
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of a self-directed replication system for label-free and real-time sensing of repair glycosylases with zero background†
Li-juan
Wang‡
,
Ying-ying
Lu‡
and
Chun-yang
Zhang
 *
*
College of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Shandong Provincial Key Laboratory of Clean Production of Fine Chemicals, Shandong Normal University, Jinan 250014, China. E-mail: cyzhang@sdnu.edu.cn; Fax: +86-531-82615258; Tel: +86-531-86186033
First published on 26th November 2019
Abstract
Genomic DNA damage and repair are involved in multiple fundamental biological processes, including metabolism, disease, and aging. Inspired by the natural repair mechanism in vivo, we demonstrate for the first time the construction of a self-directed replication system for label-free and real-time sensing of repair glycosylases with zero background. The presence of DNA glycosylase can catalyze the excision repair of the damaged base, successively autostarting the self-directed replication through recycling polymerization extension and strand-displacement DNA synthesis for the generation of exponentially amplified dsDNAs. The resultant dsDNA products can be label-free and real-time monitored with SYBR Green I as the fluorescent indicator. Owing to the high efficiency of self-directed exponential replication and the absolute zero background resulting from the efficient inhibition of nonspecific amplification induced by multiple primer-dependent amplification, this strategy exhibits high sensitivity with a detection limit of 1 × 10−8 U μL−1in vitro and 1 cell in vivo, and it can be further used to screen inhibitors, quantify DNA glycosylase from diverse cancer cells, and even monitor various repair enzymes by simply changing the specific damaged base in the DNA template. Importantly, this assay can be performed in a label-free, real-time and isothermal manner with the involvement of only a single type of polymerase, providing a simple, robust and universal platform for repair enzyme-related biomedical research and clinical therapeutics.
Introduction
Genomic DNA, constituted of Watson–Crick-pairing bases, is selected over the course of evolution as the ideal carrier for preserving and transmitting the genetic information.1 However, the genomic DNAs are frequently damaged by various exogenous and endogenous factors, creating ∼105 lesions (e.g., modified bases, abasic sites, DNA adducts, DNA strand breaks, intra- and inter-strand crosslinks) in a single cell per day.2,3 Persistent DNA damage may severely induce base substitutions, insertions, deletions and chromosomal rearrangements, causing genomic instability, premature ageing, developmental disorders and carcinogenesis progression.4,5 Cells have evolved multiple protection mechanisms to specially repair a wide range of DNA lesions.6 Among these, the base-excision repair (BER) pathway is the most important repair mechanism, repairing a variety of DNA damages arising from oxidation, alkylation, methylation, deamination, and hydrolysis reactions.7–9 DNA glycosylase represents one of the most important repair enzymes, responsible for initiating the first step of the BER pathway through cleaving the N-glycosidic bond between the damaged base and the DNA backbone.10,11 The dysregulation of DNA glycosylases is closely linked to multiple human diseases including neurodegeneration, immunodeficiency, hypoalbuminemia, lymphomas, leukaemias, xeroderma pigmentosum, cockayne syndrome, trichothiodystrophy, and cancers (e.g., lung, breast, gastric, gallbladder, bladder, orolaryngeal, and colorectal cancers).12–15To understand the functions of DNA glycosylases in DNA repair and to facilitate clinical diagnosis and drug discovery, great efforts have been put into the development of DNA glycosylase assays. In principle, the quantification of DNA glycosylases can be achieved in two modes: one is through the measurement of the released damaged bases, and the other is through the quantification of DNA products with apurinic/apyrimidinic (AP) sites. Conventional DNA glycosylase assays including high-performance liquid chromatography (HPLC),16 mass spectrometry (MS),17 and enzyme-linked immunosorbent assays (ELISA)13 are mainly based on the former mode. Besides their laborious procedures, HPLC and MS usually suffer from high background signals due to the generation of artificial damaged bases during sample collection and preparation, while ELISA may underestimate the actual damaged base level owing to the loss of samples during multi-step washing. The latter mode can overcome these limitations by directly detecting DNA products with AP sites, driving the development of colorimetric,18 electrochemical,19 and fluorescent20–24 methods. Despite their improved performances, each method has its own limitations for practical applications. For example, the colorimetric assay requires the time-consuming preparation of gold nanoparticle (AuNP) probes.18 The electrochemical assay needs cumbersome immobilization of capture probes on solid supports and complicated preparation of graphene-deposited electrodes.19 The fluorescence assays based on quantum dot (QD) nanosensor and DNA repair-responsive molecular beacons enable effective detection of DNA glycosylase activities,20,21 but they involve complicated manipulations. The introduction of enzymes such as exonuclease III (Exo III),22 lambda exonuclease (λ exo)23 and endonuclease IV (Endo IV)24 for target signal amplification has greatly improved the detection sensitivity, but they suffer from high cost for the preparation of fluorophore- and quencher-labeled probes, and low specificity and high background caused by the nonspecific nuclease digestion. Therefore, the development of simple, cost-effective, specific and sensitive methods for the DNA glycosylase assay is highly desirable.
Recently, a series of enzyme-based exponential amplification approaches have been developed for sensitive quantification of low-abundance targets, including polymerase chain reaction (PCR),25,26 branched rolling circle amplification (RCA),27 exponential isothermal amplification reaction (EXPAR),28–30 and ligase chain reaction (LCR).30,31 PCR is a standard enzymatic amplification technique based on thermal cycle-mediated DNA amplification with the involvement of long assay time and precise thermal cycling.25,26 Branched RCA27 and EXPAR28–30 are isothermal enzymatic amplification techniques with the requirement of multiple tool enzymes (i.e., polymerases and nickases), complex operation steps, and high experimental cost. In addition, PCR,25,26 branched RCA27 and EXPAR28–30 are two or one primer-dependent amplification techniques with the disadvantage of cross-contamination from nonspecific amplification. LCR is based on thermostable DNA ligase-mediated repetitive cycles of ligation of adjacent hybridized DNA probes,30,31 but the analysis of LCR products is always challenged by electrophoresis separation. Alternatively, loop-mediated isothermal amplification (LAMP) is a new generation of nucleic acid amplification technique based on autocycling strand displacement DNA synthesis (SDS) with the involvement of only a single type of DNA polymerase, achieving ∼109 copies accumulated from less than 10 copies of input template under isothermal conditions within 1 h.32 In comparison with other nucleic acid amplification techniques, LAMP can efficiently inhibit the nonspecific amplification to achieve low background by using four or six specific primers to recognize multiple distinct regions of a double-stranded DNA (dsDNA) template.33 Owing to its distinct advantages of simplicity, rapidity, good specificity and high sensitivity, LAMP has substituted the standard PCR for specific detection of DNA sequences,34 telomerase activity,35 single nucleotide polymorphisms,36 viruses and pathogenic microorganisms.37 In this research, taking advantage of the natural repair mechanism in vivo and the intrinsic superiorities of loop-mediated amplification, we demonstrate for the first time the construction of a self-directed replication system for highly sensitive and specific sensing of repair glycosylases with zero background. In this strategy, the whole reaction can be performed in one tube with the involvement of only a single type of polymerase under isothermal conditions, and the output signal can be monitored in a label-free and real-time manner with a wide dynamic range. Moreover, this method can be used for the screening of potential inhibitors, the quantification of DNA glycosylase from diverse cancer cells, and even the monitoring of various repair enzymes by simply changing the specific damaged base in the DNA template.
