 18 nm (Fig. 1a and S1c†). X-ray diffraction (XRD) has confirmed the successful synthesis of CaO2 (Fig. S2a†). To obtain the Fe–GA/CaO2@PCM NPs, PCMs were utilized to co-encapsulate Fe–GA NPs and CaO2 NPs via a resolidification method. With the modification of lecithin and DSPE-mPEG, the Fe–GA/CaO2@PCM NPs exhibited excellent dispersibility and stability. The final capacity of CaO2 and Fe in Fe–GA/CaO2@PCM NPs was determined to be 8.1% and 12.7% (w/w) characterized by inductively coupled plasma mass spectrometry (ICP-MS), respectively. As revealed by the SEM and TEM images in Fig. 1b, c and S2b,† Fe–GA/CaO2@PCM NPs exhibit a cubic-like morphology with uniform distribution. Interestingly, upon laser irradiation, the NPs were melted down and the morphology turned out to be chaotic. Meanwhile, the diameters of the Fe–GA/CaO2@PCM NPs were decreased from 130 nm to 55 nm with laser irradiation (Fig. 1d). The sharply decreased diameter further confirms the thermal responsive profile of Fe–GA/CaO2@PCM NPs.
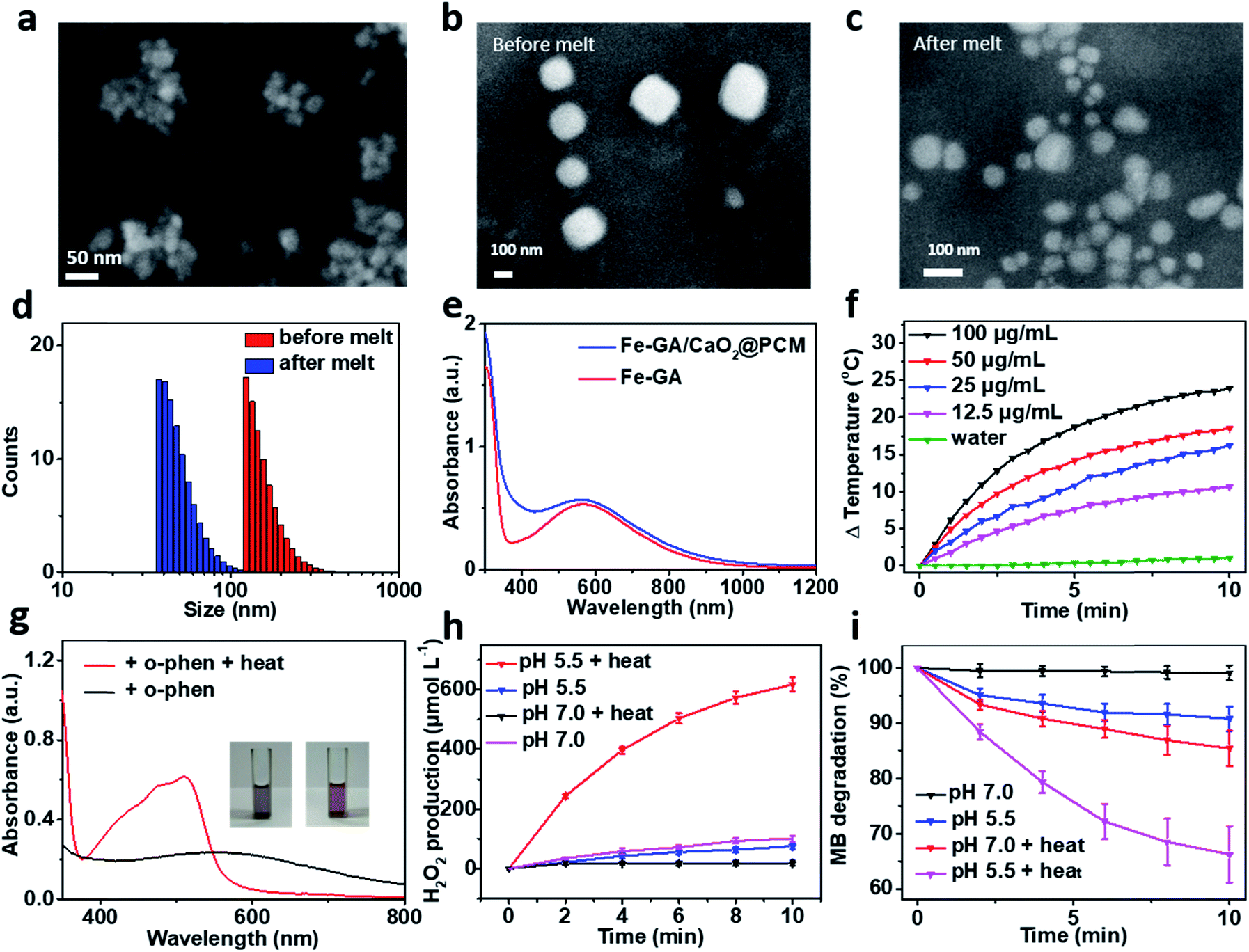 | ||
| Fig. 1 (a) SEM image of CaO2 NPs. SEM image of Fe–GA/CaO2@PCM NPs (b) before and (c) after melting. (d) Size distribution of Fe–GA/CaO2@PCM NPs before and after melting. (e) UV-vis absorption spectra of Fe–GA/CaO2@PCM NPs and Fe–GA. (f) Photothermal heating curve of different concentrations of Fe–GA/CaO2@PCM NPs (0–100 μg mL−1). (g) UV-vis absorption spectra of Fe–GA/CaO2@PCM NPs added with o-phen to detect Fe2+. (h) H2O2 generation from CaO2@PCM (100 μg mL−1) under different conditions. (i) MB degradation caused by Fe–GA/CaO2@PCM NP Fenton reaction under different conditions. | ||
Owing to the encapsulation of Fe–GA NPs, Fe–GA/CaO2@PCM NPs exhibited high absorbance in the near infrared region (NIR) (Fig. 1e). The photothermal performance of Fe–GA/CaO2@PCM NPs was measured at different concentrations with 808 nm laser irradiation. Compared with water, the temperature of Fe–GA/CaO2@PCM NPs increased rapidly even at a low concentration (Fig. 1f). Meanwhile, Fe–GA/CaO2@PCM NPs still maintained good photothermal performance even after cycles of photothermal heating and cooling, presenting great photostability (Fig. S3a†). Calculated using the equation in Fig. S3b,† the photothermal conversion efficiency (η) of Fe–GA/CaO2@PCM NPs (100 μg mL−1) is as high as 49.8%. Moreover, the photothermal effects of Fe–GA@PCM NPs, CaO2@PCM NPs and Fe–GA/CaO2@PCM NPs irradiated with a constant power density (1.0 W cm−2, 10 min) were compared. As shown in Fig. S3c,† there were no significant differences between Fe–GA@PCM NPs and Fe–GA/CaO2@PCM NPs, while the temperature increase of CaO2@PCM NPs was similar to that of water, demonstrating that the PTT effect was contributed by Fe–GA nanoparticles.
