
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBrønsted acid catalysis of photosensitized cycloadditions†
Evan M.
Sherbrook‡
 a,
Hoimin
Jung‡
bc,
Dasol
Cho
bc,
My-Hyun
Baik
*bc and
Tehshik P.
Yoon
*a
a,
Hoimin
Jung‡
bc,
Dasol
Cho
bc,
My-Hyun
Baik
*bc and
Tehshik P.
Yoon
*a
aDepartment of Chemistry, University of Wisconsin–Madison, 1101 University Avenue, Madison, WI 53706, USA. E-mail: tyoon@chem.wisc.edu
bDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, South Korea. E-mail: mbaik2805@kaist.ac.kr
cCenter for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, South Korea
First published on 2nd December 2019
Abstract
Catalysis is central to contemporary synthetic chemistry. There has been a recent recognition that the rates of photochemical reactions can be profoundly impacted by the use of Lewis acid catalysts and co-catalysts. Herein, we show that Brønsted acids can also modulate the reactivity of excited-state organic reactions. Brønsted acids dramatically increase the rate of Ru(bpy)32+-sensitized [2 + 2] photocycloadditions between C-cinnamoyl imidazoles and a range of electron-rich alkene reaction partners. A combination of experimental and computational studies supports a mechanism in which the Brønsted acid co-catalyst accelerates triplet energy transfer from the excited-state [Ru*(bpy)3]2+ chromophore to the Brønsted acid activated C-cinnamoyl imidazole. Computational evidence further suggests the importance of driving force as well as geometrical reorganization, in which the protonation of the imidazole decreases the reorganization penalty during the energy transfer event.
Introduction
The influence of Lewis acids on the reactivity of excited-state organic molecules has been the subject of extensive investigation. Seminal studies by Lewis and coworkers established that photoreactions of conjugated enones can be dramatically impacted by strong oxophilic Lewis acids, resulting in both increased reaction efficiency and altered product distributions.1 Recently, several groups, including those of Bach2 and Meggers,3 have developed a number of highly enantioselective photoreactions that use Lewis acid catalysts to control the dynamics of excited-state conjugated carbonyl compounds. Much of this work has relied upon the concept of “chromophore activation”, in which the coordination of Lewis acids to carbonyl compounds results in a bathochromatic shift in their absorption spectrum.4 We recently reported an alternate approach in which Lewis acids catalyze excited-state photoreactions by increasing the rate of Dexter energy transfer between an appropriate triplet sensitizer and an enone substrate (Scheme 1A).5 This effect is a consequence either of a decrease in the energy of the enone triplet excited state (i.e., an increase in thermodynamic driving force) or as a lowering of the absolute energies of the Frontier molecular orbitals (i.e., an increase in orbital overlap).6 We have proposed that the impact of Lewis acids on triplet sensitization reactions may therefore be quite general and applicable to the design of a wide range of synthetically useful excited-state photoreactions.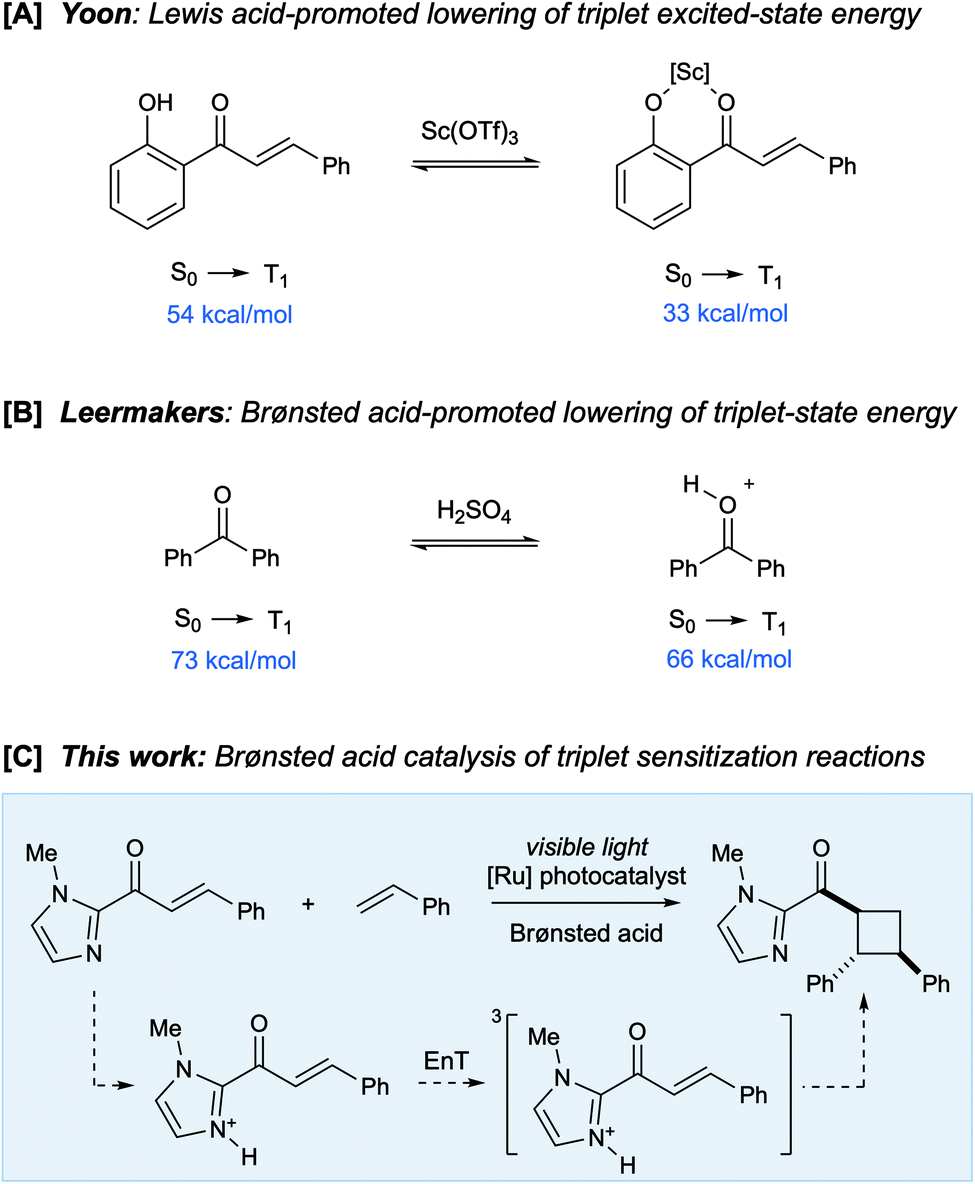 | ||
| Scheme 1 Strategies for Lewis and Brønsted acid-catalyzed generation of excited states. | ||
