
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploiting the versatile alkyne-based chemistry for expanding the applications of a stable triphenylmethyl organic radical on surfaces†
J. Alejandro
de Sousa‡
 ae,
Francesc
Bejarano‡
a,
Diego
Gutiérrez
a,
Yann R.
Leroux
b,
Ewa Malgorzata
Nowik-Boltyk
c,
Tobias
Junghoefer
c,
Erika
Giangrisostomi
d,
Ruslan
Ovsyannikov
d,
Maria Benedetta
Casu
*c,
Jaume
Veciana
a,
Marta
Mas-Torrent
a,
Bruno
Fabre
*b,
Concepció
Rovira
*a and
Núria
Crivillers
*a
ae,
Francesc
Bejarano‡
a,
Diego
Gutiérrez
a,
Yann R.
Leroux
b,
Ewa Malgorzata
Nowik-Boltyk
c,
Tobias
Junghoefer
c,
Erika
Giangrisostomi
d,
Ruslan
Ovsyannikov
d,
Maria Benedetta
Casu
*c,
Jaume
Veciana
a,
Marta
Mas-Torrent
a,
Bruno
Fabre
*b,
Concepció
Rovira
*a and
Núria
Crivillers
*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Networking Research Center on Bioengineering Biomaterials and Nanomedicine (CIBER-BBN), Campus de la UAB, 08193 Bellaterra, Spain. E-mail: ncrivillers@icmab.es
bUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes)-UMR 6226, F-35000 Rennes, France
cInstitute of Physical and Theoretical Chemistry, University of Tübingen, 72076 Tübingen, Germany
dHelmholtz-Zentrum Berlin für Materialien und Energie (HZB), Albert-Einstein-Str 15, 12489 Berlin, Germany
eLaboratorio de Electroquímica, Departamento de Química, Facultad de Ciencias, Universidad de los Andes, 5101 Mérida, Venezuela
First published on 20th November 2019
Abstract
The incorporation of terminal alkynes into the chemical structure of persistent organic perchlorotriphenylmethyl (PTM) radicals provides new chemical tools to expand their potential applications. In this work, this is demonstrated by the chemical functionalization of two types of substrates, hydrogenated SiO2-free silicon (Si–H) and gold, and, by exploiting the click chemistry, scarcely used with organic radicals, to synthesise multifunctional systems. On one hand, the one-step functionalization of Si–H allows a light-triggered capacitance switch to be successfully achieved under electrochemical conditions. On the other hand, the click reaction between the alkyne-terminated PTM radical and a ferrocene azide derivative, used here as a model azide system, leads to a multistate electrochemical switch. The successful post-surface modification makes the self-assembled monolayers reported here an appealing platform to synthesise multifunctional systems grafted on surfaces.
Introduction
The impact of (bio)molecular surface modification in different fields of research is supported by the wide variety of applications that derive from it. The goal of the functionalization can be either transferring specific molecular properties to the substrate, i.e. by grafting (multi)functional molecular systems, or generating novel interfaces, for example to promote cell-adhesion, (bio)sensing or to inhibit undesired reactions, among others.1–7 The molecular modification of surfaces has also been a determining factor in the emerging field of molecular (spin)electronic devices.8 In this context, among the extensive library of functional molecules investigated, optically-, redox- and magnetically-active stable organic radicals are very attractive.9 Nowadays, these materials have attracted much interest and their exploitation as components of molecular functional materials is boosting a new generation of devices for applications in OLEDs,10,11 energy storage and conversion,12,13 molecular spintronics,14 imaging,15,16 sensors17 and memory devices.18–20 It has been shown that for the device implementation, in many cases their immobilization onto surfaces is needed.21To this end, the formation of stable self-assembled monolayers (SAMs) on substrates typically employed for the fabrication of electronic components, such as silicon or gold, is a well-established strategy.22,23 The hybrid system robustness, and hence its potential applicability, is equally dependent on the stability of the molecular entity as well as on the stability of the molecule–substrate interface. For this, beyond the search for transferring a specific molecular functionality to the surface (mainly magnetically-, redox- or optically-active), great efforts have been focused on: (i) the search for new robust anchoring groups and (ii) the post-grafting modification of SAMs. Terminal alkynes have appeared as an appealing alternative to the thiol group as they spontaneously and covalently react with gold.24,25 Besides the stability of the C–Au bond, the high directionality of the σ-bonded C–Au has been demonstrated to provide higher conductance at the single-molecule level,26 which makes terminal alkynes very attractive for molecular electronics. Interestingly, these functional groups can also be used to covalently react with hydrogen-terminated silicon (Si–H) surfaces.27–29 Concerning post-grafting modification, preparing monolayers with exposed functional end groups which can react with other (bio)molecular systems is of great importance to expand the applicability of these surfaces. Among the several chemical strategies reported to perform interfacial reactions on SAMs,30 the so-called “click chemistry” has been one of the most employed, in particular the 1,3-dipolar cycloaddition of azides and alkynes to form 1,2,3 triazoles.31–34 Hence, alkynes can be considered as very versatile groups to be employed for the modification and post-modification of substrates.