Benefiting from PCM protection, Fe–GA/CaO2@PCM NPs exhibit excellent thermal responsiveness. To detect the release of Fe–GA, o-phenanthroline (o-phen) is utilized as an indicator, since it can form an orange-red complex with Fe2+ at pH 2–9. As shown in Fig. 1g, when the Fe–GA/CaO2@PCM NPs were placed at room temperature (25 °C), there was no significant change in the colour of the solution. Interestingly, a rapid change in the colour could be observed when the Fe–GA/CaO2@PCM NPs were under irradiation because of the melting of the PCM. The typical absorbance at 500 nm further demonstrated the thermal responsive release of Fe–GA. Next, the thermal responsive release of CaO2 and H2O2 generation under different conditions was investigated. Considering the reaction between Fe–GA and H2O2, CaO2@PCM NPs were chosen for the following experiment. KI, which could react with H2O2 to generate I3−, was used as the probe to detect H2O2 produced by CaO2 under different pH values by UV-vis measurements (Fig. S3d†). As shown in Fig. 1h, when the temperature of CaO2@PCM solution was heated to about 50 °C, the amount of produced H2O2 was calculated to be as high as 617 μmol L−1 under acidic conditions, which is far more than that in the solid tumors (less than 100 μmol L−1). However, no significant generation of H2O2 was found neither in the group of CaO2@PCM under neutral conditions with or without heating nor in the group of CaO2@PCM under acidic conditions without heating.
The above results demonstrated that the H2O2 self-supply behavior of CaO2@PCM could only be observed under acidic conditions with heating. Based on this, the Fenton effect of Fe–GA/CaO2@PCM NPs under different conditions was studied. The generation of ˙OH was demonstrated by electron paramagnetic resonance (EPR, Fig. S3e†) and a typical 1:2:2:1 signal could be observed in the EPR spectrum. Meanwhile, methylene blue (MB) was also used to detect the generation of ˙OH, for the absorption of MB at 660 nm decreases by reacting with ˙OH. With laser irradiation, the absorbance of MB decreased rapidly under acidic conditions, compared with the other groups (Fig. 1i), which is consistent with the results of H2O2 generation. Furthermore, in vitro thermal response release of Ca2+ was investigated by ICP-MS. A burst release of Ca2+ could be realized under 50 °C (Fig. S3f†).
The in vitro combined therapeutic efficiency of Fe–GA/CaO2@PCM NPs was evaluated. First, HeLa cells were incubated with CaO2@PCM NPs, Fe–GA@PCM NPs and Fe–GA/CaO2@PCM NPs at different concentrations for 24 h. There was no significant decrease in cell viability even at a high concentration (Fig. 2a and S4a†), indicating the great biocompatibility of the nanoparticles and the Wax-Sealed function of the PCM. Remarkably, when exposed to 808 nm laser irradiation, the cell viability in the group of Fe–GA/CaO2@PCM NPs was much lower than that in the group of Fe–GA@PCM NPs (Fig. 2b) with the same concentration of Fe. The results might be caused by the melting of the PCM under laser irradiation which triggered the release of CaO2, thus producing a large amount of H2O2, which could further react with released Fe ions to generate toxic ˙OH. The propidium iodide (PI) and calcein AM staining assay in Fig. S4b† has further demonstrated the cytotoxicity of Fe–GA/CaO2@PCM NPs.