Results and discussion
DNA glycosylase is an important repair enzyme superfamily, and it catalyzes the first step of the BER pathway to counter a wide range of DNA damages.38 Based on distinct catalysis functions, DNA glycosylases can be divided into two classes: monofunctional and bifunctional DNA glycosylases. Monofunctional DNA glycosylase has only glycosylase activity, and it catalyzes the hydrolysis of the glycosidic bond to release the damaged base. Bifunctional DNA glycosylases have both glycosylase and AP lyase activities, and they not only excise the glycosidic bond to generate an AP site, but also hydrolytically cleave the 5′ phosphodiester bond at the AP site to create a single nucleotide incision.38 The general BER mechanism is shown in Fig. 1. In genomic DNA, DNA glycosylase can nonspecifically bind DNA duplexes, and subsequently conduct one-dimensional sliding or three-dimensional hopping on them to efficiently search for the damaged bases.39 Upon finding the damaged base, DNA glycosylase can flip the damaged nucleotide 180° to approach its active site, and subsequently catalyzes the hydrolysis of the C1–N glycosidic bond between the damaged base and the deoxyribose to generate an AP site. The resultant AP site can be cleaved by AP endonuclease through hydrolyzing the 5′ phosphodiester bond, leaving 5′-phosphoryl (PO4) and 3′-hydroxyl (OH) termini, followed by the cooperation of a series of repair enzymes (e.g., AP endonuclease, DNA polymerase, and DNA ligase) to complete the entire repair process.38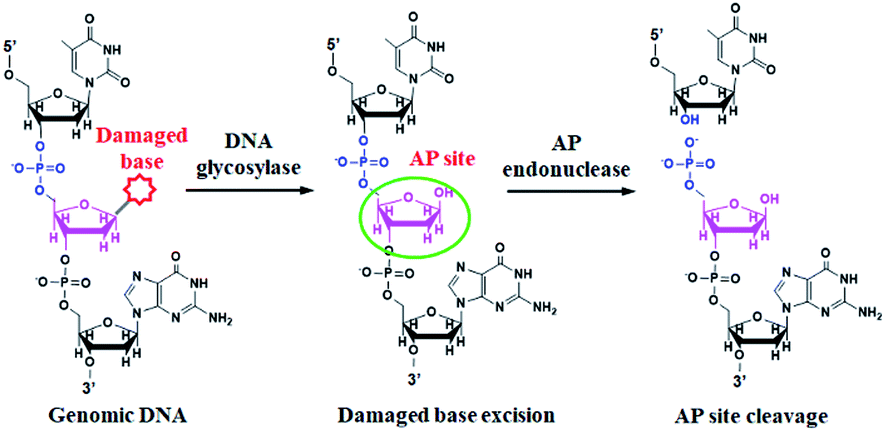 | ||
| Fig. 1 Mechanism of DNA glycosylase-catalyzed damaged base-excision repair through the BER pathway. DNA glycosylase can excise the specific damaged base (red color) to generate an AP site (pink color). The AP site will be cleaved by AP endonuclease to generate the 5′-PO4 and 3′-OH termini. | ||
The principle of the self-directed replication system for the glycosylase assay is illustrated in Scheme 1. As a proof of concept, human 8-oxoguanine-DNA glycosylase (hOGG1) is used as a model target. The hOGG1 is a kind of bifunctional DNA glycosylase and is responsible for the repair of 8-oxoG, a biomarker of oxidative damages arising from reactive oxygen species (ROS).40 In this strategy, a stem-loop DNA template and four liner primer probes (i.e., forward inner and outer primers (FIP and FOP) and backward inner and outer primers (BIP and BOP)) are ingeniously designed. The stem-loop DNA template containing a damaged 8-oxoG functions as both the catalytic substrate for hOGG1 and the template for strand-displacement polymerization. Four linear primer probes (i.e., FIP, BIP, FOP and BOP) are designed to mediate the self-directed exponential replication. As shown in Scheme 1A, in the presence of hOGG1, the damaged 8-oxoG in the DNA template is specially recognized and efficiently excised through the BER mechanism (Fig. 1), generating a nucleotide gap and simultaneously unfolding the loop structure of the DNA template. With the addition of two-pair primers and Bst DNA polymerase, FIP will initially hybridize with F2c in the unfolded DNA template to induce the polymerization extension. Meanwhile, FOP (a few bases shorter and lower in concentration than FIP) can hybridize with FOPc in the unfolded DNA template to initiate the strand displacement DNA synthesis (SDS), generating a dsDNA (I) and simultaneously releasing a single-strand DNA (ssDNA) that can form a stem-loop structure at the 5′ end through the hybridization between F1 and F1c (i.e., the one-stem-loop DNA contains F2 in the loop). Similarly, BIP and BOP can hybridize with B2c and BOPc in the resultant one-stem-loop DNA side by side, and successively initiate the polymerization extension and SDS, producing a dsDNA (II) and releasing an ssDNA that can form a double stem-loop structure at both 5′ and 3′ ends through the hybridization of B1 and B1c, F1 and F1c (i.e., the double-stem-loop DNA (I) contains F2c and B2 in the loops). Notably, once the double-stem-loop DNAs are formed, the self-directed exponential replication is initiated at a constant temperature. As shown in Scheme 1B, the formed double-stem-loop DNA (I) can initiate the self-primed polymerization extension at the 3′ end to form a one-stem-loop DNA with only F2c in the loop. Then FIP will hybridize with the F2c in the loop to prime the SDS, producing a dsDNA intermediate which contains one loop with B2c. The resultant dsDNA will subsequently initiate the self-primed polymerization extension and SDS, generating a one-stem-loop DNA (I) which contains a loop with B2c and simultaneously releasing a double-stem-loop DNA (II) which contains F2 and B2c in the loops, respectively. Importantly, BIP can hybridize with B2c in the loop of one-stem-loop DNA (I) to repeatedly initiate the SDS and self-primed polymerization extension for recycling amplification (I), producing abundant one-stem-loop DNAs with various stem lengths (Fig. S1†). Meanwhile, the released double-stem-loop DNA (II) can initiate the self-primed polymerization and SDS to form a one-stem-loop DNA with only B2c in the loop, and BIP can subsequently hybridize with the B2c in the loop to initiate SDS, producing a dsDNA intermediate which contains one loop with F2c. The resultant dsDNA will subsequently initiate the self-primed polymerization extension and SDS, generating a one-stem-loop DNA (II) which contains a loop with F2c and simultaneously releasing a double-stem-loop DNA (I) which contains F2c and B2 in the loops, respectively. The FIP can hybridize with F2c in the loop of one-stem-loop DNA (II) to initiate the recycling amplification (II) for the generation of abundant stem-loop DNAs, and the FIP can hybridize with F2c in the loop of the newly formed double-stem-loop DNA (I) to autostart a new cycle of polymerization extension, SDS and stem-loop DNA generation. Eventually, cycles of self-directed replication processes lead to the exponential amplification of DNA and the generation of large amounts of dsDNA products with different lengths. The resultant products can be label-free and real-time monitored with SYBR Green I (a fluorescent dye for selectively staining dsDNA) as the indicator for quantifying DNA glycosylase activity. In comparison with the reported DNA glycosylase assays,13,16–18,20–23 this strategy has significant advantages: (1) this assay enables the real-time monitoring of DNA glycosylase activity with a wide dynamic range, (2) the self-directed autocycling polymerization and SDS can result in the exponential DNA amplification, (3) the four primers can recognize multiple distinct regions of the DNA template to inhibit the nonspecific amplification, achieving the zero background, (4) the assay reaction is implemented in one tube with involvement of only a single type of polymerase under isothermal conditions, and the output signal can be detected in a label-free manner, greatly reducing the assay costs.