We wondered if there might be a similarly general effect of protic acids on triplet sensitization processes. The impact of Brønsted acids on the structure and dynamics of organic compounds is often analogous to that of Lewis acid catalysts, but comparatively little is known about the influence of Brønsted acids on the outcome of excited-state organic photoreactions.4,7 In analogy to the effect of Lewis acid catalysis, the protonation of α,β-unsaturated carbonyl compounds can cause a bathochromatic shift in their absorption spectra; Zalewski and Dunn reported that the absorption of benzaldehyde, crotonaldehyde, and cyclopentenone each exhibited a significant red-shift upon addition of sulfuric acid.8 Subsequently, the effect of Brønsted acids on photochemical E/Z isomerizations9 and rearrangements10 of α,β-unsaturated carbonyl compounds was investigated in detail.
In 1971, Leermakers reported an intriguing study showing that Brønsted acids could alter the energy of a carbonyl compound's triplet excited state. The phosphorescence of benzophenone was observed to be significantly red-shifted in strongly acidic media compared to non-polar solvents (Scheme 1B), with a concomitant 200-fold increase in its excited state lifetime.11 This suggested to us that Brønsted acids might catalyze triplet energy transfer processes from transition metal photosensitizers in a fashion similar to Lewis acids. If so, this finding would significantly broaden the diversity of binding interactions and co-catalyst structures that could be exploited to modulate the reactivity of excited-state organic compounds. Herein, we report the first example of an organic photoreaction enabled by Brønsted acid-catalyzed triplet energy transfer. Moreover, a computational analysis of this reaction reveals a new mode of catalysis involving acid-catalyzed optimization of the excited state geometry for vertical Dexter energy transfer.
Results and discussion
As a starting point for our investigations, we examined triplet sensitization reactions of C-cinnamoyl imidazole 1. The selection of this model substrate was premised on several considerations. First, the imidazole nitrogen is sufficiently basic that we expected it to interact strongly with a variety of Brønsted acids in relatively non-polar media. Second, we speculated that the adjacency of the imidazolyl unit to the reactive enone moiety might exert a significant influence on the electronic structure of the excited state. Finally, 2-acyl imidazoles have attracted interest as substrates for a number of synthetic methods because of the ease with which the imidazoyl unit can be cleaved to reveal carboxylic acid derivatives in good yields.12The results of our initial investigations are outlined in Table 1. When an acetonitrile solution of 2-cinnamoyl-1-methylimidazole (1) with 10 equiv. of styrene13 is irradiated with visible light in the presence of Ru(bpy)3Cl2, the crossed [2 + 2] cycloadduct forms relatively slowly, affording 11% yield of 2 after 16 h (entry 1). Interestingly, the addition of 20 mol% of a variety of Brønsted acids results in an increase in the rate of the cycloaddition, and furthermore, stronger acids provide higher yields of the cycloadduct (entries 2–5), as expected in an acid-catalyzed process. Among the acids screened, p-TsOH resulted in the highest yield, affording 2 in 75% yield after 16 h of irradiation (entry 5). Control experiments conducted in the absence of Ru(bpy)3Cl2 indicate that a photocatalyst is strictly required for this reaction to occur and that the observed increase in reactivity is not the result of acid catalysis alone (entries 6 and 7). Thus, these studies reveal a synergistic effect arising from the combination of the photocatalyst and the acid co-catalyst in this transformation. Finally, an experiment conducted in CH2Cl2 afforded similar yields, indicating that the cycloaddition does not require the use of acetonitrile as a more polar solvent.14