 | ||
| Fig. 2 (a) Dark cytotoxicity of Fe–GA/CaO2@PCM NPs and Fe–GA@PCM NPs on HeLa cells. (b) Cytotoxicity of HeLa cells of Fe–GA/CaO2@PCM NPs and Fe–GA@PCM NPs at different concentrations under laser irradiation. *P < 0.001. (c) Intracellular hydroxyl radical detection with the DCFH-DA probe. (d) Mitochondrial membrane potential staining with the JC-1 probe. (e) Intracellular Ca2+ detection with the Fluo-4 AM probe. Scale bars: 100 μm. | ||
To study the intracellular action mechanism of Fe–GA/CaO2@PCM NPs as the H2O2 self-supplier to kill cancer cells, 2,7-dichlorofluorescein diacetate (DCFH-DA) staining assay was utilized to confirm the generation of hydroxyl radicals (Fig. 2c). Without laser irradiation, HeLa cells in the groups of Fe–GA/CaO2@PCM and Fe–GA@PCM exhibited weak green fluorescence. When the cells were exposed to laser irradiation, no obvious green fluorescence could be observed in Fe–GA@PCM, which might be owing to the limited content of intracellular H2O2. In contrast, strong green fluorescence was observed in cells treated with an Fe–GA/CaO2@PCM + laser, indicating the generation of a large amount of hydroxyl radicals in cells. According to the reaction of CaO2 under acidic conditions, not only H2O2 but also Ca2+ could be burst generated upon laser irradiation. The excessive Ca2+ in the cells will damage intracellular proteins and nucleic acids, as well as organelles especially mitochondria, leading to cell death. Therefore, the intracellular generation of Ca2+ was further investigated. Consistent with the results in ROS detection, strong green fluorescence can be detected only in the group of Fe–GA/CaO2@PCM + laser, demonstrating the burst release of Ca2+ (Fig. 2e). The mitochondrion, acting as an indispensable organelle in cell energy conversion and apoptosis, is vulnerable to excessive Ca2+ and reactive oxygen species. To characterize mitochondrial damage, the JC-1 fluorescence probe was employed to detect the mitochondrial membrane potential (MMP) of cells. Compared with the groups of the Fe–GA/CaO2@PCM or Fe–GA@PCM without irradiation and Fe–GA@PCM with irradiation, fluorescence in the Fe–GA/CaO2@PCM + laser turned from red to green (Fig. 2d), indicating that MMP declined and cellular apoptosis occurred under such a therapy. These results demonstrate that Fe–GA/CaO2@PCM acts as an efficient H2O2 self-supplier with great potential in combined PTT/CDT.
To realize more accurate therapy and reduce damage on normal tissues, imaging guidance is beneficial to trace the nanoparticles and provide the information on tumors. Herein, taking advantage of the absorbance of Fe–GA in the NIR, photoacoustic (PA) imaging was conducted after intravenously injected with Fe–GA/CaO2@PCM NPs. PA signals in the tumor site appeared at 2 h and gradually enhanced with time (Fig. 3a). Meanwhile, IR780 (a commonly used fluorescent dye) was encapsulated within Fe–GA/CaO2@PCM NPs to explore the biodistribution of Fe–GA/CaO2@PCM NPs (Fig. 3b). 24 h after intravenous injection of Fe–GA/CaO2@PCM NPs, the main organs and tumors of HeLa tumor-bearing mice were obtained for ex vivo fluorescence imaging. A strong fluorescence signal could be observed in the tumor site, confirming the high tumor uptake via the EPR effect. Meanwhile, the blood circulation study was carefully carried out by detecting the concentration of the Fe ion content in blood samples by ICP-MS. The obtained data were fitted to a time-dependent concentration curve using a two-compartment model, in which the diffusion half-time and elimination half-time were calculated to be 0.6 ± 0.14 h and 11.55 ± 1.43 h, respectively (Fig. 3c). Benefiting from the high tumor accumulation and long blood circulation, Fe–GA/CaO2@PCM NPs show great potential for application in cancer therapy.
