
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular polymerization provides non-equilibrium product distributions of imine-linked macrocycles†
Michael J.
Strauss
 ,
Austin M.
Evans
,
Ioannina
Castano
,
Rebecca L.
Li
and
William R.
Dichtel
*
,
Austin M.
Evans
,
Ioannina
Castano
,
Rebecca L.
Li
and
William R.
Dichtel
*
Department of Chemistry, Northwestern University, Evanston, IL, 60208 USA. E-mail: wdichtel@northwestern.edu
First published on 9th January 2020
Abstract
Supramolecular polymerization of imine-linked macrocycles has been coupled to dynamic imine bond exchange within a series of macrocycles and oligomers. In this way, macrocycle synthesis is driven by supramolecular assembly, either into small aggregates supported by π–π interactions, or high-aspect ratio nanotubes stabilized primarily by electrostatic and solvophobic interactions. For the latter, supramolecular polymerization into nanotubes restricts imine exchange, thereby conferring chemical stability to the assemblies and their constituent macrocycles. Competition in the formation and component exchange among macrocycles favored pyridine-2,6-diimine-linked species due to their rapid synthesis, thermodynamic stability, and assembly into high-aspect ratio nanotubes under the reaction conditions. In addition, the pyridine-containing nanotubes inhibit the formation of similar macrocycles containing benzene-1,3-diimine-linkages, presumably by disrupting their assembly and templation. Finally, we exploit rapid imine exchange within weak, low-aspect ratio macrocycle aggregates to carry out monomer exchange reactions to macrocycles bearing pyridine moieties. Once a pyridine-containing dialdehyde has exchanged into a macrocycle, the macrocycle becomes capable of nanotube formation, which dramatically slows further imine exchange. This kinetic trap provides chemically diverse macrocycles that are not attainable by direct synthetic methods. Together these findings provide new insights into coupling supramolecular polymerization and dynamic covalent bond-forming processes and leverages this insight to target asymmetric nanotubes. We envision these findings spurring further research efforts in the synthesis of nanostructures with designed and emergent properties.
Introduction
Supramolecular polymers are a compelling platform to design nanostructures with diverse functionality, long range order, and dynamic properties that are not attainable via traditional covalent polymerization.1–5 Due to the promise of accessing materials with these sought-after properties, the last decade of research has seen an emergence in novel supramolecular polymerization strategies such as ‘sergeant and soldier’ chirality amplification,6–9 living supramolecular polymerization,10–14 and supramolecular (co)polymerization.5,15–18 While these strategies allow access to diverse nanostructures, they all employ a general two-step process in which the building blocks are first isolated as a unimolecular species and polymerized in a second synthetic step. In this process, polymerization is typically induced by altering the solvent composition19–23 or changing the temperature of a monomer solution.5,24–27 Using this approach, the chemical structure of the building blocks remain fixed, and diverse nanostructures emerge from the order in which they assemble. However, supramolecular polymerization under conditions in which the monomers can also undergo structural changes can target diverse nanostructures that are kinetically stabilized as a function of supramolecular assembly.28–30Using the traditional two-step approach, we have previously isolated imine-linked macrocycles derived from aromatic dialdehydes and a bifunctional aryl amine (DAPB); and studied their aptitude to undergo acid-mediated supramolecular polymerization into high-aspect ratio nanotubes.31,32 In the case of macrocycles derived from simple aromatic dialdehydes such as terephthaldehyde and isophthalaldehyde (IDA, MC 1), high concentrations of CF3CO2H (>2000 equiv) were required to protonate the imine linkages and drive assembly, while lower acid concentrations catalyzed macrocycle hydrolysis.31,32 However, including pyridine moieties (MC 2), which are more basic than the imine linkages allowed macrocycle assembly to occur via electrostatic attractions upon pyridinium formation, even in the presence of sub-stoichiometric acid loadings.31 Because of the low concentrations of CF3CO2H needed for supramolecular polymerization, we hypothesized that pyridine-containing macrocycles would form nanotubes during their covalent synthesis. In this way, the process of supramolecular polymerization and imine-exchange amongst pyridine containing species may interact and influence each other.
Furthermore, we hypothesized that the relative strengths of non-covalent interactions supporting the assemblies of MC 1 and MC 2 would have profound effects on their kinetic stability and ability to undergo monomer exchange. Under this hypothesis, the relatively weak π–π interactions supporting MC 1, coupled with the inherent reactivity of IDA and 2,6-pyridinedicarboxaldehyde (DFP), would enable monomer exchange to macrocycles bearing pyridine moieties. This exchange would unlock nanotube formation and provide non-symmetric macrocycles that are inaccessible by direct synthetic methods (Fig. 1).
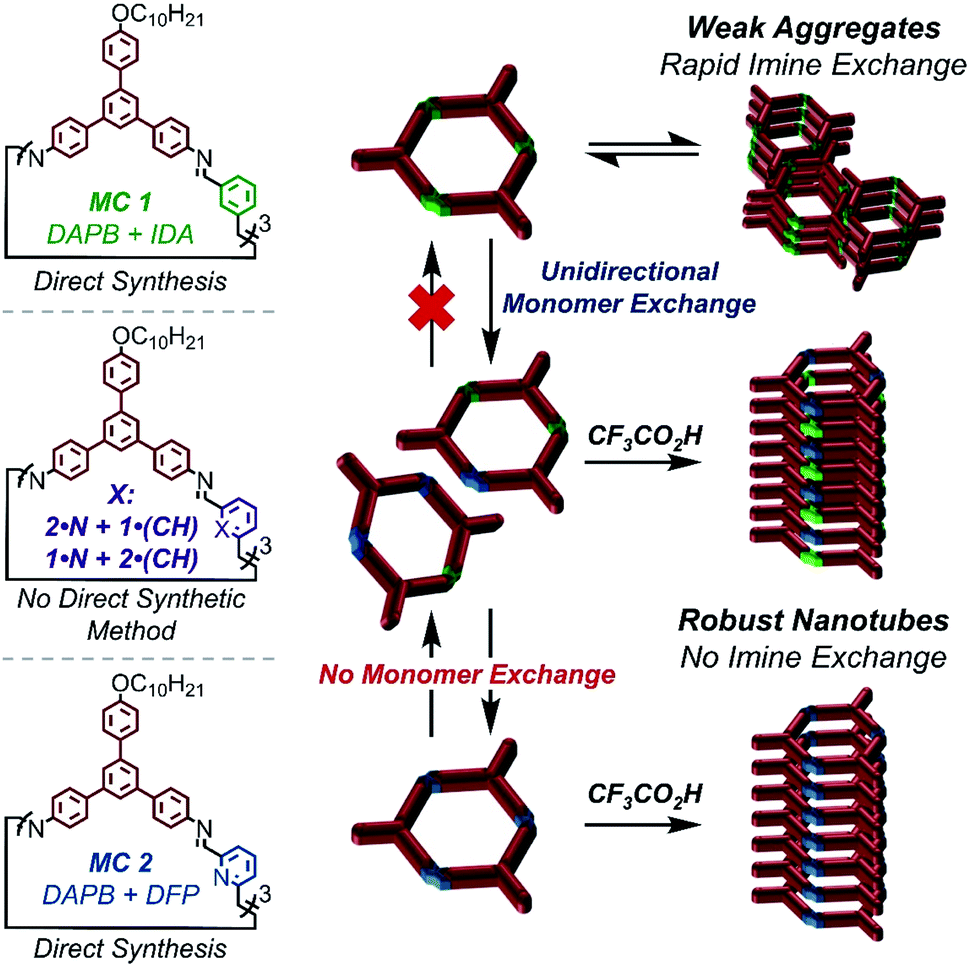 | ||
| Fig. 1 Impacts of pyridine moiety incorporation on macrocycles aptitude to undergo acid-mediated supramolecular polymerization, and the impacts of supramolecular polymerization on imine dynamics. | ||
Results and discussion
The reactivity of DFP along with the assembly of MC 2 into nanotubes under reaction relevant conditions accelerates the formation of MC 2 relative to MC 1 by at least two orders of magnitude (Fig. 2A). The assembly processes of nanotube formation of charged macrocycles and π–π aggregation of neutral macrocycles that each drive macrocyclization cause the relative rates of macrocycle formation to be correlated to the emergence of an X-ray diffraction (XRD) signal. Tracing the emergence of the predominant nanotube diffraction feature via time-resolved XRD (TR-XRD) demonstrates that the formation of MC 2 occurs before the first data point was obtained (2.5 min), while the formation of MC 1 takes over two hours (Fig. 2B and E). The results of TR-XRD were validated using gel-permeation chromatography (GPC), in which MC 2 demonstrated a single narrow elution band after 2 minutes of reaction time. However, in the case of MC 1, 2 minutes of reaction time yielded linear polymer as the major product, with a small secondary peak corresponding to the target macrocycle. At longer times, the GPC signal of the MC 1 experiment narrowed, indicating successful macrocycle formation (see ESI†), which is consistent with our previous study on how neutral macrocycles form via linear polyimines.33 Characterization of the resulting macrocycles by matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) revealed single peaks corresponding to the target macrocycles (Fig. S4 and S24†). Furthermore, analysis of the final states of each macrocyclization reaction by atomic force microscopy (AFM), scanning electron microscopy (SEM), and transmission electron microscopy (TEM) demonstrated that MC 2 forms high aspect ratio nanotubes, while MC 1 yield ill-defined aggregates, which is consistent with previous reports (Fig. 2C, D, F and G) (see ESI†).31,33 The assembly of MC 2 into nanotubes under conditions typical for its synthesis was also observed in the presence of other acids (HCl, p-toluenesulfonic acid, and methanesulfonic acid) capable of protonating the pyridine ring (Fig. S40†). Contributing factors to the observed rate acceleration are the inductive nature of the pyridine ring, which increase the rate of imine condensation to yield acyclic products, and supramolecular polymerization, which drives imine-exchange of these undesired intermediates to yield MC 2.34–37 A small molecule study in which DFP was condensed with aniline under conditions typical for macrocycle synthesis yielded 2,6-diiminophenylpyridine within the first five minutes of reaction time, which is an order of magnitude faster than the reaction of aniline with IDA (Fig. S114–S119†). The above experiments demonstrate that MC 2 assembles into nanotubes under conditions typical for its synthesis and that its linkages form more rapidly than those of MC 1. These two factors explain the rapid and highly selective formation of MC 2, and we designed follow-up experiments to further explore this interplay.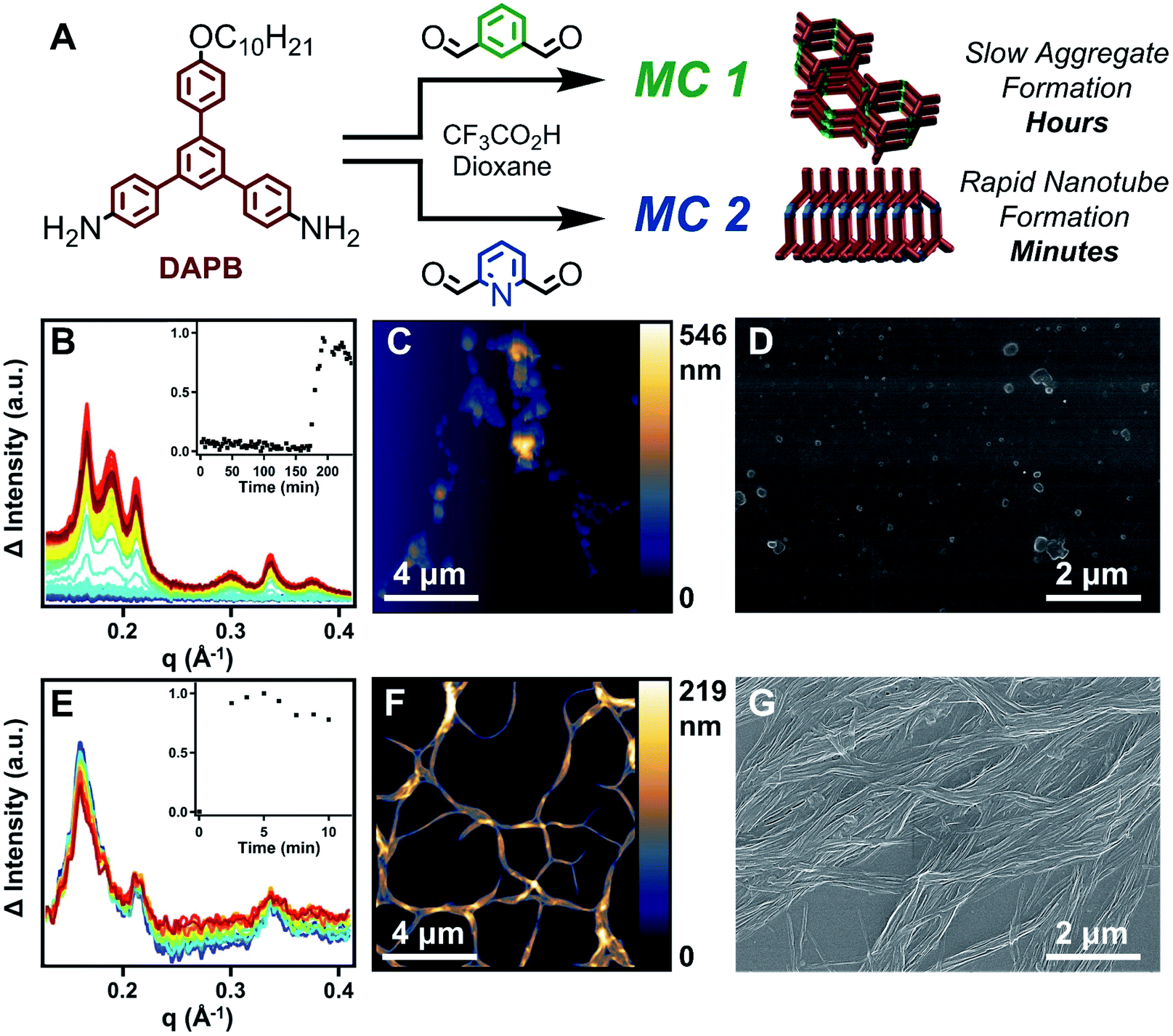 | ||
| Fig. 2 Probing the kinetics of the assembly processes that govern the formation of MC 1 and MC 2. (A) Scheme of macrocycle formation. (B) TR-XRD patterns from 0 to 240 minutes depicting the formation and assembly of MC 1. (Inset) Normalized integration of the diffraction signal with respect to time. (C) Atomic force micrograph of the aggregates resulting from the formation of MC 1. (D) Scanning electron micrograph of the aggregates resulting from the formation of MC 1. (E) TR-XRD patterns from 0 to 10 minutes depicting the rapid formation and assembly of MC 2. Slight decreases in intensity were observed due to X-ray beam damage of the resulting nanotubes. (Inset) Normalized integrations of the diffraction signal with respect to time. (F) Atomic force micrograph of the nanotubes resulting from the formation of MC 2. (G) Scanning electron micrograph of the nanotubes resulting from the formation of MC 2. | ||
The formation of MC 2 dominates a competition experiment in which both dialdehydes compete for a limited number of amine nucleophiles (Fig. 3A). By combining 1 equiv of DAPB with 1 equiv each of DFP and IDA, MC 2 is formed in high yield while reaction of IDA with small quantities of DAPB yields acyclic oligomers. In order to ensure that the formation of a small population of MC 1 was not limited by availability of the acid catalyst, the reaction was run at an elevated acid loading (10 equiv). The products of this competition reaction were characterized by GPC, MALDI-MS, and 1H NMR spectroscopy. GPC analysis indicated the predominant formation of a single macrocyclic product, as judged by the narrow peak shape whose retention time matched purified samples of MC 2, along with relatively weak signals corresponding to other oligomeric species (Fig. 3B, purple trace). MALDI-MS analysis of the same product mixture indicated a strong signal corresponding to the mass of MC 2, along with small oligomers containing IDA and DAPB species that did not react to form macrocycle (Fig. 3C and S82–S86†). No signal corresponding to MC 1 was observed. 1H NMR spectroscopy of the crude products also indicated the selective formation of MC 2. Similar to the MALDI-MS analysis, no signals in the NMR spectrum corresponding to MC 1 were observed (Fig. S87†). The formation of nanotubes under the reaction conditions was confirmed by AFM, SEM, and TEM; demonstrating that small oligomers containing IDA moieties did not interrupt the assembly of MC 2 (Fig. S89–S91†). Lastly, the presence of assembled nanotubes in the reaction solution prior to workup was confirmed by in situ XRD, which yielded a pattern comparable to the direct synthesis of MC 2 (Fig. S92†). Finally, a small molecule competition study in which IDA (1 equiv), DFP (1 equiv), and aniline (2 equiv) were condensed under conditions typical for macrocycle synthesis exclusively yielded 2,6-diiminophenylpyridine, whereas unreacted IDA remained in solution (Fig. S115 and S116†). Collectively, these results indicate that pyridine-containing macrocycles have two factors that favor their formation relative to benzene-containing derivatives. First, DFP forms imines more rapidly than IDA. Second, pyridine-containing macrocycles undergo supramolecular polymerization as they form, which further drives macrocycle formation over acyclic products. The corresponding process for IDA macrocycles requires 103 higher acid concentrations, such that only more weakly bound assemblies are present during their synthesis.
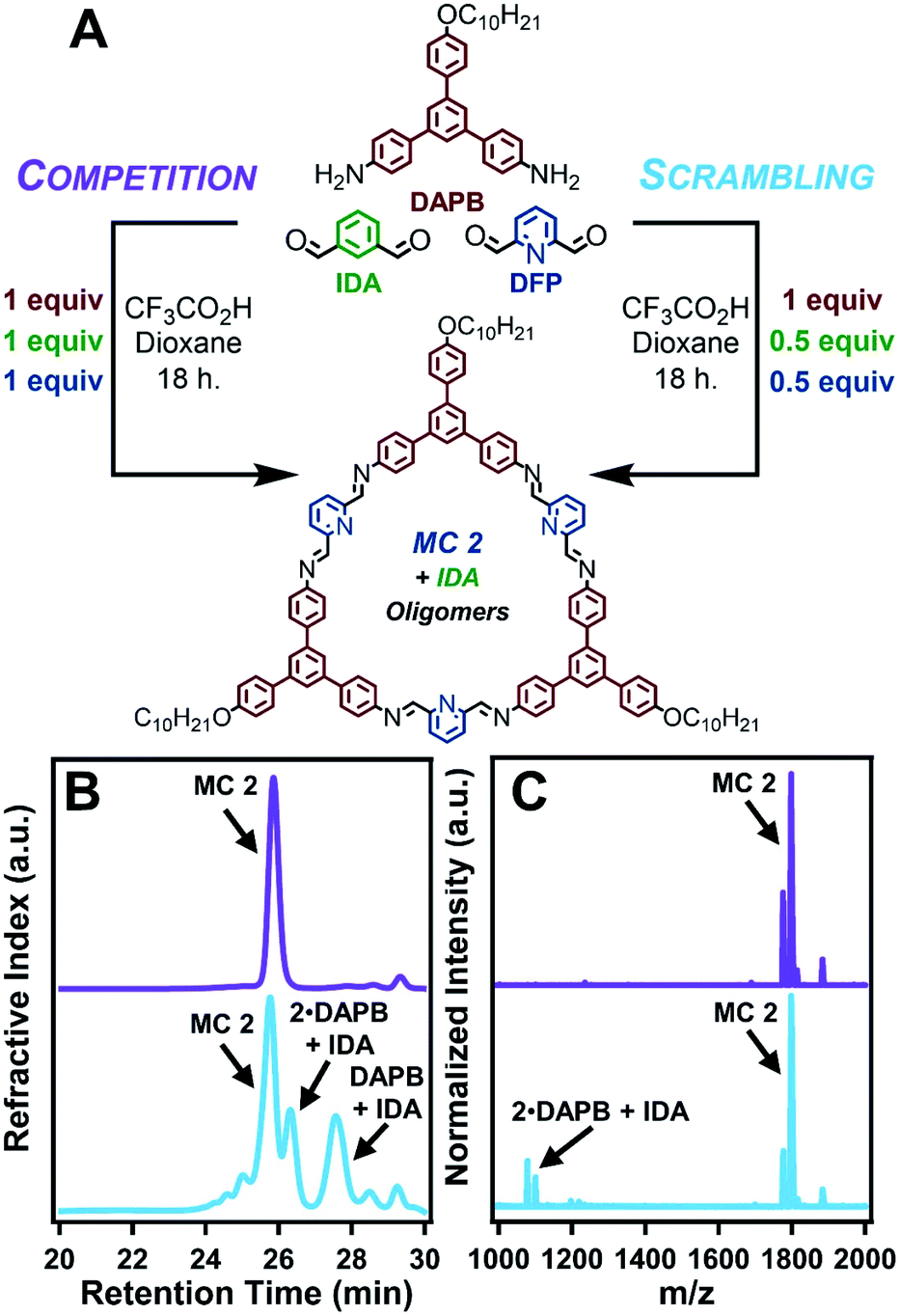 | ||
| Fig. 3 Probing the interplay of kinetic preference and chemical stability of MC 2 through competition and scrambling experiments. (A) Scheme of the competition and scrambling experiments. (B) Representative gel permeation chromatograms of the competition experiment (purple) and the scrambling experiment (blue). (C) Representative MALDI-MS spectra of the results of the competition experiment (purple) and the scrambling experiment (blue). MC 2 was observed as the [M + H]+, [M + Na]+, and [M + K]+ adducts. | ||
The most surprising finding of a scrambling experiment was that MC 1 was not formed in detectable amounts, despite there being sufficient DAPB to form a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of MC 1 and MC 2. This finding suggests that the formation and/or assembly of MC 2 interrupts the templation process required to form MC 1. A scrambling reaction between DAPB (1 equiv), DFP (0.5 equiv), and IDA (0.5 equiv) resulted in the selective formation of MC 2 and small IDA-containing oligomers (Fig. 3A). In contrast, both pyridine-2,6-diimine and benzene-1,3-diimine species are formed in the presence of aniline under the same conditions (see ESI†). The GPC trace of the scrambling reaction indicated the presence of macrocyclic and oligomeric species. MALDI-MS of the reaction indicated that only MC 2 was formed, and that all identifiable peaks corresponding to oligomers were IDA-containing species (Fig. 3B, C and S63–S68†). Furthermore, 1H NMR spectroscopy of the reaction mixture showed resonances corresponding to MC 2, but not MC 1, as well as oligomers containing IDA (Fig. S70†). Isolation of MC 2 by precipitation into CH2Cl2 resulted in an isolated macrocycle yield of 72–94% with respect to DFP, corresponding to half of the available DAPB reacting to yield macrocycles (Table S3†). Similar to the direct synthesis of MC 2 and the previous competition experiment, AFM, SEM, and TEM images confirmed the formation of nanotubes that drive macrocycle formation (Fig. S72–S74†). Lastly, the in situ XRD pattern of the scrambling reaction demonstrates an extended structure analogous to the direct synthesis of MC 2 (Fig. S75†). These combined findings indicate that the scrambling experiment, conducted at 25 mM DAPB and 12.5 mM each of DFP and IDA, forms MC 2 with no IDA incorporation, as well as no evidence MC 1 formation. In contrast, when 12.5 mM of DAPB and 12.5 mM of IDA are reacted in the absence of DFP, MC 1 is formed in high yield. This suppression of MC 1 formation in the presence of MC 2 was also observed at four other starting concentrations (12.5 mM, 8.5 mM, 6.40 mM, and 5.10 mM with respect to DAPB) of the scrambling experiment (see ESI†). The results of the scrambling experiment, combined with small molecule experiments and concentration dependent controls, suggest that MC 2 nanotubes disrupt formation of MC 1.
:1 mixture of MC 1 and MC 2. This finding suggests that the formation and/or assembly of MC 2 interrupts the templation process required to form MC 1. A scrambling reaction between DAPB (1 equiv), DFP (0.5 equiv), and IDA (0.5 equiv) resulted in the selective formation of MC 2 and small IDA-containing oligomers (Fig. 3A). In contrast, both pyridine-2,6-diimine and benzene-1,3-diimine species are formed in the presence of aniline under the same conditions (see ESI†). The GPC trace of the scrambling reaction indicated the presence of macrocyclic and oligomeric species. MALDI-MS of the reaction indicated that only MC 2 was formed, and that all identifiable peaks corresponding to oligomers were IDA-containing species (Fig. 3B, C and S63–S68†). Furthermore, 1H NMR spectroscopy of the reaction mixture showed resonances corresponding to MC 2, but not MC 1, as well as oligomers containing IDA (Fig. S70†). Isolation of MC 2 by precipitation into CH2Cl2 resulted in an isolated macrocycle yield of 72–94% with respect to DFP, corresponding to half of the available DAPB reacting to yield macrocycles (Table S3†). Similar to the direct synthesis of MC 2 and the previous competition experiment, AFM, SEM, and TEM images confirmed the formation of nanotubes that drive macrocycle formation (Fig. S72–S74†). Lastly, the in situ XRD pattern of the scrambling reaction demonstrates an extended structure analogous to the direct synthesis of MC 2 (Fig. S75†). These combined findings indicate that the scrambling experiment, conducted at 25 mM DAPB and 12.5 mM each of DFP and IDA, forms MC 2 with no IDA incorporation, as well as no evidence MC 1 formation. In contrast, when 12.5 mM of DAPB and 12.5 mM of IDA are reacted in the absence of DFP, MC 1 is formed in high yield. This suppression of MC 1 formation in the presence of MC 2 was also observed at four other starting concentrations (12.5 mM, 8.5 mM, 6.40 mM, and 5.10 mM with respect to DAPB) of the scrambling experiment (see ESI†). The results of the scrambling experiment, combined with small molecule experiments and concentration dependent controls, suggest that MC 2 nanotubes disrupt formation of MC 1.
MC 1 formation was also inhibited in the presence of a pure sample of MC 2, further validating that MC 2 nanotubes prevent the self-templation necessary for MC 1 formation. Furthermore, MC 2 was stable to the reaction conditions, despite the presence of free aldehydes, amines, and acid catalyst. Independently synthesized MC 2 nanotubes were added to a solution of DAPB (1 equiv), IDA (1 equiv), and CF3CO2H (10 equiv) and left undisturbed for 3 days (Fig. 4A). Analysis of the reaction by GPC yielded results analogous to the scrambling reaction in which several elution bands were observed, corresponding to discrete macrocycles and imine-linked oligomers (Fig. 4B). Subsequent analysis by MALDI-MS demonstrated that MC 2 was retained throughout the process by the lack of oligomers containing DFP moieties as well as no evidence of IDA moieties exchanging into MC 2. The MALDI-MS spectrum contained signals corresponding to IDA-containing oligomers with no DFP incorporation, consistent with the results of the scrambling reactions (Fig. 4C). These findings demonstrate that either free MC 2 or nanotubes comprised of MC 2 disrupt the formation of MC 1. This inhibition may result from the association of IDA-containing oligomers with pyridinium moieties within the MC 2 nanotubes, thereby hampering the reactivity of the oligomeric species, but a specific mechanism of inhibition remains unclear. These experiments and previous studies point to a templation process driving macrocycle formation, which is disrupted in the presence of monomeric or assembled MC 2 species (Fig. 2B).
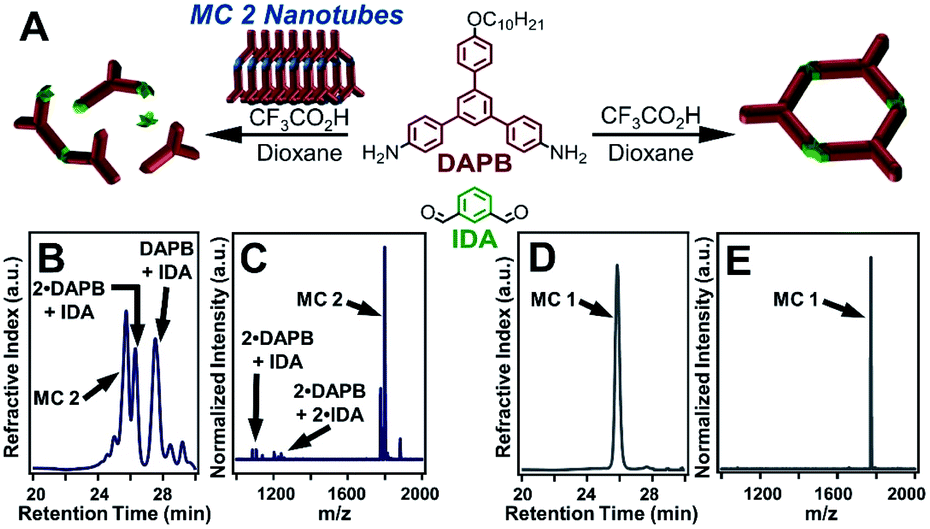 | ||
| Fig. 4 Control experiment demonstrating the effects of MC 2 nanotubes on the selective synthesis of MC 1. (A) Scheme depicting the control experiment in which previously synthesized MC 2 nanotubes are combined with DAPB, IDA, and CF3CO2H under conditions which in the absence of MC 2 nanotubes selectively yields MC 1. (B) Gel permeation chromatogram of the inhibited MC 1 synthesis. (C) MALDI-MS spectra of the results of the inhibited MC 1 synthesis depicting the recovery of MC 2, with no hydrolysis artifacts, and oligomers containing IDA moieties. MC 2 was observed as the [M + H]+, [M + Na]+, and [M + K]+ adducts. (D) Representative gel permeation chromatogram of the direct synthesis of MC 1. (E) Representative MALDI-MS spectra of the direct synthesis of MC 1. | ||
Monomer exchange experiments demonstrate that imines within acid-mediated nanotube assembles are far less dynamic than those in weak assemblies or monomeric species. Due to the stability of the imine linkages of MC 2, through a combination of supramolecular polymerization and the inherent chemistry of pyridine-2,6-diimine moieties, attempts to exchange its DFP moieties for IDA moieties failed. A previously synthesized sample of MC 2 was resuspended in 1,4-dioxane with IDA (10 equiv), and excess CF3CO2H (Scheme S6†). The reaction mixture was sonicated and held at room temperature for 3 days (Fig. 5A). Analysis of the monomer exchange product by GPC confirmed the formation of discrete macrocycles (Fig. S93†). However, analysis of the solution by MALDI-MS showed full recovery of MC 2, with no incorporation of IDA (Fig. 5D). 1H NMR spectroscopy of product of the attempted monomer exchange demonstrates recovery of MC 2 with no resonances corresponding to IDA-containing species (Fig. 5C). Based on the lack of monomer exchange as demonstrated by MALDI-MS and 1H NMR, we hypothesized that upon exposure to CF3CO2H, MC 2 assembled into nanotubes which, along with the inherent stability of pyridine-2,6-diimines, prevented reaction of the imine linkages. This hypothesis was supported by AFM, SEM, and TEM, which depicted the formation of nanotubes akin to the direct synthesis of MC 2 (Fig. S97–S99†). Lastly, the in situ XRD pattern of the failed monomer exchange matches well with that of the direct MC 2 synthesis (Fig. S100†). Collectively, the inability to exchange the DFP moieties out of nanotubes assembled from MC 2 highlights the chemical persistence and kinetic stability of the imine linkages within the protonation driven molecular assembly.32
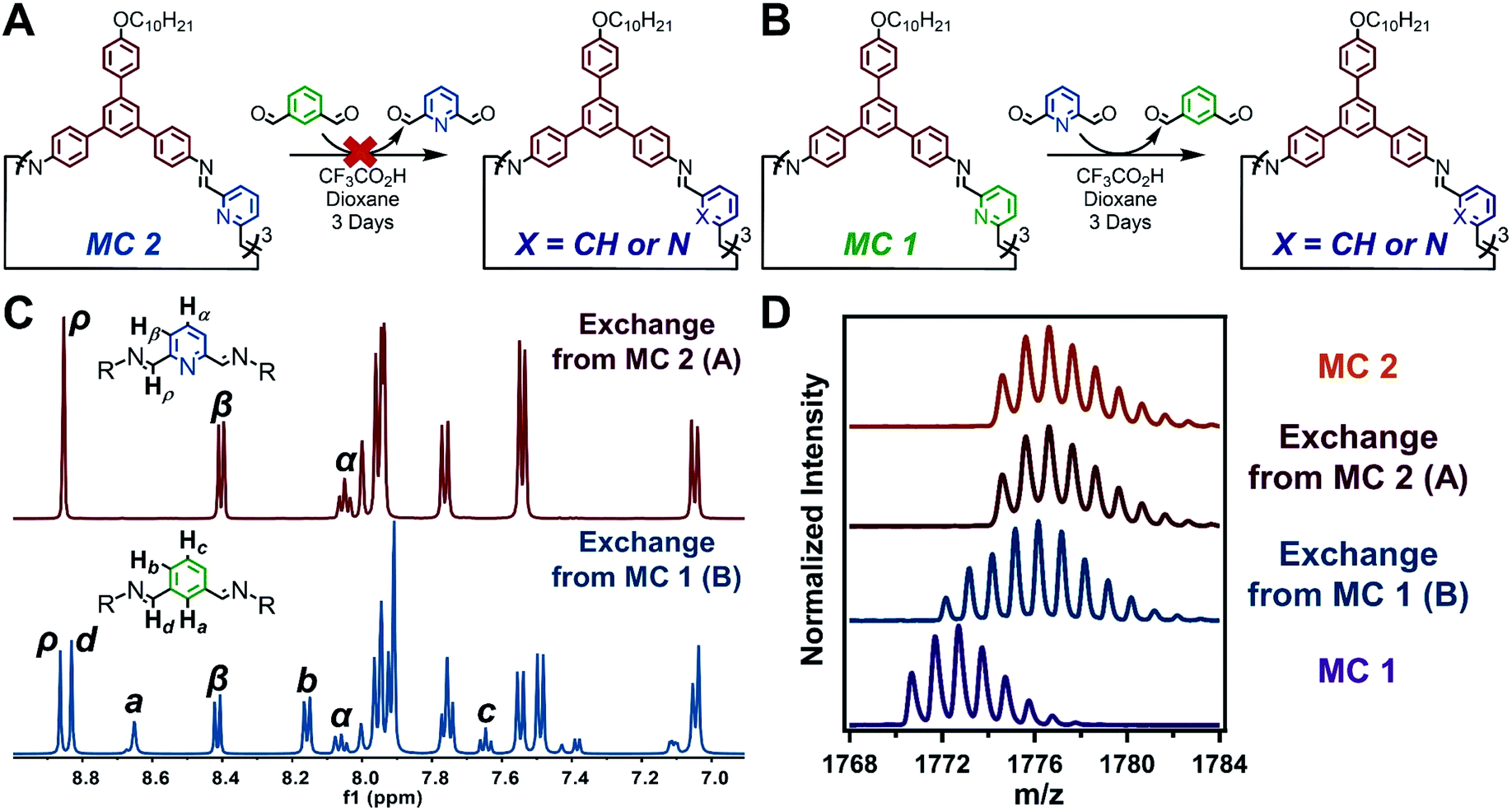 | ||
| Fig. 5 Monomer exchange of imine-linked macrocycles. (A) Monomer exchange of MC 2 with IDA. (B) Monomer exchange of MC 1 with DFP. (C) 1H NMR spectra of macrocycles resulting from each monomer exchange. (D) MALDI-MS comparison of the macrocycles resulting from each monomer exchange. | ||
Although IDA does not exchange into MC 2 nanotubes, it readily exchanges into macrocycles linked by other substituted isophthalaldehydes. Macrocycles were synthetized using a 5-bromoisophthalaldehyde monomer, which assemble similar to MC 1 (Scheme S8, Fig. S111 and S112†). Monomer exchange of the 5-bromoisophthalaldehyde linked macrocycles with IDA resulted in the formation of macrocyclic species, as evident by the narrow elution band in the corresponding GPC trace (Scheme S9 and Fig. S113†). MALDI-MS depicts scrambling of the linkages, such that peaks were observed corresponding to macrocycles containing 0, 1, 2, or 3,5-bromoisophthalaldehyde moieties (Fig. S114†). These observations indicate that IDA can exchange into macrocycles that contain similar linkages. IDA's inability to exchange into MC 2 under similar conditions arises either from the increased stability of the pyridine-2,6-diimine moiety or the kinetic persistence of macrocycles in acid-mediated assemblies.
When the previous exchange experiment was run in reverse, DFP was able to exchange into MC 1 (Fig. 5B). However, the major product of the exchange is a macrocycle containing only one pyridine-2,6-diimine moiety, despite DFP being used in 10-fold excess with respect to MC 1. MALDI-MS of the products revealed the presence of macrocycles containing a mixture of IDA and DFP subunits (Fig. 5D). The spectrum clearly shows no evidence of remaining MC 1 or the fully exchanged MC 2. However, the relative amounts of singly and doubly exchanged macrocycles are not clear. 1H NMR spectroscopy of the reaction indicated that 86% of the macrocycles contained a single pyridine moiety and 14% contained two pyridine moieties (Fig. 5C and S106†). Given the rapid and complete formation of MC 2 from DAPB and DFP, even in the presence of IDA, coupled with the thermodynamic preference for pyridine-2,6-diimine linkages; the selective formation of singly and doubly exchanged macrocycles under these conditions strongly suggest that incorporation of a single pyridine unit drives nanotube formation in the presence of CF3CO2H and results in a kinetic trap en route to the thermodynamically favored MC 2. Indeed, nanotubes were observed in these exchange experiments by AFM, SEM, and TEM (Fig. S107–S109†). The results of the three monomer exchange experiments highlight the diminished reactivity of imine linkages within acid-mediated assemblies. Furthermore, supramolecular polymerization into nanotubes served as a kinetic trap in the full conversion of MC 1 to MC 2, which allows access to non-symmetric macrocycles.
Conclusions
In conclusion, we have developed a system in which supramolecular polymerization is coupled to dynamic covalent bond-forming processes in the synthesis of imine-linked macrocycles. We have demonstrated that the formation of MC 2 is kinetically favored relative to MC 1, and its imine linkages are stabilized as a function of acid-mediated supramolecular polymerization and the inherent chemistry of the linkage itself. These three factors led to the selective synthesis of MC 2 dominating a competition experiment with MC 1. Additionally, the mere presence of nanotubes assembled from MC 2 proved to interrupt the synthesis of MC 1, presumably by disrupting the self-templation that guides its selective synthesis. Lastly, monomer exchange experiments demonstrated that once a pyridine-containing dialdehyde exchanged into MC 1 macrocycle, the macrocycle became capable of nanotube formation, which dramatically slowed further imine exchange, and resulted in the kinetic trapping of chemically diverse macrocycles not attainable by direct synthetic methods. These findings highlight the complex interplay of covalent and non-covalent synthesis that can give rise to complex dynamic reaction networks and stimuli responsive materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Army Research Office through the Multidisciplinary University Research Initiative (MURI; W911NF-15-1-04477, to W. R. D.). M. J. S. was supported by the National Science Foundation (NSF) through the Graduate Research Fellowship Program (GRFP) under Grant No. (DGE-1842165). A. M. E. was supported by the NSF through the GRFP under Grant No. (DGE-1324585). I. C. was supported by the NSF through the GRFP under Grant No. (DGE-1842165). I. C. is partially supported by the Ryan Fellowship and the International Institute for Nanotechnology. This work made use of the Integrated Molecular Structure Education and Research Center (IMSERC) at Northwestern University, which has received support from the NSF (CHE-1048773), the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF; NNCI-1542205), the State of Illinois, and the International Institute for Nanotechnology (IIN). This work also made use of the Scanned Probe Imaging and Development (SPID), and the Electron Probe Instrumentation Center (EPIC), facilities of Northwestern University's Atomic and Nanoscale Characterization Experiment Center (NUANCE), which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF; ECCS-1542205); the MRSEC program (NSF; DMR-1720139) at the Materials Research Center; the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois. This work was also supported by the Northwestern University Keck Biophysics Facility and a Cancer Center Support Grant (NCI CA060553). Parts of this work were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS) at Argonne National Lab. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facilities operated for the DOE Office of Science by Argonne National Laboratory under Grant No. (DGE-1324585). We acknowledge Prof. Julia Kalow for the use of her GPC instrument. We acknowledge Prof. Doug Philp for helpful discussions in the preparation of this manuscript.References
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- M. H. Bakker, C. C. Lee, E. W. Meijer, P. Y. W. Dankers and L. Albertazzi, ACS Nano, 2016, 10, 1845–1852 CrossRef CAS PubMed.
- J. López-Andarias, M. J. Rodriguez, C. Atienza, J. L. López, T. Mikie, S. Casado, S. Seki, J. L. Carrascosa and N. Martín, J. Am. Chem. Soc., 2015, 137, 893–897 CrossRef PubMed.
- B. Adelizzi, A. Aloi, A. J. Markvoort, H. M. M. Ten Eikelder, I. K. Voets, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 7168–7175 CrossRef CAS PubMed.
- M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606–611 CrossRef CAS PubMed.
- L. J. Prins, P. Timmerman and D. N. Reinhoudt, J. Am. Chem. Soc., 2001, 123, 10153–10163 CrossRef CAS PubMed.
- F. Helmich, C. C. Lee, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 16753–16755 CrossRef CAS PubMed.
- N. Hosono, A. R. A. Palmans and E. W. Meijer, Chem. Commun., 2014, 50, 7990–7993 RSC.
- R. D. Mukhopadhyay and A. Ajayaghosh, Science, 2015, 349, 241–242 CrossRef CAS PubMed.
- J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS PubMed.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188 CrossRef CAS PubMed.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS PubMed.
- A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2015, 8, 10 CrossRef PubMed.
- W. Zhang, W. Jin, T. Fukushima, A. Saeki, S. Seki and T. Aida, Science, 2011, 334, 340–343 CrossRef CAS PubMed.
- D. Görl, X. Zhang, V. Stepanenko and F. Würthner, Nat. Commun., 2015, 6, 7009 CrossRef PubMed.
- P. A. Rupar, L. Chabanne, M. A. Winnik and I. Manners, Science, 2012, 337, 559–562 CrossRef CAS PubMed.
- H. Qiu, Z. M. Hudson, M. A. Winnik and I. Manners, Science, 2015, 347, 1329–1332 CrossRef CAS PubMed.
- Q. Lin, G.-F. Gong, Y.-Q. Fan, Y.-Y. Chen, J. Wang, X.-W. Guan, J. Liu, Y.-M. Zhang, H. Yao and T.-B. Wei, Chem. Commun., 2019, 55, 3247–3250 RSC.
- K. Venkata Rao, D. Miyajima, A. Nihonyanagi and T. Aida, Nat. Chem., 2017, 9, 1133 CrossRef CAS PubMed.
- C. Kulkarni, P. A. Korevaar, K. K. Bejagam, A. R. A. Palmans, E. W. Meijer and S. J. George, J. Am. Chem. Soc., 2017, 139, 13867–13875 CrossRef CAS PubMed.
- R. S. Johnson, T. Yamazaki, A. Kovalenko and H. Fenniri, J. Am. Chem. Soc., 2007, 129, 5735–5743 CrossRef CAS PubMed.
- Y. Liu, Y. Jia, E. Zhu, L. Liu, Y. Qiao, G. Che and B. Yin, New J. Chem., 2017, 41, 11060–11068 RSC.
- P. A. Korevaar, T. F. A. de Greef and E. W. Meijer, Chem. Mater., 2014, 26, 576–586 CrossRef CAS.
- D. van der Zwaag, P. A. Pieters, P. A. Korevaar, A. J. Markvoort, A. J. H. Spiering, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 12677–12688 CrossRef CAS PubMed.
- J. S. Valera, R. Gómez and L. Sánchez, Small, 2018, 14, 1870012 CrossRef.
- S. Engel, D. Spitzer, L. L. Rodrigues, E.-C. Fritz, D. Straßburger, M. Schönhoff, B. J. Ravoo and P. Besenius, Faraday Discuss., 2017, 204, 53–67 RSC.
- J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A. Stuart, J. J.-P. Peyralans and S. Otto, Science, 2010, 327, 1502–1506 CrossRef CAS PubMed.
- R. Nguyen, L. Allouche, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2009, 48, 1093–1096 CrossRef CAS PubMed.
- R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2008, 4, 19 CrossRef PubMed.
- M. J. Strauss, D. Asheghali, A. M. Evans, R. L. Li, A. D. Chavez, C. Sun, M. L. Becker and W. R. Dichtel, Angew. Chem., Int. Ed., 2019, 58(41), 14708–14714 CrossRef CAS PubMed.
- C. Sun, M. Shen, A. D. Chavez, A. M. Evans, X. Liu, B. Harutyunyan, N. C. Flanders, M. C. Hersam, M. J. Bedzyk, M. O. de la Cruz and W. R. Dichtel, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8883–8888 CrossRef CAS PubMed.
- A. D. Chavez, A. M. Evans, N. C. Flanders, R. P. Bisbey, E. Vitaku, L. X. Chen and W. R. Dichtel, Chem.–Eur. J., 2018, 24, 3989–3993 CrossRef CAS PubMed.
- N. Giuseppone and J.-M. Lehn, Chem.–Eur. J., 2006, 12, 1715–1722 CrossRef CAS PubMed.
- C. Godoy-Alcántar, A. K. Yatsimirsky and J.-M. Lehn, J. Phys. Org. Chem., 2005, 18, 979–985 CrossRef.
- S. Kulchat, M. N. Chaur and J.-M. Lehn, Chem.–Eur. J., 2017, 23, 11108–11118 CrossRef CAS PubMed.
- D. Schultz and J. R. Nitschke, Chem.–Eur. J., 2007, 13, 3660–3665 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization. See DOI: 10.1039/c9sc05422g |
| This journal is © The Royal Society of Chemistry 2020 |