|
|
|||
|---|---|---|---|
| Entry | Photocatalyst | Acid | Yielda,b [d.r.] |
| a Yields determined by 1H NMR spectroscopy using phenanthrene as internal standard. b Diastereomeric ratios determined by 1H NMR analysis of crude reaction mixtures. Major diastereomer shown. c Reaction conducted in CH2Cl2 instead of CH3CN. | |||
| 1 | Ru(bpy)3Cl2 | None | 11% [2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1] :1] |
| 2 | Ru(bpy)3Cl2 | CH3CO2H | 17% [2:1] |
| 3 | Ru(bpy)3Cl2 | ClCH2CO2H | 28% [2:1] |
| 4 | Ru(bpy)3Cl2 | CF3CO2H | 68% [2:1] |
| 5 | Ru(bpy) 3 Cl 2 | p-TsOH |
75% [2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1] :1]
|
| 6 | None | None | 0 |
| 7 | None | p-TsOH | Trace |
| 8c | Ru(bpy)3(PF6)2 | p-TsOH | 70% [2:1] |
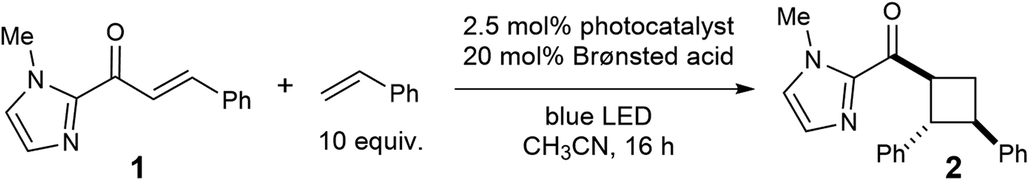
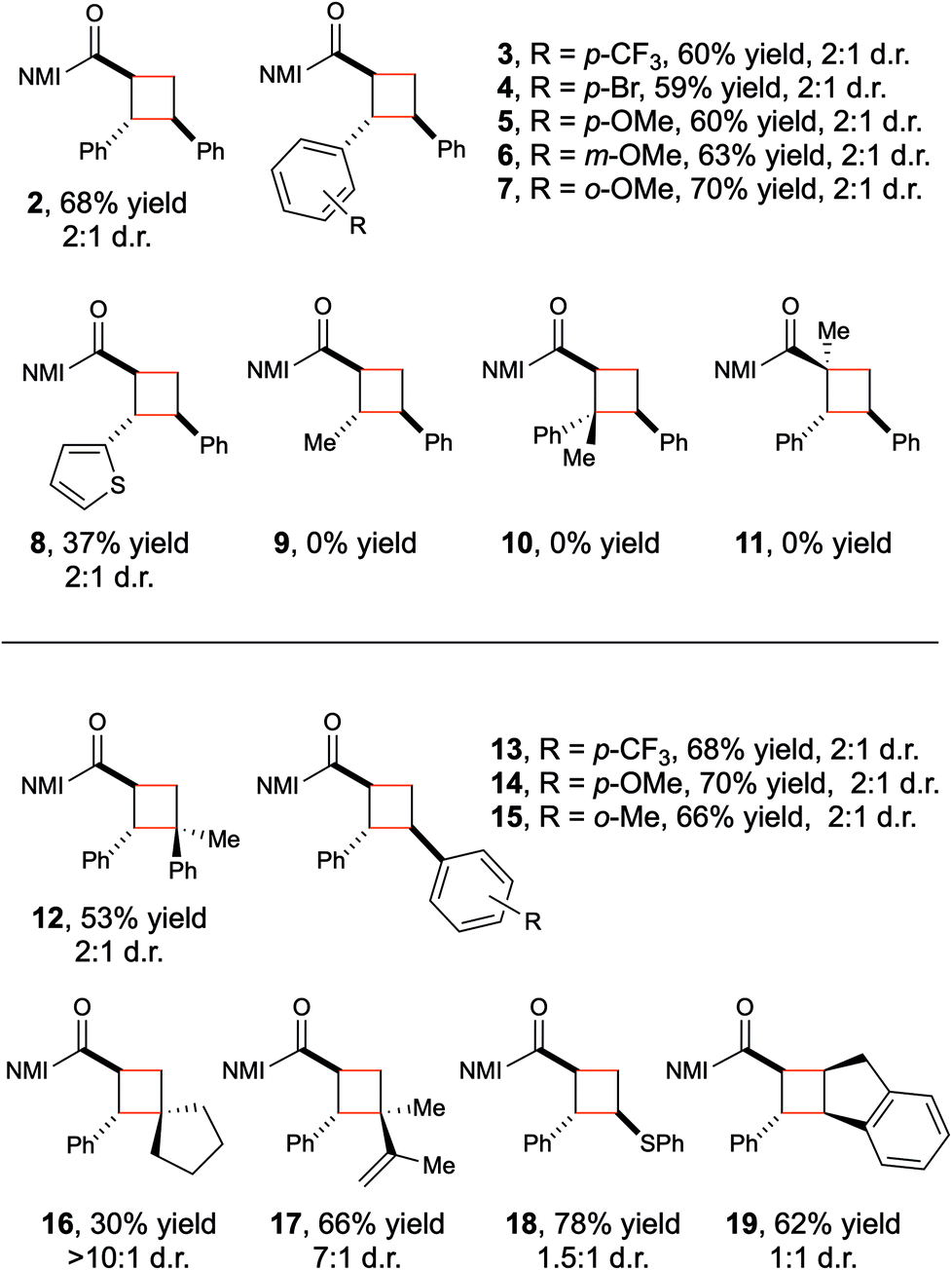
With optimized conditions for Brønsted acid-catalyzed photocycloaddition in hand, the generality of the reaction was tested (Table 2). A variety of electron-rich and electron-poor β-aryl enones (2–7) demonstrate good reactivity. A thiophene-substituted enone reacts sluggishly (8) but illustrates a modest tolerance for heterocycles, provided they are not so basic that they compete with imidazole for protonation. Structurally analogous C-crotonoyl imidazoles, however, generate none of the corresponding cycloadduct (9). This is consistent with the significantly higher triplet energy (computed, ∼58 kcal mol−1) of the less-conjugated π system; we speculate that the organic triplet is never formed in the initial photosensitization step. Trisubstituted olefins (10–11) also are poor substrates for this reaction. While these can generate organic triplets, the subsequent [2 + 2] cycloaddition is likely slowed by the increased steric bulk of the enone, and decay to the ground state becomes competitive.15
| a Reactions conducted using 1 equiv. 2-acyl imidazole, 10 equiv. alkene coupling partner, 2.5 mol% Ru(bpy)3Cl2, and 20 mol% p-TsOH in MeCN, irradiating with a blue LED for 24 h. Isolated yields reported. Diastereomer ratios determined by 1H NMR. NMI = 2-(N-methylimidazolyl). |
|---|
|
The alkene reaction partner in this transformation reacts from its ground state, and the scope with respect to this component is consequently somewhat broader. A range of acceptor styrenes including 1,1-disubstituted (12), electron-poor (13), electron-rich (14), and sterically hindered (15) styrenes all react efficiently. The scope is not limited to styrenes: electron-rich aliphatic alkenes such as methylenecyclopentane give modest yields of a single diastereomer of an unusual spirocyclic cyclobutane (16). In addition, both 2,3-dimethylbutadiene (17) and phenyl vinyl sulfide (18) also undergo cycloaddition in high yield, broadening the diversity of cyclobutane products that can be accessed through this approach. Finally, although trans-β-methylstyrene failed to return the desired cycloadduct, indene provided a reasonable yield of the cyclobutane product (19).