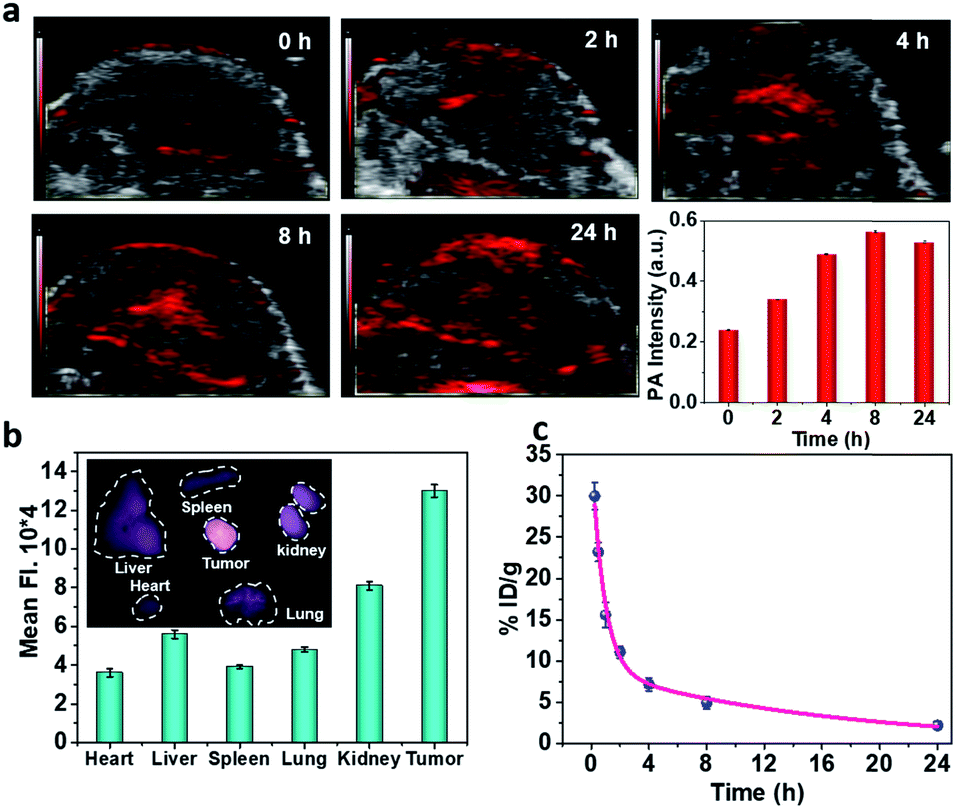 | ||
| Fig. 3 (a) In vivo PA images of HeLa xenograft mice and quantification of the PA signal several time points after i.v. injection with Fe–GA/CaO2@PCM NPs. (b) Ex vivo fluorescence images of major organs and tumors 24 h after injection and quantification of the fluorescence signal. (c) Concentration–time curve of Fe–GA/CaO2@PCM NPs in mice after i.v. injection. The concentration refers to the Fe ion content detected by ICP-MS. | ||
To evaluate PTT & CDT synergistic treatment effects of Fe–GA/CaO2@PCM NPs in vivo, nude mice bearing HeLa tumors were divided into 4 groups randomly: (i) i.v. injected with saline (200 μL). (ii) i.v. injected with Fe–GA/CaO2@PCM NPs (200 μL, 1.5 mg kg−1) and without laser irradiation. (iii) i.v. injected with Fe–GA@PCM NPs (200 μL, 1.5 mg kg−1) and irradiated with an 808 nm laser. (iv) i.v. injected with Fe–GA/CaO2@PCM NPs (200 μL, 1.5 mg kg−1) and irradiated with an 808 nm laser. All the illumination was given 6 h after injection with a constant power density (1.0 W cm−2, 20 min). The temperature changes in the tumor site under laser irradiation were detected with an infrared camera (Fig. 4a and b). The temperature in groups of Fe–GA/CaO2@PCM NPs and Fe–GA@PCM NPs rapidly increased to about 47.5 °C, which was sufficient for the melting of the PCM to trigger the release of CaO2. During the treatment period, the tumor volume change of each group was recorded with a digital caliper every two days (Fig. 4c). No noticeable tumor growth inhibition effect was observed in group ii, which might due to the perfect protection of PCM. When laser irradiation was added, the hyperthermia effect generated by Fe–GA@PCM NPs induced a partial anticancer effect. Remarkably, owing to the self-sufficient H2O2 enhanced CDT by thermal responsive Fe–GA/CaO2@PCM, tumor growth in the mice treated with Fe–GA/CaO2@PCM NPs plus laser irradiation was completely ablated without recurrence in the period of therapy (Fig. 4d, e and S5a†). The high efficacy of combined PTT/CDT was also confirmed using the haematoxylin and eosin (H&E) staining tumor slices after different treatments (Fig. 4f). Consistent with the tumor growth, compared with the other three groups, the most severe damage was observed in the tumors treated with Fe–GA/CaO2@PCM NPs plus laser irradiation. To further study the ability of Fe–GA/CaO2@PCM NPs to self-supply H2O2 under laser irradiation, an ex vivo DCFH-DA staining assay was conducted to confirm ˙OH generation. Weak green fluorescence could be detected in the control group or tumor treated with Fe–GA/CaO2@PCM NPs. A slightly enhanced fluorescence signal was observed in tumors treated with Fe–GA@PCM NPs plus laser irradiation, which might be owing to the released Fe–GA reacted with the endogenous H2O2 to generate ˙OH. Significantly, strong green fluorescence appeared in the tumors treated with Fe–GA/CaO2@PCM NPs plus laser irradiation owing to the on-demand generation of a large amount of H2O2 (Fig. 4g and h). The results demonstrate that Fe–GA/CaO2@PCM NPs could serve as a H2O2 self-sufficient nanoplatform for enhanced PTT/CDT. Meanwhile, there was no significant decrease in the mouse body weights during the therapeutic period (16 days), suggesting no acute toxicity of the Fe–GA/CaO2@PCM NPs (Fig. S5b†). H&E staining slices of major organs and tissues revealed no obvious damage, further indicating the biosafety of Fe–GA/CaO2@PCM NPs (Fig. S6†).