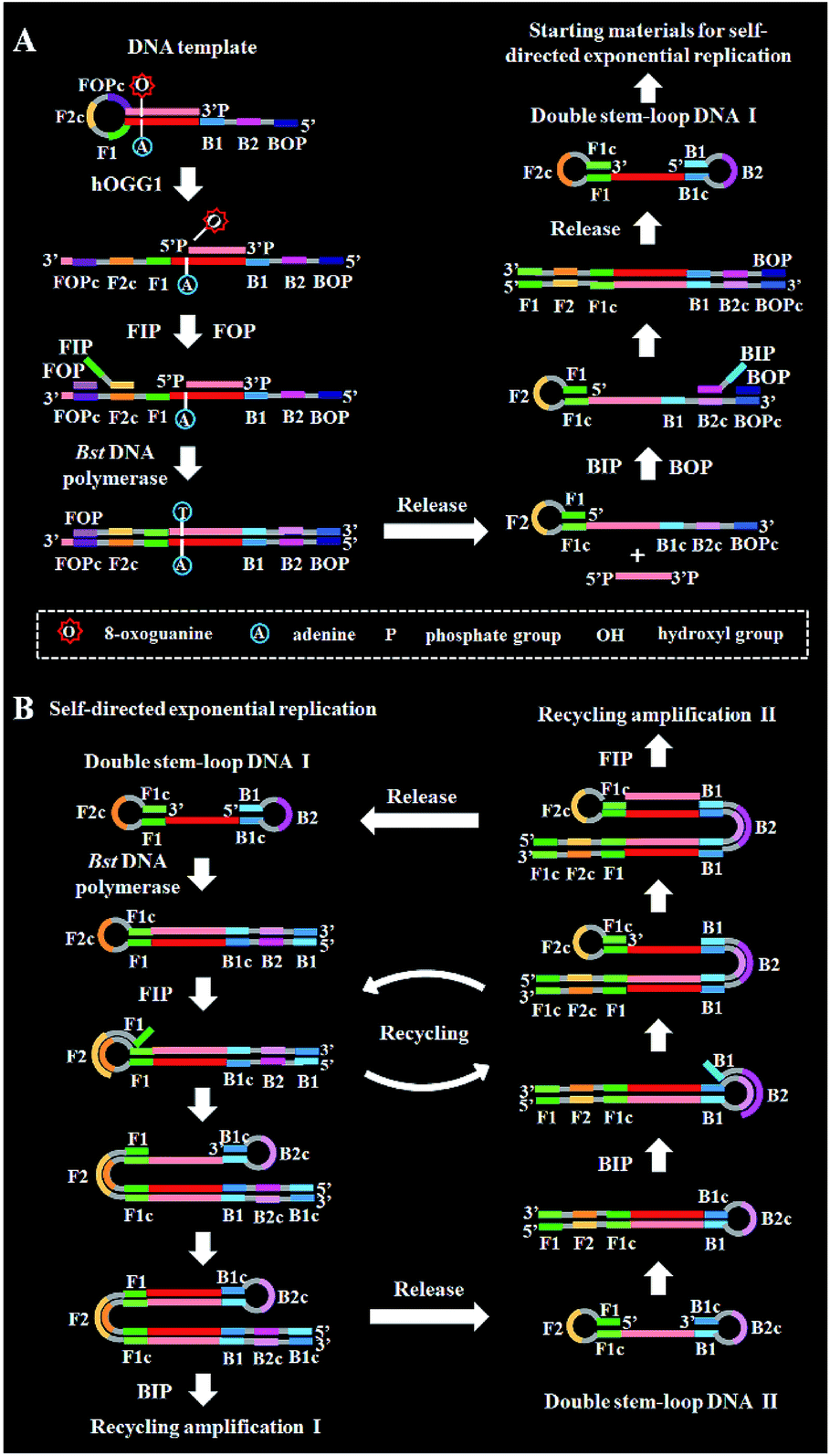 | ||
| Scheme 1 Schematic illustration of the construction of a self-directed replication system for the repair glycosylase assay. This strategy contains two consecutive steps: (A) DNA repairing-driven formation of double-stem-loop DNAs for initiating the self-directed exponential replication, (B) self-directed exponential replication for the generation of dsDNA products. | ||
The proposed DNA glycosylase assay is dependent on the successful unfolding of the stem-loop DNA templates by DNA glycosylase-catalyzed damaged base repair to initiate the self-directed exponential replication. To evaluate whether hOGG1 excises 8-oxoG to induce loop unfolding of the DNA template, we employed 12% denaturing PAGE to analyze the excision products with a silver staining kit as the fluorescent indicator. As shown in Fig. 2A, three characteristic bands of 148, 132 and 15 nt, which are exactly the sizes of the DNA template (148 nt, Fig. 2A, lane 1), the longer excision product (132 nt, Fig. 2A, lane 3) and the shorter excision product (15 nt), are detected in the presence of the hOGG1 + DNA template (Fig. 2A, lane 2), indicating that hOGG1 can efficiently excise the damaged 8-oxoG through the BER mechanism to generate a nucleotide gap in the DNA template, which results in the subsequent cleavage of the DNA template to produce two DNA fragments (i.e. a longer excision product (132 nt) and a shorter excision product (15 nt)). In contrast, only one original band of the DNA template (148 nt) is observed in the presence of only the DNA template (Fig. 2A, lane 1), indicating no occurrence of excision reaction. We further analyze the amplification products after the addition of two-pair primers (i.e., FIP and FOP, BIP and BOP). As shown in Fig. 2B, except for the original bands of FIP (46 nt, Fig. 2B, lane 2), BIP (45 nt, Fig. 2B, lane 1), FOP (17 nt, Fig. 2B, lane 5) and BOP (17 nt, Fig. 2B, lane 5), multiple bands in a ladder-like pattern are detected in the presence of hOGG1 (Fig. 2B, lane 3), indicating that self-directed exponential replication is initiated to generate abundant stem-loop DNAs and dsDNA intermediates with different lengths. Moreover, none of the characteristic ladder-like bands is observed in the absence of hOGG1 (Fig. 2B, lane 4), suggesting no occurrence of amplification reaction. To further investigate the feasibility of the proposed strategy, we performed real-time fluorescence monitoring of the whole amplification reaction with SYBR Green I as the fluorescent indicator. As shown in Fig. 2C, in the presence of hOGG1, the real-time fluorescence signal increases in a sigmoidal fashion (Fig. 2C, red curve), indicating that hOGG1-catalyzed 8-oxoG-excision repair can successfully induce the unfolding of DNA templates and the subsequent self-directed exponential replication, while in the control group without hOGG1, an absolute zero-background signal is observed even after a long reaction time of 120 min (Fig. 2C, black curve). Such an absolute zero background may be ascribed to (1) the high accuracy of hOGG1-catalyzed 8-oxoG-excision repair, (2) the efficient inhibition of nonspecific amplification by the high specificity of multiple primer-dependent amplification, (3) the high selectivity of SYBR Green I towards the dsDNA.