The functionalization of SiO2-free silicon and gold substrates with electrical and/or light triggered molecules has been pursued with the aim of obtaining switchable surfaces. In the case of silicon, unlike metals, its electronic properties can be finely tuned by modifying the density and the nature of the charge carriers (electrons and holes) under light illumination, which can be used as a second gate for the tuning of the properties of the modified surface.29
In this work, by exploiting the rich chemistry offered by alkynes and the high chemical and thermal stability of perchlorotriphenylmethyl (PTM) radicals,35 the redox properties of a PTM radical bearing one and two terminal alkyne groups (Fig. 1) have been dually exploited as: (i) a capacitance switch on Si–H triggered by light and, (ii) as an organic radical-based platform to be further modified by click chemistry giving rise to a multistate electrochemical switch. In both cases, a particular focus has been placed on the optimization of the experimental conditions to ensure the integrity of the radical and the terminal alkyne.
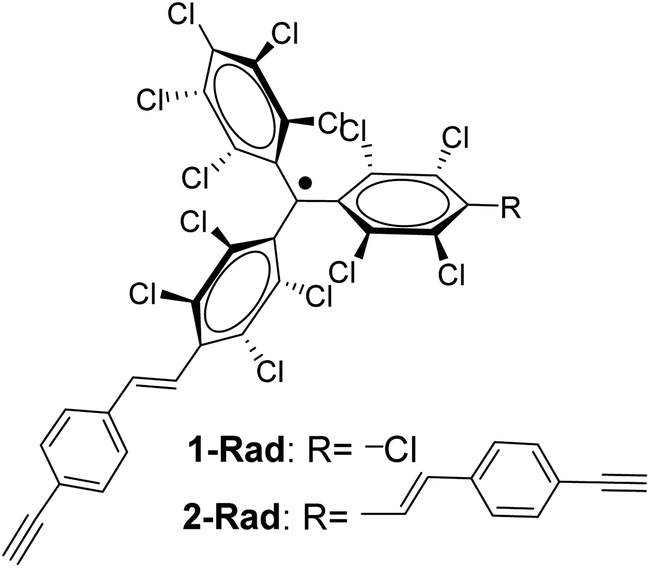 | ||
| Fig. 1 Chemical structure of the PTM radicals employed in this work. | ||
Results and discussion
Functionalization of Si–H surfaces with PTM radicals
Interfacing technologically important semiconducting surfaces, such as silicon, with high-quality, and stable redox-active films has appeared as a promising strategy toward functional devices for charge storage. Remarkably, by taking advantage of the fact that the charge transfer characteristics of silicon can be either inhibited or activated upon light illumination, this substrate constitutes a relevant platform for the development of photochemically switchable systems.29,36,37 Then, silicon itself can be used as a light-controlled gate to turn ON/OFF the electronic communication with the grafted electroactive centers, in this case the PTM radicals. With this aim, we exploit herein the reactivity of the alkyne terminated PTM (1-Rad) to chemically modify SiO2-free p-type Si–H substrates, forming robust Si–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. To date, the functionalization of Si–H surfaces has been achieved with numerous electroactive molecules, among them are ferrocene, quinones, tetrathiafulvalenes (TTF) and metallic complexes.29 However, to the best of our knowledge, before this work, only one example of an organic radical grafted on Si–H has been reported. In that case, the grafting was done through a two-step approach, to avoid the reactivity of the unpaired electron with the silicon surface bonds, thus leading to a loss of the radical character.38 Herein, the Si–H functionalization was carried out in a single step by the hydrosilylation reaction of molecule 1-Rad through the terminal acetylene group leading to the covalently bound PTM-terminated monolayer (SAM-1-Rad-Si) (Fig. 2a).