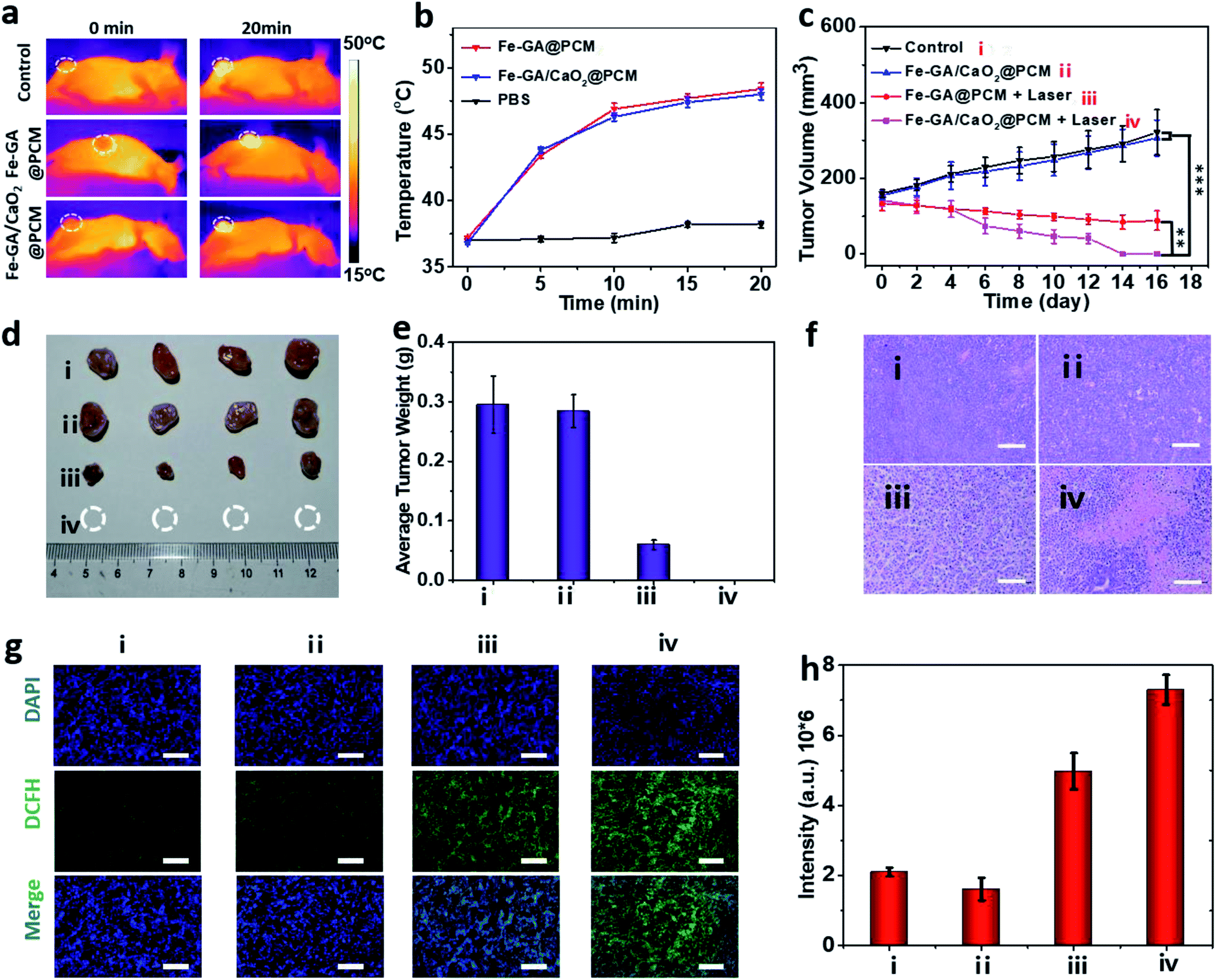 | ||
| Fig. 4 (a) IR thermal images of mice bearing HeLa tumors irradiated with an 808 nm laser (1.0 W cm−2, 20 min). (b) The temperature changes in the tumor site under laser irradiation. (c) Tumor volume curves of each group: **P < 0.01 and ***P < 0.01. (d) Resected tumors of each group on day 16. (e) Average tumor weight of mice in each group after 16 days. (f) H&E stained images of tumor sections from each treatment group. Scale bars: 100 μm. (g) Immunofluorescence staining of DCFH-DA to detect intratumoral hydroxyl radicals after different treatments. Scale bars: 50 μm. (h) Semiquantitative analysis of fluorescence signals with DCFH-DA immunofluorescence staining. | ||
Conclusions
In summary, we developed an intelligent nanoplatform by co-encapsulating Fe–GA and CaO2 in organic PCMs, and their application as a thermal responsive CDT agent with self-supplied H2O2. PCMs, as the protective layer, could be melted by the hyperthermia induced by Fe–GA under laser irradiation, thus releasing Fe–GA and CaO2. In acidic TMEs, CaO2 could serve as a H2O2 supplier to generate a large amount of H2O2 and Ca2+ as well. The overproduced H2O2 could be converted into toxic ˙OH by Fe–GA, achieving an enhanced CDT effect. Meanwhile, the burst release of Ca2+ in cancer cells further caused damage to mitochondria and led to cell apoptosis. Owing to the modification of polyethylene glycol, the Fe–GA/CaO2@PCM NPs exhibited a long blood circulation and high tumor uptake. With the imaging-guidance, in vivo combined PTT/CDT was carried out and the interlock performance of Fe–GA/CaO2@PCM NPs enabled a superior tumor-suppressive effect under laser irradiation without inducing noticeable damage to normal tissues or organs. Compared with other methods to improve the intratumoral concentration of H2O2, the work presented an intelligent and much safer nanoplatform with thermal and acidic TME responsiveness. It is worthy to expect that the development of smart types of multifunctional agents with high-performance may provide new possibilities for the potential clinical translation of PTT/CDT.Experimental section
Synthesis of Fe–GA nanoparticles
100 mg of polyvinylpyrrolidone (PVP) was added into 10 mL DI water under sonication at 25 °C. Then, 0.1 mL of FeCl2 aqueous solution (200 mg mL−1) was mixed with the PVP solution under vigorous stirring. 20 mg of GA was dispersed in 1 mL of water for use. After 30 min of stirring, the FeCl2–PVP mixture was mixed with GA solution and stirred overnight. After being filtered with the PP microporous membrane (0.22 μm), the resultant product was stored in a 4 °C refrigerator for the following use.Synthesis of CaO2 nanoparticles
45 mL of PEG-200 and 1 mL of CaCl2 aqueous solution (0.2 g mL−1) were added into a round bottom flask and stirred for 15 minutes. After adding 0.55 mL of ammonia solution (1.0 M), the mixture was stirred for 10 minutes. Then, 1 mL 30% H2O2 solution was slowly added into the mixture using a syringe. After stirring for 6 hours, a clear transparent liquid was obtained. After slowly adding 4 mL of NaOH solution (1.0 mol L−1) with stirring, the liquid turned into white turbid liquid. The final products were obtained by centrifugation with DI water and ethanol. CaO2 nanoparticles were stored in ethanol at 4 °C.Preparation of PCMs
PCMs were synthesized according to previous studies.29 In brief, 40 mg of oleic acid and 140 mg of 1-hexadecanol were added into 10 mL absolute ethanol. After sonication, PCMs were obtained and stored at 4 °C for use.Preparation of Fe–GA/CaO2@PCM nanoparticles
Fe–GA/CaO2@PCM NPs were formed by a resolidification method. In brief, 0.4 mL CaO2 ethanol solution (2 mg mL−1) and 0.4 mL of PCM ethanol solution (18 mg mL−1) were mixed and used as solution 1.6 mg DSPE-mPEG-MW2000 and 20 mg of L-α-lecithin were added into 10 mL of Fe–GA aqueous solution (1 mg mL−1) and used as solution 2. Under sonication at 50 °C, solution 1 was added into solution 2 and then rapidly cooled in an ice bath. The purple dispersion of Fe–GA/CaO2@PCM NPs was obtained after dialyzing against DI water for 24 h. The concentration of iron and calcium was determined by inductively coupled plasma mass spectrometry (ICP-MS).In vitro H2O2 production