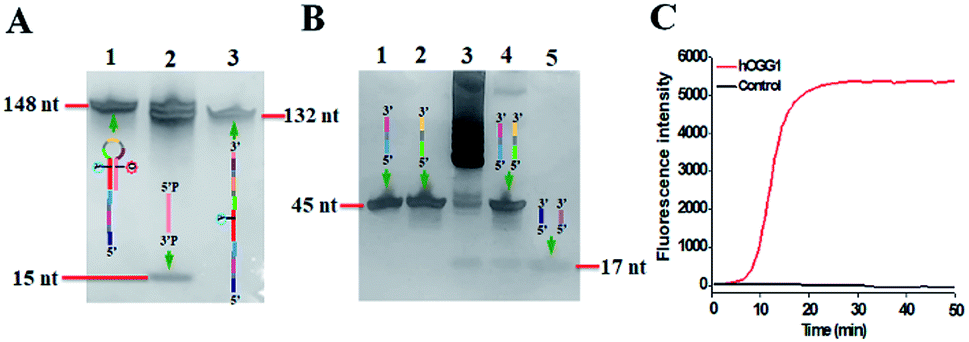 | ||
| Fig. 2 (A) PAGE analysis of the products of hOGG1-catalyzed 8-oxoG-excision reaction under different conditions. Lane 1, in the presence of DNA template; lane 2, in the presence of hOGG1 + DNA template; lane 3, the synthesized excision product. (B) PAGE analysis of the products of self-directed exponential replication reaction. Lane 1, the synthesized BIP; lane 2, the synthesized FIP; lane 3, in the presence of hOGG1; lane 4, without hOGG1; lane 5, the synthesized primers FOP and BOP. (C) Real-time fluorescence monitoring of the amplification reaction in the presence (red line) and absence (black line) of hOGG1. The color images marked in (A) and (B) are the fragments in Scheme 1 corresponding to the bands. 0.25 U μL−1 hOGG1 was used in this experiment. | ||
Under optimum experimental conditions (Fig. S2–S4†), we investigated the assay sensitivity by measuring the fluorescence intensity in response to different concentrations of hOGG1. As shown in Fig. 3A, in response to the increasing concentration of hOGG1 from 1.0 × 10−8 to 0.25 U μL−1, the real-time fluorescence signal increases in a sigmoidal fashion with the conversion of the unfolded hairpin templates from the single-stranded to the partially double-stranded DNA duplexes. The point of inflection (POI) is used for the quantification of hOGG1 activity, and it is defined as the time corresponding to the maximum slope in the sigmoidal amplification curve.41 As shown in Fig. 3B, a good linear relationship is obtained between the POI value and the logarithm of hOGG1 concentration over a large dynamic range of 7 orders of magnitude from 1.0 × 10−8 to 0.25 U μL−1. The regression equation is POI = 3.5 + 10.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log10C with a correlation coefficient of 0.9913, where C is the concentration of hOGG1 (U μL−1). The detection limit is directly measured to be 1.0 × 10−8 U μL−1. Notably, an absolute zero-background signal is observed in the control group without hOGG1 (Fig. 3A, black curve). The sensitivity of the proposed method has been improved by 4 orders of magnitude compared with that of the colorimetric assay based on DNA–AuNP probes (7.0 × 10−4 U μL−1),18 220.0-fold compared with that of single-molecule counting-based fluorescence assay (2.2 × 10−6 U μL−1),21 180.0-fold compared with that of a single QD-based fluorescent nanosensor (1.8 × 10−6 U μL−1),20 and is comparable to those of exonuclease (i.e., Exo III and λ exo)-directed cycling signal amplification-based fluorescence assays (1.0 × 10−9 and 3.0 × 10−9 U μL−1).22,23 The improved sensitivity can be attributed to the high efficiency of DNA repairing-driven self-directed exponential replication and the zero background signal resulting from the efficient inhibition of nonspecific amplification by the high specificity of multiple primer-dependent amplification.
log10C with a correlation coefficient of 0.9913, where C is the concentration of hOGG1 (U μL−1). The detection limit is directly measured to be 1.0 × 10−8 U μL−1. Notably, an absolute zero-background signal is observed in the control group without hOGG1 (Fig. 3A, black curve). The sensitivity of the proposed method has been improved by 4 orders of magnitude compared with that of the colorimetric assay based on DNA–AuNP probes (7.0 × 10−4 U μL−1),18 220.0-fold compared with that of single-molecule counting-based fluorescence assay (2.2 × 10−6 U μL−1),21 180.0-fold compared with that of a single QD-based fluorescent nanosensor (1.8 × 10−6 U μL−1),20 and is comparable to those of exonuclease (i.e., Exo III and λ exo)-directed cycling signal amplification-based fluorescence assays (1.0 × 10−9 and 3.0 × 10−9 U μL−1).22,23 The improved sensitivity can be attributed to the high efficiency of DNA repairing-driven self-directed exponential replication and the zero background signal resulting from the efficient inhibition of nonspecific amplification by the high specificity of multiple primer-dependent amplification.
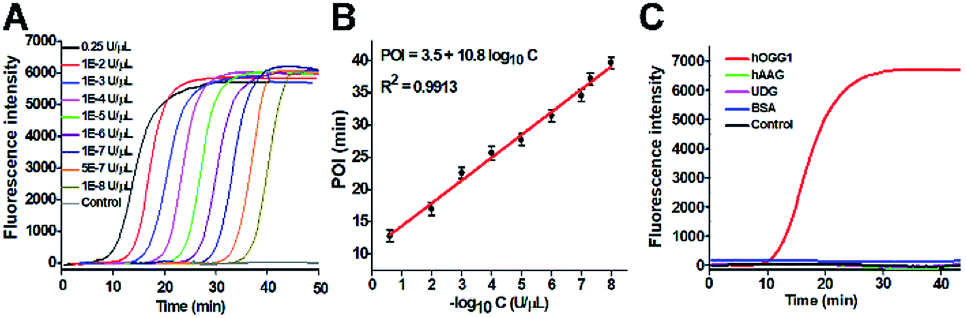 | ||
| Fig. 3 (A) Real-time fluorescence curves in response to different concentrations of hOGG1. (B) Linear relationship between the POI value and the logarithm of hOGG1 concentration. (C) Real-time fluorescence curves in response to 0.1 U μL−1 hOGG1 (red line), 0.1 U μL−1 hAAG (green line), 0.1 U μL−1 UDG (pink line), 0.1 g L−1 BSA (blue line), and the control group with only reaction buffer (black line), respectively. The error bars represent standard deviations of three independent experiments. | ||
DNA glycosylases are a superfamily of enzymes involved in the BER pathway, and they encompass a large group of members. It is a great challenge to specifically discriminate one DNA glycosylase from other family members. To evaluate the selectivity of the proposed assay, we used other members of the DNA glycosylase family (e.g., human alkyladenine DNA glycosylase (hAAG)21,42 and uracil-DNA glycosylase (UDG))43 and an irrelevant protein (e.g., bovine serum albumin (BSA))20 as the interferences. hAAG and UDG can only locate and excise the alkylated purines and the mismatched uracil, but they cannot cleave the DNA template used in this research.21,43 BSA cannot recognize and remove the damaged 8-oxoG in the DNA template.20 As shown in Fig. 3C, in the presence of hOGG1, the real-time fluorescence signal increases in a sigmoidal fashion (Fig. 3C, red curve). In contrast, an absolute zero background signal is observed in the presence of hAAG (Fig. 3C, green curve), UDG (Fig. 3C, pink curve), BSA (Fig. 3C, blue curve), and the control group with only reaction buffer (Fig. 3C, black curve), respectively. Such high specificity may be ascribed to the high selectivity of hOGG1-catalyzed 8-oxoG-excision repair. These results demonstrate that the proposed method can specifically discriminate hOGG1 from the interfering proteins including other DNA glycosylase members.
To investigate the capability of the proposed strategy for inhibitor screening, we used CdCl2 as the inhibitor model.20,21 CdCl2 is a classic inhibitor of DNA glycosylases and it can efficiently inactivate the enzyme activity through competitively occupying the active site of hOGG1 that binds to the DNA substrate containing the corresponding DNA lesion.44 Because this method involves Bst DNA polymerase, the effect of CdCl2 upon this enzyme should be evaluated. As shown in Fig. S5,† CdCl2 has no obvious effect on the activity of Bst DNA polymerase. We measured the real-time fluorescence signals (Fig. 4A) and the corresponding POI values (inset of Fig. 4B) in response to the increasing concentrations of CdCl2. The relative activity of hOGG1 can be measured according to the linear equation in Fig. 3B. As shown in Fig. 4B, the relative activity of hOGG1 decreases with the increasing concentration of CdCl2 from 0 to 100 μM, and the half maximal inhibition (IC50) of hOGG1 is determined to be 8.86 μM, consistent with that measured by the single QD-based fluorescent nanosensor (10.93 μM).20 This result suggests that the proposed method can be applied for the screening of potential DNA glycosylase inhibitors.