One reason for the selection of 2-acyl imidazoles as substrates for this study was the ease with which the imidazolyl ketone moiety can be cleaved under mild conditions: alkylation of the imidazole nitrogen enables facile acyl substitution of the resulting N-heterocyclic carbene leaving group.12 An operationally convenient, one-pot version of this protocol enabled the cleavage of the imidazolyl group, affording good yields of the corresponding cyclobutyl carboxylic esters and amides from a variety of sterically varied alcohols and amines (Scheme 2).
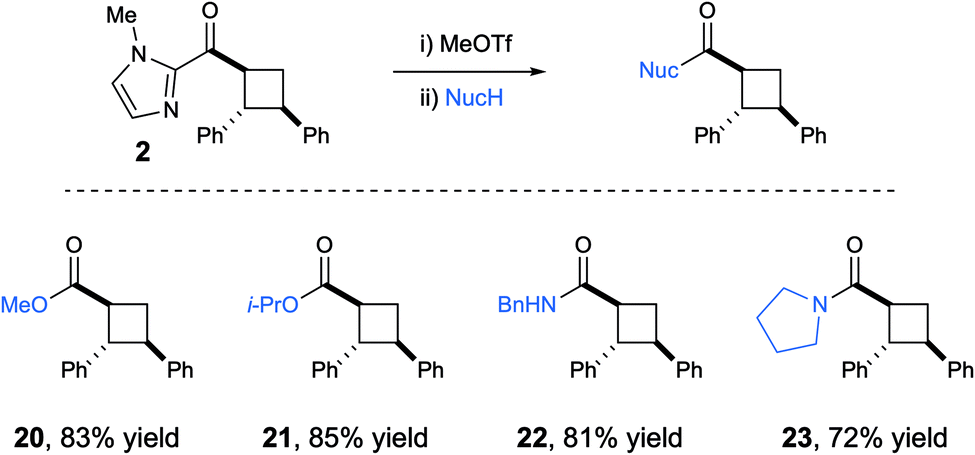 | ||
| Scheme 2 Cleavage of imidazoyl cycloadducts by acyl substitution. | ||
To support the contention that the Brønsted acid co-catalyst accelerates photocatalytic triplet energy transfer, we conducted the steady-state Stern–Volmer luminescence quenching experiments summarised in Fig. 1. These studies show that the emission of [Ru*(bpy)3]2+ is not impacted by the addition of p-TsOH, and that it is only modestly quenched in the presence of cinnamoyl imidazole 1 alone, consistent with the slow rate of background photocycloaddition. Luminescence quenching is, however, significantly increased upon the addition of p-TsOH and 1. An analysis of these Stern–Volmer plots indicates that the rate constant for quenching of [Ru*(bpy)3]2+ by 1 increases by an order of magnitude in the presence of 1 equiv. of p-TsOH (1.16 × 106 M−1 s−1 to 1.24 × 107 M−1 s−1). These data establish that the Brønsted acid co-catalyst has a significant effect on the rate of the key photocatalytic activation step.
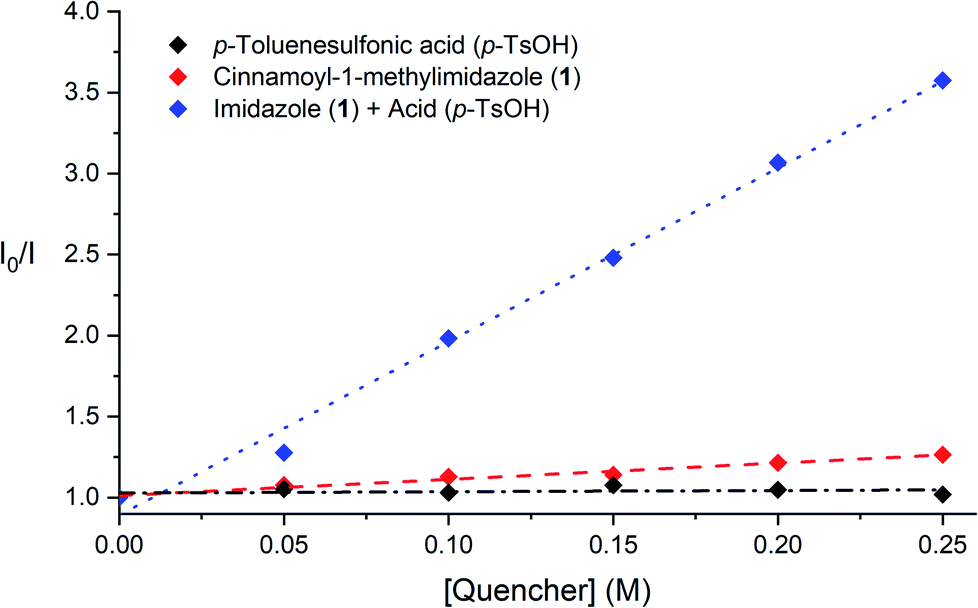 | ||
| Fig. 1 Steady-state Stern–Volmer quenching studies of 2-acyl imidazole 1 in the presence and absence of p-TsOH. | ||
In order to investigate whether the key photocatalytic step involves electron transfer (photoredox catalysis) or energy transfer (photosensitization), we next examined the influence of the Brønsted acid co-catalyst on the electrochemistry of the substrate. The cyclic voltammogram of cinnamoyl imidazole 1 exhibits an irreversible reduction feature whose half-wave potential was estimated to be −1.5 V vs. SCE (see ESI, Fig. S4†), well outside of the range of the excited-state [Ru*(bpy)3]2+ photocatalyst (−0.81 V). Addition of p-TsOH to this solution results in a significantly more complex voltammogram, in which most positive reduction wave features a half-wave potential of approximately −0.5 V. Thus, while these data strongly suggest that the background reaction in the absence of TsOH arises via energy transfer (Table 1, entry 1), they do not enable us to entirely rule out the possibility that one-electron photoreduction of protonated 1 could be responsible for product formation in the presence of the Brønsted acid co-catalyst.
We found, however, that the electron-poor, ligand-modified Ru(II) complex Ru(deeb)3(PF6)2 also successfully catalyzes this photocycloaddition in similar yield and identical diastereoselectivity to the optimized conditions (Scheme 3A). Its significantly less-reducing excited state (−0.28 V vs. SCE) is not sufficient for the reduction of the substrate, even in the presence of Brønsted acid. However, its triplet energy (ET = 45 kcal mol−1) is quite similar to that reported for Ru(bpy)3Cl (ET = 46 kcal mol−1).16 Similarly, the well-studied organic triplet sensitizer benzil (ET = 54 kcal mol−1) is a good catalyst for the [2 + 2] cycloaddition and affords the same ratio of diastereomeric cycloadducts with or without acid co-catalyst (Scheme 3B). In contrast, several one-electron chemical reductants and oxidants failed to produce any observable trace of [2 + 2] products (Scheme 3C). Collectively, these experiments support the hypothesis that this reaction occurs through a Brønsted acid-catalyzed triplet energy transfer process, and that an electron transfer mechanism is unlikely to be relevant to the formation of the cycloadduct.
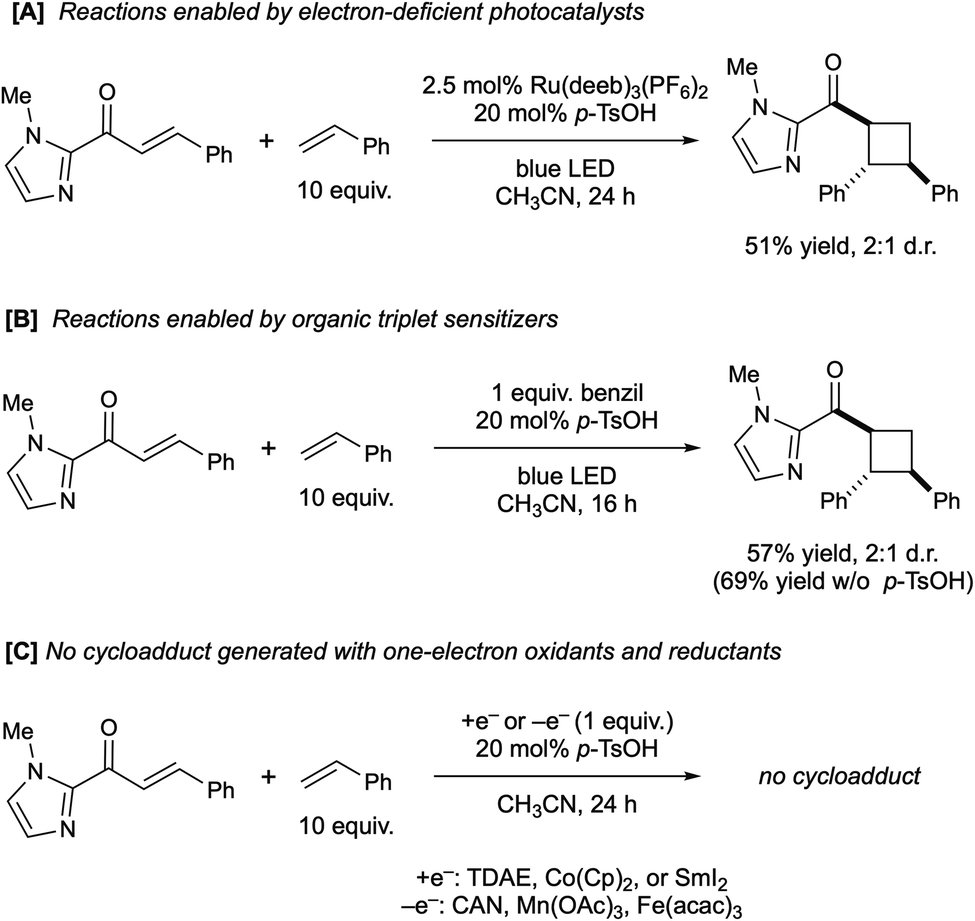 | ||
| Scheme 3 Control studies supporting a triplet energy transfer mechanism. | ||
To understand the significant acceleration of the rate of energy transfer by this Brønsted acid co-catalyst, we further interrogated the role of the acid using density-functional theory (DFT) calculations. As shown in Fig. 2a, the triplet energy for 1 was estimated to be 47.6 kcal mol−1. Similar to the Lewis-acid-catalyzed processes reported previously, a decrease in the triplet energy is seen upon activation by a Brønsted acid, but the magnitude of this stabilization is only 3.1 kcal mol−1. Previously, we observed a shift of ∼20 kcal mol−1 upon coordination of Sc3+. Moreover, protonation of 1 changes the thermodynamics of the triplet energy transfer with 3Ru(bpy)32+ from somewhat endergonic to essentially thermoneutral. This again contrasts to the Lewis acid effects we had previously observed, in which coordination of a Lewis acids rendered the energy transfer notably exergonic. We wondered, therefore, if Brønsted acid-based stabilization of the energy of 31H+ might be insufficient to account for the significant rate increase that we observe in the energy transfer event, and if an analysis of the kinetic barrier might be more revealing.
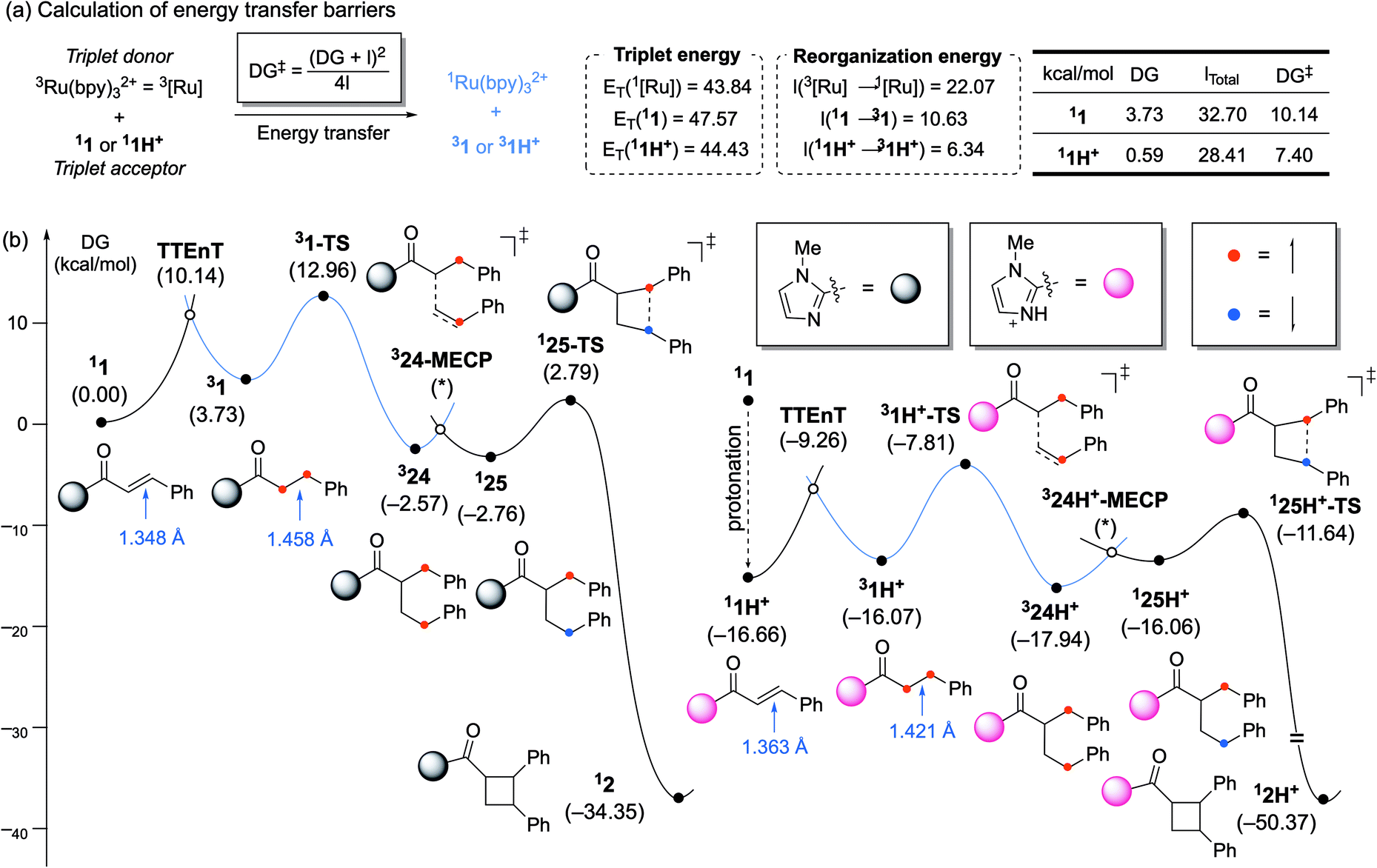 | ||
| Fig. 2 (a) Calculation of energy transfer barriers and key parameters. Gibbs energy profile of [2 + 2] photocycloaddition (b, left) without and (b, right) with Brønsted acid co-catalyst. | ||
Dexter energy transfer can be understood as a double electron exchange between the triplet photosensitizer and the acceptor molecule,17 resulting in a singlet ground state photocatalyst and a triplet excited state acceptor. The barrier for this process can be approximated using Marcus theory as:
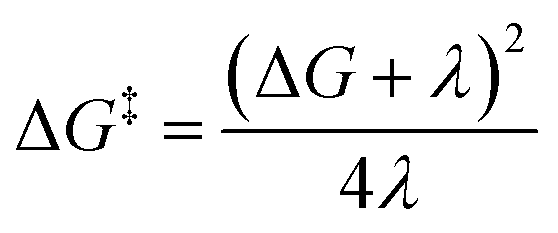 | (1) |
The thermodynamic driving force (ΔG) is the energy difference between the triplet donor and triplet acceptor (+3.73 kcal mol−1 with 1 and +0.59 kcal mol−1 with 1H+, respectively). The reorganization energy (λ) is related to the structural distortion cost needed for the energy transfer and can be estimated by the singlet state energy in the triplet state geometry (see ESI, Table S7†). Using eqn (1), these two parameters allow for the energy transfer barrier (ΔG‡) to be evaluated. This approach has been extensively used to estimate the barriers of both single electron transfer18 and energy transfer19 processes.
The reorganization energy of 3Ru(bpy)32+ was found to be 22.1 kcal mol−1. Similarly, the reorganization energies for 1 and 1H+ were calculated to be 10.6 and 6.3 kcal mol−1, respectively (see ESI†). Upon excitation, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the substrate elongates as the π* orbital becomes occupied. The CC bond of 11 lengthens by 0.110 Å from 1.348 to 1.458 Å upon entering its triplet state. Protonated substrate 11H+ shows a smaller elongation of only 0.058 Å, meaning that protonation minimizes the structural change during excitation to reduce the reorganization penalty. From these values, the Marcus energy transfer barriers are estimated to be 10.1 kcal mol−1 for 11 and 7.4 kcal mol−1 for 11H+, in good qualitative agreement with the observation that Brønsted acids substantially enhance Dexter energy transfer. These calculations show that two features of protonation contribute sizably to the lowering of the energy transfer barrier: (a) the thermodynamic driving force, ΔG, which increases from 3.7 kcal mol−1 to 0.6 kcal mol−1, and (b) reorganization energy, λ, which decreases from 10.6 kcal mol−1 to 6.3 kcal mol−1. Numerically, the change in ΔG lowers the barrier by 1.7 kcal mol−1, whereas the reduction of λ is responsible for 1.1 kcal mol−1 of the overall 2.8 kcal mol−1 that the barrier has been lowered.
C double bond of the substrate elongates as the π* orbital becomes occupied. The CC bond of 11 lengthens by 0.110 Å from 1.348 to 1.458 Å upon entering its triplet state. Protonated substrate 11H+ shows a smaller elongation of only 0.058 Å, meaning that protonation minimizes the structural change during excitation to reduce the reorganization penalty. From these values, the Marcus energy transfer barriers are estimated to be 10.1 kcal mol−1 for 11 and 7.4 kcal mol−1 for 11H+, in good qualitative agreement with the observation that Brønsted acids substantially enhance Dexter energy transfer. These calculations show that two features of protonation contribute sizably to the lowering of the energy transfer barrier: (a) the thermodynamic driving force, ΔG, which increases from 3.7 kcal mol−1 to 0.6 kcal mol−1, and (b) reorganization energy, λ, which decreases from 10.6 kcal mol−1 to 6.3 kcal mol−1. Numerically, the change in ΔG lowers the barrier by 1.7 kcal mol−1, whereas the reduction of λ is responsible for 1.1 kcal mol−1 of the overall 2.8 kcal mol−1 that the barrier has been lowered.
Fig. 2b shows the computed full reaction energy profiles for the reactions of the non-protonated C-cinnamoyl imidazole (1 to 2) and that of the protonated analogues (1H+ to 2H+). The energy profile was drawn based on the general mechanism of [2 + 2] photocycloadditions via triplet states.3b,20 As the mechanism of this reaction has been extensively described in previous studies, its details are not discussed here. The non-protonated substrate (11) can transform into its triplet state via energy transfer with 10.1 kcal mol−1 barrier (Fig. 2b). The α-carbon of cinnamoyl imidazole undergoes C–C coupling with β-carbon of styrene with 9.2 kcal mol−1 barrier to afford intermediate 324, which possesses two α spins on each benzylic position. 324 can convert to open-shell singlet species 124 by traversing the minimum energy crossing point (MECP) and this process facilitates a remaining diradical recombination easily only with 5.6 kcal mol−1 barrier. The protonation of the imidazole group by p-TsOH is calculated to be thermodynamically preferred by 16.7 kcal mol−1 (Fig. 2c). This protonated C-cinnamoyl imidazole species 11H+ is much more effective in triplet energy transfer, which only has a barrier of 7.6 kcal mol−1, as explained above. The subsequent processes follows the mechanism seen for the non-protonated substrate with similar barriers. Thus the most significant impact of the Brønsted acid co-catalyst indeed appears to be on the rate of the energy transfer event.
Conclusions
We have demonstrated that Brønsted acids have a significant impact on the rate of triplet sensitization of basic heteroaryl organic substrates and have used this insight to develop a new method for the [2 + 2] cycloaddition of C-cinnamoyl imidazoles with electron-rich alkenes. These experiments show that Brønsted acids, like Lewis acids, can be used to modulate the reactivity of electronically excited organic intermediates. A straightforward application of Marcus theory revealed that the key to lowering the energy transfer barrier by as much as 3 kcal mol−1 lies in significantly reducing the reorganization energy as well as the increasing thermodynamic driving force. Protonation reduces the degree of elongation that the CC bond must undergo during substrate photosensitization, which in turn enhances the rate of energy transfer and the resulting cycloaddition. The importance of both thermodynamic driving force and reorganization energy on the rate of triplet sensitization reactions is not well appreciated in the design and analysis of synthetic organic photochemical transformations. We speculate that this factor could be an important feature that could be exploited to design and understand a wide range of photoreactions that involve Dexter energy transfer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the NIH (GM127545). M.-H. B. acknowledges the Institute for Basic Science in Korea for financial support (IBS-R10-A1). H. J. is grateful to the National Research Foundation of Korea (NRF) for the global PhD fellowship (NRF-2019H1A2A1076213).Notes and references
- (a) F. D. Lewis, D. K. Howard and J. D. Oxman, J. Am. Chem. Soc., 1983, 105, 3344 CrossRef CAS; (b) F. D. Lewis, S. L. Quillen, P. D. Hale and J. D. Oxman, J. Am. Chem. Soc., 1988, 110, 1261 CrossRef CAS.