H2O2 can react with KI to generate I3− which resulted in an absorption peak at 350 nm; the standard curve can be used to quantitatively analyze the hydrogen peroxide concentration in the solution. The CaO2@PCM NPs were divided into three groups (n = 3): (i) CaO2@PCM NPs dispersed in DI water (pH 7.0, 100 μg mL−1) at 25 °C. (ii) CaO2@PCM NPs dispersed in PBS buffer (pH 5.5, 100 μg mL−1) at 25 °C. (iii) CaO2@PCM NPs dispersed in PBS buffer (pH 5.5, 100 μg mL−1) at 50 °C. 20 μL of KI (0.1 M) aqueous solution was added to each group. The UV-vis absorption spectra of solution were measured every 2 minutes to calculate the concentration of generated H2O2.Fenton effect
Methylene blue was used to detect the hydroxyl radicals generated by the Fenton reaction. The Fe–GA/CaO2@PCM NPs were divided into four groups (n = 3): (i) Fe–GA/CaO2@PCM NPs dispersed in DI water (pH 7.0, 100 μg mL−1) at 25 °C. (ii) Fe–GA/CaO2@PCM NPs dispersed in DI water (pH 7.0, 100 μg mL−1) at 50 °C. (iii) Fe–GA/CaO2@PCM NPs dispersed in PBS buffer (pH 5.5, 100 μg mL−1) at 25 °C. (iv) Fe–GA/CaO2@PCM NPs dispersed in PBS buffer (pH 5.5, 100 μg mL−1) at 50 °C. The decrease of absorption at 660 nm of each group was recorded every 2 minutes to quantitatively analyze the degradation of methylene blue, which represented the production of hydroxyl radicals.Photothermal effect
Photothermal heating curves under 808 nm laser irradiation (1.0 W cm−2, 10 min) of different nanoparticles (Fe–GA@PCM, Fe–GA/CaO2@PCM and CaO2@PCM) and different concentrations (100, 50, 25, 12.5 and 0 μg mL−1) of Fe–GA/CaO2@PCM NPs were measured by using an FLIR infrared camera. Furthermore, Fe–GA/CaO2@PCM NPs were tested under 5 photothermal cycles with 808 nm laser irradiation (1.0 W cm−2, 10 min) to validate the photostability.Thermal response release of Ca2+
To investigate the thermal responsive release of Ca2+, 3 mL of Fe–GA/CaO2@PCM (100 μg mL−1) was dialyzed at 50 °C and 25 °C in 30 mL H2O, respectively. The liquid outside the dialysis bag was collected every 5 min under continuous stirring. The Ca content was detected by ICP-MS.In vitro cytotoxicity
HeLa cells were seeded into 96-well plates and incubated for 24 h under standard conditions (37 °C and 5% CO2). The medium was replaced by different concentrations of Fe–GA/CaO2@PCM NP and Fe–GA@PCM NP medium solutions. For dark toxicity, the cells were incubated in the dark for 24 h, and for phototoxicity, the cells were irradiated with an 808 nm laser (3 min, 1.0 W cm−2) and were incubated for 12 h. 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assays were conducted to measure the relative cell viability.Intracellular detection of ˙OH
A 2,7-dichlorodi-hydrofluorescein diacetate (DCFH-DA, Sigma-Aldrich) probe was used to detect the intracellular hydroxyl radicals produced by the Fenton effect according to the standard protocol. The HeLa cells were divided into four groups with different treatments: (i) added with 20 μg mL−1 of Fe–GA/CaO2@PCM NPs in the dark; (ii) added with 20 μg mL−1 of Fe–GA/CaO2@PCM NPs with 808 nm laser irradiation (1.0 W cm−2, 10 min); (iii) added with 20 μg mL−1 of Fe–GA@PCM NPs in the dark; (iv) added with 20 μg mL−1 of Fe–GA@PCM NPs with 808 nm laser irradiation (1.0 W cm−2, 10 min).Detection of mitochondrial membrane potential
The JC-1 fluorescent probe was employed to detect the changes of mitochondrial membrane potential. HeLa cells were seeded in a 6-well plate and incubated in the dark for 24 h under standard conditions (37 °C and 5% CO2). Fe–GA/CaO2@PCM NPs (20 μg mL−1) and Fe–GA@PCM NPs (20 μg mL−1) were added in the medium and incubated for 6 h, respectively. The illumination group was irradiated with an 808 nm laser at 1.0 W cm−2 for 10 min. 2 h after incubation, the cells were stained with JC-1 for 40 min. After washing with PBS buffer 3 times, the fluorescence of JC-1 was observed with an inverted fluorescence microscope.Intracellular detection of Ca2+
Fluo-4 AM is a commonly used fluorescent probe for detecting the intracellular concentration of Ca2+. HeLa cells were seeded in a 6-well plate. After being incubated at 37 °C for 24 h, the cells were stained with Fluo-4 AM and DAPI according to the standard method. The fluorescence was then detected using a fluorescence microscope.In vivo imaging
Photoacoustic (PA) imaging and in vivo fluorescence imaging were used to observe the enrichment of nanoparticles in the tumor site after intravenous injection. For PA imaging, after being i.v. injected with Fe–GA/CaO2@PCM NPs, the tumor regions of mice were imaged with a Visual sonics Vevo 2100 LAZR system at different time points (0, 2, 4, 8 and 24 h) with 808 nm laser excitation.Biodistribution and blood circulation study
To explore the biodistribution of Fe–GA/CaO2@PCM NPs, the fluorescent dye IR780 was added into Fe–GA/CaO2@PCM NPs to provide near-infrared fluorescence. After being intravenously injected with the above Fe–GA/CaO2@PCM NPs, the major organs and tumors of HeLa xenograft mice were taken out 24 h later, and the fluorescence intensity was detected with a Fluor Vivo 200 (INDEC Biosystems, USA) with 760 nm for excitation to analyze the biodistribution of the nanoparticles. To study the blood circulation of nanoparticles in vivo, mice were intravenously injected with Fe–GA/CaO2@PCM NPs (Fe injection dose: 1.5 mg kg−1), and blood samples were taken out at several time points (0, 0.25, 0.5, 1.0, 1.5, 3.0, 6.0, 9.0, and 24 h). The concentration of Fe in the blood was determined by ICP.In vivo tumor therapy
Nude mice bearing HeLa tumors were divided into 4 groups randomly (n = 4): (i) i.v. injected with saline (200 μL); (ii) i.v. injected with Fe–GA/CaO2@PCM NPs (200 μL, 1.5 mg kg−1) without irradiation; (iii) i.v. injected with Fe–GA@PCM NPs (200 μL, 1.5 mg kg−1) and irradiated with an 808 nm laser (1.0 W cm−2, 20 min); (iv) i.v. injected with Fe–GA/CaO2@PCM NPs (200 μL, 1.5 mg kg−1) and irradiated with an 808 nm laser (1.0 W cm−2, 20 min). The temperature changes in the tumor site were detected with an infrared camera. The tumor sizes and weight of mice were recorded every 2 days for 16 days. After treatment, tumors and other main organs of mice were taken out for hematoxylin and eosin (H&E) staining to study systemic toxicity.In vivo ROS staining
Tumor-bearing mice received the same treatment as the therapy group. Tumors were immediately taken out after treatment for cryosection. With DCFH-DA staining, the production of hydroxyl radicals in tumors was observed by fluorescence microscopy.Ethical statement
All animal experiments were performed according to the NIH guidelines for the care and use of laboratory animals. All the experiments were approved by the School of Pharmaceutical Science in Nanjing Tech University.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (61525402, 61775095, 51803091, and 61935004), Jiangsu Provincial Key Research and Development Plan (BE2017741), and Six Talent Peak Innovation Team in Jiangsu Province (TD-SWYY-009).References
- Z. Tang, Y. Liu, M. He and W. Bu, Angew. Chem., 2019, 58, 946 CrossRef CAS PubMed.
- H. Lin, Y. Chen and J. Shi, Chem. Soc. Rev., 2018, 47, 1938 RSC.
- H. Ranji-Burachaloo, P. Gurr, D. Dunstan and G. Qiao, ACS Nano, 2018, 12, 11819 CrossRef CAS PubMed.
- M. Huo, L. Wang, Y. Chen and J. Shi, Nat. Commun., 2017, 8, 357 CrossRef.
- A. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121 CrossRef CAS.
- Y. Dai, Z. Yang, S. Cheng, Z. Wang, R. Zhang, G. Zhu, Z. Wang, B. Yung, R. Tian, O. Jacobson, C. Xu, Q. Ni, J. Song, X. Sun, G. Niu and X. Chen, Adv. Mater., 2018, 30, 1704877 CrossRef.
- T. Li, J. Zhou, L. Wang, H. Zhang, C. Song, J. de la Fuente, Y. Pan, J. Song, C. Zhang and D. Cui, Adv. Healthcare Mater., 2019, 8, 1900192 CrossRef PubMed.
- L. Lin, T. Huang, J. Song, X. Ou, Z. Wang, H. Deng, R. Tian, Y. Liu, J. Wang, Y. Liu, G. Yu, Z. Zhou, S. Wang, G. Niu, H. Yang and X. Chen, J. Am. Chem. Soc., 2019, 141, 9937 CrossRef CAS PubMed.
- L. Lin, J. Song, L. Song, K. Ke, Y. Liu, Z. Zhou, Z. Shen, J. Li, Z. Yang, W. Tang, G. Niu, H. Yang and X. Chen, Angew. Chem., 2018, 57, 4902 CrossRef CAS.
- Y. Liu, W. Zhen, L. Jin, S. Zhang, G. Sun, T. Zhang, X. Xu, S. Song, Y. Wang, J. Liu and H. Zhang, ACS Nano, 2018, 12, 4886 CrossRef CAS.
- B. Ma, S. Wang, F. Liu, S. Zhang, J. Duan, Z. Li, Y. Kong, Y. Sang, H. Liu, W. Bu and L. Li, J. Am. Chem. Soc., 2019, 141, 849 CrossRef CAS.
- P. Ma, H. Xiao, C. Yu, J. Liu, Z. Cheng, H. Song, X. Zhang, C. Li, J. Wang, Z. Gu and J. Lin, Nano Lett., 2017, 17, 928 CrossRef CAS PubMed.
- M. Valko, K. Jomova, C. Rhodes, K. Kuca and K. Musilek, Arch. Toxicol., 2016, 90, 1 CrossRef CAS PubMed.
- L. Zhang, S. Wan, C. Li, L. Xu, H. Cheng and X. Zhang, Nano Lett., 2018, 18, 7609 CrossRef CAS.
- Z. Zhou, J. Song, R. Tian, Z. Yang, G. Yu, L. Lin, G. Zhang, W. Fan, F. Zhang, G. Niu, L. Nie and X. Chen, Angew. Chem., 2017, 56, 6492 CrossRef CAS.
- Q. Chen, L. Feng, J. Liu, W. Zhu, Z. Dong, Y. Wu and Z. Liu, Adv. Mater., 2016, 28, 7129 CrossRef CAS.
- P. Agostinis, K. Berg, K. Cengel, T. Foster, A. Girotti, S. Gollnick, S. Hahn, M. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250 CrossRef PubMed.
- V. Estrella, T. Chen, M. Lloyd, J. Wojtkowiak, H. Cornnell, A. Ibrahim-Hashim, K. Bailey, Y. Balagurunathan, J. Rothberg, B. Sloane, J. Johnson, R. Gatenby and R. Gillies, Cancer Res., 2013, 73, 1524 CrossRef CAS PubMed.
- Z. Wang, Y. Zhang, E. Ju, Z. Liu, F. Cao, Z. Chen, J. Ren and X. Qu, Nat. Commun., 2018, 9, 3334 CrossRef.
- H. Ranji-Burachaloo, A. Reyhani, P. Gurr, D. Dunstan and G. Qiao, Nanoscale, 2019, 11, 5705 RSC.
- M. Zhang, R. Song, Y. Liu, Z. Yi, X. Meng, J. Zhang, Z. Tang, Z. Yao, Y. Liu, X. Liu and W. Bu, Chem, 2019, 5, 2171 CAS.
- X. Liu, Y. Liu, J. Wang, T. Wei and Z. Dai, ACS Appl. Mater. Interfaces, 2019, 11, 23065 CrossRef CAS.
- T. Souho, L. Lamboni, L. Xiao and G. Yang, Biotechnol. Adv., 2018, 36, 1928 CrossRef CAS.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2 CrossRef CAS PubMed.
- V. Biju, Chem. Soc. Rev., 2014, 43, 744 RSC.
- S. Kelkar and T. Reineke, Bioconjug. Chem., 2011, 22, 1879 CrossRef CAS PubMed.
- Y. Dai, J. Su, K. Wu, W. Ma, B. Wang, M. Li, P. Sung, Q. Shen, Q. Wang and Q. Fan, ACS Appl. Mater. Interfaces, 2019, 11, 10540 CrossRef CAS.
- Q. Li, L. Sun, M. Hou, Q. Chen, R. Yang, L. Zhang, Z. Xu, Y. Kang and P. Xue, ACS Appl. Mater. Interfaces, 2019, 11, 417 CrossRef CAS.
- G. Liu, S. Zhang, Y. Shi, X. Huang, Y. Tang, P. Chen, W. Si, W. Huang and X. Dong, Adv. Funct. Mater., 2018, 28, 1804317 CrossRef.
- Q. Sun, F. He, H. Bi, Z. Wang, C. Sun, C. Li, J. Xu, D. Yang, X. Wang, S. Gai and P. Yang, Chem. Eng. J., 2019, 362, 679 CrossRef CAS.
- Y. Yuan, N. Zhang, W. Tao, X. Cao and Y. He, Renewable Sustainable Energy Rev., 2014, 29, 482 CrossRef CAS.
- Z. Yuan, S. Qu, Y. He, Y. Xu, L. Liang, X. Zhou, L. Gu, Y. Gu and H. Chen, Biomater. Sci., 2018, 6, 3219 RSC.
- X. Li, J. Kim, J. Yoon and X. Chen, Adv. Mater., 2017, 29, 1606857 CrossRef.
- L. An, C. Yan, X. Mu, C. Tao, Q. Tian, J. Lin and S. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 28483 CrossRef CAS.
- Q. Jin, W. Zhu, D. Jiang, R. Zhang, C. Kutyreff, J. Engle, P. Huang, W. Cai, Z. Liu and L. Chen, Nanoscale, 2017, 9, 12609 RSC.
- Y. Wang, J. Zhang, C. Zhang, B. Li, J. Wang, X. Zhang, D. Li and S. Sun, ACS Sustainable Chem. Eng., 2018, 7, 994 CrossRef.
- J. Zeng, M. Cheng, Y. Wang, L. Wen, L. Chen, Z. Li, Y. Wu, M. Gao and Z. Chai, Adv. Healthcare Mater., 2016, 5, 772 CrossRef CAS.
- Q. Chen, D. Huo, H. Cheng, Z. Lyu, C. Zhu, B. Guan and Y. Xia, Adv. Healthcare Mater., 2018, 8, 1801113 CrossRef.
- L. Liu, Y. Zhang, W. Qiu, L. Zhang, F. Gao, B. Li, L. Xu, J. Fan, Z. Li and X. Zhang, Small, 2017, 13, 1701621 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05506a |
| This journal is © The Royal Society of Chemistry 2020 |