
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle molecule observation of hard–soft-acid–base (HSAB) interaction in engineered Mycobacterium smegmatis porin A (MspA) nanopores†
Sha
Wang
 ab,
Jiao
Cao
ab,
Wendong
Jia
ab,
Weiming
Guo
ab,
Shuanghong
Yan
ab,
Yuqin
Wang
ab,
Panke
Zhang
a,
Hong-Yuan
Chen
*a and
Shuo
Huang
*ab
ab,
Jiao
Cao
ab,
Wendong
Jia
ab,
Weiming
Guo
ab,
Shuanghong
Yan
ab,
Yuqin
Wang
ab,
Panke
Zhang
a,
Hong-Yuan
Chen
*a and
Shuo
Huang
*ab
aState Key Laboratory of Analytical Chemistry for Life Sciences, School of Chemistry and Chemical Engineering, Nanjing University, 210023, Nanjing, China. E-mail: hychen@nju.edu.cn; shuo.huang@nju.edu.cn
bChemistry and Biomedicine Innovation Center (ChemBIC), Nanjing University, 210023, Nanjing, China
First published on 10th December 2019
Abstract
In the formation of coordination interactions between metal ions and amino acids in natural metalloproteins, the bound metal ion is critical either for the stabilization of the protein structure or as an enzyme co-factor. Though extremely small in size, metal ions, when bound to the restricted environment of an engineered biological nanopore, result in detectable perturbations during single channel recordings. All reported work of this kind was performed with engineered α-hemolysin nanopores and the observed events appear to be extremely small in amplitude (∼1–3 pA). We speculate that the cylindrical pore restriction of α-hemolysin may not be optimal for probing extremely small analytes. Mycobacterium smegmatis porin A (MspA), a conical shaped nanopore, was engineered to interact with Ca2+, Mn2+, Co2+, Ni2+, Zn2+, Pb2+ and Cd2+ and a systematically larger event amplitude (up to 10 pA) was observed. The measured rate constant suggests that the coordination of a single ion with an amino acid follows hard–soft-acid–base theory, which has never been systematically validated in the case of a single molecule. By adjusting the measurement pH from 6.8 to 8.0, the duration of a single ion binding event could be modified with a ∼46-fold time extension. The phenomena reported suggest MspA to be a superior engineering template for probing a variety of extremely small analytes, such as monatomic and polyatomic ions, small molecules or chemical intermediates, and the principle of hard–soft-acid–base interaction may be instructive in the pore design.
1. Introduction
Coordination interactions between amino acids and metal ions play crucial roles in structural and functional aspects of natural metalloproteins and metal activating proteins.1–3 Undesired binding of heavy metal ions, such as lead, cadmium or mercury, with protein residues results in irreversible damage to protein functions.4,5 Conventional techniques such as NMR6,7 and spectroscopic titration,8,9 which provides indirect and static measurements with which probing the interactions of metal ions with amino acids suffers from the absence of direct and dynamic observation of the binding kinetics in a single molecule.Biological nanopores, a group of transmembrane porins for single molecule sensing, can be genetically engineered with atomic precision to mimic a reactive site in natural metalloproteins, which binds metal ions. The reversible interaction of the metal ion with a designed reactive site of a biological nanopore could be monitored by single channel recording.10–13 Pioneered by Bayley et al.,10 direct ion sensing11,12 has been performed with a series of α-hemolysin (α-HL) mutants. All reported binding events consistently appear to be small in amplitude (1–3 pA)10–12 and therefore barely detectable using a patch clamp amplifier. Although it is rational to assume that a single monatomic ion may be too small to be probed by a nanopore, we speculate that a pore geometry different from the cylindrical shape in α-HL may be more optimal. To the best of our knowledge, however, this approach has not yet been examined with any other biological nanopores.
In principle, any biological nanopore could be engineered to probe the binding of a single ion.14 Rationally, a narrow pore restriction is advantageous in that binding of a single ion would produce a more noticeable event against a background of thermal noise. The single ion binding site is best designed on a rigid restriction substrate, such as a β-barrel, which is advantageous in minimizing perturbations resulting from a floppy pore lumen. Moreover, a wider pore vestibule, which allows more ionic flow through the pore, would generally maximize the amplitude of an event. This suggests that an optimal pore geometry should be mechanically rigid and geometrically conical with the reactive site at the smaller end, on top of the pore restriction. It is possible that this conical configuration could also minimize signal perturbations from non-specific binding of the analyte to amino acid residues distant from the pore restriction, and this significantly simplifies the task of site directed mutagenesis.
Mycobacterium smegmatis porin A (MspA),15 which is a conical shaped nanopore containing a rigid restriction composed of β-barrels, is an optimum candidate. Its geometric advantage, which produces a high spatial resolution when a strand of DNA is examined, has been recognized previously,16 but only in the context of nanopore sequencing.17 Engineered MspA mutants have been optimized for nanopore sequencing, but MspA mutants of other kinds have been reported only rarely. In this paper, three MspA mutants, which have histidine, cysteine or aspartic acid placed at site 91 in a monomeric subunit, were prepared as previously reported.18 When compatible analyte ions were applied, MspA mutants report noticeable events during single channel recordings. The recorded event from binding of a single ion has reached ∼10 pA in amplitude, which is considerably greater than that measured with α-HL mutants.10–12 We believe this work is the first report of MspA being used as a nano-reactor to probe binding of single monatomic ions. It suggests that by taking advantage of its unique pore geometry, MspA may be advantageous to examine a variety of other small analytes such as polyatomic ions and chemical intermediates.
Nanopore sensing of Zn2+, Cd2+, Co2+, Ag+ and Ni2+ has previously been demonstrated with a series of precisely designed α-HL mutants,10–13 but the general principles of ion–amino acid interactions in a nanopore have not been investigated. Many ensemble results involving coordination can be explained by the theory of hard–soft-acid–base (HSAB) which was first proposed in 1963 by Pearson19 with a principle that “hard acids prefer to coordinate to hard bases and soft acids to soft bases”. We have confirmed that interactions between pores and ions also follow hard–soft-acid–base (HSAB) theory, but on a single molecule scale. HSAB theory may be useful in the prediction of other unreported ion–amino acid interactions that may be probed using a nanopore sensor. For example, Pb2+, which is a borderline softer acid ion and has never been directly probed by any engineered nanopores, was confirmed to interact with both histidine and cysteine residues, from testing with the corresponding MspA mutants. Other than the principle suggested in HSAB theory, ion–amino acid interactions were also monitored to be dependent on the pH of the measurement environment, which could be tuned with a 46-fold extension in the dwell time of an event when the pH is adjusted slightly.
2. Results and discussion
2.1. Experimental section
Unlike biomacromolecules,20 analytes such as monatomic ions or small molecules are too small to be directly examined by a nanopore. However, the introduction of a reversible analyte–pore interaction, which chemically confines the analyte so that a signal perturbation could be observed during single channel recording, forms the basis of sensing. Three engineered MspA mutants were prepared and characterized as previously reported.18 For simplicity, these mutants were respectively named MspA-D (D93N/D90N/D118R/D134R/E139K), MspA-H (D93N/D91H/D90N/D118R/D134R/E139K) and MspA-C (D93N/D91C/D90N/D118R/D134R/E139K) (ESI Methods, Fig. S1–S3†) throughout the paper. The charge distribution plots of all MspA octamers were generated using PyMOL (Fig. S4†). All single ion binding assays were performed similarly as reported.21,22 Briefly, the cis and the trans compartments, which respectively are filled with 500 μL electrolyte buffer (1 M NaCl, 10 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), pH 7.4), are separated using a self-assembled lipid membrane composed of 1,2-diphytanoyl-sn-glycero-3-phosphocholine, DPhPC (Avanti Polar Lipids, Alabaster, AL, USA). The electrolytes in the cis and the trans compartments were each in contact with a pair of Ag/AgCl electrodes and monitored using a patch clamp amplifier (Axon 200B, Molecular Devices, Thermofisher, Waltham, MA, USA). Conventionally, the side which is electrically grounded is defined as the cis side and the opposing side as the trans side. MspA nanopores were initially placed in the cis side which is spontaneously inserted into the membrane. With a single nanopore inserted, the measurement was performed when the analyte ions were placed in the trans side and a +100 mV applied voltage was continuously applied. I–V characteristics were obtained for all three MspA mutants, of which MspA-D and MspA-C demonstrate a larger open pore conductance and a noticeable rectification effect, whereas, MspA-H appears to be slightly less conductive with no clear rectification effect (Fig. S5†). When recorded with a continuously applied potential without the addition of any analyte, MspA-C produces a much less noisy open pore current than MspA-D and MspA-H (Fig. S6†). All single ion binding assays in this paper were performed with this configuration (Fig. 1a), unless otherwise stated. All MspA mutants are homo-octameric, which means that the reactive sites were identically altered and simultaneous binding from multiple ions is possible. All measurements were performed with low concentrations of analyte ions, in which only single ion binding events were observed.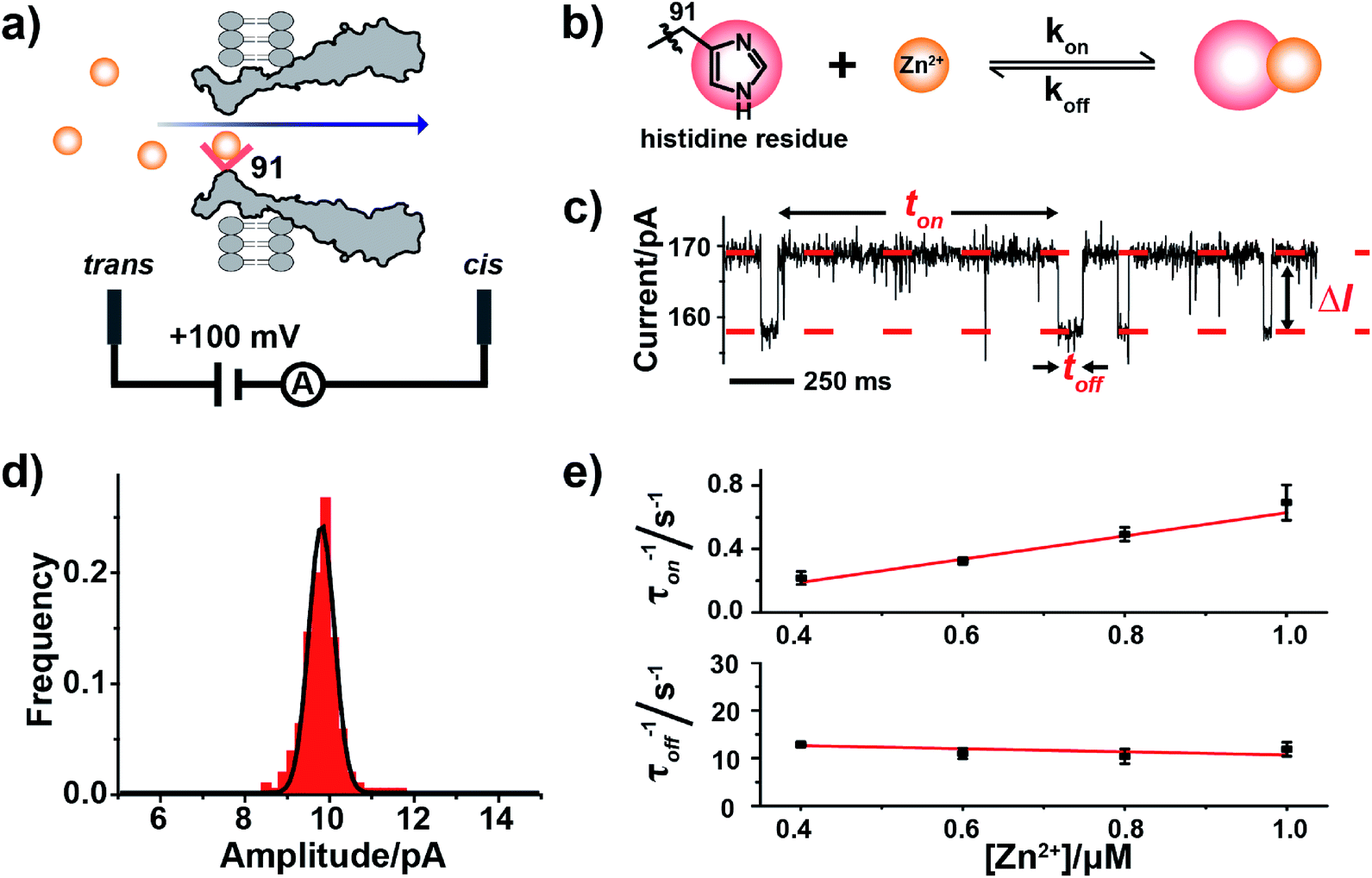 | ||
| Fig. 1 Single molecule observation of Zn2+ binding with MspA-H. (a) A schematic diagram of Zn2+ binding with MspA-H. The histidine residue (red sphere) of MspA-H binds reversibly with individual Zn2+ ions (yellow sphere). The MspA-H pore was inserted in the bilayer (ESI Methods†). Zn2+ was added to the trans compartment. The single channel recording was performed with a buffer of 1 M NaCl, 10 mM HEPES, pH 7.4 and a +100 mV applied potential. (b) The model of Zn2+ binding to the histidine residue. (c) A representative current trace of Zn2+ binding to MspA-H. Zn2+ was added to the trans compartment with a final concentration of 1.0 μM. ton and toff represent inter-event interval (unbound state) and dwell time (bound state) of Zn2+ binding events, respectively. ΔI is the current difference between the open pore and the blockage event level. (d) The event-amplitude (ΔI = 9.8 ± 0.7 pA) histograms with a Gaussian fitting for Zn2+ binding events. (e) Reciprocals of the mean inter-event intervals (τon) and the mean dwell times of Zn2+ (τoff) versus the final Zn2+ concentration in trans. The values of τon and τoff were derived from the results of single exponential fittings (Fig. S9†). The association rate kon = (1/τonc) and the dissociation rate koff = (1/τoff) were derived accordingly. | ||
2.2. Probing Zn2+ binding with MspA-H
Zinc, an element essential for life, plays a critical role in the structural and catalytic aspects of proteins. Zinc may coordinate in a protein with a histidine, a cysteine, an aspartic acid, a glutamic acid residue or their spatial combinations.23 Probing of binding of Zn2+ to a series of α-HL mutants10,11,13 has been previously attempted but the reported Zn2+ binding events consistently appeared to be small in amplitude (1–3 pA).10,11To confirm our hypothesis that a conical pore geometry may increase the event amplitude resulting from binding of a single Zn2+ ion, which reversibly interacts with the imidazole group of histidine (Fig. 1b), Zn2+ was treated as a model analyte to be probed by MspA-H. During single channel recording, MspA-H stays open with an open pore current of 161.1 ± 4.0 pA (N = 30, Table S1†) when recorded with an electrolyte buffer of 1 M NaCl, 10 mM HEPES, pH 7.4 and a +100 mV voltage applied. The addition of Zn2+ to the trans compartment with a 1 μM final concentration immediately resulted in the appearance of resistive pulse events with a highly consistent blockage amplitude (ΔI) (Fig. 1c). Statistics (N = 206) of ΔI show a monodisperse distribution measuring 9.8 ± 0.7 pA (Fig. 1d), derived from the corresponding Gaussian fitting results. These resistive pulse events disappeared when ethylenediaminetetraacetic acid (EDTA) was further added to trans to a 1 mM final concentration (Fig. S7†), suggesting that these blockage events result from binding of individual Zn2+ ions to the pore restriction.
The measurements were systematically performed with a Zn2+ concentration of 0.4–1.0 μM in the trans (Fig. S8†). The corresponding mean interevent interval (τon) and dwell time (τoff), derived from the fitting results (Fig. S9†), indicate a strong concentration dependence. The reciprocal of the mean interevent interval (τon) is proportional to the Zn2+ concentration, which is consistent with a bimolecular model, in which 1/τon = kon[Zn2+]. In contrast, the reciprocal of the mean dwell time (τoff) of the complex is independent of the Zn2+ concentration (Fig. 1e), which is consistent with a unimolecular dissociation mechanism (1/τoff = koff). These results further confirm that the observed ∼10 pA event results from Zn2+ binding to the pore restriction. The observed event amplitude, which is approximately 3–10 times of that reported with α-HL mutants (1–3 pA),11,12 confirms our hypothesis that MspA is optimal in single ion sensing by producing a larger event amplitude.
Besides the conclusion that a conical shaped nanopore reports single ion binding events with a higher event amplitude, other questions that could be subsequently raised include whether similar measurements could be performed with other divalent ions, which residues could be placed around a pore restriction and how they differ in their binding kinetics.
2.3. The binding kinetics of metal ions to MspA-H
According to HSAB theory,19 chemical groups such as the imidazole of histidine, the carboxylate of aspartic acid or the sulfhydryl of cysteine are considered to be either Lewis bases or lone pair electron donors. In contrast, metal ions such as Ca2+, Ni2+ or Cd2+ are considered to be Lewis acids or lone pair electron acceptors. The acid–base interactions follow the general principle that hard prefers hard and soft prefers soft. Specifically, the imidazole group of a histidine residue, as placed in the restriction of MspA-H, is a borderline base (Fig. 2a) and binds preferentially to borderline acid ions such as Co2+, Ni2+, Zn2+ or Pb2. In principle, it may weakly interact with soft or hard ions.24 Similar experiments were performed with MspA-H with a variety of easily accessible divalent ions, including Ca2+, Mn2+, Co2+, Ni2+, Zn2+, Pb2+ and Cd2+ as trial analytes. Experimentally, Ca2+ doesn't show any detectable events even at a final concentration of 1 mM in trans (Fig. S10†). However, binding of Mn2+ to MspA-H shows a clear increase in the baseline noise on top of the open pore current (Fig. S11†), which indicates that a weak interaction between Mn2+ and MspA-H could be established and loosely monitored but not clearly resolved. This is expected as Mn2+ is a softer hard acid ion, close to a borderline acid.25 Other softer ions, such as Co2+, Ni2+, Zn2+ or Pb2+, which belong to borderline acid ions, report discreetly resolvable events when bound to MspA-H (Fig. S12†). Cd2+, which is the softest acid ion among the trial analytes, reports short-resident spiky events.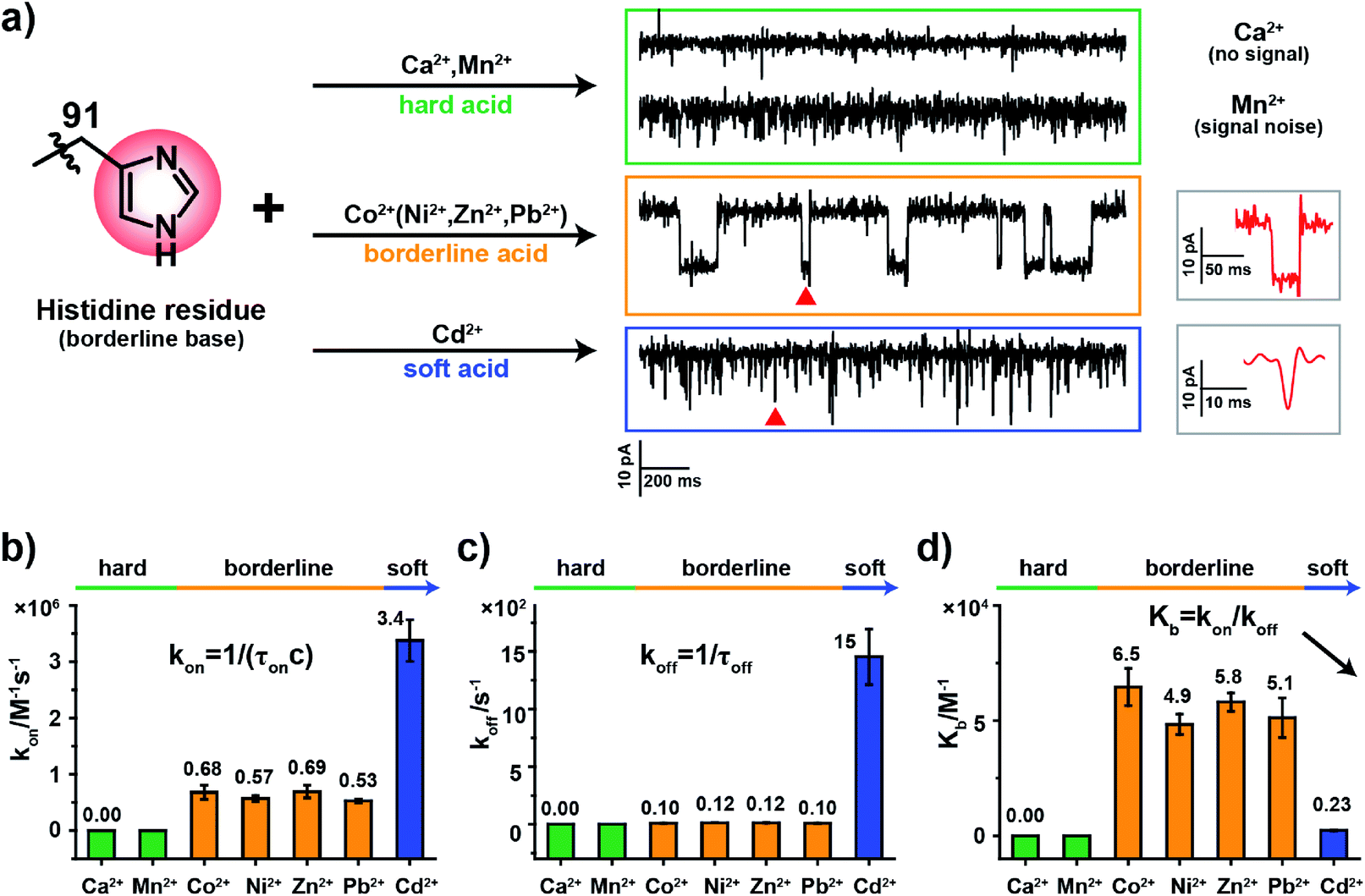 | ||
| Fig. 2 The coordination kinetics of a histidine residue (borderline base) with different metal ions. (a) Representative current traces of MspA-H interacting with different metal ions. Briefly, Ca2+ produced no events, Mn2+ produced only background noise and Co2+ generated clear blockage events. Similar results, as observed with Co2+, were observed with Ni2+, Zn2+ and Pb2+; short spiky signals were generated from Cd2+. (b) A comparison of kon (association constant) from the interaction between the histidine residue and different metal ions. (c) A comparison of koff (dissociation constant) from the interaction between the histidine residue with different metal ions. (d) A comparison of the equilibrium binding constant (Kb). Kb was calculated according to the equation Kb = (kon/koff). The imidazole group of a histidine residue, which is a borderline base, interacts strongly with borderline ions (Co2+, Ni2+, Zn2+, and Pb2+) but however interacts weakly with hard ions (Mn2+) or soft ions (Cd2+). According to the Kb value, the single molecule observation is systematically consistent with the theory of hard–soft-acid–base (HSAB). All data were acquired with the buffer of 1 M NaCl, 10 mM HEPES, pH 7.4 and a +100 mV applied potential. Error bars were derived from three independent experiments (N = 3). | ||
Though the blockage amplitude appears to be similar between Co2+, Ni2+, Zn2+ and Pb2+(Table S2†), by comparing their rate constants kon and koff (Fig. 2b–d and Table S2†), it can be clearly seen that Cd2+ binds to and detaches from a histidine residue extremely rapidly. On the other hand, Co2+, Ni2+, Zn2+ and Pb2+ bind to and detach from a histidine residue with a slower and similar rate. The binding strengths can be compared according to their equilibrium binding constant Kb, defined as Kb = kon/koff. The derived Kb value follows the order Co2+ ≈ Ni2+ ≈ Zn2+ ≈ Pb2+ > Cd2+ > Mn2+ > Ca2+, which suggests that Co2+, Ni2+, Zn2+ and Pb2+ all bind to MspA-H strongly. Cd2+ and Mn2+ however bind to MspA-H weakly and Ca2+ doesn't report any binding signal. These results confirm that the imidazole residue of histidine, which is a borderline base, selectively binds to borderline acids such as Co2+, Ni2+, Zn2+ and Pb2+, as monitored at the single molecule scale by nanopore measurements. Pb2+, which is a hazardous ion, harmful to human health, has not to the best of our knowledge been directly sensed by any engineered nanopores (Video S1†). Incompatible ions in this assay, such as Ca2+, Mn2+ or Cd2+, may bind to nanopores with compatible reactive residues, as suggested by HSAB theory.
2.4. The binding kinetics of metal ions to MspA-D
Hard divalent ions, such as Ca2+, Mg2+ or Mn2+, are widely recognized as critical components in natural metalloproteins or metal activating enzymes. However, previous attempts at single ion sensing by nanopores with hard divalent ions have never been reported. Conformational change of calmodulin is regulated by simultaneous binding of calcium to aspartic acid, glutamic acid and peptide carbonyl.26,27 The exonuclease domain of a phi29 DNA polymerase requires Mg2+, which binds to aspartic acid residues within the protein, as a cofactor.28 Binding of Ca2+ or Mg2+ to these metal activating proteins suggests that these hard divalent ions prefer to bind to hard bases such as the carboxylate oxygen.MspA-D, which has an aspartic acid residue placed in the 91st site of a monomeric subunit, was prepared as described in ESI Methods† and used to test how hard divalent ions may be probed by a nanopore. Before the addition of any metal ions, the open pore current of MspA-D measured was 245.7 ± 5.8 pA (N = 13) when recorded with 1 M NaCl, 10 mM HEPES, pH 7.4 and a +100 mV applied voltage (Table S1†).
Experimentally, no detectable single ion binding event was observed with the addition of Ca2+, Mn2+, Co2+, Zn2+, Pb2+ or Cd2+ to trans when measured with MspA-D (Fig. S13 and S14†). It is expected for Co2+, Zn2+, Pb2+ or Cd2+, which are either borderline acids or soft acids and thus don't interact strongly with the aspartic acid residue. However, hard acid ions, such as Ca2+ or Mn2+, also report no detectable events, which indicates that interactions between a single Ca2+ or Mn2+ ion and a single aspartic acid residue are not strong enough to cause reports of sensing events. Unexpectedly however, Ni2+ reports spiky signals with no defined blockage amplitude (Fig. S14e†), which is an exception that cannot be explained by HSAB theory.
Even with an unexplained exception for Ni2+, these observations suggest that hard divalent ions such as Ca2+ or Mn2+may not strongly bind to a single amino acid when placed in a nanopore. According to HSAB theory, hard acid–hard base interactions tend to be more electrostatic and thus have a weaker bond strength than the soft acid–soft base interactions, which are more covalent. As observed in natural metal activating enzymes, simultaneous binding of multiple reactive residues closely packed around the pore restriction may be required to form a reactive cluster so that a strong binding strength may be achieved. However, a laborious screening effort would be needed to achieve such an optimum spatial distribution of these amino acids in a pore design. On the other hand, weak interactions of Ca2+ with amino acid residues of a protein pore may be advantageous when the nanopore measurement is performed with optical single channel recordings29,30 or DiffusiOptoPhysiology,31 in which Ca2+ is critical in the generation of the fluorescence readout and less undesired perturbations resulting from Ca2+ interacting with the pore may need to be taken into account.
2.5. The binding kinetics of metal ions to MspA-C
To examine how different metal ions interact with the softer sulfhydryl group of cysteine, similar metal ion binding assays were carried out with MspA-C. Before the addition of any metal ions, the open pore current of MspA-C measures 222.0 ± 6.8 pA (N = 24) when recorded with 1 M NaCl, 10 mM HEPES, 0.4 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP), pH 7.4 and a +100 mV applied voltage (Table S1†). The larger open pore current in comparison with that of MspA-H may result more from the smaller size of the sulfhydryl group around the pore restriction of MspA-C than from the imidazole group around that of MspA-H. The addition to the buffer of TCEP, which prevents the formation of disulfide bonds within the pore restriction, is performed in all metal ion sensing assays whenever MspA-C is used.The sulfhydryl group of a cysteine residue, which is a sulfur-containing ligand belonging to a soft base, binds preferentially to softer acid ions.32–34 The experimental results showed resolvable binding events when a soft acid (Cd2+) or softer borderline ions (Zn2+ and Pb2+) were used as the analyte ions (Fig. 3a). However, no events were observed from hard acid ions such as Ca2+, Mn2+ or other harder borderline acid ions such as Co2+ or Ni2+ even when 1 mM final concentrations of these ions were placed in trans (Fig. S15†). Binding events of Zn2+, Pb2+ or Cd2+ to MspA-C produce easily recognizable event characteristics, which differ in their blockage amplitude, shape and binding kinetics (Table S3†). This is different from the case of MspA-H, to which binding of Zn2+, Pb2+, Co2+ or Ni2+ results in almost indistinguishable event amplitudes and binding rates. Specifically, binding of Pb2+ to MspA-C produces unique cluster shaped events (Fig. S16 and Video S2†), which is different from that of any other ions being tested and is useful in identifying Pb2+ with a high reliability. To extract the dwell time, each event cluster resulted from Pb2+ binding was counted as an individual event as demonstrated in Fig. S16.† According to the statistical results, the event dwell time of Cd2+, Pb2+ and Zn2+ measures ∼555 ms, 200 ms and 109 ms, respectively, from which it can be observed that Cd2+ exhibits the longest mean dwell time (Fig. S17†). It is worth noticing that binding of Pb2+ to MspA-H (Fig. S12j and Video S1†) appears different from that reported by MspA-C. This indicates that Pb2+, which is a borderline acid, interacts with either an imidazole or a sulfhydryl group; however, clearly distinguishable event characteristics can be easily recognized when different binding chemistry was happening.
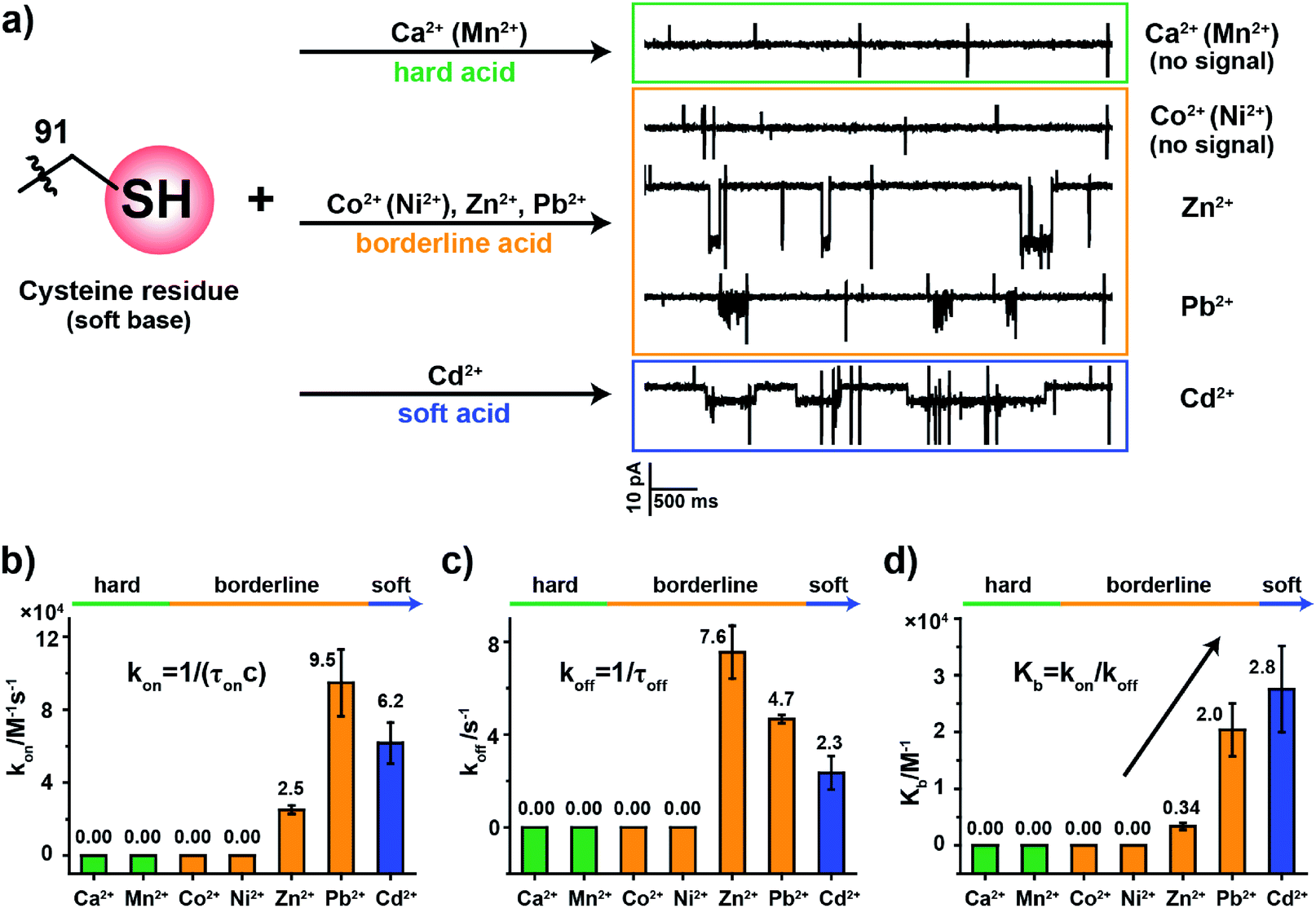 | ||
| Fig. 3 The coordination kinetics of a cysteine residue with different metal ions. (a) Representative current traces of MspA-C interacting with different metal ions. The sulfhydryl group of a cysteine residue is a soft base and sulfur-containing ligand which interacts strongly with softer borderline acids or soft acid ions (Zn2+, Pb2+, and Cd2+) but produces no events with hard ions (Ca2+/Mn2+) or borderline ions (Co2+/Ni2+). (b) A comparison of kon (association constant) from the interaction between the cysteine residue and different metal ions. (c) A comparison of koff (dissociation constant) from the interaction between the cysteine residue with different metal ions. (d) A comparison of Kb (equilibrium binding constants). Kb values were calculated according to Kb = (kon/koff). All data were acquired with the buffer of 1 M NaCl, 10 mM HEPES, 0.4 mM TCEP, pH 7.4 and a +100 mV applied potential. Error bars were based on three independent nanopore experiments (N = 3). | ||
According to the derived rate constants (Table S3†), it was observed that Zn2+ has the slowest association rate kon (Fig. 3b) and the fastest dissociation rate koff (Fig. 3c) when binding to a sulfhydryl group. Consequently, it has the minimum equilibrium binding constant Kb (Fig. 3d), which is derived from Kb = kon/koff. Among the ions which report binding events to MspA-C (Cd2+, Zn2+ and Pb2+), the Kb value indicates that Cd2+ reports the strongest binding to cysteine. This result is also consistent with HSAB theory, in which the Cd2+ is the softest of the selected ions.19 This comparison also explains another ensemble observation that cysteine-rich Zn2+-binding sites in proteins are weakly protected against heavy metals such as Cd2+ and Pb2+ ions.35
The above demonstration confirms that the MspA-C mutants selectively bind to softer target ions. The highly discriminatory event amplitude and shape, absent in MspA-H, may indicate that a more polarizable interaction between a soft acid and a soft base is more advantageous in reporting distinguishable signals when different ions bind to the pore restriction. However, further investigations are in progress in our separate, follow-up studies.
2.6. The effect of pH on coordination between the cysteine residue and metal ions
HSAB theory qualitatively suggests a general binding affinity between ions and amino acid residues. However, these coordination interactions are also affected by other factors, such as temperature,36 steric hindrance37 and the pH38 of the reaction environment. The pH value of the measurement buffer, which can be easily and precisely manipulated during the nanopore measurement, can finely adjust the protonation state of the amino acid side residue around the pore restriction. Consequently, the coordination strength may also be finely adjusted.The pKa of the imidazole group of histidine is 6.0 and that of the sulfhydryl group of cysteine is 8.2.39 When measured in an aqueous buffer of pH 7.4, an imidazole group, such as that in the MspA-H mutant, is largely deprotonated. However, the sulfhydryl group from a MspA-C mutant should be dynamically changing between a sulfhydryl and a thiolate, which makes the reactive site possibly sensitive to environment pH and may be monitored during a nanopore assay. Though Pb2+, Zn2+ and Cd2+ report positive events when bound to MspA-C, we took Zn2+ and Cd2+ as model analytes to probe this pH effect in order to avoid complications from analyzing the cluster shaped events produced by Pb2+ binding.
Experimentally, the nanopore assay was performed similarly to that demonstrated in Fig. 3. However, the pH value of the measurement buffer was adjusted to 6.8, 7.4 and 8.0. As the pH increases from 6.8 to 8.0, the dwell time of metal ions was systematically extended (Fig. 4a and c). The statistical results indicated that the mean dwell time of Zn2+ and Cd2+ binding has been extended for ∼10 and 46 times respectively (Fig. 4b and d); however the event amplitude and shape remain unchanged (Fig. 4a, c, Tables S4 and S5†). Other detailed experimental results are displayed in Fig. S18, S19, Video S3 and S4.† However, pH has little effect on the association rate kon as summarized in Tables S4 and S5.†
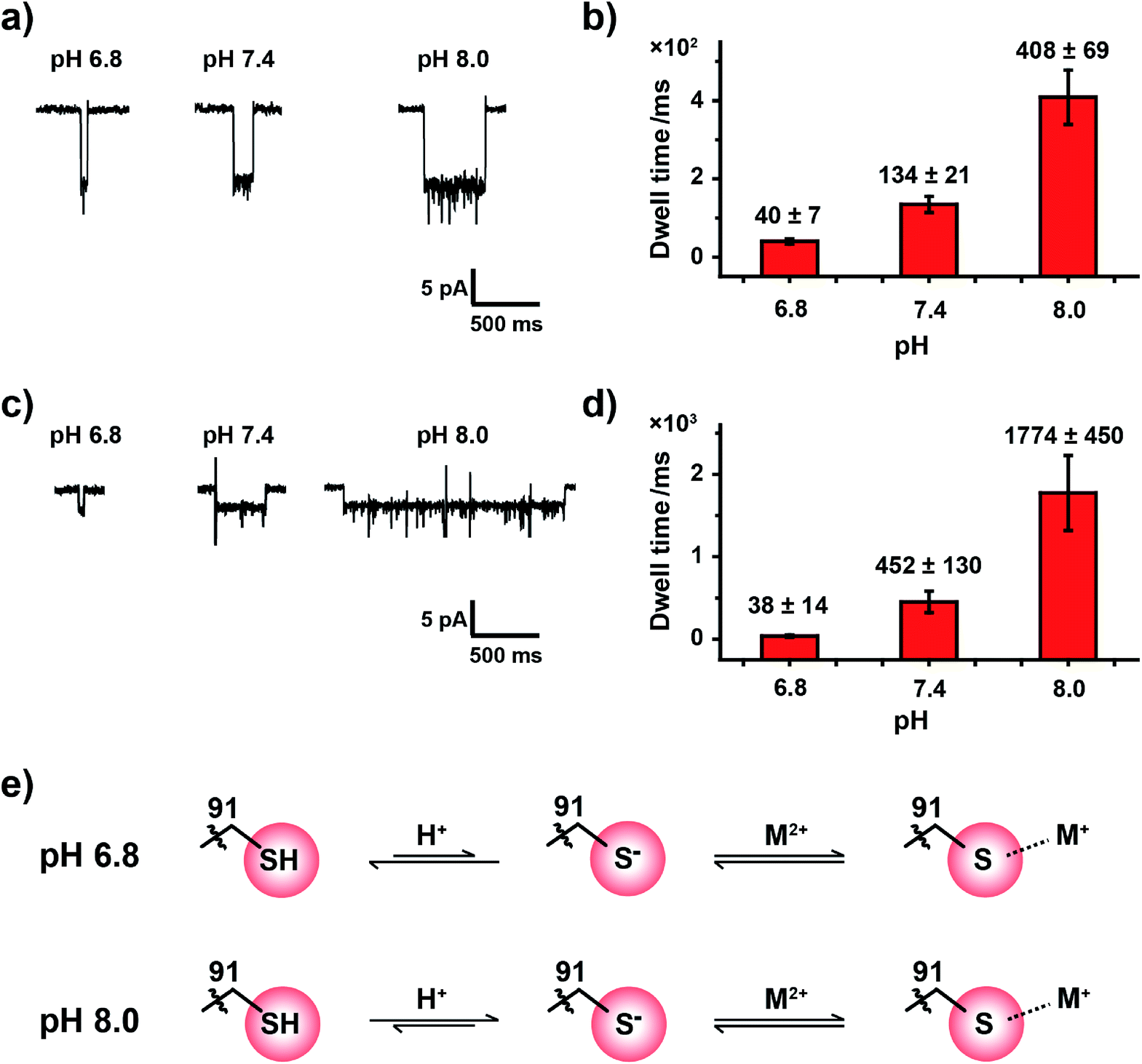 | ||
| Fig. 4 Tuning the coordination bond strength between the cysteine residue and metal ions with pH. (a) Representative events of Zn2+ binding to a cysteine residue with a buffer pH of 6.8, 7.4 and 8.0. (b) The mean dwell times (τoff) of Zn2+ binding events when acquired at different pH values, as demonstrated in (a). (c) Representative events of Cd2+ binding to a cysteine residue with a buffer pH of 6.8, 7.4 and 8.0. (d) The mean dwell times (τoff) of Cd2+ binding events when acquired at different pHs, as demonstrated in (c). Error bars were based on three independent experiments (N = 3). (e) The proposed coordination mechanism. The cysteine residue is considered to be deprotonated when bound to metal ions. At a lower pH (6.8), more protons would compete with the thiolate–metal ion complex to make the complex readily dissociable; in contrast, the thiolate–metal ion complex is more stable at a higher pH (8.0). The above demonstrated results were acquired with the buffer of 1 M NaCl, 10 mM HEPES, 0.4 mM TCEP and a +100 mV continuous applied bias . Zn2+ was added to trans with a 30 μM final concentration. | ||
It is generally agreed that a metal ion binds to a thiolate group, which is the deprotonated form of cysteine thiol.40 Although the protonation state of the cysteine thiol can't be directly probed from a nanopore measurement, tuning of the pH from 6.8 to 8.0 finely modulates the competition between the metal ion and the proton in the vicinity of the reactive site, and this subsequently modulates the dwell time of a bound single ion (Fig. 4e). However, measurements at a pH value below 6.8 result in the disappearance of single ion binding events while measurements at a pH value above 8.0 result in spontaneous pore closure, which prohibits long term continuous measurements. Nonetheless, to the best of our knowledge we believe that this is the first single molecule investigation on metal ion–thiol/thiolate interaction when probed at different protonation states of the cysteine.
2.7. Prospects
The work demonstrated above is not without limitations. All MspA mutants used in this paper are homomeric, which means that all eight subunits were altered identically. To avoid complications from simultaneous binding of multiple ions, all results in this work were carried out with a relatively low analyte concentration. A heterooctameric MspA nanopore which contains reactive and unreactive subunits in a 1 + 7 form may be prepared as reported,10 but the subsequent protein purification is technically challenging. Alternatively, all eight subunits of MspA may be genetically chained into a single unit.41 However, difficulties in pore oligomerization or an extremely low expression yield may be encountered. The monomeric OmpG nanopore42,43 may be promising, despite the fact that its pore geometry may or may not be sensitive enough to report a single ion binding event. The reported 10 pA event amplitude from binding of a single ion to MspA, which represents a <5% percentage blockage, indicates that the restriction dimension of MspA may be further genetically or chemically shrunk to boost its sensitivity with extremely small analytes. Similar to previous reports using engineered α-HL mutants,11,12 all rate constants were measured in a nano-confined pore cavity where the electroosmotic flow or the electric field may affect the measured binding constants. This may be overcome by the recently reported electrode-free nanopore sensing platform DiffusiOptoPhysiology.31 According to HSAB theory and the demonstrated results, hard acid ions such as Ca2+ and Mn2+, which don't interact strongly with amino acids belonging to soft or borderline bases and barely interact with amino acids belonging to hard bases, may be monitored with difficulty in any combinatorial placement of natural amino acids within the pore lumen. However, this limitation may be overcome by the introduction of unnatural amino acids,44 chemical modifications with a high specificity45 or protein phosphorylation46 around the pore restriction, so that a compatible ligand may be precisely placed.According to HSAB theory, other soft acid ions, which contain Au(I), Au(III) and Pt(II) atoms, should interact with corresponding soft base residues such as cysteine,47 methionine,48,49 selenocysteine50 or selenomethionine.51 Trials from our group have successfully validated these interactions in corresponding nanopore mutants and will be discussed with more details in separate studies.
3. Conclusions
In summary, we report the first demonstration of an MspA nanopore monitoring binding of a variety of single monatomic ions. With its conical geometry, MspA reports a systematically higher event amplitude than α-HL mutants. This suggests that MspA may have advantages in probing a variety of other small analytes such as polyatomic ions, small molecules or chemical intermediates. Such experiments have not been demonstrated with MspA to date and could be complementary to its well-known use in nanopore sequencing. Measurements with a combination of MspA mutants and different analyte ions suggest that the theory of HSAB, which is based on observations in ensembles, was systematically validated in single molecules. Metal ions exhibited coordination preference for different amino acid residues with a principle of “hard to hard and soft to soft”. Though never having been directly probed by an engineered nanopore, Pb2+, which is a softer borderline acid, was monitored binding to a histidine or a cysteine residue in corresponding MspA mutants, consistent with HSAB theory. The coordination interaction between the analyte ion and the pore restriction was also sensitively modulated with pH, with which up to a 46-fold extension in the event duration was demonstrated by simply adjusting the pH from 6.8 to 8.0. This is also the first single molecule investigation on the pH effect of coordination between metal ions and a sulfhydryl group. The results in this paper may be inspiring and instructive in the design of MspA mutants to probe a wider variety of small analytes or serve as a single molecule tool in the investigation on the properties of natural metalloproteins or metal activating enzymes. The principle of hard–soft-acid–base interaction may be also instructive in the pore design so that a variety of metal embedded porins (metalloporins) may be formed as a new class of biological nanopore sensors, mimicking natural metalloproteins.Conflicts of interest
The authors declare no conflicts of interests.Acknowledgements
The authors would like to thank Academician/Prof. Zijian Guo (Nanjing University), Prof. Jin Zhao (Nanjing University), Prof. Yi Lu (Nanjing University) and Prof. Yuncong Chen (Nanjing University) for discussions on the coordination chemistry and Prof. Hagan Bayley (University of Oxford), Prof. Shaolin Zhu (Nanjing University) and Prof. Congqing Zhu (Nanjing University) for other inspiring discussions. This work was supported by the National Natural Science Foundation of China (Grant No. 21327902, Grant No. 21675083, Grant No. 91753108, and Grant No. 31972917), Fundamental Research Funds for the Central Universities (Grant No. 020514380142 and No. 020514380174), State Key Laboratory of Analytical Chemistry for Life Science (Grant No. 5431ZZXM1804 and Grant No. 5431ZZXM1902), 1000 Plan Youth Talent Program of China, Program for high-step Entrepreneurial and Innovative Talents Introduction of Jiangsu Province, Technology Innovation Fund Program of Nanjing University, and Excellent Research Program of Nanjing University (Grant No. ZYJH004).References
- K. J. Waldron and N. J. Robinson, Nat. Rev. Microbiol., 2009, 7, 25–35 CrossRef CAS PubMed.
- C. Liu and H. Xu, J. Inorg. Biochem., 2002, 88, 77–86 CrossRef CAS.
- T. Dudev and C. Lim, Annu. Rev. Biophys., 2008, 37, 97–116 CrossRef CAS PubMed.
- D. H. Petering, M. Huang, S. Moteki and C. F. Shaw III, Mar. Environ. Res., 2000, 50, 89–92 CrossRef CAS PubMed.
- M. J. Tamas, S. K. Sharma, S. Ibstedt, T. Jacobson and P. Christen, Biomolecules, 2014, 4, 252–267 CrossRef PubMed.
- C. Hureau, Y. Coppel, P. Dorlet, P. L. Solari, S. Sayen, E. Guillon, L. Sabater and P. Faller, Angew. Chem., Int. Ed., 2009, 48, 9522–9525 CrossRef CAS PubMed.
- S. Parthasarathy, F. Long, Y. Miller, Y. Xiao, D. McElheny, K. Thurber, B. Ma, R. Nussinov and Y. Ishii, J. Am. Chem. Soc., 2011, 133, 3390–3400 CrossRef CAS PubMed.
- B. A. Krizek, D. L. Merkle and J. M. Berg, Inorg. Chem., 1993, 32, 937–940 CrossRef CAS.
- D. Hebenstreit and F. Ferreira, Allergy, 2005, 60, 1208–1211 CrossRef CAS PubMed.
- O. Braha, B. Walker, S. Cheley, J. J. Kasianowicz, L. Song, J. E. Gouaux and H. Bayley, Chem. Biol., 1997, 4, 497–505 CrossRef CAS PubMed.
- O. Braha, L.-Q. Gu, L. Zhou, X. Lu, S. Cheley and H. Bayley, Nat. Biotechnol., 2000, 18, 1005–1007 CrossRef CAS PubMed.
- L. S. Choi, T. Mach and H. Bayley, Biophys. J., 2013, 105, 356–364 CrossRef CAS PubMed.
- J. J. Kasianowicz, D. L. Burden, L. C. Han, S. Cheley and H. Bayley, Biophys. J., 1999, 76, 837–845 CrossRef CAS PubMed.
- Y. L. Ying, C. Cao and Y. T. Long, Analyst, 2014, 139, 3826–3835 RSC.
- M. Faller, M. Niederweis and G. E. Schulz, Science, 2004, 303, 1189–1192 CrossRef CAS PubMed.
- T. Z. Butler, M. Pavlenok, I. M. Derrington, M. Niederweis and J. H. Gundlach, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20647–20652 CrossRef CAS PubMed.
- E. A. Manrao, I. M. Derrington, A. H. Laszlo, K. W. Langford, M. K. Hopper, N. Gillgren, M. Pavlenok, M. Niederweis and J. H. Gundlach, Nat. Biotechnol., 2012, 30, 349–353 CrossRef CAS PubMed.
- Y. Wang, S. Yan, P. Zhang, Z. Zeng, D. Zhao, J. Wang, H. Chen and S. Huang, ACS Appl. Mater. Interfaces, 2018, 10, 7788–7797 CrossRef CAS PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- Z. T. Zhu, R. P. Wu and B. L. Li, Chem. Sci., 2019, 10, 1953–1961 RSC.
- S. Yan, X. Li, P. Zhang, Y. Wang, H.-Y. Chen, S. Huang and H. Yu, Chem. Sci., 2019, 10, 3110–3117 RSC.
- Y. Wang, K. M. Patil, S. H. Yan, P. K. Zhang, W. M. Guo, Y. Q. Wang, H. Y. Chen, D. Gillingham and S. Huang, Angew. Chem., Int. Ed., 2019, 58, 8432–8436 CrossRef CAS PubMed.
- T. Kochanczyk, A. Drozd and A. Krezel, Metallomics, 2015, 7, 244–257 RSC.
- L. r. Rulíšek and J. Vondrášek, J. Inorg. Biochem., 1998, 71, 115–127 CrossRef.
- J. Liang and J. W. Canary, Angew. Chem., Int. Ed., 2010, 49, 7710–7713 CrossRef CAS PubMed.
- Y. S. Babu, J. S. Sack, T. J. Greenhough, C. E. Bugg, A. R. Means and W. J. Cook, Nature, 1985, 315, 37–40 CrossRef CAS PubMed.
- Y. S. Babu, C. E. Bugg and W. J. Cook, J. Mol. Biol., 1988, 204, 191–204 CrossRef CAS PubMed.
- A. Bernad, L. Blanco, J. Lázaro, G. Martín and M. Salas, Cell, 1989, 59, 219–228 CrossRef CAS.
- A. J. Heron, J. R. Thompson, B. Cronin, H. Bayley and M. I. Wallace, J. Am. Chem. Soc., 2009, 131, 1652–1653 CrossRef CAS PubMed.
- S. Huang, M. Romero-Ruiz, O. K. Castell, H. Bayley and M. I. Wallace, Nat. Nanotechnol., 2015, 10, 986–991 CrossRef CAS PubMed.
- Y. Q. Wang, Y. Wang, X. Y. Du, S. H. Yan, P. K. Zhang, H. Y. Chen and S. Huang, Sci. Adv., 2019, 5, eaar3309 CrossRef PubMed.
- Q. Lu and C. Miller, Science, 1995, 268, 304–307 CrossRef CAS PubMed.
- M. Perez-Garcia, N. Chiamvimonvat, E. Marban and G. Tomaselli, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 300–304 CrossRef CAS PubMed.
- G. M. Preston, J. S. Jung, W. B. Guggino and P. Agre, J. Biol. Chem., 1993, 268, 17–20 CAS.
- T. Dudev and C. Lim, Chem. Rev., 2003, 103, 773–788 CrossRef CAS PubMed.
- A. S. Bastug, S. E. Goz, Y. Talman, S. Gokturk, E. Asil and E. Caliskan, J. Coord. Chem., 2011, 64, 281–292 CrossRef CAS.
- A. Raja, V. Rajendiran, P. Uma Maheswari, R. Balamurugan, C. A. Kilner, M. A. Halcrow and M. Palaniandavar, J. Inorg. Biochem., 2005, 99, 1717–1732 CrossRef CAS PubMed.
- H. Sigel and D. B. McCormick, Acc. Chem. Res., 1970, 3, 201–208 CrossRef CAS.
- R. Bischoff and H. Schlueter, J. Proteomics, 2012, 75, 2275–2296 CrossRef CAS PubMed.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 2002, 124, 6759–6766 CrossRef CAS PubMed.
- M. Pavlenok, I. M. Derrington, J. H. Gundlach and M. Niederweis, PLoS One, 2012, 7, e38726 CrossRef CAS PubMed.
- M. A. Fahie, B. Yang, M. Mullis, M. A. Holden and M. Chen, Anal. Chem., 2015, 87, 11143–11149 CrossRef CAS PubMed.
- T. Zhuang and L. K. Tamm, Angew. Chem., Int. Ed., 2014, 53, 5897–5902 CrossRef CAS PubMed.
- J. Lee and H. Bayley, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13768–13773 CrossRef CAS PubMed.
- M. M. Haugland, S. Borsley, D. F. Cairns-Gibson, A. Elmi and S. L. Cockroft, ACS Nano, 2019, 13, 4101–4110 CrossRef CAS PubMed.
- C. T. Walsh, S. Garneau-Tsodikova and G. J. Gatto Jr, Angew. Chem., Int. Ed., 2005, 44, 7342–7372 CrossRef CAS PubMed.
- P. P. Corbi, E. E. Castellano, F. Cagnin and A. C. Massabni, J. Chem. Crystallogr., 2007, 37, 91–95 CrossRef CAS.
- A. V. Vujačić, J. Z. Savić, S. P. Sovilj, K. Mészáros Szécsényi, N. Todorović, M. Ž. Petković and V. M. Vasić, Polyhedron, 2009, 28, 593–599 CrossRef.
- T. Zimmermann, M. Zeizinger and J. V. Burda, J. Inorg. Biochem., 2005, 99, 2184–2196 CrossRef CAS PubMed.
- T. Shoeib, D. W. Atkinson and B. L. Sharp, Inorg. Chim. Acta, 2010, 363, 184–192 CrossRef CAS.
- A. A. Isab and A. P. Arnold, J. Coord. Chem., 1985, 14, 73–77 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05260g |
| This journal is © The Royal Society of Chemistry 2020 |