- (a) H. Guo, E. Herdtweck and T. Bach, Angew. Chem., Int. Ed., 2010, 49, 7782 CrossRef CAS PubMed; (b) R. Brimioulle, H. Guo and T. Bach, Chem.–Eur. J., 2012, 18, 7552 CrossRef CAS PubMed; (c) R. Brimioulle and T. Bach, Science, 2013, 342, 840 CrossRef CAS PubMed; (d) R. Brimioulle and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 12921 CrossRef CAS PubMed; (e) S. Stegbauer, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 14593 CrossRef CAS PubMed.
- (a) X. Huang, T. R. Quinn, K. Harms, R. D. Webster, L. Zhang, O. Wiest and E. Meggers, J. Am. Chem. Soc., 2017, 139, 9120 CrossRef CAS PubMed; (b) N. Hu, H. Jung, Y. Zheng, J. Lee, Z. Ullah, X. Xie, K. Harms, M.-H. Baik and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 6242 CrossRef CAS PubMed.
- C. Brenninger, J. D. Jolliffe and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 14338 CrossRef CAS PubMed.
- (a) T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391 CrossRef CAS PubMed; (b) Z. D. Miller, B. J. Lee and T. P. Yoon, Angew. Chem., Int. Ed., 2017, 56, 11891 CrossRef CAS PubMed.
- M. E. Daub, H. Jung, B. J. Lee, J. Won, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 9543 CrossRef CAS PubMed.
- (a) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS PubMed; (b) R. F. Childs, Res. Chem. Intermed., 1980, 3, 285 CrossRef CAS.
- (a) R. I. Zalewski and G. E. Dunn, Can. J. Chem., 1968, 46, 2469–2470 CrossRef CAS; (b) R. I. Zalewski and G. E. Dunn, Can. J. Chem., 1969, 47, 2263 CrossRef CAS.
- (a) R. F. Childs, E. F. Lund, A. G. Marshall, W. J. Morrisey and C. V. Rogerson, J. Am. Chem. Soc., 1976, 98, 5924 CrossRef CAS; (b) R. F. Childs, B. Duffey and A. Mika-Gibala, J. Org. Chem., 1984, 49, 4352 CrossRef CAS; (c) R. F. Childs, M. Mahendran, C. Blackburn and G. Antoniadis, Can. J. Chem., 1988, 66, 1355 CrossRef CAS.
- D. G. Cornell and N. Filipescu, J. Org. Chem., 1977, 42, 3331 CrossRef CAS.
- R. Rusakowicz, G. W. Byers and P. A. Leermakers, J. Am. Chem. Soc., 1971, 93, 3263 CrossRef.
- (a) D. H. Davies, N. A. Haire, J. Hall and E. H. Smith, Tetrahedron, 1992, 48, 7839 CrossRef CAS; (b) D. A. Evans, H.-J. Song and K. R. Fandrick, Org. Lett., 2006, 8, 3351 CrossRef CAS PubMed; (c) E. L. Tyson, E. P. Farney and T. P. Yoon, Org. Lett., 2012, 14, 1110 CrossRef CAS PubMed.
- We found that a large excess of styrene was required for optimal yields because of the competitive rate at which 1 undergoes [2 + 2] homodimerization.
- Ru(bpy)3(PF6)2 was used for this experiment because of its superior solubility in CH2Cl2 compared to the chloride salt.
- The formation of triplet excited state enones from these substrates is supported by the observation that they undergo E/Z isomerization under the reaction conditions. Thus the lack of product formation is most reasonably attributed to a slow rate of [2 + 2] cycloaddition and not to lack of energy transfer.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159 CrossRef CAS.
- J.-L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971 CrossRef PubMed.
- (a) S. F. Nelsen, S. C. Blackstock and Y. Kim, J. Am. Chem. Soc., 1987, 109, 677 CrossRef CAS; (b) G. M. Anderson, I. Cameron, J. A. Murphy and T. Tuttle, RSC Adv., 2016, 6, 11335 RSC; (c) A. J. Smith, A. Young, S. Rohrbach, E. F. O'Connor, M. Allison, H.-S. Wang, D. L. Poole, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2017, 56, 13747 CrossRef CAS PubMed; (d) J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, L. E. A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402 CrossRef CAS PubMed; (e) V. K. Singh, C. Yu, S. Badgujar, Y. Kim, Y. Kwon, D. Kim, J. Lee, T. Akhter, G. Thangavel, L. S. Park, J. Lee, P. C. Nandajan, R. Wannemacher, B. Milián-Medina, L. Lüer, K. S. Kim, J. Gierschner and M. S. Kwon, Nat. Catal., 2018, 1, 794 CrossRef CAS.
- (a) J. E. Subotnik, J. Vura-Weis, A. J. Sodt and M. A. Ratner, J. Phys. Chem. A, 2010, 114, 8665 CrossRef CAS PubMed; (b) C. J. Suess, J. D. Hirst and N. A. Besley, J. Comput. Chem., 2017, 38, 1495 CrossRef CAS PubMed.
- M. Zhu, C. Zheng, X. Zhang and S.-L. You, J. Am. Chem. Soc., 2019, 141, 2636 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04822g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |