
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formyl-selective deuteration of aldehydes with D2O via synergistic organic and photoredox catalysis†
Jianyang
Dong
,
Xiaochen
Wang
,
Zhen
Wang
,
Hongjian
Song
,
Yuxiu
Liu
 and
Qingmin
Wang
*
and
Qingmin
Wang
*
State Key Laboratory of Elemento-Organic Chemistry, Research Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, People's Republic of China. E-mail: wangqm@nankai.edu.cn
First published on 4th December 2019
Abstract
Formyl-selective deuteration of aldehydes is of high interest for labeling purposes and for optimizing properties of drug candidates. Herein, we report a mild general method for formyl-selective deuterium labeling of aldehydes with D2O, an inexpensive deuterium source, via a synergistic combination of light-driven, polyoxometalate-facilitated hydrogen atom transfer and thiol catalysis. This highly efficient, scalable reaction showed excellent deuterium incorporation, a broad substrate scope, and excellent functional group tolerance and selectivity and is therefore a practical method for late-stage modification of synthetic intermediates in medicinal chemistry and for generating libraries of deuterated compounds.
Introduction
Deuterium labeling has a range of applications, including in the investigation of reaction mechanisms1 and the analysis of drug absorption, distribution, metabolism, and excretion,2 as well as in nuclear magnetic resonance spectroscopy3 and mass spectrometry.4 In recent years, interest in the incorporation of deuterium atoms into patented drugs and drug candidates to enhance their metabolism and pharmacokinetic properties has burgeoned.5 In 2017, the US Food and Drug Administration approved the first deuterated drug, deutetrabenazine (Austedo),6 and the increasing demand for new deuterium-labeled drugs has motivated the development of efficient deuteration methods.7Aromatic aldehydes are often used as building blocks for pharmaceutical synthesis owing to their versatile reactivity;8 indeed, they can be rapidly transformed to other compounds via C–C and C–X bond forming reactions. The development of an efficient protocol for constructing formyl-deuterated aromatic aldehydes can be expected to increase the availability of deuterated lead compounds. Aromatic aldehydes selectively labeled at the formyl position are traditionally produced from the corresponding esters by means of reduction with LiAlD4 followed by oxidation,9 from the corresponding amides by reaction with deuterated Schwartz's reagent (obtained from LiAlD4),10 from aryl halides via Pd/Rh-cocatalyzed reductive carbonylation,11 or from carboxylic acids via deoxygenative deuteration with synergistic photoredox and organic catalysis (Scheme 1A).12 In terms of atom- and step-economy, the ideal protocol for preparing deuterated aromatic aldehydes would be direct hydrogen isotope exchange (HIE) at the formyl C–H bond. In fact, a few protocols for Ir- and Ru-catalyzed HIE at the formyl moiety have been reported, although there is an intrinsic difficulty in controlling the reactivity of the aryl ring moiety and formyl moiety (Scheme 1B).13 Moreover, late-stage introduction of deuterium into structurally complex aldehydes remains a challenge. Given the importance of deuterated aldehydes, we were interested in developing a method for efficient, atom- and step-economical HIE with the goal of achieving formyl-selective labeling of aldehydes with D2O, which is an inexpensive deuterium source. In addition, we wanted the new HIE method to be useful for late-stage introduction of deuterium into structurally complex aldehydes.
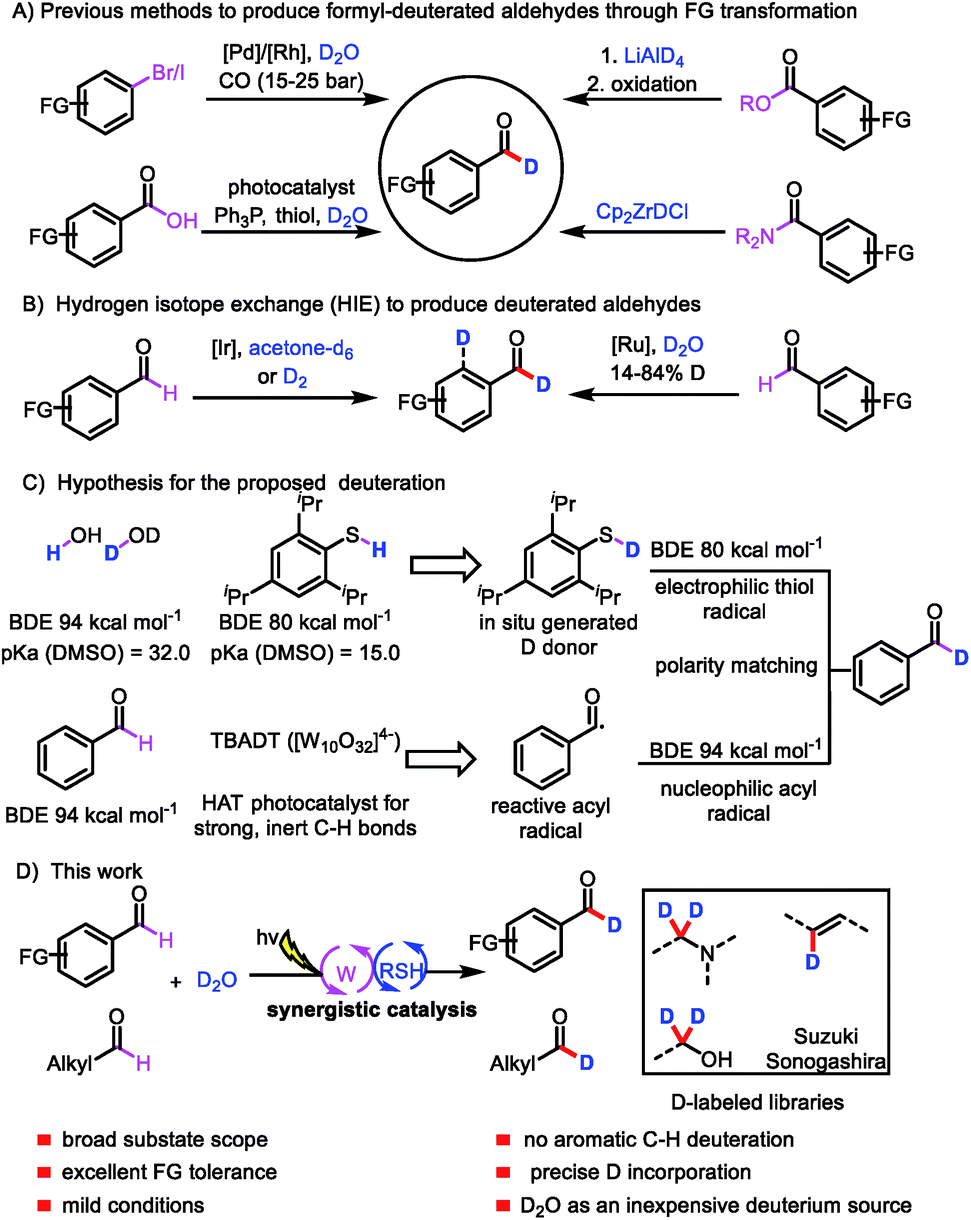 | ||