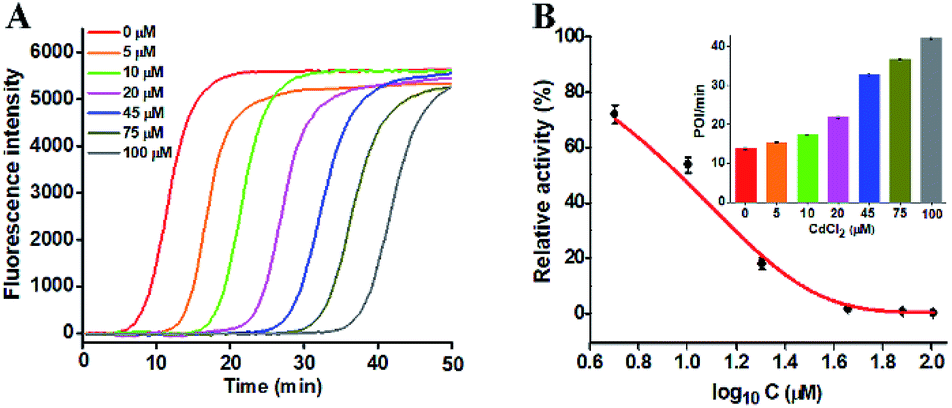 | ||
| Fig. 4 (A) Real-time fluorescence curves in response to different concentrations of CdCl2. (B) Variance of the relative activity of hOGG1 with the logarithm of CdCl2 concentration. Inset shows the POI values of the real-time fluorescence curves in response to the different concentrations of CdCl2. The error bars represent standard deviations of three independent experiments. | ||
To demonstrate the potential applications of the proposed strategy, we used the human lung adenocarcinoma cell line (A549 cells) as a model. First, we analyzed the cellular hOGG1 level with the enzyme-linked immunosorbent assay (ELISA). A distinct white-to-yellow color change is observed in the presence of nucleus and whole cell extracts, respectively, but no visible color change is observed in the presence of cytoplasm extracts (Fig. 5A). We then quantified the corresponding optical densities (O.D.) at 450 nm. Only a low O.D. is detected in response to the cytoplasm extract (Fig. 5A, green column) and the control group (i.e., the nuclear extracts without hOGG1) (Fig. 5A, black column). In contrast, a high O.D. is detected in response to nucleus (Fig. 5A, red column) and whole cell extracts (Fig. 5A, blue column), respectively. The values of relative O.D. in response to cytoplasm, nucleus and whole cell extracts are 3.1, 96.4 and 97.8-fold higher than that in response to the control group (inset of Fig. 5A), respectively. These results indicate that hOGG1 is mainly located in the nucleus of human cells.45,46 We further measured the same amount of cellular extracts using the proposed method (Fig. 5B). Distinct real-time fluorescence signals in a sigmoidal fashion are observed in response to the nucleus extracts (Fig. 5B, red curve), which can be separated completely from those obtained in response to cytoplasm extracts (Fig. 5B, blue curve) and the control group (i.e., the nuclear extracts without hOGG1) (Fig. 5B, black curve). These results (Fig. 5B) are consistent with those obtained by ELISA (Fig. 5A), suggesting that the proposed method can be used for complex sample analysis. In addition, we used the proposed method to measure hOGG1 activity in the nucleus extracts from different numbers of A549 cells (Fig. 5C and D). The corresponding POI values are linearly dependent on the logarithm of the A549 cell numbers in the range from 1 to 1000 (inset of Fig. 5D). The regression equation is POI = 38.4–6.4log10N with a correlation coefficient of 0.9857, where N is the number of A549 cells. Notably, most previously reported methods are not suitable for the detection of hOGG1 activity in cellular samples due to their limited sensitivity and specificity.13,16–18,22 Moreover, the detection limit of the proposed method is directly measured to be 1 cancer cell, which is much higher that of the single-molecule counting-based fluorescence assay (9 cells),21 single QD-based fluorescent nanosensor (5 cells),20 and λ exo-assisted recycling amplification-based fluorescence assay (3 cells).23 These results clearly demonstrate that the proposed method can be applied for accurate detection of hOGG1 activity in crude cell extracts with high sensitivity.
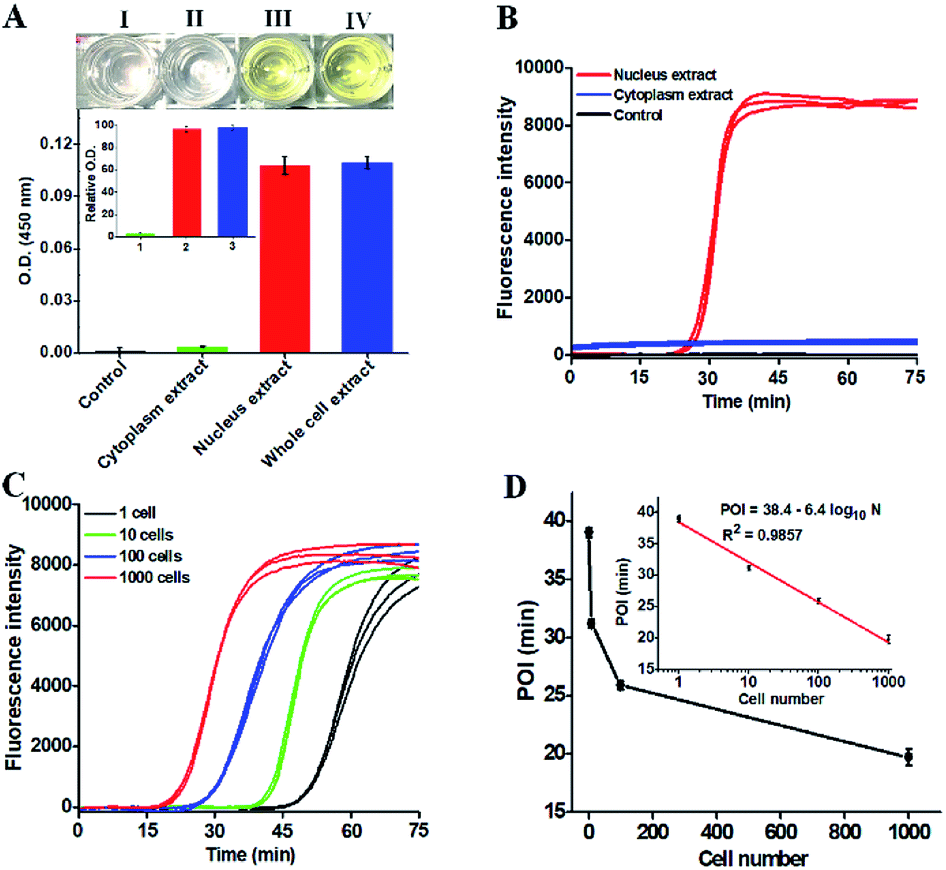 | ||
| Fig. 5 (A) ELISA analysis of hOGG1 in A549 cells. Color changes in response to the control (I), cytoplasm (II), nucleus (III) and whole cell extracts (IV), respectively, and the variance of O.D. in response to the control, cytoplasm, nucleus and whole cell extracts, respectively. Inset shows the relative O.D. values in response to the cytoplasm, nucleus and whole cell extracts, respectively. (B) Real-time fluorescence curves in response to the control, cytoplasm and nucleus extracts from 1000 A549 cells, respectively. (C) Real-time fluorescence curves in response to different numbers of A549 cells. (D) Linear relationship between the POI value and the logarithm of the A549 cell number. Each curve represents the average measurement of three independent experiments. The error bars represent standard deviations of three independent experiments. | ||
Reliability and generality are two critical factors for a new biosensing platform with practical applications. We applied the proposed strategy to measure DNA glycosylase in various cancer cell lines including lung cancer cell (A549 cells), cervical carcinoma cell (HeLa cells), breast adenocarcinoma cell (MCF-7 cells), and normal epithelial mammary cell (MCF-10A cells). In the presence of cancer cell lines (i.e., A549 cells, HeLa cells, and MCF-7 cells), distinct real-time fluorescence signals in a sigmoidal fashion are observed in response to the nucleus extracts equivalent to 5000 cells (Fig. 6A, red, green and blue curves). In contrast, in the presence of a normal cell line (i.e., MCF-10A cells) and the control groups (i.e., the nuclear extracts from the respective cancer cell without hOGG1), absolute zero background signals are detected (Fig. 6A, black, brown, purple, orange, and grey curves), respectively. In addition, the real-time fluorescence signal in response to the mixture of MCF-10A and A549 cells (Fig. 6A, pink curve) is same as that in response to only A549 cells (Fig. 6A, red curve), with same POI values being obtained (Fig. 6B, pink and red columns), suggesting the capability of the proposed method for accurate quantification of hOGG1 activity from different cancer cells. Importantly, the proposed strategy may provide a general platform for the detection of various DNA repair enzymes such as hOGG1, hAAG,21,42 UDG43 and TDG47 by simply changing the specific damaged base in DNA templates (see the ESI, Table S2†). As shown in Fig. 6C, the well-defined real-time fluorescence signals in a sigmoidal fashion are detected in response to hOGG1 (Fig. 6C, red curve), hAAG (Fig. 6C, blue curve), UDG (Fig. 6C, pink curve), and TDG (Fig. 6C, cyan curve), respectively, and the corresponding POI values can be accurately measured as well (Fig. 6D). These results clearly demonstrate that the proposed method may provide a robust and universal platform for real-time monitoring of DNA damage-related repair enzymes in complex environments.
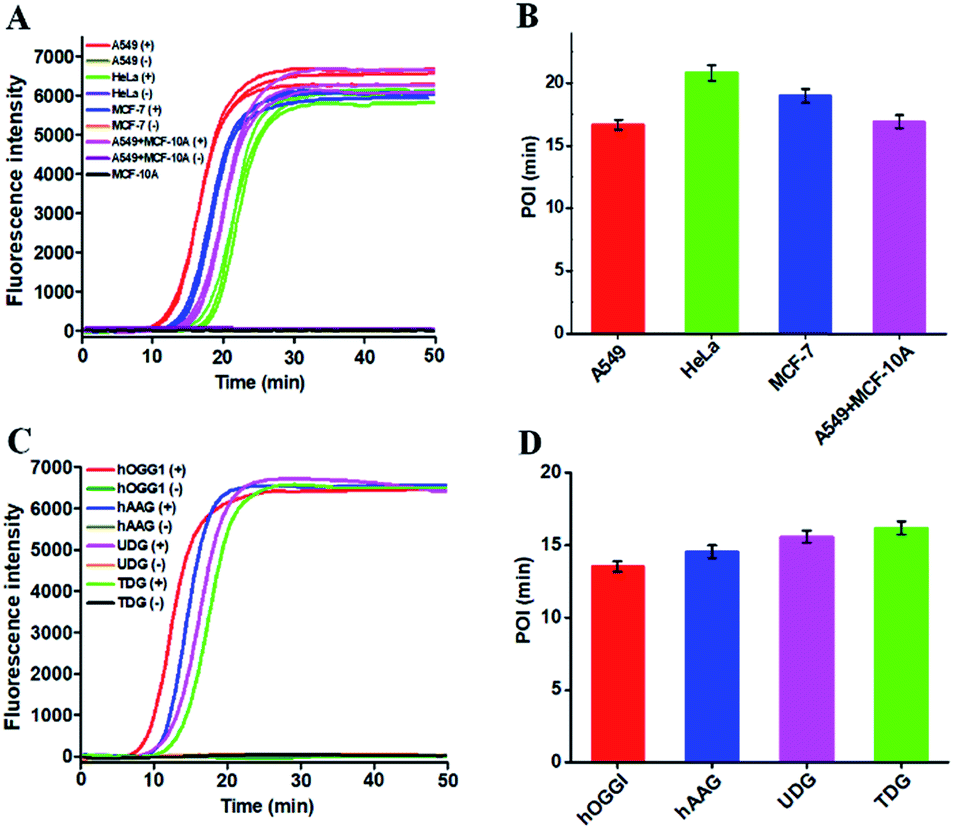 | ||
| Fig. 6 (A) Real-time fluorescence curves in response to 5000 cancer cells including A549 cells, HeLa cells, MCF-7 cells, A549 + MCF-10A cells, MCF-10A cells and control groups (the nuclear extracts from the respective cancer cell without hOGG1), respectively. (B) Measurement of POI values of the real-time fluorescence curves in (A). (C) Real-time fluorescence curves in response to 0.25 U μL−1 hOGG1, 0.25 U μL−1 hAAG, 0.25 U μL−1 UDG, 0.25 U μL−1 TDG and control groups (the reaction solution with the specific DNA template), respectively. (D) Measurement of POI values of the real-time fluorescence curves in (C). Each curve represents the average measurement of three independent experiments. The error bars represent standard deviations of three independent experiments. | ||
Conclusions
Since the “mechanistic studies of DNA repair” discovery was awarded the Nobel Prize for Chemistry in 2015, the research about DNA damage and repair has become a flourishing frontier.48 By taking advantage of the natural repair mechanism in vivo and the intrinsic superiorities of loop-mediated amplification, we construct for the first time the self-directed replication system for highly specific and sensitive detection of repair glycosylases with zero background. This strategy can be performed in one tube with the involvement of only a single type of polymerase under isothermal conditions, and the output signal can be detected in a label-free and real-time manner with a wide dynamic range. Owing to the high specificity of hOGG1-catalyzed 8-oxoG-excision repair, the high efficiency of self-directed exponential replication and the absolute zero background resulting from the efficient inhibition of nonspecific amplification induced by multiple primer-dependent amplification, the proposed method can detect hOGG1 with a detection limit of 1 × 10−8 U μL−1 and a large dynamic range of 7 orders of magnitude, and it can even quantify cellular hOGG1 activity from 1 cancer cell, superior to the reported hOGG1 assays.13,16–18,20–23 Moreover, this method can be applied to discriminate hOGG1 from the interfering enzymes, screen inhibitors, quantify DNA glycosylase activity from diverse cancer cells, and even monitor various repair enzymes by simply changing the specific damaged base in the DNA template. This strategy has significant advantages of simple operation, low cost, good specificity, high sensitivity, multiplexability and generality, providing a facile, robust and universal platform for DNA repair enzyme-related research and holding great potential in biomedical research, clinical diagnosis and cancer therapeutics.Experimental section
Chemicals and materials
Human 8-oxoguanine DNA glycosylase 1 (hOGG1), 10× NEBuffer 2 (500 mM sodium chloride (NaCl), 100 mM trizma hydrochloride (Tris–HCl), 100 mM magnesium chloride (MgCl2), 10 mM DL-dithiothreitol (DTT), pH 7.9), 10 mg mL−1 bovine serum albumin (BSA), human apurinic/apyrimidinic endonuclease (APE1), 10× NEBuffer 4 (500 mM potassium acetate, 200 mM Tris–acetate, 100 mM magnesium acetate, 10 mM DTT, pH 7.9), human alkyladenine DNA glycosylase (hAAG), uracil-DNA glycosylase (UDG), Bst DNA polymerase (large fragment), 10× ThermoPol reaction buffer (200 mM Tris–HCl, 100 mM ammonium sulfate ((NH4)2SO4), 100 mM potassium chloride (KCl), 20 mM magnesium sulfate (MgSO4), 1% Triton X-100, pH 8.8), and the deoxynucleotide (dNTP) solution set were purchased from New England Biolabs (Beverly, MA, USA). Thymine DNA glycosylase (TDG) was bought from R&D System (Minneapolis, Minnesota, USA). Chromium(II) chloride (CdCl2), bovine serum albumin (BSA), MgCl2, ethylenediaminetetraacetic acid (EDTA), and Tris–HCl solution (pH 8.0) were obtained from Sigma Aldrich Company (St Louis, MO, USA). SYBR Green I (10000×) was obtained from Life Technologies (Carlsbad, CA, USA). Human lung adenocarcinoma cell line (A549 cells), cervical carcinoma cell (HeLa cells), breast adenocarcinoma cell (MCF-7 cells), and normal epithelial mammary cell (MCF-10A cells) were bought from Cell Bank, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). All HPLC-purified oligonucleotides (Tables S1 and S2†) were synthesized by Takara Biotechnology Co. Ltd (Dalian, China). Ultrapure water involved in all experiments was prepared using a Millipore filtration system (Millipore, Milford, MA, USA).
DNA repairing-driven self-directed exponential replication
First, all the synthesized oligonucleotides were dissolved in 1× Tris–EDTA buffer (10 mM Tris, 1 mM EDTA, pH 8.0) for preparing the stock solutions. Second, the DNA templates were incubated in the hybridization buffer (10 mM Tris–HCl, 1.5 mM MgCl2, pH 8.0) at 95 °C for 5 min, followed by slowly cooling to room temperature to perfectly form the stem-loop structure. Third, 0.5 μL of the prepared DNA templates (10 μM) were added into the excision reaction solution (10 μL) containing different concentrations of hOGG1, 2 μL of 10× NEBuffer 2, and 2 μL of BSA (10×), incubated at 37 °C for 40 min to perform the excision reaction catalyzed by hOGG1. Fourth, the amplification reaction solution (25 μL) was added into the above reaction system containing 6 U of Bst DNA polymerase, 250 μM dNTPs, 500 nM forward inner primer (FIP), 500 nM backward inner primer (BIP), 125 nM forward outer primer (FOP), 125 nM backward outer primer (BOP), and 3.5 μL of 10× ThermoPol reaction buffer, and incubated at 65 °C for 40 min to carry out the self-directed exponential replication reaction.Real-time fluorescence measurement and gel electrophoresis
The real-time fluorescence was monitored using a BIO-RAD CFX Connect TM Real-Time System (Hercules, CA, USA), and the fluorescence intensity was simultaneously measured at intervals of 30 s. SYBR Green I was used as the fluorescent indicator. For analyzing the excision and the amplification products, 12% denaturing polyacrylamide gel electrophoresis (PAGE) was performed in 1× TBE buffer (9 mM Tris–HCl, 9 mM boric acid, 0.2 mM EDTA, pH 7.9) at 110 V constant voltage for 50 min at room temperature. After electrophoresis, the gels were stained using a silver staining kit (Tiandz Inc., Beijing, China), and further imaged with a ChemiDoc MP Imaging System (Hercules, CA, USA).Inhibition assay
Different concentrations of CdCl2 were incubated with 0.25 U μL−1 hOGG1 at 37 °C for 10 min, followed by the addition of 2 μL of 10× NEBuffer 2 and 500 nM DNA templates into the reaction mixture (20 μL) and incubation at 37 °C for 40 min. Then, 10 μL of the above reaction products were added into the amplification reaction solution (25 μL) containing 6 U of Bst DNA polymerase, 250 μM dNTPs, 500 nM FIP, 500 nM BIP, 125 nM FOP, 125 nM BOP, 3.5 μL of 10× ThermoPol reaction buffer, and 3.5 μL of SYBR Green I (10×) for hOGG1 assay according to the procedures described above.Cell culture and preparation of cellular extracts
Human lung adenocarcinoma cell (A549 cells), cervical carcinoma cell (HeLa cells), and breast adenocarcinoma cell (MCF-7 cells) were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and 1% penicillin-streptomycin (PS, Gibco, USA) in a 5% CO2 incubator at 37 °C. Human normal epithelial mammary cells (MCF-10A cells) were maintained in DMEM with 20 ng mL−1 epidermal growth factor (EGF, Gibco, USA) and 100 ng mL−1 cholera toxin (Gibco, USA) in a humidified chamber containing 5% CO2 at 37 °C. Cancer cells were collected during the exponential phase of growth, and were subsequently lysed with a nuclear extract kit (Active Motif, Carlsbad, CA, USA) according to the manufacturer's protocol. The obtained hOGG1 enzyme in the crude nuclear extracts was immediately subjected to the hOGG1 activity assay. In addition, the nuclear extracts were mixed with the rabbit anti-hOGG1 polyclonal antibody (ZIKER-3687R, ZIKER Bio, Shenzhen, China) to delete hOGG1 enzyme, which were used as the negative controls for the measurement of hOGG1 in A549 cells, HeLa cells, MCF-7 cells, and A549 cells + MCF-10A cells (Fig. S6†).ELISA and western blotting analysis
After the collection of A549 cells, the hOGG1 enzyme was extracted from the cytoplasm, nucleus, and whole cell, respectively, with the same nuclear extract kit according to the procedures described above. The obtained supernatants from different parts of the A549 cell were analyzed by using an ELISA kit (ZK-H2550) (ZIKER Bio, Shenzhen, China). The optical densities (O.D.) were quantified at a wavelength of 450 nm by using a SpectraMax i3× multi-mode microplate reader (Molecular Devices, San Jose, CA, USA). The relative O.D. was calculated based on eqn (1):| Relative O.D. = O.D.t/O.D.c | (1) |
For western blotting analysis, the rabbit anti-hOGG1 polyclonal antibody was used against hOGG1 expressed in different cancer cells. Cancer cells were collected, and hOGG1 enzymes were extracted from A549 cells, HeLa cells, and MCF-7 cells, respectively, with the above nuclear extraction kit. The obtained supernatants were analyzed by western blotting. With actin (GB12001, Servicebio, Wuhan, China) as the internal reference protein, the expression level of hOGG1 was evaluated with a western blot detection kit (E-IR-R304A) (Elabscience, Wuhan, China). The immune complexes were measured using an excellent chemiluminescent substrate detection kit (E-BC-R347) (Elabscience, Wuhan, China), and the protein strips were displayed on the X-ray film. The intensities of strips were detected through densitometric scanning using an Epson V300 scanner (Epson, Suwa, Japan) and further quantified using Alpha Ease FC software (Alpha Innotech, San Leandro, CA, USA).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21527811, 21735003, and 21705097), and the Award for Team Leader Program of Taishan Scholars of Shandong Province, China.Notes and references
- O. D. Schärer, Angew. Chem., Int. Ed., 2003, 42, 2946–2974 CrossRef.
- S. H. Han, S.-H. Hahm, A. H. V. Tran, J. W. Park, J. H. Chung, G. T. Park and Y. S. Han, Bull. Korean Chem. Soc., 2015, 36, 2451–2457 CrossRef CAS.
- C.-H. Leung, H.-J. Zhong, H.-Z. He, L. Lu, D. S.-H. Chan and D.-L. Ma, Chem. Sci., 2013, 4, 3781–3795 RSC.
- S. P. Jackson and J. Bartek, Nature, 2009, 461, 1071 CrossRef CAS.
- H. Park and S. B. Park, Chem. Sci., 2019, 10, 3449–3458 RSC.
- N. J. Curtin, Nat. Rev. Cancer, 2012, 12, 801 CrossRef CAS PubMed.
- O. D. Schärer and J. Jiricny, BioEssays, 2001, 23, 270–281 CrossRef.
- D. K. O'Flaherty, A. Patra, Y. Su, F. P. Guengerich, M. Egli and C. J. Wilds, Chem. Sci., 2016, 7, 4896–4904 RSC.
- S. Kavoosi, B. Sudhamalla, D. Dey, K. Shriver, S. Arora, S. Sappa and K. Islam, Chem. Sci., 2019, 10, 10550–10555 RSC.
- P. L. McKibbin, A. Kobori, Y. Taniguchi, E. T. Kool and S. S. David, J. Am. Chem. Soc., 2012, 134, 1653–1661 CrossRef CAS.
- T. Fu, L. Liu, Q.-L. Yang, Y. Wang, P. Xu, L. Zhang, S. Liu, Q. Dai, Q. Ji, G.-L. Xu, C. He, C. Luo and L. Zhang, Chem. Sci., 2019, 10, 7407–7417 RSC.
- T. Paz-Elizur, M. Krupsky, S. Blumenstein, D. Elinger, E. Schechtman and Z. Livneh, J. Natl. Cancer Inst., 2003, 95, 1312–1319 CrossRef CAS.
- L. Ma, H. Chu, M. Wang, D. Shi, D. Zhong, P. Li, N. Tong, C. Yin and Z. Zhang, Cancer Sci., 2012, 103, 1215–1220 CrossRef CAS.
- M. D'Errico, E. Parlanti, B. Pascucci, P. Fortini, S. Baccarini, V. Simonelli and E. Dogliotti, Free Radical Biol. Med., 2017, 107, 278–291 CrossRef.
- J. Fukae, M. Takanashi, S.-i. Kubo, K.-i. Nishioka, Y. Nakabeppu, H. Mori, Y. Mizuno and N. Hattori, Acta Neuropathol., 2005, 109, 256–262 CrossRef CAS PubMed.
- D. Li, P. F. Firozi, W. Zhang, J. Shen, J. DiGiovanni, S. Lau, D. Evans, H. Friess, M. Hassan and J. L. Abbruzzese, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2002, 513, 37–48 CrossRef CAS.
- D. Nikolić, Anal. Biochem., 2010, 396, 275–279 CrossRef.
- Z. Wu, Z.-K. Wu, H. Tang, L.-J. Tang and J.-H. Jiang, Anal. Chem., 2013, 85, 4376–4383 CrossRef CAS.
- F. Jiao, P. Qian, Y. Qin, Y. Xia, C. Deng and Z. Nie, Talanta, 2016, 147, 98–102 CrossRef CAS.
- L.-j. Wang, F. Ma, B. Tang and C.-y. Zhang, Anal. Chem., 2016, 88, 7523–7529 CrossRef CAS.
- J. Hu, M.-h. Liu, Y. Li, B. Tang and C.-y. Zhang, Chem. Sci., 2018, 9, 712–720 RSC.
- X. Wang, T. Hou, T. Lu and F. Li, Anal. Chem., 2014, 86, 9626–9631 CrossRef CAS.
- Y. Zhang, C.-c. Li, B. Tang and C.-y. Zhang, Anal. Chem., 2017, 89, 7684–7692 CrossRef CAS.
- L.-j. Wang, Z.-Y. Wang, Q. Zhang, B. Tang and C.-y. Zhang, Chem. Commun., 2017, 27, 3878–3881 RSC.
- C.-Y. Yu, B.-C. Yin, S. Wang, Z. Xu and B.-C. Ye, Anal. Chem., 2014, 86, 7214–7218 CrossRef CAS PubMed.
- H. Wang, H. Wang, X. Duan, Y. Sun, X. Wang and Z. Li, Chem. Sci., 2017, 8, 3635–3640 RSC.
- Y.-p. Zeng, J. Hu, Y. Long and C.-y. Zhang, Anal. Chem., 2013, 85, 6143–6150 CrossRef CAS.
- F. Ma, Y. Yang and C.-y. Zhang, Anal. Chem., 2014, 86, 6006–6011 CrossRef CAS.
- Y. Sun, Y. Sun, W. Tian, C. Liu, K. Gao and Z. Li, Chem. Sci., 2018, 9, 1344–1351 RSC.
- F. Su, L. Wang, Y. Sun, C. Liu, X. Duan and Z. Li, Chem. Sci., 2015, 6, 1866–1872 RSC.
- Z.-Y. Wang, L.-j. Wang, Q. Zhang, B. Tang and C.-y. Zhang, Chem. Sci., 2017, 9, 1330–1338 RSC.
- N. Tomita, Y. Mori, H. Kanda and T. Notomi, Nat. Protoc., 2008, 3, 877 CrossRef CAS.
- B. Li, X. Chen and A. D. Ellington, Anal. Chem., 2012, 84, 8371–8377 CrossRef CAS.
- H. Tani, T. Teramura, K. Adachi, S. Tsuneda, S. Kurata, K. Nakamura, T. Kanagawa and N. Noda, Anal. Chem., 2007, 79, 5608–5613 CrossRef CAS.
- H. Wang, H. Wang, C. Liu, X. Duan and Z. Li, Chem. Sci., 2016, 7, 4945–4950 RSC.
- N. Nakamura, K. Ito, M. Takahashi, K. Hashimoto, M. Kawamoto, M. Yamanaka, A. Taniguchi, N. Kamatani and N. Gemma, Anal. Chem., 2007, 79, 9484–9493 CrossRef CAS.
- H. Iseki, S. Kawai, N. Takahashi, M. Hirai, K. Tanabe, N. Yokoyama and I. Igarashi, J. Clin. Microbiol., 2010, 48, 2509–2514 CrossRef CAS.
- J. T. Stivers and Y. L. Jiang, Chem. Rev., 2003, 103, 2729–2760 CrossRef CAS PubMed.
- C. M. Crenshaw, K. Nam, K. Oo, P. S. Kutchukian, B. R. Bowman, M. Karplus and G. L. Verdine, J. Biol. Chem., 2012, 287, 24916–24928 CrossRef CAS.
- S. Boiteux and J. P. Radicella, Arch. Biochem. Biophys., 2000, 377, 1–8 CrossRef CAS.
- H. Jia, Z. Li, C. Liu and Y. Cheng, Angew. Chem., Int. Ed., 2010, 49, 5498–5501 CrossRef CAS PubMed.
- C.-c. Li, W.-x. Liu, J. Hu and C.-y. Zhang, Chem. Sci., 2019, 10, 8675–8684 RSC.
- L.-j. Wang, M. Ren, Q. Zhang, B. Tang and C.-y. Zhang, Anal. Chem., 2017, 89, 4488–4494 CrossRef CAS PubMed.
- D. O. Zharkov and T. A. Rosenquist, DNA Repair, 2002, 1, 661–670 CrossRef CAS.
- M. Saki and A. Prakash, Free Radical Biol. Med., 2017, 107, 216–227 CrossRef CAS.
- V. A. Bohr, Free Radical Biol. Med., 2002, 32, 804–812 CrossRef CAS.
- C. Chen, D. Zhou, H. Tang, M. Liang and J. Jiang, Chem. Commun., 2013, 49, 5874–5876 RSC.
- J. H. McKerrow, Nat. Prod. Rep., 2015, 32, 1610–1611 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04738g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |