
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of circularly polarized luminescence through excited-state symmetry breaking and interbranched exciton coupling in helical push–pull organic systems†
Kais
Dhbaibi
 ah,
Ludovic
Favereau
*a,
Monika
Srebro-Hooper
*b,
Cassandre
Quinton
a,
Nicolas
Vanthuyne
c,
Lorenzo
Arrico
d,
Thierry
Roisnel
a,
Bassem
Jamoussi
e,
Cyril
Poriel
a,
Clément
Cabanetos
f,
Jochen
Autschbach
g and
Jeanne
Crassous
*a
ah,
Ludovic
Favereau
*a,
Monika
Srebro-Hooper
*b,
Cassandre
Quinton
a,
Nicolas
Vanthuyne
c,
Lorenzo
Arrico
d,
Thierry
Roisnel
a,
Bassem
Jamoussi
e,
Cyril
Poriel
a,
Clément
Cabanetos
f,
Jochen
Autschbach
g and
Jeanne
Crassous
*a
aUniv Rennes, CNRS, ISCR – UMR 6226, ScanMAT – UMS 2001, F-35000 Rennes, France. E-mail: ludovic.favereau@univ-rennes1.fr; jeanne.crassous@univ-rennes1.fr
bFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Krakow, Poland. E-mail: srebro@chemia.uj.edu.pl
cAix Marseille University, CNRS, Centrale Marseille, iSm2, Marseille, France
dDipartimento di Chimica e Chimica Industriale, University of Pisa, via Moruzzi 13, 56124, Pisa, Italy
eDepartment of Environmental Sciences, Faculty of Meteorology, Environment and Arid Land Agriculture, King Abdulaziz University, 21589 Jeddah, Saudi Arabia
fMOLTECH-Anjou, CNRS UMR 6200, University of Angers, 2 Bd Lavoisier, 49045 Angers, France
gDepartment of Chemistry, University at Buffalo, State University of New York, Buffalo, NY 14260, USA
hUniversity of Gabès, Faculty of Science of Gabès, Zrig, 6072 Gabès, Tunisia
First published on 20th November 2019
Abstract
π-Helical push–pull dyes were prepared and their (chir)optical properties were investigated both experimentally and computationally. Specific fluorescent behaviour of bis-substituted system was observed with unprecedented solvent effect on the intensity of circularly polarized luminescence (CPL, dissymmetry factor decreasing from 10−2 to 10−3 with an increase in solvent polarity) that was linked to a change in symmetry of chiral excited state and suppression of interbranched exciton coupling. The results highlight the potential of CPL spectroscopy to study and provide a deeper understanding of electronic photophysical processes in chiral π-conjugated molecules.
Introduction
Circularly polarized (CP) luminescence (CPL) has attracted significant attention due to its potential applications in the fields of (chir)optoelectronics (stereoscopic displays, organic light-emitting diodes (OLED), optical information processing, etc.) as well as in bio-imaging and chiral sensing.1 For a long time, lanthanide complexes have been the chiral molecular class of choice for studying CPL phenomena due to their (formally Laporte forbidden) f → f transitions, which provide them with large values (more than 1) luminescence dissymmetry factor glum = 2(IL − IR)/(IL + IR).2 In recent years, however, (small) chiral organic molecules have been gaining more and more interest as potential emitters thanks to their tuneable photophysical and chiroptical properties, and their relatively simple integration in optoelectronic devices such as CP-OLEDs, chiral photovoltaics and transistors.2d,3 Indeed, this class of CPL emitters often exhibits superior luminescence quantum yields than lanthanide complexes owing to their electric dipole-allowed transitions. The involvement of such electronic transitions results however simultaneously in much lower glum values for organic systems, ranging from 10−4 to 10−2, which hampers their use as emissive materials in chiroptoelectronic applications.1d,f,g,4 Comprehensive synthetic guidelines for designing efficient chiral emitters are therefore currently needed to enable further developments in this field. Of particular importance here is identifying and understanding electronic factors that govern the glum at the molecular level, which still remains a crucial challenge for CPL dyes.4b,c,5 The potential of CPL spectroscopy to investigate the chirality of the corresponding excited state is indeed largely unexplored in comparison with unpolarized fluorescence spectroscopy of achiral molecules.6 For instance, solvent effects usually influence the emission and intensity wavelength (solvatochromism) through electronic interactions with the excited state.7 For chiral emitters, such effects are rarely taken into consideration and associated glum is commonly defined only for one arbitrary solvent,1f,4c thus limiting the potential of CPL spectroscopy to provide unique information relating features of the chiral excited state to the intensity of polarized emission.8 Consequently, a deep understanding of excited-state chirality is crucial from a fundamental point of view, as it may help to develop more efficient CPL emitters for CP-OLED, bio-imaging and chiral sensing where interactions between the chiral dye and its surroundings play a crucial role.To investigate this hitherto unexplored aspect of CPL, we focus our attention on π-helical push–pull systems such as the newly synthesized mono-naphthalimide helicene 1 and the bis-naphthalimide helicene 2, previously used in organic photovoltaic devices (Fig. 1).3l These are ideal candidates to explore innovative CPL-emitter designs based on intramolecular charge-transfer (ICT) transitions owing to the electron donor and electron acceptor abilities of the helicene and naphthalimide fragments, respectively.9 As we report herein, the chiral multipolar acceptor (A)–π–donor (D) 1 and A–π–D–π–A 2 derivatives exhibit intense electronic circular dichroism (ECD) responses in the visible spectral region along with high fluorescence quantum yields and intense CPL signals, up to 80% and glum = 10−2, respectively. Interestingly, while both compounds show similar solvatochromism, only 2 displays a modulation of CPL intensity with an increase in solvent polarity, showing the importance of the branching effect in such chiral push–pull dyes. The specific emission behaviour of 2vs. 1 was rationalized based on both experimental and computational characterizations, and related mainly to a change in symmetry of the emitting S1 state and suppression of exciton coupling between individual helicene → naphthalimide ICT transitions upon increasing the polarity of the environment.
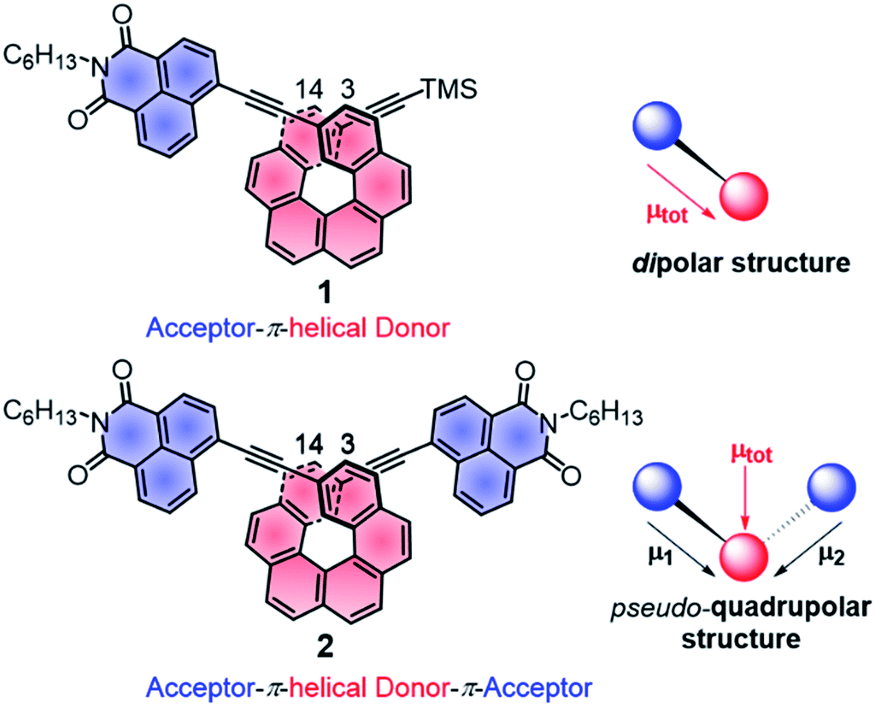 | ||
| Fig. 1 Chemical structures of chiral A–π–D P-1 and A–π–D–π–A P-2 with schematic representations of the local and total permanent electric dipole moments μ. TMS: trimethylsilyl, C6H13: n-hexyl. | ||
Results and discussion
Synthesis and structural characterization
Enantiopure P-1 and P-2 were synthesized following the previously reported strategy10 that involves a deprotection reaction of P-2,15-bis-(trimethylsilyl-ethynyl)[6]helicene (P-H6(TMS)2) to obtain partially (P-H6a) and fully (P-H6b) unprotected ethynylhelicene derivatives (Scheme 1).11 This statistical mixture was then directly engaged in a final Sonogashira coupling with an excess of 6-bromo-2-hexyl-1H-benzoisoquinoline-1,3(2H)-dione (NPhBr),3l to afford P-1 and P-2 within the same reaction in 85–90% yield for both compounds (see ESI†). M-1 and M-2 were obtained in a similar way using M-2,15-bis-(trimethylsilyl-ethynyl)[6]helicene (M-H6(TMS)2) as a starting material.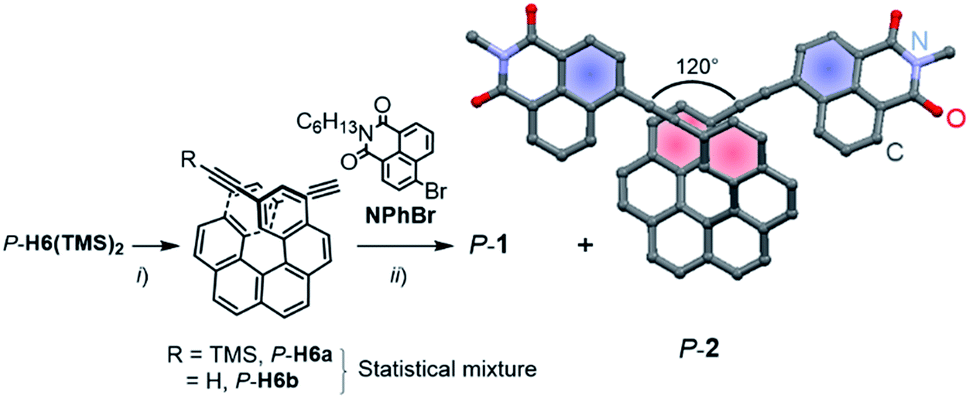 | ||
| Scheme 1 Synthesis of enantiopure naphthalimide–helicene derivatives P-1 and P-2. TMS: trimethylsilyl, C6H13: n-hexyl. Reaction conditions: (i) TBAF, CHCl3; (ii) Pd(PPh3)4, CuI, Et3N/toluene, 50 °C, NPhBr, 85% (P-1) and 90% (P-2). X-ray crystal structure of P-2 (hexyl chains on the imide fragments have been omitted for clarity). | ||
Characteristic signatures of both the [6]helicene and naphthalimide units were identified in the 1H NMR spectra of 1 and 2, which also showed the typical differences between C1- and C2-symmetric structures with two distinct signals (doublet of doublets) at 7.56 and 7.28 ppm assigned to H3 and H14 (Fig. 1) for 1 and only one signal (doublet) at 7.29 ppm for these protons in 2 (see also ESI†). Single crystals of P-2 were obtained by slow diffusion of pentane vapours into CH2Cl2 solution. The compound crystallized in the non-centrosymmetric monoclinic P21 space group and displayed helicity (dihedral angle between the two terminal helicenic rings) of 37.77–47.91° (two molecules in the asymmetric unit), which is in the range of classical carbo[6]helicenes.9d Both naphthalimide fragments are oriented towards the helix, (i.e. with the aromatic ring not directly linked with the ethynyl bridge being in cis position with respect to the helicene) and show a dihedral angle of 7.2° with the corresponding terminal helicene phenyl ring. This should ensure a strong electronic communication between the two units (vide infra). The angle between each naphthalimide-ethynyl arms was estimated to be around 120° (Scheme 1) and allows to consider 2 as a quasi-quadrupolar A–π–D–π–A chiral compound.
UV-vis spectroscopy
UV-vis absorption spectra of 1 and 2 were recorded in CH2Cl2 and showed similar features but are noticeably different from those observed for their helicenic and naphthalimide precursors (Fig. 2a and S7 in the ESI†). Both compounds displayed three main bands with two higher-energy ones centered at ca. 300 and 360 nm that relate to absorption maxima of H6(TMS)2 and NPhBr, respectively, but are bathochromically shifted and more intense, and with an additional, strong, broad intensity at lower-energy, centered at 410 nm. Such modifications found in 1 and 2 compared to UV-vis spectra of precursors clearly reflect an extended π-electronic structure and push–pull effects within these helical naphthalimide systems (vide infra). Although no red-shift of the low-energy part of the spectrum was observed for 2vs.1 (indicating lack of electronic conjugation through the whole helix in 2) grafting two electron acceptor groups on the [6]helicene unit strongly affects absorption intensity in this spectral region leading to molar extinction coefficients for 2 twice as high as for 1 (ε = 3.2 × 104 and ε = 6.5 × 104 M−1 cm−1 at 410 nm for 1 and 2, respectively, Fig. 2a). The absorption band at ca. 300 nm, assigned mainly to the helicene fragment (vide infra), appears slightly red-shifted by about 10 nm for 2 as compared to 1, which confirms extension of helical π-electron system upon introduction of the second naphthalimide fragment. As shown in Fig. 2a and ESI (Fig. S8 and S9†), 1 and 2 exhibit almost no solvatochromism in their ground state. Only optical transitions between 370–450 nm display some slight shifts upon changing the solvent polarity, presumably due to their significant charge-transfer character (vide infra). However, these spectral changes are rather minor and exclusively observed in non-polar cyclohexane where both 1 and 2 exhibit a structured blue-shifted response with two distinct maxima, at 396 and 420 nm, equal in intensity (ε = 4.1 × 104 and 7.2 × 104 M−1 cm−1 for 1 and 2, respectively). This all indicates that naphthalimide–helicene derivatives 1 and 2 have moderate electronic dipole moments in their ground state.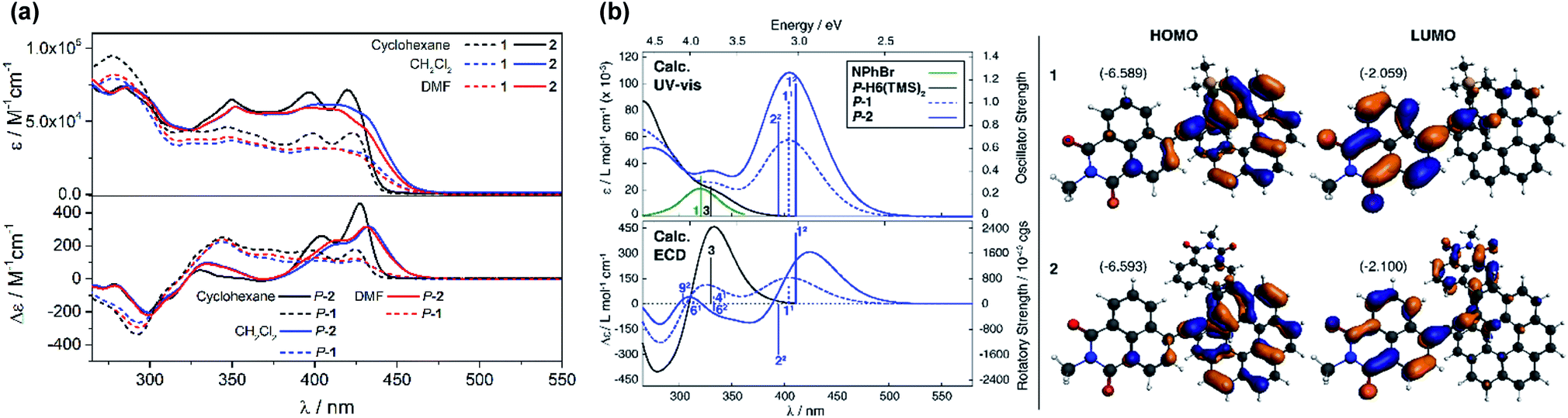 | ||
| Fig. 2 (Panel a) Experimental UV-vis (top) and ECD (bottom) spectra of 1 (dashed lines) and 2 (solid lines) measured in cyclohexane (black), CH2Cl2 (blue) and DMF (red) at 298 K (∼10−5 M). (Panel b) Comparison of the simulated UV-vis and ECD spectra of 1 and 2 with H6(TMS)2 and NPhBr. No spectral shift has been applied. Calculated excitation energies along with oscillator and rotatory strengths indicated as ‘stick’ spectra. Numbered excitations (N1 for 1 and N2 for 2) correspond to those analyzed in detail. Isosurfaces (±0.03 au) of frontier MOs of 1 and 2. Values listed in the parentheses are the corresponding orbital energies, in eV. See also ESI.† | ||
Further characterizations of the electronic and photophysical properties of 1 and 2 were then obtained with the help of quantum-chemical calculations for truncated systems (with n-hexyl groups replaced by methyls) using (time-dependent) Kohn–Sham theory, (TD)KS = (TD)DFT.12 All computational details along with the full set of theoretical results are provided in the ESI.† Conformational analyses (BP/SV(P)) performed for 1 and 2 revealed existence of two and three low-energy nearly isoenergetic conformers, respectively, that can be thus assumed to be present roughly in equal amounts at room temperature. These rotamer structures differ in the relative orientations of the naphthalimide group(s) and the helicene moiety (Fig. S18†) but demonstrate overall very similar electronic features with (i) the dipolar A–π–D and pseudo-quadrupolar A–π–D–π–A character for 1 and 2, respectively, and (ii) efficient π-conjugation between electron acceptor naphthalimide group(s) and electron donor helicene moiety via the alkynyl bridge in both 1 and 2. Indeed, calculated electronic dipole moment vectors in 1 and 2 are oriented as expected for the dipolar and pseudo-quadrupolar structures (Fig. S28 and S29†). Moreover, the frontier molecular orbitals (MOs) in 1 and 2 are consistent with the push–pull character of the dyes. Indeed, the occupied MOs span over the whole helicene fragment, the ethynyl bridges and partially also naphthalimide units, while the unoccupied ones are predominantly centered at the naphthalimide fragments but also delocalized over the adjacent helicene's rings via the alkynyl (Fig. 2b and ESI†). The simulated (BHLYP/SV(P), CH2Cl2 continuum solvent model), Boltzmann-averaged UV-vis absorption spectra (Fig. 2b) are in good agreement with the experimental ones (Fig. 2a). In particular, the appearance of the additional low-energy absorption bands in naphthalimide–helicene derivatives compared to their precursors H6(TMS)2 and NPhBr along with the significant increase in the absorption intensity observed for 2vs.1 are correctly reproduced by theory and linked to push–pull character of the dyes. In line with the A–π–D and A–π–D–π–A electronic structures of 1 and 2, an MO-pair analysis of the dominant excitations calculated in the low-energy spectral regions assigns the additional absorption at 420 nm in 1 and 2 to mainly intramolecular charge-transfer π–π* excitations from the helicene core to the naphthalimide group(s). In the case of 1, the band originates from the lowest-energy excitation no. 11 (calculated at 405 nm) that involves HOMO, HOMO−2 and LUMO (Fig. 2b and ESI†). For 2, two excitations with sizeable oscillator strength were found in this spectral range, no. 12 and 22 calculated at 411 and 395 nm, respectively, that correspond to transitions from HOMO, HOMO−1, and HOMO−2 to LUMO and LUMO+1. Note that for both 1 and 2 occupied MOs involved in these excitations represent distinct π-orbitals of the alkynyl-helicene electron system with some contributions from the naphthalimide, while unoccupied MOs mainly extend over the naphthalimide-alkynyl group with LUMO and LUMO+1 in 2 representing in-phase and out-of-phase linear combinations of the naphthalimide substituents' LUMO. Accordingly, increase in the intensity of the band for 2 is due to exciton-coupling (EC) interactions between these ICT states, similar to what we previously noticed for push–pull helicenic systems based on strongly electron acceptor tetracyanobutadienes.13 Visible helicene → naphthalimide charge-transfer character can also be noticed for higher-energy excitations including those calculated around 300–330 nm with large contributions from the helicene-centered π–π* transitions (see ESI†). All this clearly supports the conclusions drawn from experimental optical observations.
Cyclic voltammetry
Electrochemical properties of 1 and 2 were examined via cyclic voltammetry measurements that resulted in potentials of ca. +1.4 V and −1.2 V vs. SCE for both compounds (Fig. S17 and Table S2†), assigned respectively to the oxidation of the helicene core and reduction of the naphthalimide unit(s).14 While only one quasi-reversible reduction process was recorded for both systems, confirming a weak electronic interaction between the two naphthalimide units through the π-conjugated helix in 2, bis-substituted naphthalimide–helicene derivative shows a slightly more positive oxidation potential than its mono-substituted analogue (ΔEOx = 80 mV), which is consistent with a decrease in the electronic density on the helicene core upon introduction of the second (electron deficient) naphthalimide group.ECD spectroscopy
P- and M-1 and P- and M-2 compounds displayed electronic circular dichroism (ECD) with expected mirror-image relationships. Their ECD spectral signatures are strongly modified compared to those observed for P- and M-carbo[6]helicene derivatives (Fig. 2a).15 For instance, P-1 in CH2Cl2 solution exhibits an intense negative band (Δε = −290 M−1 cm−1) at 293 nm that is 14 nm red-shifted compared to P-H6(TMS)2, a large positive band between 320 and 450 nm with a maximum at 340 nm (Δε = +240 M−1 cm−1), and three less intense peaks in the lowest-energy region (Δε = +150, +127, and +125 M−1 cm−1 at 370, 410, and 425 nm, respectively). P-2 shows a similar high-energy negative ECD band at 300 nm (Δε = −220 M−1 cm−1) that is only slightly less intense and red-shifted (by ca. 5 nm) compared to P-1. Its positive ECD signal between 320 and 370 nm becomes however much less intense and dips even slightly below zero (Δε = +90 M−1 cm−1 at 335 nm and −5 M−1 cm−1 at 370 nm vs. Δε = +230 M−1 cm−1 at 340 nm and +126 M−1 cm−1 at 370 nm, for P-2vs. P-1), while the region between 370 and 460 nm is now dominated by two very intense positive bands (Δε = +200 and +300 M−1 cm−1 at 410 and 430 nm, respectively), which are ca. 10 nm red-shifted in comparison to P-1. This significant increase in low-energy ECD intensity between P-1 and P-2 is reminiscent of what we reported for two other push–pull [6]helicene-based compounds bearing either diketopyrrolopyrrole (DPP) dyes10 or tetracyanobutadiene (TCBD) electron withdrawing groups.13 Their bis-substituted derivatives exhibit a chiral excitonic coupling originating from the helical arrangement of the two weakly interacting electronic dipoles of the π–π* excitations within a DPP dimer and of ICT helicene → TCBD excitations, respectively. In the present case, the naphthalimide group may act as both an achiral organic dye and an electron acceptor unit, affording a new example of chiral exciton coupling in helicenic systems that leads to one of the largest ECD signal at the molecular level in the visible region (vide infra).Similarly to UV-vis absorption, changing the solvent polarity has rather minor effect on ECD spectra for both 1 and 2, except for non-polar cyclohexane in which a 5 nm blue-shift of two lowest-energy bands along with their intensity increase were observed relative to peaks measured in more polar solvents (e.g. for P-2, Δε = +260 and +450 M−1 cm−1 at 404 and 426 nm, respectively, Fig. 2a, S10 and S11†). To provide a more quantitative characterization of the solvent effects for the examined systems, dissymmetry factors gabs for all the studied solvents were then calculated and plotted (Fig. S12 and S13†). Their values range from 3.8 × 10−3 to 4.5 × 10−3 for P-1 and from 7.0 × 10−3 to 9.5 × 10−3 for P-2 at ca. 430 nm, which confirms the small impact of the polarity of the environment on the chiroptical ground-state responses of P-1 and P-2. This is in line with the dipole moments calculated for both 1 (of 7–8 D) and 2 (of 6.5–7.5 D) that maintain the same spatial orientation and demonstrate only slight increase in magnitude with increase in the solvent polarity (Table S13 and Fig. S29†). Note also that among computationally examined solvents (cyclohexane, CH2Cl2, and DMF) the most pronounced change in the ground-state dipole moments of 1 and 2 upon increase in the solvent polarity is observed for cyclohexane vs. dichloromethane that may rationalize the most significant differences between spectra recorded in cyclohexane and those measured in remaining (more polar) solvents.
As shown in Fig. 2 and ESI,† the simulated (BHLYP/SV(P), CH2Cl2 continuum solvent model) ECD spectra of P-1 and P-2 agree well with the experimental results. In particular, the calculations correctly reproduced (i) the presence of positive ECD signal in the spectral region where the parent helicene absorbs, i.e. around 325 nm, for both P-1 and P-2, with a decrease in the signal's intensity for the latter compound, and (ii) the appearance of low-energy intense positive ECD band along with its red-shift and strong increase in intensity for P-2vs. P-1. As expected and aforementioned above, the former band indeed originates from the predominantly helicene-centered π–π* excitations (no. 41 computed at 333 nm and 61 at 314 nm for 1 and no. 92 at 304 nm for 2 in Fig. 2b, see also ESI†) that involve, however, also ICT transitions from the helicene's π-electron system to the naphthalimide group(s), whereas the low-energy band is attributed to intense lowest-energy excitation (no. 11 (405 nm) and 12 (411 nm)) of purely helicene → naphthalimide ICT character. In the case of 2, this first excitation is accompanied by analogous helicene → naphthalimide ICT excitation no. 22 (395 nm) that reveals similar energy along with comparable, although opposite-sign, rotatory strength value. In consequence, the appearance of a strong bisignate pair of bands in the low-energy part of the computed ECD spectrum of 2 is observed. The bisignate signature and significant enhancement of the long-wavelength ECD intensity for 2vs.1 along with the donor → acceptor ICT character of underlying excitations clearly resemble the case of helicene–TCBD derivatives, and based on its analysis presented in ref. 13, the ECD-intense low-energy excitations of 2, 12 and 22, can also be treated as the exciton couplet arising from coupling between transitions from helicene's π-orbitals to the π*-orbital localized at either one of the naphthalimide groups. The effect is clearly enhanced with respect to TCBD-functionalized helicenes which may be due to a more efficient π-conjugation between electron acceptor substituents and helicene moiety occurring in 2 promoted by highly conjugated and aromatic structure of the naphthalimide dye. The intensity of the band originating from the negative couplet component for P-2 appears to be additionally increased by a presence of excitation no. 62 (333 nm) that also reveals sizable negative rotatory strength value, which may be responsible for the substantial overestimation of the calculated ECD response at around 350 nm for this compound compared to the experimental results. Furthermore, it is worth noting that the experimental intensity of this band (see Fig. S11†) seems to decrease with the polarity of the solvent which may indicate suppression of exciton-coupling interactions between the ICT states in 2 in polar solvents.
Unpolarized luminescence and excited-state symmetry breaking
Fluorescence responses of 1 and 2 appear rather independent on the number of naphthalimide electron acceptor units, showing only a slight blue-shift of the spectra for 2vs.1, and, contrary to the UV-vis absorption and ECD, display significant positive solvatochromism with typical (and similar for both compounds) red-shift of the emission spectra with an increase in the solvent polarity (Table 1 and Fig. S14†). For example, and as illustrated in Fig. 3a for 2, the structured emission in cyclohexane with two distinct bands at 436 and 462 nm becomes less structured and slightly red-shifted in toluene (λmax = 459 nm), while further gradual increase in the polarity of the solvent results in a broad emission profile with its maximum located from 496 nm in THF to 565 nm in DMF. Such pronounced red-shifts of fluorescence spectra and no obvious shift of the absorption in various solvents lead to a significant increase in the Stokes shifts with increasing solvent polarity for 1 and 2 that is consistent with the formation of ICT-type excited state with a much larger dipole moment than in the ground state for both compounds. As presented in Fig. 3b, the solvent-dependent fluorescence behaviour of 1 and 2 overall follows well the Lippert–Mataga relationship between Stokes shift Δν (in cm−1) and solvent orientational polarizability Δf values (eqn (1) and (2)):6a,16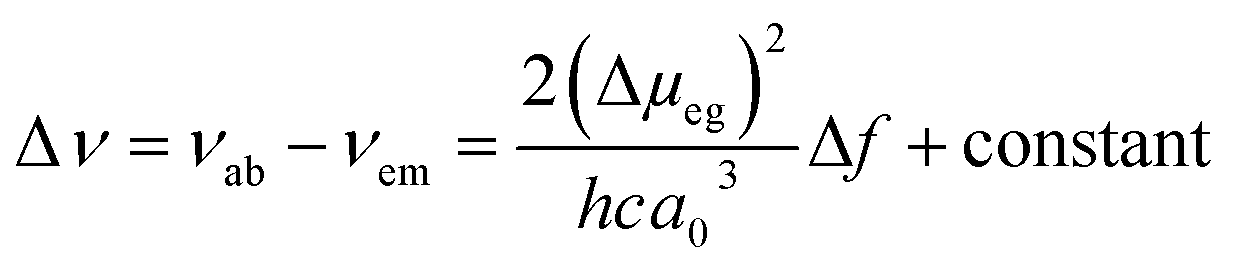 | (1) |
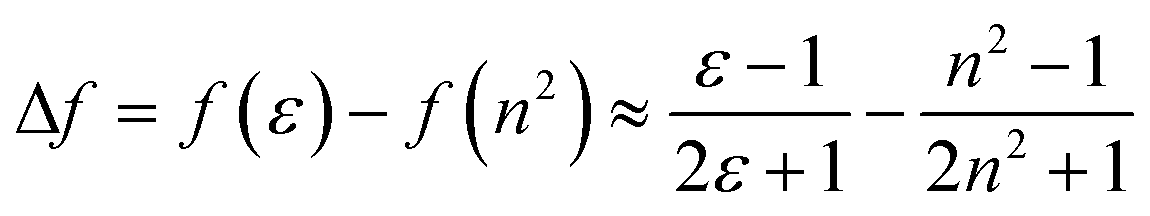 | (2) |
| Solvent | D–π–A 1 | A–π–D–π–A 2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λ abs (nm) | λ em (nm) | Δνa (cm−1) | ϕ fluo (%) | τ fluo (ns) | |glum| (×10−3) | λ abs (nm) | λ em (nm) | Δνa (cm−1) | ϕ fluo (%) | )τFluoc (ns] | |glum| (×10−3) | |
| a Stokes shift. b Absolute quantum yield (error ± 10%), measured using an integrating sphere. c Fluorescence lifetime (error ± 5%), only the main component decay is given (weight in parenthesis). | ||||||||||||
| CyH | 420 | 436 | 870 | 70 | 2.1 | 3.2 | 420 | 436 | 1030 | 45 | 2.4 (91%) | 9.5 |
| Tol | 428 | 457 | 1480 | 85 | 3.4 | 2.4 | 430 | 453 | 1180 | 65 | 2.8 | 6.5 |
| THF | 424 | 503 | 3700 | 80 | 4.4 | 2.0 | 423 | 497 | 3520 | 75 | 4.3 | 4.0 |
| CHCl3 | 431 | 509 | 3560 | 80 | 4.0 | 2.0 | 432 | 503 | 3270 | 70 | 4.2 | 4.0 |
| CH2Cl2 | 427 | 527 | 4440 | 75 | 4.7 | 1.8 | 427 | 520 | 4000 | 70 | 4.3 (96%) | 3.1 |
| Acetone | 424 | 548 | 5340 | 65 | 5.7 | 1.8 | 429 | 539 | 4790 | 60 | 5.7 | 2.8 |
| DMF | 427 | 576 | 6060 | 55 | 6.2 | 1.8 | 433 | 562 | 5450 | 55 | 6.3 (97%) | 2.5 |
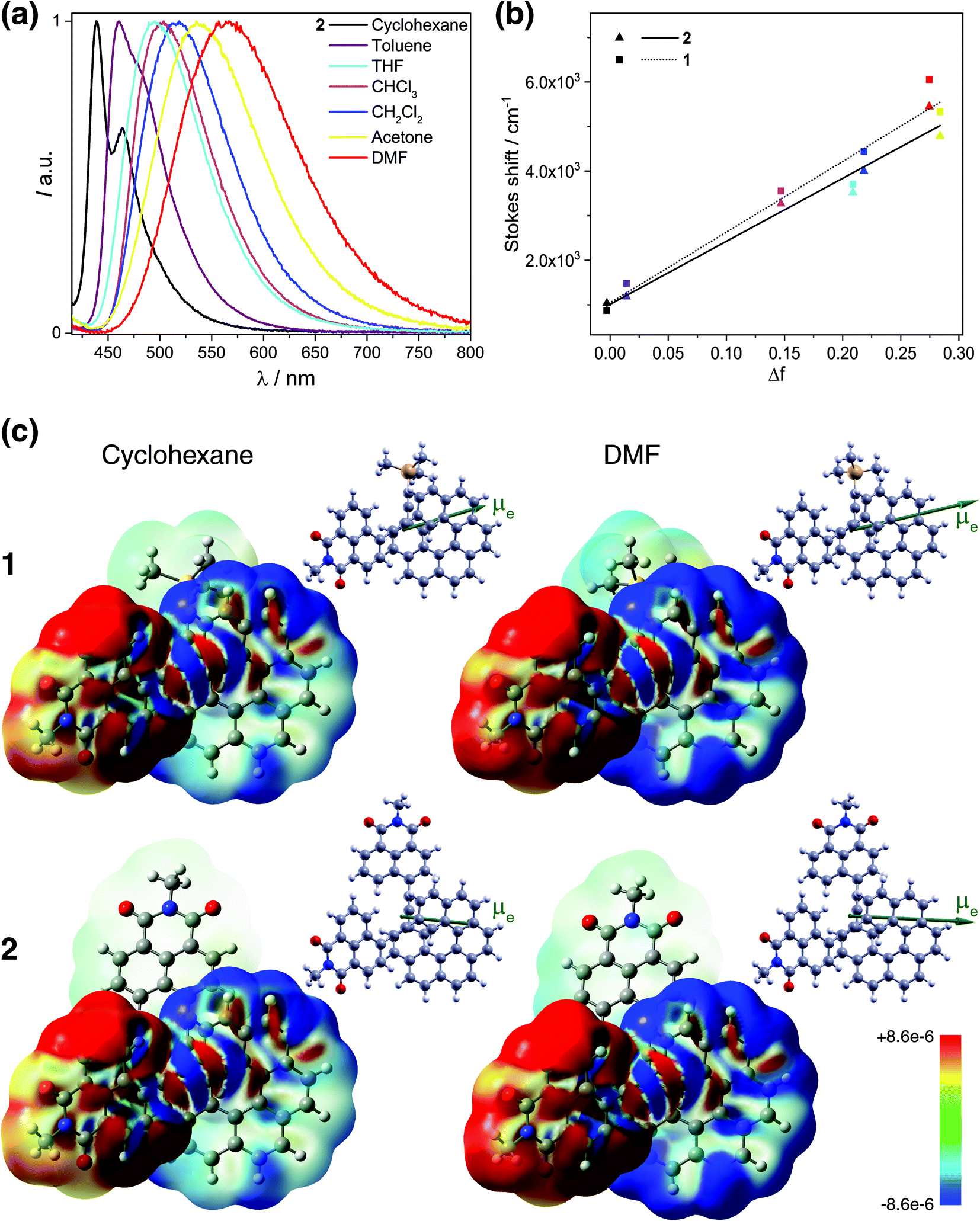 | ||
| Fig. 3 (Panel a) Emission of 2 in cyclohexane (black), toluene (purple), THF (sky blue), CHCl3 (pink), CH2Cl2 (blue), acetone (yellow), and DMF (red) at 298 K (∼10−5 M). (Panel b) Lippert–Mataga plots for 1 (dotted line) and 2 (solid line) (see also Table S12†). (Panel c) Difference density between the S0 ground state and S1 excited state, Δρ = ρg − ρe, color-mapped on ρg (±0.0003 au) for 1 and 2 in cyclohexane and DMF. Alongside, the corresponding S1 excited-state dipole moment vectors (with origin located at the center of nuclear charge, scaled by a factor of 0.5, pointing from the negative to the positive pole of the dipole) are shown. TDDFT BHLYP/SV(P) continuum solvent model calculations. See also ESI.† | ||
These results indicate that the observed spectral shifts in solution are determined by dipole–dipole type interactions between the helical fluorophore and solvent molecules rather than by specific solute–solvent interactions such as hydrogen bonding. More interesting, the plots show nearly identical behaviour with large and similar slope values for both 1 and 2 suggesting that fluorescence in these compounds stems from an excited state of essentially the same electronic nature and is accompanied by large values of the effective dipole moment change Δμeg (Table S12†). Such large slopes appear to reflect relatively small Stokes shifts of 1 and 2 in cyclohexane and toluene, which, along with the structured luminescence profile and the symmetry observed between the absorption and emission spectra, suggests a smaller reorganization before emission in non-polar solvents (i.e. weaker charge-transfer character of S1 excited states) for these systems. Further luminescence characterizations, including fluorescence quantum yields (ϕfluo) and lifetime decays measurements, also confirm a similar photophysical behaviour of 1 and 2 (Table 1). Namely, both compounds display moderate to intense emission efficiencies in the different solvents with ϕfluo up to 85% and 75% for 1 and 2, respectively. Fluorescence lifetimes of 1 and 2 increase from ca. 2.5 ns in cyclohexane to 6.3 ns in DMF, which also evidence the impact of polarity on the charge-transfer excited-state dynamics.
All these experimental observations are well corroborated by TDDFT emission modeling employing state-specific solvation for cyclohexane (ε = 2.0), CH2Cl2 (ε = 8.9), and DMF (ε = 37.2) (see Computational details in the ESI†). The calculations nicely reproduced the solvent-dependent fluorescence behaviour of 1 and 2 with theoretical emission maxima very close to the experimental ones (Tables S10 and S11†). Values of the computed S1 electronic dipole moments visibly increase with the solvent polarity, from ca. 16 and 13 D in cyclohexane to 24 and 21 D in DMF for 1 and 2, respectively, and are strongly enhanced (ca. 2–3 times) compared to those in S0, confirming that the excited state of these compounds is more polar than the ground state (Table S13, Fig. S29 and S30†). More importantly, a significant change in the orientation of μe vector compared to that of μg is observed for 2 toward that observed for 1 and typical for dipolar structure (Fig. 3a, S29 and S30†). Additionally, the magnitude of the effective dipole moment change Δμeg is large and almost the same for both compounds (Table S14†). The calculations also enabled to confirm fundamentally identical nature of the emitting excited state in 1 and 2. In Fig. 3c, the difference densities between the ground state and the excited state for 1 and 2 are presented with their negative (red)/positive (blue) values corresponding to outflow/inflow of electron density accompanying S1 → S0 fluorescence transition (see also Fig. S32†). It is clearly seen that S1 for both 1 and 2 exhibits the same intramolecular naphthalimide → helicene CT characteristics with involvement of only one naphthalimide unit in the case of 2 due to the excited-state symmetry-breaking effect (vide infra).17 Such localization is consistent with the observed ‘dipolar-like’ orientation of μe vector and rationalizes similar photophysical behaviour of both naphthalimide–helicenes. The difference density plots also reveals noticeable increase in the charge-transfer character of S1 excited states (more electron density transferred from naphthalimide group to helicene moiety) for 1 and 2 with the increase in the polarity of the solvent correlating well with the corresponding rise of μe values and higher stabilization of the excited state by polar solvents reflected in the larger red-shift of fluorescence spectra in such environments. The analysis of S1 → S0 emission in terms of individual MO pairs (Fig. S31 and Table S15†) shows that the process mainly corresponds to LUMO → HOMO transition with LUMO localized predominantly on the naphthalimide-alkynyl group and HOMO spanning over the helicene-alkynyl fragment and partially naphthalimide unit. While isosurface of LUMO hardly changes with the polarity of the solvent, for HOMO less/more electron density is observed on naphthalimide/helicene when going from cyclohexane to DMF, in agreement with the increase in the CT character of S1 for 1 and 2 in more polar environment.
Summarizing, unpolarized luminescence measurements and calculations for mono- and bis-substituted naphthalimide–helicene derivatives 1 and 2 reveal a similar evolution of their first excited state with the solvent polarity, even though the latter may be viewed in its ground state as pseudo-quadrupolar A–π–D–π–A structure with a slightly smaller resultant electronic dipole moment than in 1. Such behaviour has been reported for numerous multibranched achiral push–pull molecules,17 and rationalized in terms of a symmetry-breaking in excited state due to structural fluctuations via vibrational relaxation and/or solvation effects. The reported computations demonstrate essentially the same character of S1 excited state for 2 in both non-polar and polar solvents (and also in gas-phase) indicating that the observed localization of the excitation is predominantly induced by nuclear relaxation that involves mainly planarization of the naphthalimide units with respect to the corresponding terminal helicene phenyl ring (leading consequently to increase in naphthalimide–helicene π-conjugation) upon excited-state geometry optimization (Fig. S26†). Note that similar excited-state localization for an A–π–D–π–A-type system was reported and described in terms of a Frenkel exciton model for example in ref. 17a. Note also that Vauthey et al. have recently been able to directly visualize such excited-state symmetry-breaking process using ultra-fast time-resolved infrared spectroscopy, thus providing fundamental understanding of this phenomenon and opening new perspectives for multipolar π-conjugated systems in optoelectronic applications and photochemistry.17d,e,18
CPL and interbranched excition coupling
To further investigate the impact of solvatochromism on the chiral excited states of 1 and 2, the corresponding CPL spectra were recorded in the same set of solvents used in the unpolarized luminescence study (Fig. 4, S15 and S16†). As can be seen, both compounds display intense, mirror-image CPL signals for P and M enantiomers with similar red-shifts upon increasing the polarity of the solvent, in agreement with unpolarized emission. Interestingly, while P-1 exhibits almost constant CPL intensity in all solvents (glum ∼ 2 × 10−3), P-2 shows an unexpected three-fold decrease of intensity when going from non-polar to polar environment (Fig. 5 and Table 1). Namely, the intense structured CPL spectrum for P-2 in cyclohexane (glum = 9.5 × 10−3) becomes broader in toluene (glum = 6.5 × 10−3), and unstructured and weakened in THF and CHCl3 (glum = 4.0 × 10−3). For more polar solvents (CH2Cl2, acetone, and DMF) a further decrease in intensity of the P-2 CPL signal (with glum values of ca. 3.0 × 10−3) was observed.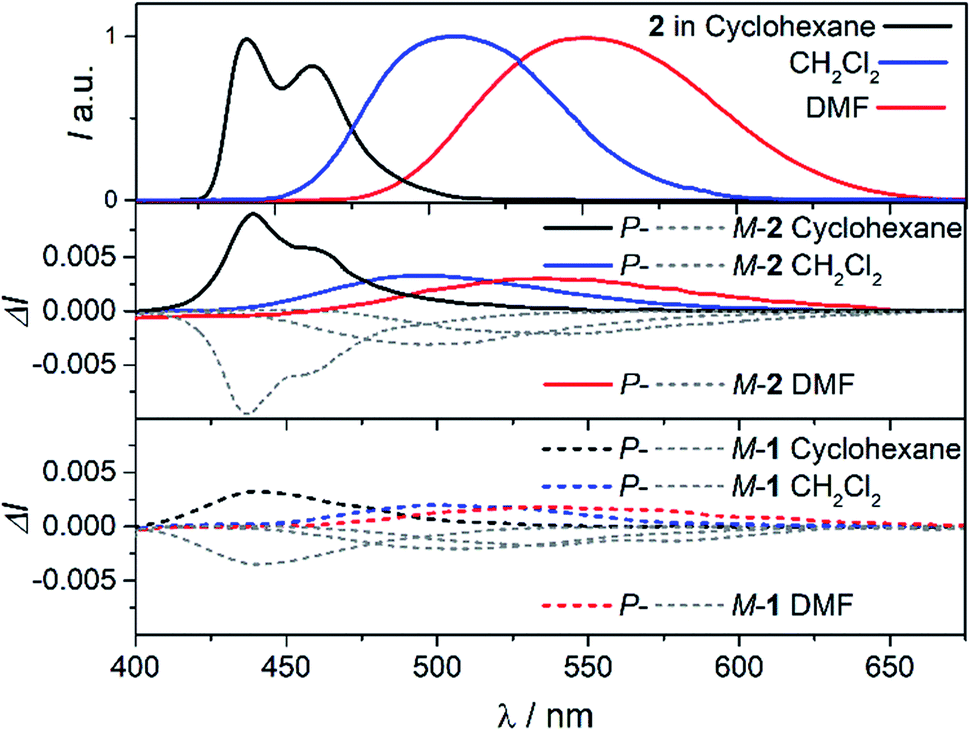 | ||
| Fig. 4 CPL spectra of 2 (middle panel) and 1 (bottom panel) in cyclohexane (black), CH2Cl2 (blue), and DMF (red) at 298 K (∼10−5 M). For a comparison, fluorescence spectra of 2 are presented in the top panel (the corresponding spectra for 1 are not shown as they are similar to those for 2). See ESI† for a full set of recorded spectra. | ||
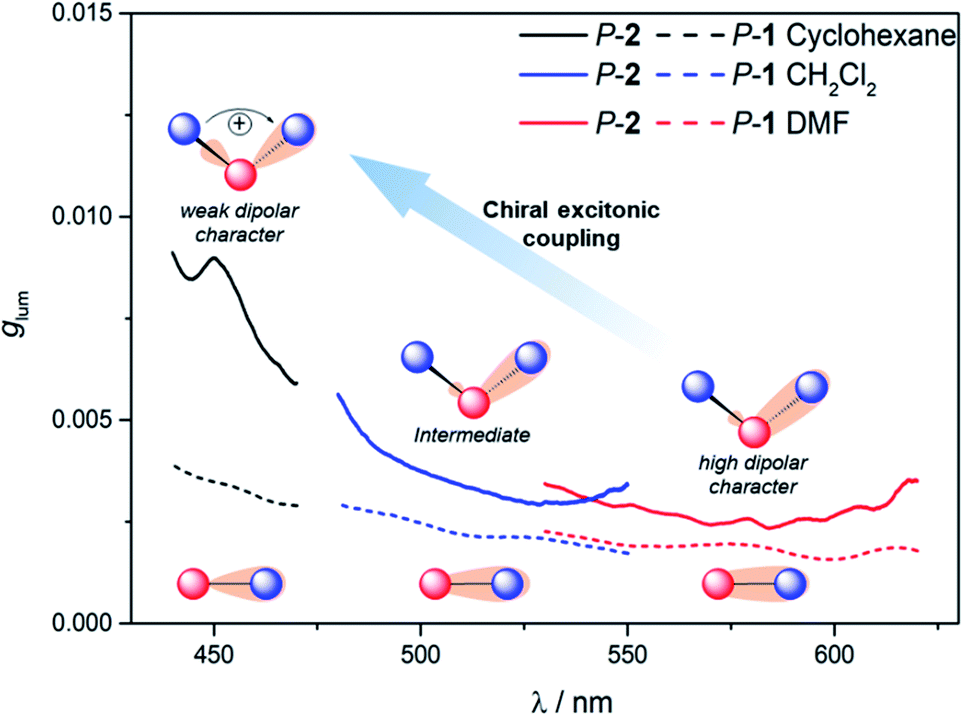 | ||
| Fig. 5 Emission dissymmetry factors glum of P-1 (dashed lines) and P-2 (solid lines) in cyclohexane (black), CH2Cl2 (blue), and DMF (red) at 298 K (∼10−5 M) along with illustration of the effects underlying the observed trends. | ||
The effect of the solvation on CPL intensity is also visible in the computed results that show almost constant rotatory strength R and the corresponding glum values for S1 → S0 fluorescence transition in different solvents for 1, and their noticeable (although clearly underestimated comparing to the experiment) increase when changing the polarity of the solvent from CH2Cl2 and DMF to cyclohexane for 2 (Tables S10 and S11†). Keeping in mind that R is a function of the magnitudes of the underlying electric d and magnetic m transition dipole moments and the angle θ between their vectors, this enhancement can be traced back to an increase in the value of m and a more beneficial orientation factor observed in 2 for cyclohexane vs. more polar solvents (Table S15 and Fig. S33†). This solvent-dependent modulation of m and θ along with the chiral exciton coupling between individual helicene → naphthalimide ICT transitions established in the low-energy region of the UV-vis and ECD spectra for 2 (with S0 → S1 and S0 → S2 constituting the exciton couplet) indicate that its unprecedented for organic CPL emitters behaviour can also be linked to the corresponding coupling in the excited state with the resulting lower-energy couplet's component (emitting S1 state) undergoing localization on one naphthalimide branch due to nuclear relaxation. This seems to be further supported by overall strong enhancement of the CPL intensity for 2vs.1 visible in the experiment following similar trends in the UV-vis and ECD signals. In the presence of an intense reaction field imposed by the polar solvents the electrostatic interaction between electric transition dipoles (each connected with either one of the electron acceptor unit) is suppressed,17i affecting magnetic transition dipoles and their relative orientations, and, as a consequence, a decrease in the CPL intensity is observed.
Finally, it is thus worth emphasizing that 2 exhibits in cyclohexane one of the highest glum values reported to date for (small) organic molecules in solution,1f,4c,19 which highlights the benefits of chiral exciton coupling strategy to enhance the chiroptical properties of helicene-based dyes.
Conclusions
We have synthesized chiral push–pull A–π–D and A–π–D–π–A organic CPL emitters based on the functionalization of enantiopure [6]helicene with naphthalimide chromophores. Unpolarized and polarized characterizations revealed much more intense ECD signal (Δε = +456 M−1 cm−1 at 430 nm) and CPL activity with glum up to 10−2 for the bis-substituted P-2 than for its mono-substituted analogue P-1 (Δε = +179 M−1 cm−1 at 430 nm, glum < 3 × 10−3), arising from an intramolecular chiral exciton coupling effect. Being highly luminescent (ϕfluo up to 0.85), these chiral emitters display also significant solvatochromism, which strongly impacts the intensity of polarized emission for 2, with a pronounced decrease of glum when going from non-polar cyclohexane (glum = 10−2) to polar dimethylformamide solvent (glum = 3 × 10−3), despite the overall similar fluorescent behaviour of both 1 and 2. This was associated with a symmetry breaking of the emitting S1 state in 2 adopting fundamentally identical nature as in 1 and suppression of exciton coupling between individual helicene → naphthalimide ICT transitions upon increasing the solvent polarity. These unprecedented results highlight the potential of CPL spectroscopy to investigate and characterize electronic features of luminescent states in organic chiral π-conjugated systems and to provide a deeper understanding of the underlying photophysical effects that may open new directions for designing novel efficient CPL emitters.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the Ministère de l'Education Nationale, de la Recherche et de la Technologie, the Centre National de la Recherche Scientifique (CNRS). K. D. is grateful for financial support from the University of Gabès, the University of Rennes 1, and Campus France. J. A. thanks the National Science Foundation (CHE-1855470) for financial support and the Center for Computational Research (CCR) at the University at Buffalo for computational resources. C. C. thanks the region pays de la loire for the “SAMOA” project. The PRISM core facility (Biogenouest©, UMS Biosit, Université de Rennes 1 – Campus de Villejean – 35043 RENNES Cedex, FRANCE) is acknowledged for the NMR characterizations and ECD measurements. Dr Y. Molard (ISCR, Rennes) is thanked for luminescence quantum yield measurements and Dr P.-A. Bouit (ISCR, Rennes) for fruitful discussions. Prof. L. Di Bari and Dr M. Górecki are warmly thanked for their assistance in CPL measurements.References
- (a) F. S. Richardson and J. P. Riehl, Chem. Rev., 1977, 77, 773–792 CrossRef CAS; (b) J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1–16 CrossRef CAS; (c) Chirality at the Nanoscale, Nanoparticles, Surfaces, Materials and more, ed. D. Amabilino, Wiley-VCH, 2009 Search PubMed; (d) J. Kumar, T. Nakashima and T. Kawai, J. Phys. Chem. Lett., 2015, 6, 3445–3452 CrossRef CAS PubMed; (e) B. Kunnen, C. Macdonald, A. Doronin, S. Jacques, M. Eccles and I. Meglinski, J. Biophotonics, 2015, 8, 317–323 CrossRef PubMed; (f) E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem.–Eur. J., 2015, 21, 13488–13500 CrossRef PubMed; (g) J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef.
- (a) F. Zinna and L. Di Bari, Chirality, 2015, 27, 1–13 CrossRef CAS PubMed; (b) S. Shuvaev, E. A. Suturina, K. Mason and D. Parker, Chem. Sci., 2018, 9, 2996–3003 RSC; (c) S. Shuvaev, M. A. Fox and D. Parker, Angew. Chem., Int. Ed., 2018, 57, 7488–7492 CrossRef CAS PubMed; (d) F. Zinna, M. Pasini, F. Galeotti, C. Botta, L. Di Bari and U. Giovanella, Adv. Funct. Mater., 2017, 27, 1603719 CrossRef.
- (a) M. Li, S. H. Li, D. Zhang, M. Cai, L. Duan, M. K. Fung and C. F. Chen, Angew. Chem., Int. Ed. Engl., 2018, 57, 2889–2893 CrossRef CAS PubMed; (b) J. Han, S. Guo, J. Wang, L. Wei, Y. Zhuang, S. Liu, Q. Zhao, X. Zhang and W. Huang, Adv. Opt. Mater., 2017, 5, 1700359 CrossRef; (c) T.-Y. Li, Y.-M. Jing, X. Liu, Y. Zhao, L. Shi, Z. Tang, Y.-X. Zheng and J.-L. Zuo, Sci. Rep., 2015, 5, 14912 CrossRef CAS PubMed; (d) J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS PubMed; (e) S. Feuillastre, M. Pauton, L. Gao, A. Desmarchelier, A. J. Riives, D. Prim, D. Tondelier, B. Geffroy, G. Muller, G. Clavier and G. Pieters, J. Am. Chem. Soc., 2016, 138, 3990–3993 CrossRef CAS PubMed; (f) M. Schulz, M. Mack, O. Kolloge, A. Lutzen and M. Schiek, Phys. Chem. Chem. Phys., 2017, 19, 6996–7008 RSC; (g) J. Gilot, R. Abbel, G. Lakhwani, E. W. Meijer, A. P. H. J. Schenning and S. C. J. Meskers, Adv. Mater., 2010, 22, E131–E134 CrossRef CAS PubMed; (h) X. Shang, I. Song, H. Ohtsu, Y. H. Lee, T. Zhao, T. Kojima, J. H. Jung, M. Kawano and J. H. Oh, Adv. Mater., 2017, 29, 1605828 CrossRef; (i) Y. Yang, B. Rice, X. Shi, J. R. Brandt, R. Correa da Costa, G. J. Hedley, D. M. Smilgies, J. M. Frost, I. D. W. Samuel, A. Otero-de-la-Roza, E. R. Johnson, K. E. Jelfs, J. Nelson, A. J. Campbell and M. J. Fuchter, ACS Nano, 2017, 11, 8329–8338 CrossRef CAS PubMed; (j) Y. Yang, R. C. da Costa, M. J. Fuchter and A. J. Campbell, Nat. Photonics, 2013, 7, 634–638 CrossRef CAS; (k) F. Song, Z. Xu, Q. Zhang, Z. Zhao, H. Zhang, W. Zhao, Z. Qiu, C. Qi, H. Zhang, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam, Z. Zhao, A. Qin, D. Ma and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1800051 CrossRef; (l) P. Josse, L. Favereau, C. Shen, S. Dabos-Seignon, P. Blanchard, C. Cabanetos and J. Crassous, Chem.–Eur. J., 2017, 23, 6277–6281 CrossRef CAS PubMed; (m) N. Y. Kim, J. Kyhm, H. Han, S. J. Kim, J. Ahn, D. K. Hwang, H. W. Jang, B.-K. Ju and J. A. Lim, Adv. Funct. Mater., 2019, 29, 1808668 CrossRef; (n) M. Schulz, F. Balzer, D. Scheunemann, O. Arteaga, A. Lützen, S. C. J. Meskers and M. Schiek, Adv. Funct. Mater., 2019, 29, 1900684 CrossRef; (o) F. Zinna, U. Giovanella and L. D. Bari, Adv. Mater., 2015, 27, 1791–1795 CrossRef CAS PubMed.
- (a) H. Maeda and Y. Bando, Pure Appl. Chem., 2013, 85, 1967 CAS; (b) N. Chen and B. Yan, Molecules, 2018, 23, 3376 CrossRef PubMed; (c) H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
- H. Tanaka, M. Ikenosako, Y. Kato, M. Fujiki, Y. Inoue and T. Mori, Commun. Chem., 2018, 1, 38 CrossRef.
- (a) J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 CrossRef; (b) B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley-VCH, Weinheim, 2001 CrossRef.
- (a) P. Suppan and N. Ghoneim, Solvatochromism, The Royal Society of Chemistry, Cambridge, 1997 Search PubMed; (b) C. Reichardt, Solvent and solvent effects in organic chemistry, Wiley-VCH, Weinheim, 3rd edn, 2002 CrossRef; (c) A. Marini, A. Muñoz-Losa, A. Biancardi and B. Mennucci, J. Phys. Chem. B, 2010, 114, 17128–17135 CrossRef CAS PubMed.
- (a) Rare examples where CPL was recorded in different solvents without intense solvatochromism effect on intensity of polarization emission effects; (b) H. Nishimura, K. Tanaka, Y. Morisaki, Y. Chujo, A. Wakamiya and Y. Murata, J. Org. Chem., 2017, 82, 5242–5249 CrossRef CAS PubMed; (c) S. Nakanishi, N. Hara, N. Kuroda, N. Tajima, M. Fujiki and Y. Imai, Org. Biomol. Chem., 2018, 16, 1093–1100 RSC.
- (a) P. Gopikrishna, N. Meher and P. K. Iyer, ACS Appl. Mater. Interfaces, 2018, 10, 12081–12111 CrossRef CAS PubMed; (b) Y. Yoshida, Y. Nakamura, H. Kishida, H. Hayama, Y. Nakano, H. Yamochi and G. Saito, CrystEngComm, 2017, 19, 3626–3632 RSC; (c) T. Kogiso, K. Yamamoto, H. Suemune and K. Usui, Org. Biomol. Chem., 2012, 10, 2934–2936 RSC; (d) Y. S. C.-F. Chen, Helicenes Chemistry: From Synthesis to Applications, Springer, Berlin, 2017 CrossRef.
- K. Dhbaibi, L. Favereau, M. Srebro-Hooper, M. Jean, N. Vanthuyne, F. Zinna, B. Jamoussi, L. Di Bari, J. Autschbach and J. Crassous, Chem. Sci., 2018, 9, 735–742 RSC.
- E. Anger, M. Srebro, N. Vanthuyne, L. Toupet, S. Rigaut, C. Roussel, J. Autschbach, J. Crassous and R. Réau, J. Am. Chem. Soc., 2012, 134, 15628–15631 CrossRef CAS PubMed.
- (a) J. Autschbach, Chirality, 2009, 21, E116–E152 CrossRef CAS PubMed; (b) M. Srebro-Hooper and J. Autschbach, Annu. Rev. Phys. Chem., 2017, 68, 399–420 CrossRef CAS PubMed.
- R. Bouvier, R. Durand, L. Favereau, M. Srebro-Hooper, V. Dorcet, T. Roisnel, N. Vanthuyne, Y. Vesga, J. Donnelly, F. Hernandez, J. Autschbach, Y. Trolez and J. Crassous, Chem.–Eur. J., 2018, 24, 14484–14494 CrossRef CAS PubMed.
- S. M. McAfee, J. R. Cann, P. Josse, P. Blanchard, C. Cabanetos and G. C. Welch, ACS Sustainable Chem. Eng., 2016, 4, 3504–3517 CrossRef CAS.
- (a) Y. Nakai, T. Mori and Y. Inoue, J. Phys. Chem. A, 2012, 116, 7372–7385 CrossRef CAS PubMed; (b) Y. Nakai, T. Mori and Y. Inoue, J. Phys. Chem. A, 2013, 117, 83–93 CrossRef CAS PubMed.
- J. Zhang, C. Zhao, H. Liu, Y. Lv, R. Liu, S. Zhang, H. Chen, G. Zhang and Z. Tian, J. Phys. Chem. C, 2015, 119, 2761–2769 CAS.
- (a) C. Katan, F. Terenziani, O. Mongin, M. H. V. Werts, L. Porrès, T. Pons, J. Mertz, S. Tretiak and M. Blanchard-Desce, J. Phys. Chem. A, 2005, 109, 3024–3037 CrossRef CAS PubMed; (b) H. Y. Woo, B. Liu, B. Kohler, D. Korystov, A. Mikhailovsky and G. C. Bazan, J. Am. Chem. Soc., 2005, 127, 14721–14729 CrossRef CAS PubMed; (c) U. Megerle, F. Selmaier, C. Lambert, E. Riedle and S. Lochbrunner, Phys. Chem. Chem. Phys., 2008, 10, 6245–6251 RSC; (d) B. Dereka, A. Rosspeintner, M. Krzeszewski, D. T. Gryko and E. Vauthey, Angew. Chem., Int. Ed., 2016, 55, 15624–15628 CrossRef CAS PubMed; (e) B. Dereka, A. Rosspeintner, Z. Li, R. Liska and E. Vauthey, J. Am. Chem. Soc., 2016, 138, 4643–4649 CrossRef CAS PubMed; (f) B. Dereka, M. Koch and E. Vauthey, Acc. Chem. Res., 2017, 50, 426–434 CrossRef CAS PubMed; (g) B. Dereka, A. Rosspeintner, R. Stężycki, C. Ruckebusch, D. T. Gryko and E. Vauthey, J. Phys. Chem. Lett., 2017, 6029–6034 CrossRef CAS PubMed; (h) B. Dereka and E. Vauthey, J. Phys. Chem. Lett., 2017, 8, 3927–3932 CrossRef CAS PubMed; (i) Y. Li, M. Zhou, Y. Niu, Q. Guo and A. Xia, J. Chem. Phys., 2015, 143, 034309 CrossRef PubMed.
- A. Aster, G. Licari, F. Zinna, E. Brun, T. Kumpulainen, E. Tajkhorshid, J. Lacour and E. Vauthey, Chem. Sci., 2019, 10, 10629–10639 RSC.
- (a) Y. Sawada, S. Furumi, A. Takai, M. Takeuchi, K. Noguchi and K. Tanaka, J. Am. Chem. Soc., 2012, 134, 4080–4083 CrossRef CAS PubMed; (b) P. Reine, A. G. Campaña, L. Alvarez de Cienfuegos, V. Blanco, S. Abbate, A. J. Mota, G. Longhi, D. Miguel and J. M. Cuerva, Chem. Commun., 2019, 55, 10685–10688 RSC; (c) C. Schaack, L. Arrico, E. Sidler, M. Gorecki, L. Di Bari and F. Diederich, Chem.–Eur. J., 2019, 25, 8003–8007 CrossRef CAS PubMed; (d) K. Nakamura, S. Furumi, M. Takeuchi, T. Shibuya and K. Tanaka, J. Am. Chem. Soc., 2014, 136, 5555–5558 CrossRef CAS PubMed; (e) Y. Hashimoto, T. Nakashima, D. Shimizu and T. Kawai, Chem. Commun., 2016, 52, 5171–5174 RSC; (f) S. Ito, K. Ikeda, S. Nakanishi, Y. Imai and M. Asami, Chem. Commun., 2017, 53, 6323–6326 RSC; (g) A. Homberg, E. Brun, F. Zinna, S. Pascal, M. Gorecki, L. Monnier, C. Besnard, G. Pescitelli, L. Di Bari and J. Lacour, Chem. Sci., 2018, 9, 7043–7052 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1894823. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05231c |
| This journal is © The Royal Society of Chemistry 2020 |