| Scheme 1 Strategies for synthesizing deuterated aldehydes; (A) previous methods to produce deuterated aldehydes through FG transformation; (B) hydrogen isotope exchange (HIE) to produce deuterated aldehydes; (C) hypothesis for the proposed deuteration; (D) our work. | ||
Visible-light-mediated photoredox catalysis has recently emerged as a method for achieving organic transformations via radical processes.14 Recently, MacMillan's group reported direct HIE at α-amino C(sp3)–H bonds via a pathway involving abstraction of a deuterium atom from a deuterium-labeled thiol catalyst by an α-amino radical.15 We hypothesized that generation of acyl radicals from aldehydes by a hydrogen atom transfer (HAT) photocatalyst in the presence of D2O and a thiol catalyst would produce deuterated aldehydes.
We envisioned that polyoxometalates, many of which possess high-energy excited states that can accomplish the desired C–H abstraction,16 would be ideal HAT catalysts for the proposed transformation. We were particularly interested in the decatungstate anion ([W10O32]4−), an efficient HAT photocatalyst that has been widely used for oxygenation, dehydrogenation, conjugate addition, and, more recently, fluorination and arylation of strong, unactivated aliphatic C–H bonds, which have bond dissociation energies (BDEs) of up to 100 kcal mol−1 (for cyclohexane).17 To our knowledge, visible-light polyoxometalate-facilitated HAT has not previously been synergistically merged with thiol catalysis. Because aldehydes have relatively low BDEs (94 kcal mol−1),12 we envisioned that such a combination of catalytic processes would afford access to a considerable variety of acyl radicals and deuterated aldehydes from abundant aldehyde feedstocks (Scheme 1C). Moreover, owing to the gaps between the BDEs of C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)H bonds (94 kcal mol−1), aryl C–H bonds (113 kcal mol−1), and S–H bonds (80–88 kcal mol−1), the decatungstate anion ([W10O32]4−) would be unable to abstract a hydrogen atom from the aryl C–H bond,17g allowing us to achieve formyl labeling without the formation of aryl-labeled by-products (Scheme 1C). In contrast, the nucleophilic acyl radical would readily undergo HAT from the thiol catalyst to form a deuterated aldehyde, owing to the gap between the BDEs of the C–H and S–H bonds and a polarity matching effect (Scheme 1C).18 We recognized that the choice of a suitable thiol catalyst would be heavily influenced by thermodynamic factors, particularly the BDE of the thiol bond relative to that of the acyl C–H bond, as well as its pKa relative to that of water. Herein, we describe a general strategy for formyl-selective deuteration of aldehydes with D2O mediated by the synergistic combination of light-driven, polyoxometalate-facilitated HAT and thiol catalysis (Scheme 1D).
O)H bonds (94 kcal mol−1), aryl C–H bonds (113 kcal mol−1), and S–H bonds (80–88 kcal mol−1), the decatungstate anion ([W10O32]4−) would be unable to abstract a hydrogen atom from the aryl C–H bond,17g allowing us to achieve formyl labeling without the formation of aryl-labeled by-products (Scheme 1C). In contrast, the nucleophilic acyl radical would readily undergo HAT from the thiol catalyst to form a deuterated aldehyde, owing to the gap between the BDEs of the C–H and S–H bonds and a polarity matching effect (Scheme 1C).18 We recognized that the choice of a suitable thiol catalyst would be heavily influenced by thermodynamic factors, particularly the BDE of the thiol bond relative to that of the acyl C–H bond, as well as its pKa relative to that of water. Herein, we describe a general strategy for formyl-selective deuteration of aldehydes with D2O mediated by the synergistic combination of light-driven, polyoxometalate-facilitated HAT and thiol catalysis (Scheme 1D).
Results and discussion
The proposed mechanism for formyl-selective deuteration of aldehydes is shown in Scheme 2. In this mechanism photoexcitation of tetrabutylammonium decatungstate (TBADT, 2) followed by intersystem crossing produces triplet excited state 3, which has a lifetime of 55 ns.19 Subsequent selective abstraction of the formyl H atom of 2-naphthaldehyde (1a) affords singly reduced decatungstate 4 and reactive nucleophilic formyl radical 5. At the same time, thiol HAT catalyst 7 undergoes exchange with D2O to give deuterated thiol 8, which serves as the source of deuterium. Because of the gap between the reported BDEs for typical formyl C–H and thiol S–H bonds, we reasoned that HAT between polarity-matched radical 5 and deuterated thiol 8 would furnish deuterated aldehyde 10a and electrophilic thiol radical 9 [C(O)–H BDE = 94 kcal mol−1; S–H BDE = 80 kcal mol−1 for 7]. The two catalytic cycles converge when reduced decatungstate 4 undergoes a single-electron transfer reaction with thiol radical 9 to regenerate active HAT photocatalyst 2 and thiol HAT catalyst 7.
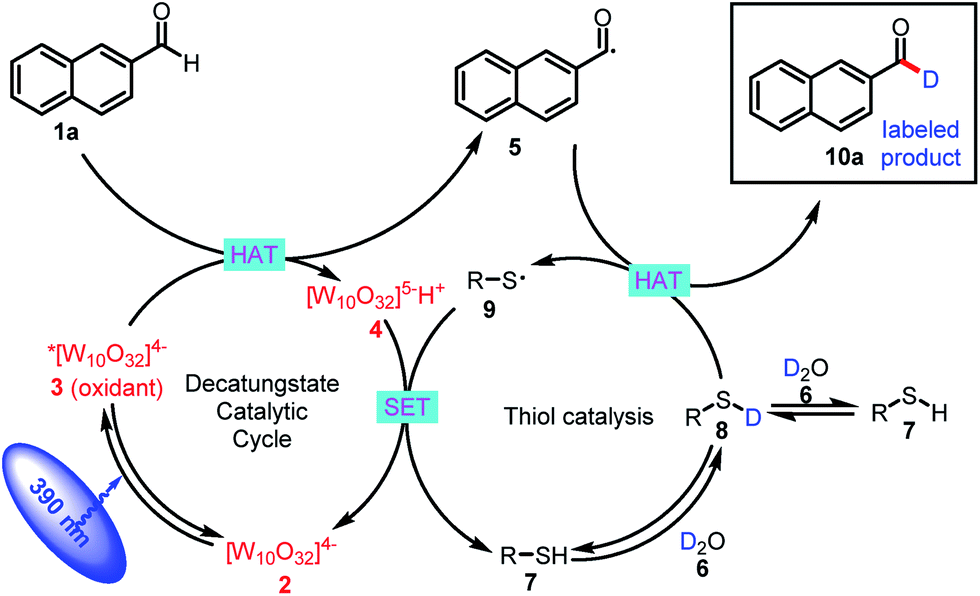 | ||
| Scheme 2 Proposed mechanism. | ||
To test our mechanistic hypothesis, we began by exploring deuteration reactions of 2-naphthaldehyde (1a) under various conditions (Table 1). First, several thiol catalysts were screened in the presence of 4 mol% of commercially available TBADT as the HAT photocatalyst in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) dichloromethane/D2O under irradiation with near-ultraviolet light (36 W 390 nm light-emitting diodes; see the ESI†). To our delight, 2-naphthaldehyde (10a) with 94% deuterium incorporation was obtained when 2,4,6-triisopropylbenzenethiol (7a, 40 mol%) was used as the thiol catalyst (entry 1). Under the same conditions, other thiols gave considerably lower deuterium incorporation percentages (entries 2–4). The use of N-methyl-2-pyrrolidinone, acetonitrile, or chloroform as the cosolvent also led to decreased deuterium incorporation (entries 5–7). Reducing the amount of TBADT or thiol 7a slightly decreased the deuterium incorporation percentage (entries 8 and 9). Control experiments showed that the reaction failed to proceed in the absence of the HAT photocatalyst, the thiol catalyst, or light (entries 10–12).
:1 (v/v) dichloromethane/D2O under irradiation with near-ultraviolet light (36 W 390 nm light-emitting diodes; see the ESI†). To our delight, 2-naphthaldehyde (10a) with 94% deuterium incorporation was obtained when 2,4,6-triisopropylbenzenethiol (7a, 40 mol%) was used as the thiol catalyst (entry 1). Under the same conditions, other thiols gave considerably lower deuterium incorporation percentages (entries 2–4). The use of N-methyl-2-pyrrolidinone, acetonitrile, or chloroform as the cosolvent also led to decreased deuterium incorporation (entries 5–7). Reducing the amount of TBADT or thiol 7a slightly decreased the deuterium incorporation percentage (entries 8 and 9). Control experiments showed that the reaction failed to proceed in the absence of the HAT photocatalyst, the thiol catalyst, or light (entries 10–12).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Deuterationb (%) |
|
a Reaction conditions, unless otherwise noted: 1a (0.3 mmol), TBADT (0.012 mmol), 7a (0.12 mmol), and 1:1 (v/v) DCM/D2O (3.0 mL) under an Ar atmosphere.
b Deuterium incorporation was determined by integration of the residual formyl proton in the 1H NMR spectrum. DCM = dichloromethane and NMP = N-methyl-2-pyrrolidinone.
|
||
| 1 | None | 94 |
| 2 | 7b instead of 7a | 63 |
| 3 | 7c instead of 7a | 71 |
| 4 | 7d instead of 7a | 78 |
| 5 | NMP instead of DCM | 13 |
| 6 | MeCN instead of DCM | 38 |
| 7 | CHCl3 instead of DCM | 62 |
| 8 | 2 mol% of TBADT | 77 |
| 9 | 20 mol% of 7a | 80 |
| 10 | No TBADT | <5 |
| 11 | No 7a | <5 |
| 12 | No light | <5 |
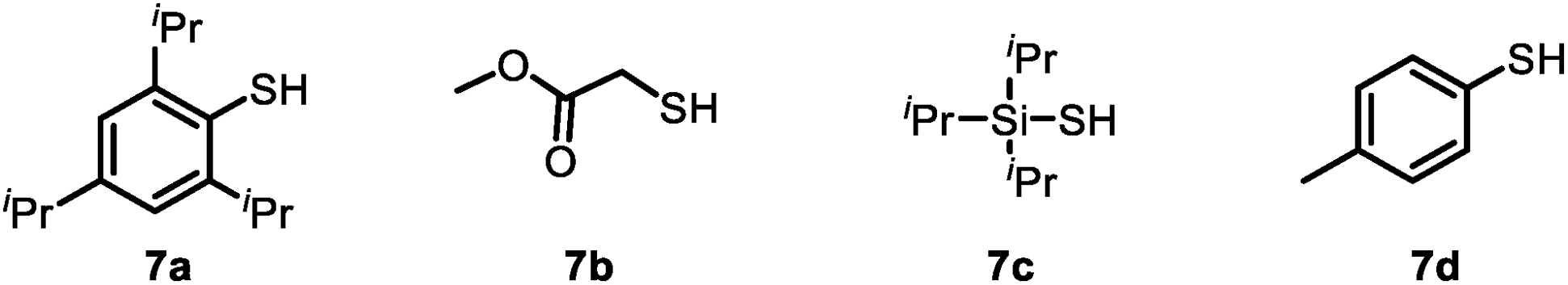
|
||
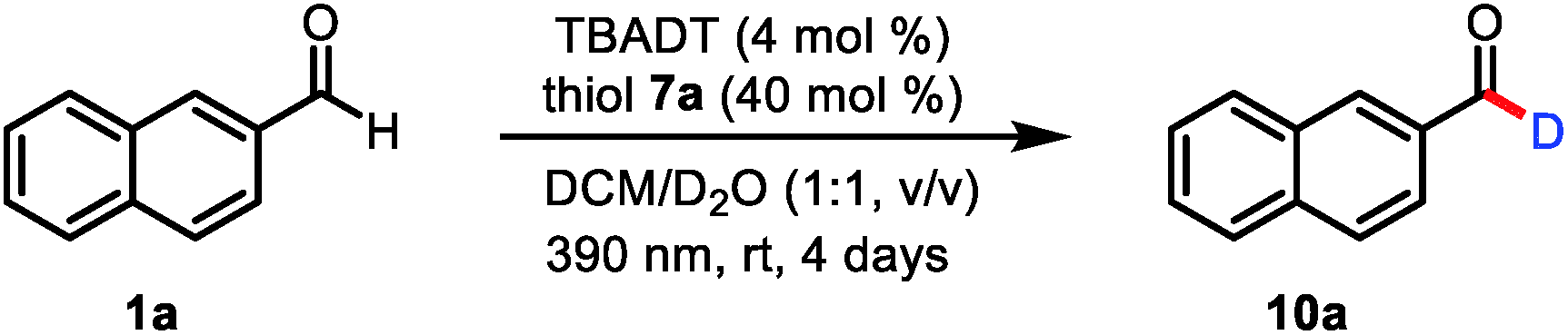
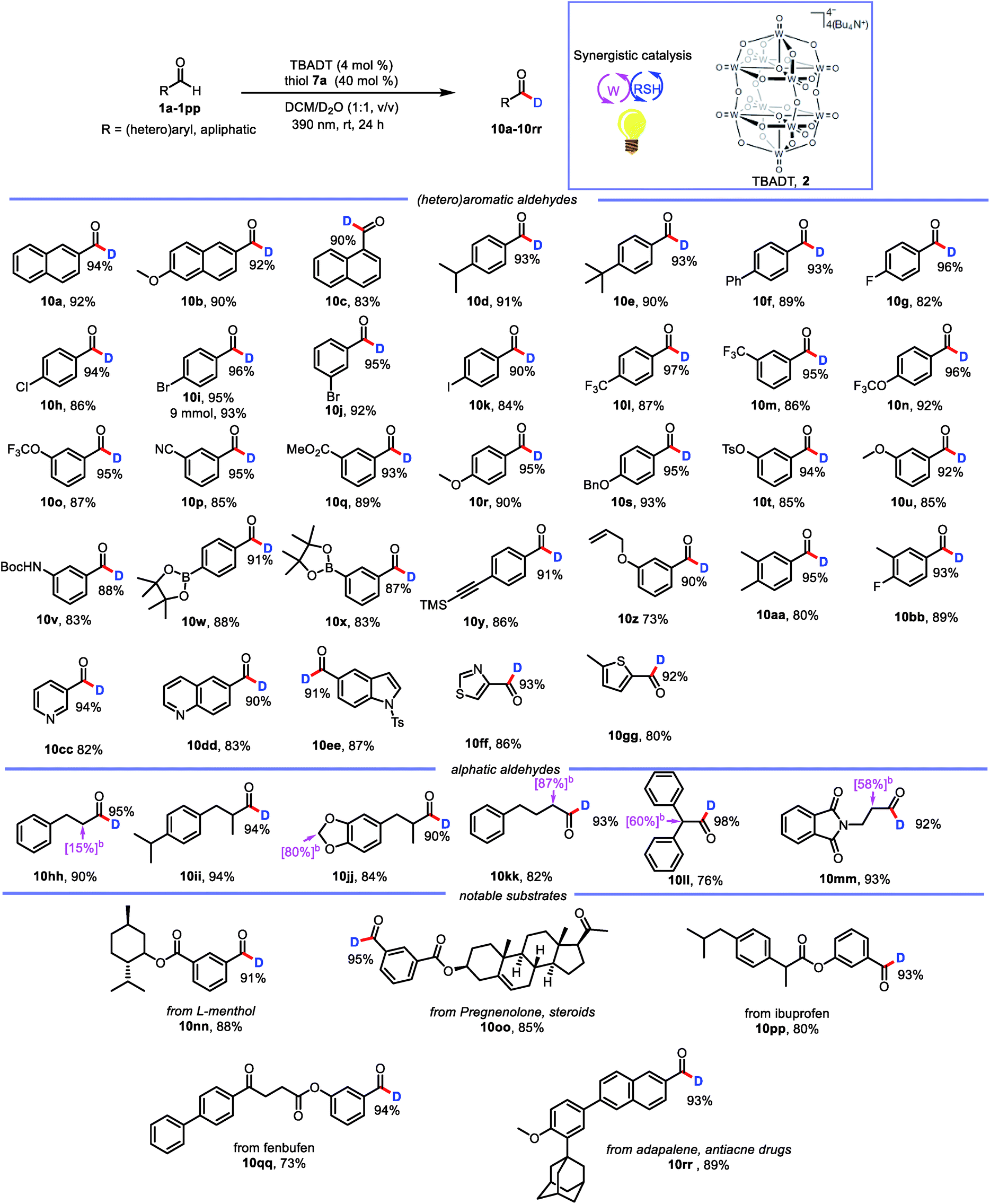
With the optimized conditions in hand, we elucidated the scope of the transformation with respect to the aldehyde. Deuterated 2-naphthaldehyde 10a was isolated in 92% yield after purification by column chromatography, indicating that decomposition and side-product formation were minimal. 6-Methoxy-2-naphthaldehyde was also a suitable substrate, giving 10b in 90% yield with 92% deuterium incorporation. 1-Naphthaldehyde gave a product (10c) with lower deuterium incorporation (90%) than 10a, presumably because the 1-position is more sterically hindered than the 2-position. We also tested benzaldehydes bearing substituents with various electronic and steric properties to obtain deuterated products 10d–10bb. Specifically, benzaldehydes with electronically neutral functional groups, such as isopropyl, t-butyl, and phenyl, worked well under the optimal conditions, giving high yields with excellent deuterium incorporation (10d–10f, 93%). Halogenated molecules account for approximately 50% of the market-leading drugs because they are less susceptible to oxidation by cytochrome P450.20 Furthermore, they offer a valuable platform for generating molecular complexity through cross-coupling reactions.21 Therefore, we were encouraged to find that benzaldehydes bearing fluoro, chloro, bromo, and iodo atoms gave the corresponding products (10g–10k) with high deuterium incorporation (90–96%). Trifluoromethyl and trifluoromethoxy groups, which have often been shown to improve the activity of pharmaceutical leads, were compatible with our protocol, showing good deuterium incorporation (10l–10o, 95–97%) regardless of their position on the benzene ring. Benzaldehydes with other electron-withdrawing groups (ester and cyano) were smoothly transformed into the corresponding deuterated aldehydes (10p and 10q). Moreover, electron-rich aldehydes were also reactive (10r–10v, 88–95% deuterium incorporation). Intriguingly, several relatively sensitive yet versatile functional groups—boronic esters (10w and 10x), an alkyne (10y), and an alkene (10z)—tolerated the deuteration conditions well, which shows the potential utility of this protocol for synthetic and medicinal chemistry applications. Polysubstituted aldehydes were also suitable substrates (10aa and 10bb). Being aware of the important role of heteroaromatic moieties in the scaffolds of pharmaceutical compounds, we were pleased to find that N- and S-heterocycles were also tolerated under our deuteration conditions (10cc–10gg). In addition, the reaction was amenable to scale up; when it was carried out on a 9 mmol scale, 10i was isolated in 93% yield with no decrease in deuterium incorporation.
In addition to aromatic aldehydes, aliphatic aldehydes are also interesting because they constitute an even greater part of the –CHO family. However, few radical-based tools for convenient modification of aliphatic aldehydes have been developed. This is particularly true for branched aldehydes, which tend to undergo decarbonylation under radical conditions.22 However, when we subjected linear and branched aldehydes to our protocol, we found that all of them underwent formyl deuteration to afford the desired products (10hh–10mm, 90–98% deuterium incorporation), and no products of CO dissociation were observed. However, a limitation of this reaction was also uncovered. That is, deuteration also takes place at the α-positions of formyl.
This excellent functional group tolerance suggested that the protocol would be useful for the synthesis of structurally complex deuterated aldehydes, and indeed we were pleased to find that deuterated aldehydes derived from menthol, pregnenolone, ibuprofen, and fenbufen derivatives (10nn–10qq) could be accessed with uncompromised reactivities. In addition, the reaction of adapalene, an antiacne drug, afforded corresponding deuterated aldehyde 10rr with high deuterium incorporation. These examples confirmed the potential utility of our protocol for practical late-stage modification of synthetic intermediates in medicinal chemistry applications (Table 2).
| a Reactions were performed on a 0.3 mmol scale, unless otherwise noted. b Some sites in compounds 10hh, 10jj, 10kk, 10ll and 10mm (shown in their structures, with the deuteration percentage in square brackets) are deuterated. Isolated yields are given. See the ESI for experimental details. Deuterium incorporation was determined by integration of the residual formyl proton in the 1H NMR spectra. |
|---|
|
Because aldehydes are versatile functional groups that undergo a wide variety of organic transformations, the protocol reported herein can be used to access libraries of deuterated compounds (Scheme 3A). For example, deuterium-labeled aldehyde 10i readily underwent reduction, reductive amination, and Horner–Wadsworth–Emmons olefination to deliver deuterated alcohol 11, deuterated amine 12,7a and β-deuterated, α,β-unsaturated ester 13. Importantly, 13 has not been accessed by direct labeling of the corresponding cinnamate ester.23 Deuterium could also be incorporated into pharmaceutical molecules and advanced materials, as indicated by the successful Suzuki coupling reaction of 10i to afford 14 and the Sonogashira coupling reaction of 10i to provide 15, which is difficult to obtain by currently available methods.
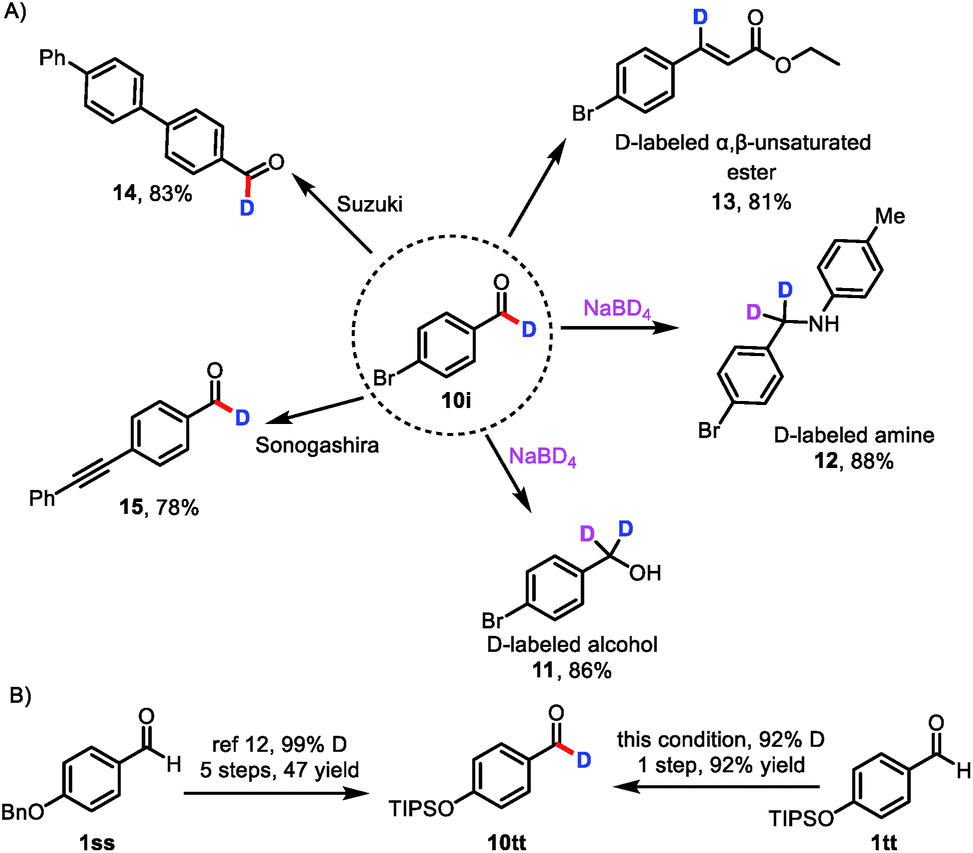 | ||
| Scheme 3 Transformations of deuterated aldehyde 10i and improved synthetic route to deuterium-labeled aldehyde 10tt; (A) 10i is used to access libraries of deuterated compounds; (B) improved synthetic route to deuterium-labeled aldehyde 10tt. | ||
In a further demonstration of the utility of this catalytic formyl-deuteration method, we used it to synthesize silyl-protected phenol 10tt, which is an intermediate in the synthesis of enantiopure d1-benzyl alcohols, in a single, high-yielding step and with a high deuterium incorporation from aldehyde 1tt (Scheme 3B).24 This method is superior to the existing route to 10tt, which employs stoichiometric quantities of reagents and requires five steps from aldehyde 1ss.24
Having explored the substrate scope and utility of the reaction, we conducted mechanistic studies to support the proposed pathway shown in Scheme 2. When radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) was present in the reaction mixture, deuterium incorporation was markedly decreased (to 10%), and the radical-trapping product 2,2,6,6-tetramethylpiperidin-1-yl-2-naphthoate (16) was isolated in 12% yield (Scheme 4A). When benzyl acrylate (17) was used as a radical scavenger, benzyl 4-(naphthalen-2-yl)-4-oxobutanoate-2-d (18) was isolated (Scheme 4B). These results suggest that acyl radical 5 was generated. When we used 4-pyridinecarboxaldehyde (1uu) as the substrate, we isolated by-product 19 (84% yield), which was generated by a radical coupling reaction of acyl radical 20 and thiol radical 21 (Scheme 4C). These experiments clearly point to a radical pathway. Furthermore, the light on/off experiments illustrated a total interruption of the reaction process in the absence of light and recuperation of reactivity on further illumination (Fig. S3†). This indicates that light was essential for this transformation, and any chain propagation process should be short-lived.
 | ||
| Scheme 4 Mechanistic studies in support of the proposed pathway; (A) TEMPO was used as radical scavenger; (B) benzyl acrylate was used as radical scavenger; (C) 4-pyridinecarboxaldehyde was used as the substrate. | ||
Conclusions
In conclusion, we have developed a mild protocol for selective direct HIE at the formyl C–H bonds of both aromatic and aliphatic aldehydes with D2O as an inexpensive deuterium source mediated by synergistic visible-light polyoxometalate-facilitated HAT catalysis and thiol catalysis. The high efficiency, broad substrate scope, excellent functional group tolerance, and mild conditions make this protocol practical for late-stage modification of pharmaceutical intermediates and for obtaining libraries of deuterated compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (21732002 and 21672117) and the PhD Candidate Research Innovation Fund of the College of Chemistry, Nankai University for generous financial support for our research.Notes and references
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 ( Angew. Chem. , 2012 , 124 , 3120 ) CrossRef CAS.
- (a) J. Helfenbein, C. Lartigue, E. Noirault, E. Azim, J. Legailliard, M. J. Galmier and J. C. Madelmont, J. Med. Chem., 2002, 45, 5806 CrossRef CAS; (b) C. S. Elmore and R. A. Bragg, Bioorg. Med. Chem. Lett., 2015, 25, 167 CrossRef CAS; (c) V. Derdau, J. Atzrodt, J. Zimmermann, C. Kroll and F. Brückner, Chemistry, 2009, 15, 10397 CrossRef CAS.
- S. E. Scheppele, Chem. Rev., 1972, 72, 511 CrossRef.
- (a) L. S. Busenlehner and R. S. Armstrong, Arch. Biochem. Biophys., 2005, 433, 34 CrossRef CAS; (b) J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744 ( Angew. Chem. , 2007 , 119 , 7890 ) CrossRef CAS.
- (a) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758 ( Angew. Chem. , 2018 , 130 , 1774 ) CrossRef CAS; (b) Y. Zhu, J. Zhou and B. Jiao, ACS Med. Chem. Lett., 2013, 4, 349 CrossRef CAS; (c) A. Katsnelson, Nat. Med., 2013, 19, 656 CrossRef CAS; (d) A. Mullard, Nat. Rev. Drug Discovery, 2016, 15, 219 CrossRef CAS PubMed.
- C. Schmidt, Nat. Biotechnol., 2017, 35, 493 CrossRef CAS PubMed.
- For selected examples of deuteration and tritiation, see: (a) L. V. A. Hale and N. K. Szymczak, J. Am. Chem. Soc., 2016, 138, 13489 CrossRef CAS PubMed; (b) J. L. Koniarczyk, D. Hesk, A. Overgard, I. W. Davies and A. McNally, J. Am. Chem. Soc., 2018, 140, 1990 CrossRef CAS PubMed; (c) X. Wang, M. H. Zhu, D. P. Schuman, D. Zhong, W. Y. Wang, L. Y. Wu, W. Liu, B. M. Stoltz and W.-B. Liu, J. Am. Chem. Soc., 2018, 140, 10970 CrossRef CAS PubMed; (d) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022 ( Angew. Chem. , 2018 , 130 , 3074 ) CrossRef CAS PubMed; (e) H. M. Xia, F. L. Zhang, T. Ye and Y. F. Wang, Angew. Chem., Int. Ed., 2018, 57, 11770 ( Angew. Chem. , 2018 , 130 , 11944 ) CrossRef CAS; (f) C. Liu, Z. Chen, C. Su, X. Zhao, Q. Gao, G. H. Ning, H. Zhu, W. Tang, K. Leng, W. Fu, B. Tian, X. Peng, J. Li, Q.-H. Xu, W. Zhou and K. P. Loh, Nat. Commun., 2018, 9, 80 CrossRef; (g) R. P. Yu, D. Hesk, N. Rivera, I. Pelczer and P. J. Chirik, Nature, 2016, 529, 195 CrossRef.
- L. N. Ferguson, Chem. Rev., 1946, 38, 227 CrossRef CAS.
- (a) E. P. K. Olsen, T. Singh, P. Harris, P. G. Andersson and R. Madsen, J. Am. Chem. Soc., 2015, 137, 834 CrossRef CAS; (b) P. W. Davies, N. Martin and N. Spencer, Beilstein J. Org. Chem., 2011, 7, 839 CrossRef CAS; (c) H. V. Adcock, E. Chatzopoulou and P. W. Davies, Angew. Chem., Int. Ed., 2015, 54, 15525 ( Angew. Chem. , 2015 , 127 , 15745 ) CrossRef CAS.
- J. T. Spletstoser, J. M. White, A. R. Tunoori and G. I. Georg, J. Am. Chem. Soc., 2007, 129, 3408 CrossRef CAS.
- M. Y. S. Ibrahim and S. E. Denmark, Angew. Chem., Int. Ed., 2018, 57, 10362 ( Angew. Chem. , 2018 , 130 , 10519 ) CrossRef CAS.
- M. Zhang, X.-A. Yuan, C.-J. Zhu and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 312 CrossRef CAS.
- (a) W. J. Kerr, M. Reid and T. Tuttle, Angew. Chem., Int. Ed., 2017, 56, 7808 ( Angew. Chem. , 2017 , 129 , 7916 ) CrossRef CAS; (b) E. S. Isbrandt, J. K. Vandavasi, W. Zhang, M. P. Jamshidi and S. G. Newman, Synlett, 2017, 28, 2851 CrossRef CAS.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS; (b) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS; (c) M. D. Kärkäs, J. A. Porco Jr and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683 CrossRef.
- Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 358, 1182 CrossRef CAS.
- C. L. Hill and C. M. Prosser-McCartha, Photosensitization and Photocatalysis Using Inorganic and Organometallic Compounds, ed. K. Kalyanasundaram and M. Gratzel, Springer, Dordrecht, 1993, pp. 307–330 Search PubMed.
- (a) R. F. Renneke and C. L. Hill, J. Am. Chem. Soc., 1986, 108, 3528 CrossRef CAS; (b) R. F. Renneke and C. L. Hill, Angew. Chem., Int. Ed. Engl., 1988, 27, 1526 CrossRef; (c) D. Ravelli, S. Protti and M. Fagnoni, Acc. Chem. Res., 2016, 49, 2232 CrossRef CAS; (d) D. M. Schultz, F. Levesque, D. A. DiRocco, M. Reibarkh, Y. Ji, L. A. Joyce, J. F. Dropinski, H. Sheng, B. D. Sherry and I. W. Davies, Angew. Chem., Int. Ed., 2017, 56, 15274 CrossRef CAS; (e) S. D. Halperin, D. Kwon, M. Holmes, E. L. Regalado, L.-C. Campeau, D. A. DiRocco and R. Britton, Org. Lett., 2015, 17, 5200 CrossRef CAS PubMed; (f) J. G. West, D. Huang and E. J. Sorensen, Nat. Commun., 2015, 6, 10093 CrossRef PubMed; (g) I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70 CrossRef CAS PubMed.
- C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79 CrossRef CAS PubMed.
- V. De Waele, O. Poizat, M. Fagnoni, A. Bagno and D. Ravelli, ACS Catal., 2016, 6, 7174 CrossRef.
- M. C. Ford and P. S. Ho, J. Med. Chem., 2016, 59, 1655 CrossRef CAS PubMed.
- (a) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC; (b) D. C. Blakemore, et al. , Nat. Chem., 2018, 10, 383 CrossRef CAS PubMed.
- (a) C. M. Jensen, K. B. Lindsay, R. H. Taaning, J. Karaffa, A. M. Hansen and T. Skrydstrup, J. Am. Chem. Soc., 2005, 127, 6544 CrossRef CAS; (b) S. A. Moteki, A. Usui, S. Selvakumar, T. Zhang and K. Maruoka, Angew. Chem., Int. Ed., 2014, 53, 11060 CrossRef CAS.
- For selected examples of direct labeling of α,β-unsaturated substrates, see: (a) W. J. Kerr, R. J. Mudd, L. C. Paterson and J. A. Brown, Chem.–Eur. J., 2014, 20, 14604 CrossRef CAS; (b) W. J. S. Lockley, Tetrahedron Lett., 1982, 23, 3819 CrossRef CAS.
- D. W. Barnett, M. S. Refaei and R. W. Curley Jr, J. Labelled Compd. Radiopharm., 2013, 56, 6 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05132e |
| This journal is © The Royal Society of Chemistry 2020 |