C bonds. To date, the functionalization of Si–H surfaces has been achieved with numerous electroactive molecules, among them are ferrocene, quinones, tetrathiafulvalenes (TTF) and metallic complexes.29 However, to the best of our knowledge, before this work, only one example of an organic radical grafted on Si–H has been reported. In that case, the grafting was done through a two-step approach, to avoid the reactivity of the unpaired electron with the silicon surface bonds, thus leading to a loss of the radical character.38 Herein, the Si–H functionalization was carried out in a single step by the hydrosilylation reaction of molecule 1-Rad through the terminal acetylene group leading to the covalently bound PTM-terminated monolayer (SAM-1-Rad-Si) (Fig. 2a).
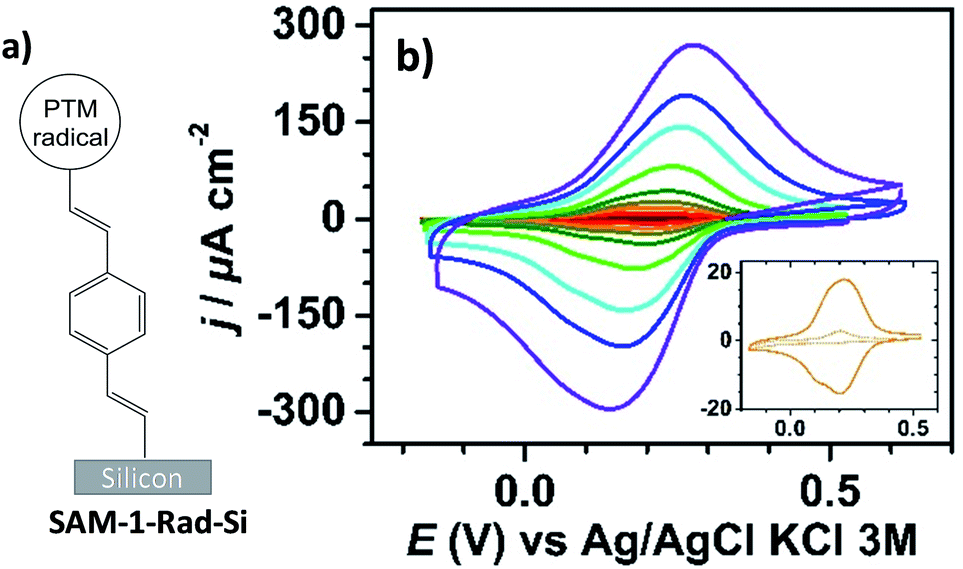 | ||
| Fig. 2 (a) Sketch of SAM-1-Rad-Si. (b) Cyclic voltammograms under red light illumination of SAM-1-Rad-Si at different scan rates (0.1, 0.2, 0.4, 0.6, 1, 2, 4, 6 and 10 V s−1, from brown to purple) in CH3CN + 0.1 M Bu4NClO4. The inset shows comparative CVs at 0.4 V s−1 in the dark (dashed line) and under illumination (solid line). | ||
Although the hydrosilylation reaction between alkynes and Si–H can be performed by either thermal39,40 or photochemical41 activation, the instability in solution under light of the radical PTM moiety excluded the possibility of using in this work such a photochemical route. Several grafting attempts were carried out by varying the concentration of the PTM radical solution, solvent, temperature and immersion time (see the experimental details in the ESI and Table S1†). PTM radical monolayers with the highest surface coverage of PTM (estimated from the electrochemical measurements, vide infra) and the lowest oxidation level of underlying silicon were obtained at 145 °C for 20 h using 1,2-dichlorobenzene (DCB) as a high-boiling solvent and a ca. 7–10 mM concentration of 1-Rad.
The SAM-1-Rad-Si monolayer was characterized by X-ray photoelectron spectroscopy, which, additionally to the chemical composition, provides information about the oxidation state of the underlying silicon (Fig. S1†). For the C 1s, two main peaks at, 285.0 and 286.6 eV, assigned to the CC and the C–Cl bonds, respectively, were observed. The Cl 2p spectrum displayed a typical doublet with both components at 200.8 (2p3/2) and 202.5 (2p1/2) eV, arising from the chlorinated phenyl rings of the PTM unit. Besides, the Si 2p spectrum was deconvoluted into two contributions at 99.2 and 99.8 eV assigned to bulk and interfacial silicon, respectively, in crystalline Si(111).42,43 Additionally, a low intense peak appeared at 103 eV, which is attributed to the unavoidable oxidation of a certain content of remaining Si–H sites by water and atmospheric oxygen, indicating that, as expected, the surface is not fully passivated by the bulky radical 1-Rad. Nevertheless, the low contribution of this peak was positively surprising considering the bulkiness of the PTM moiety.
SAM-1-Rad-Si was electrochemically characterized in the dark and under illumination through a red filter (λ > 600 nm) to avoid the possible degradation of the grafted PTM radical. As can be seen in Fig. 2b, the cyclic voltammograms (CVs) in the dark showed negligible oxidation and reduction currents (lower than 10 μA cm−2 at 10 V s−1), as expected for a semiconductor under depletion conditions,44i.e., when only a few majority charge carriers (holes in the case of p-type Si) are available for charge transfer (vide infra).
Upon illumination of SAM-1-Rad-Si, an intense reversible redox wave was observed at E°′ = 0.21 V vs. Ag/AgCl, KCl 3 M (average of the anodic Epa and cathodic Epc peak potentials) corresponding to the PTM(radical) ↔ PTM(anion) process promoted by captured photogenerated electrons (Fig. 2b). The redox response observed indicates that the radical character of the PTM remains unaltered upon grafting. Additionally, both the anodic and cathodic peak photocurrents Ipa and Ipc, corresponding to the reversible redox couple, were found to be proportional to the potential scan rate v, as expected for a surface-confined reversible redox species (Fig. S2a–c†).45 The variation of Epa and Epc with v (Fig. S2b†) enabled us to determine the apparent rate constant for electron transfer at the bound PTM center, ket,ap, using the recent theoretical model developed by Vogel et al. accounting for semiconductor diode effects.46 A value of 90 ± 20 s−1 was estimated, in accordance with the literature data reported for other silicon electrodes modified with electrochemically reversible systems.29,46
The surface coverage of attached PTM moieties was electrochemically estimated from CVs of illuminated SAM-1-Rad-Si (eqn S1†). Indeed, anodic charge integration at several scan rates (between 0.4 and 1 V s−1) resulted in an average value of (8.5 ± 0.3) × 10−11 mol cm−2, very close to the analogous SAMs on Au (see Section 2.2.2).
To obtain further insights into the light dependence of the redox process in SAM-1-Rad-Si, differential capacitance measurements were performed in the same electrolytic medium (i.e. CH3CN/Bu4NClO4). First, the flatband potential (Efb), i.e. the electrode potential for which there is no space-charge region in the semiconductor, was estimated from the commonly used Mott–Schottky plot (Csc−2vs. E, eqn S(2)†) that gives the space-charge capacitance Csc as a function of the electrode potential E under depletion conditions, i.e., the depletion of valence band holes in the space charge region of the p-type surface.47 In the dark, a linear Csc−2–E plot was obtained for potentials below 0.25 V vs. Ag/AgCl, KCl 3 M, in which the intercept and the slope of the curve enable Efb and the dopant density ND to be determined, respectively (Fig. S3†). The calculated ND value (1.2 × 1015 boron atoms cm−3) was consistent with the dopant density derived from the four-probe resistivity measurements of silicon samples, between 5 and 10 Ω cm. The extracted value of Efb was 0.25 ± 0.02 V. Based on this parameter, it can be concluded that the PTM(radical) ↔ PTM(anion) redox process occurs in a potential range wherein the semiconductor is in weak depletion. The small potential difference between E°′ and Efb (around 50 mV) explains, however, why low (but not zero) oxidation currents are observed at SAM-1-Rad-Si in the dark (inset in Fig. 2b). Much higher currents were observed under illumination because the redox process could now occur with high rate thanks to the high concentration of photogenerated minority charge carriers (i.e. electrons).
In the dark, consistent with the current response, the measured capacitance values were small and did not exceed 1 μF cm−2. In contrast, the capacitance curve under red light illumination was characterized by a much more intense capacitance peak (enhancement by a factor of ∼10) at 0.18 V, close to the formal potential of bound PTM determined by CV (Fig. 3a). This capacitance peak was clearly attributed to the charging/discharging currents associated with the oxidation/reduction of the bound PTM centers,48,49 in perfect line with previous reports on ferrocene-modified Si surfaces.36,50
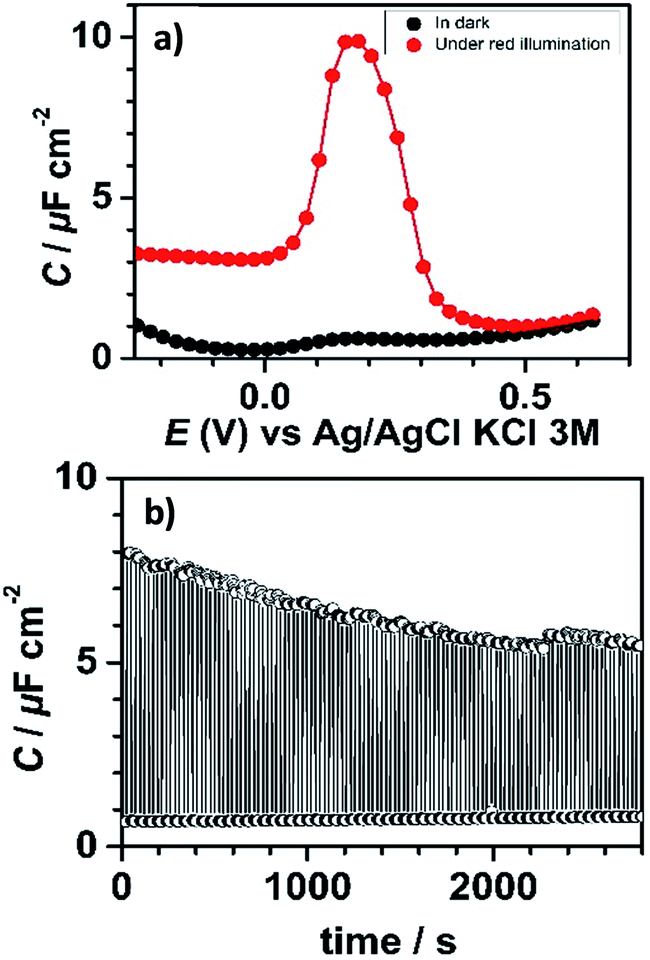 | ||
| Fig. 3 (a) Capacitance–potential curves of SAM-1-Rad-Si measured at 50 Hz in the dark and under red light illumination. (b) Capacitance–time profile at 50 Hz measured at 0.18 V during dark (OFF state, 30 s)/illumination (ON state, 30 s) switching cycles. Electrolytic solution: CH3CN + 0.1 M Bu4NClO4. | ||
This significant contrast between the two states (ON and OFF) permitted us to exploit this system as a capacitance switch and its cyclability was investigated by carrying out consecutive ON/OFF switching cycles by turning on and off the light along time (Fig. 3b). A ∼30% decrease in the maximum photocapacitance was observed along the first 30 minutes before reaching a situation of higher stability. This loss in the switching ability is believed to be caused by the gradual degradation of the electrical properties of the interface due to the oxidation of the underlying silicon. Indeed, owing to the moderately dense packing of the PTM monolayer (due to the bulky nature of the PTM head groups), traces of water and/or oxidizing species present in the electrolytic medium unavoidably penetrate through the molecular layer via defects or pinholes to react with remaining Si–H sites. Despite this, the functionalization was found to be remarkably stable.
Functionalization of PTM radical molecules via click chemistry, in solution and on the gold surface
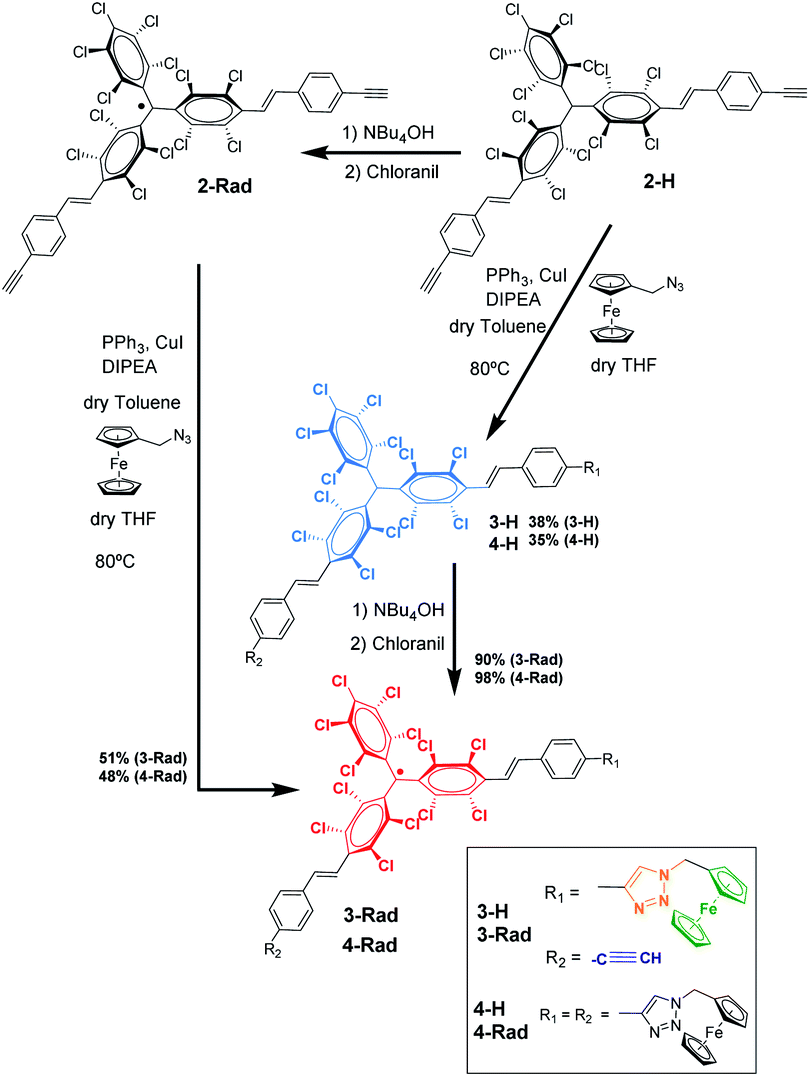 | ||
| Scheme 1 Synthetic route to the preparation of the target radical PTM–Fc dyad 3-Rad. | ||
For the synthesis of the target radical PTM–Fc dyad (3-Rad) a copper-catalyzed azide–alkyne cycloaddition (CuAAC) click chemistry strategy was envisaged.53–55 The standard conditions for the click reaction that generates in situ the copper(I) catalyst by reduction of CuSO4 using ascorbic acid or ascorbate56 could not be used here because the ascorbic acid reduces the radical to the anion that in turn is irreversibly protonated to the αH form.57 Therefore, as mentioned above, before performing the reaction on the surface, the synthetic procedure was optimized in solution. The successful synthesis of the target molecule 3-Rad with the highest reaction yield was obtained by using copper(I) iodide as a catalyst, and N-ethyldiisopropylamine (DIPEA) and triphenylphosphine as ligands (see ESI† for further details on the synthesis). As shown in Scheme 1, two routes were followed. First, the CuAAC reaction conditions starting with the non-radical precursor, αH-PTM molecules (2-H), were optimized. Both mono- and di-cycloaddition compounds, 3-H and 4-H, were obtained as products of the click reaction between the azidomethyl ferrocene and the PTM derivative with two acetylene-terminated groups (2-H) even when a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of the azide and 2-H was used. Next, the corresponding radicals 3-Rad and 4-Rad were prepared through treatment of the αH adducts, 3-H and 4-H, with tetra-n-butylammonium hydroxide (TBAOH), to generate the corresponding anions, and then the oxidation of these anions to the PTM radicals with p-chloranil. The second route consisted of directly coupling the open-shell compound, 2-Rad, with the azidomethyl ferrocene to obtain also the two cycloaddition compounds 3-Rad and 4-Rad with better yield than in the case of the non-radical derivative. All the compounds were fully characterized by spectroscopic techniques (NMR, FT-IR, and UV/vis), cyclic voltammetry and electron paramagnetic resonance for the radicals (see ESI†).
:1 molar ratio of the azide and 2-H was used. Next, the corresponding radicals 3-Rad and 4-Rad were prepared through treatment of the αH adducts, 3-H and 4-H, with tetra-n-butylammonium hydroxide (TBAOH), to generate the corresponding anions, and then the oxidation of these anions to the PTM radicals with p-chloranil. The second route consisted of directly coupling the open-shell compound, 2-Rad, with the azidomethyl ferrocene to obtain also the two cycloaddition compounds 3-Rad and 4-Rad with better yield than in the case of the non-radical derivative. All the compounds were fully characterized by spectroscopic techniques (NMR, FT-IR, and UV/vis), cyclic voltammetry and electron paramagnetic resonance for the radicals (see ESI†).
SAM-5-Rad was obtained by the on-surface post-modification of SAM-2-Rad. The determination of the Fc/PTM ratio for the SAM-5-Rad by SWV and its control were essential to (i) estimate the yield of the on-surface click reaction, (ii) improve the faradaic-background current ratio, and (iii) decrease both capacitive currents and the double layer impact. It is important to mention that there is an intrinsic error in the estimation of the Fc/PTM ratio due to the different electron transfer rates of the electrochemical processes of both redox components but the values were validated by comparison with those of SAM-3-Rad (Fig. S8†) in which the Fc/PTM ratio should be 1:1. Following the conditions used in solution, an ∼30% reaction yield (Fc vs. PTM radical) was obtained (Fig. S4 and S5†). This value was enhanced to approximately 50% when a toluene–THF mixture was changed for acetonitrile as the solvent (Fig. S6 and S7†). This gain was attributed to both the better solubility of CuI and stability of Cu(I) in solution. Other parameters such as temperature and/or reaction time did not lead to a higher Fc surface coverage. The bulkiness of the PTM moieties, which can induce certain disorder of the layer, as was observed before by STM for a previously reported thiolated PTM SAM,60 would be responsible for the yield decrease.
Fig. 4 shows the CV response of SAM-3-H, SAM-2-Rad, SAM-3-Rad and SAM-5-Rad at different scan rates. The stability of the different monolayers was demonstrated by performing consecutive voltammetric scans (Fig S9† to S14). Further, an Ipa/Ipc ratio very close to 1 and a Ipa, Ipcvs. scan rate linear relationship were determined, evidencing both the reversibility of the redox process and the confinement of the molecules on the surface (Fig. S15 and S16†). The reversible process observed at around −0.23 V (vs. Ag/AgCl, KCl 3 M) corresponds to the PTM(radical) ↔ PTM(anion) redox process while the redox signature of the Fc+/Fc couple is clearly visible at +0.60 V (vs. Ag/AgCl, KCl 3 M). It is worth noting that the redox potential of the PTM radical bound to gold was ca. 400 mV lower than that observed for SAM-1-Rad-Si under illumination. This trend is not surprising and is usually observed for electroactive molecule-modified photoelectrodes.29 In the case of silicon, the reduction of the PTM radical is easier owing to the photogenerated electron-induced activation of the redox process.61 As expected, for SAM-3-H and SAM-2-Rad, only a single redox process attributed to either Fc or the PTM radical, respectively, was observed. In the case of SAM-3-Rad and SAM-5-Rad, two redox processes were present owing to the double functionality of the grafted monolayer. Interestingly, the PTM radical surface coverage estimated for the SAM-3-Rad, which was obtained by direct Au functionalization with the PTM–Fc dyad 3-Rad, was very similar to that of the SAM-2-Rad (7.9 and 8.9 × 10−11 mol cm−2, respectively) (Table S2†). Such a result denotes that the self-organization and the reactivity of the molecules for both SAMs were quite similar. These values further support that only one of the terminal alkynes of the 2-Rad reacted with the Au surface.
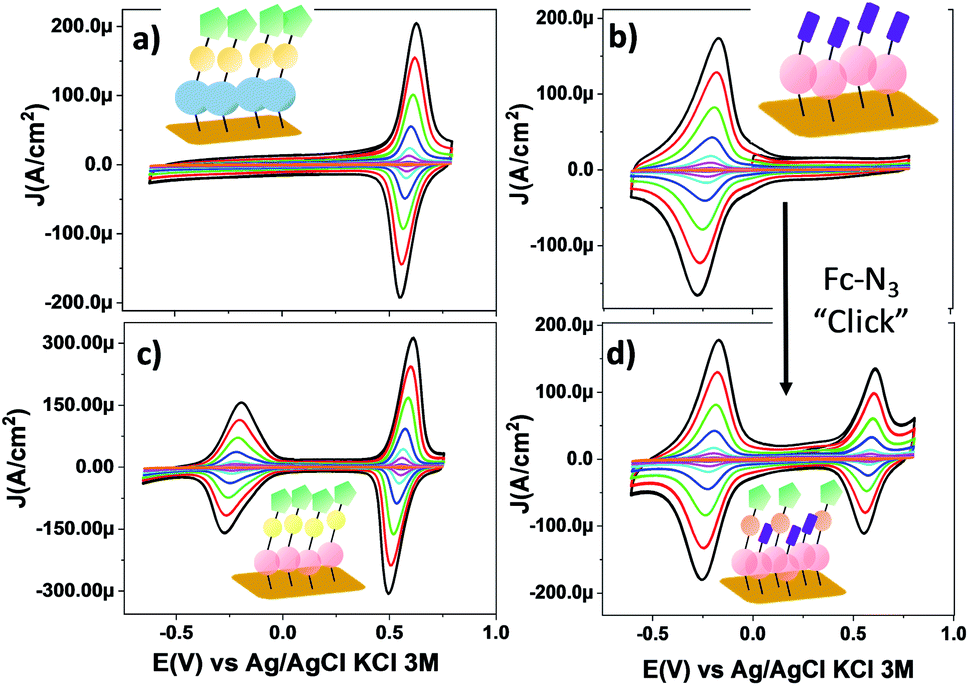 | ||
| Fig. 4 CV curves of SAMs on gold: (a) SAM-3-H, (b) SAM-2-Rad, (c) SAM-3-Rad and (d) SAM-5-Rad in CH2Cl2 + 0.2 M Bu4NPF6 at different scan rates (0.05, 0.1, 0.25, 0.5, 1, 2, 3 and 4 V s−1). The color code used for SAMs is the same as that used in Scheme 1. (Blue) PTM αH; (magenta) PTM radical; (yellow) triazole ring; (green) Fc and (violet) terminal alkyne moieties. | ||
To back up these findings, we investigated SAM-2-Rad and SAM-3-Rad by XPS62 which is an effective tool for studying organic radical thin films.63 It was found that the ratio of the integrated signal intensities of the different lines of the XPS curves agrees to a good extent with the theoretical percentages indicating that SAM-2-Rad and SAM-3-Rad were effectively obtained. The carbon concentration slightly exceeds the theoretical values (see Tables S3 and S4†). This is the usual case in samples prepared ex situ with wet-environment techniques. Molecular orbitals are highly directional and, consequently, they are perturbed if a different/new chemical interaction occurs. In this case, no deviation from the expected shapes was observed from the XPS main lines. Furthermore, a best fit procedure applied to the XPS spectra allows identifying the contributions from atomic sites having slightly different binding energies due to variations in the chemical environment.62 Several constraints based on electronegativity and bond strength were applied64,65(see ESI† for details). The comparison of the fitted results with the theoretical stoichiometry of the carbon atoms supports the successful preparation of SAM-3-Rad (Fig. S18† and Table S3†). In fact, any perturbation of the chemical environment of the carbon atoms that would hint at different chemical configurations would deliver a different best fit curve.
As depicted in the CVs shown in Fig. 4c and d, due to the presence of two electroactive moieties, an enhanced number of stable redox states can be realized, which is of interest for molecular memory devices. In particular, the PTM–Fc dyad displays two redox processes and hence, three distinct redox states, [PTManion–Fc]−, PTMradical–Fc and [PTMradical–Fc]+, in the −0.6 V to 0.8 V potential range. Such an electrochemical response prompted us to characterize SAM-3-Rad by EIS, using the applied potential as a perturbation signal and the capacitive real component as an output, characteristic of the interface at each redox state. This methodology was employed before in Fc-based and tetrathiafulvalene (TTF)-based SAMs by some of us.66,67 It is important to emphasize that the three redox states are well differentiated in a narrow and stable potential window which makes the system very appealing.68 The measurements were acquired in the frequency range from 100 kHz to 0.1 Hz with a 20 mV amplitude and the applied potentials were −0.5, 0.3 and 0.7 V. These values were chosen from the CV results to eliminate the contribution of the faradaic processes and thus to only consider the capacitive behaviour of the double layer. The Nyquist plots showed a common shape for a SAM-based interface with one semicircle without the diffusional process (Fig. S17†). The difference between the [PTManion–Fc]−, PTMradical–Fc and [PTMradical–Fc]+ species was evidenced in the Cole–Cole plot, where the imaginary capacitance (Cim) is plotted vs. the real capacitance(Cre)48 (Fig. 5a). The obtained Cre values (indicated in the plot) were: Cre1 (PTManion–Fc) = 6.5 μF cm−2, Cre2 (PTMradical–Fc) = 8.0 μF cm−2 and Cre3 (PTMradical–Fc+) = 12.8 μF cm−2.
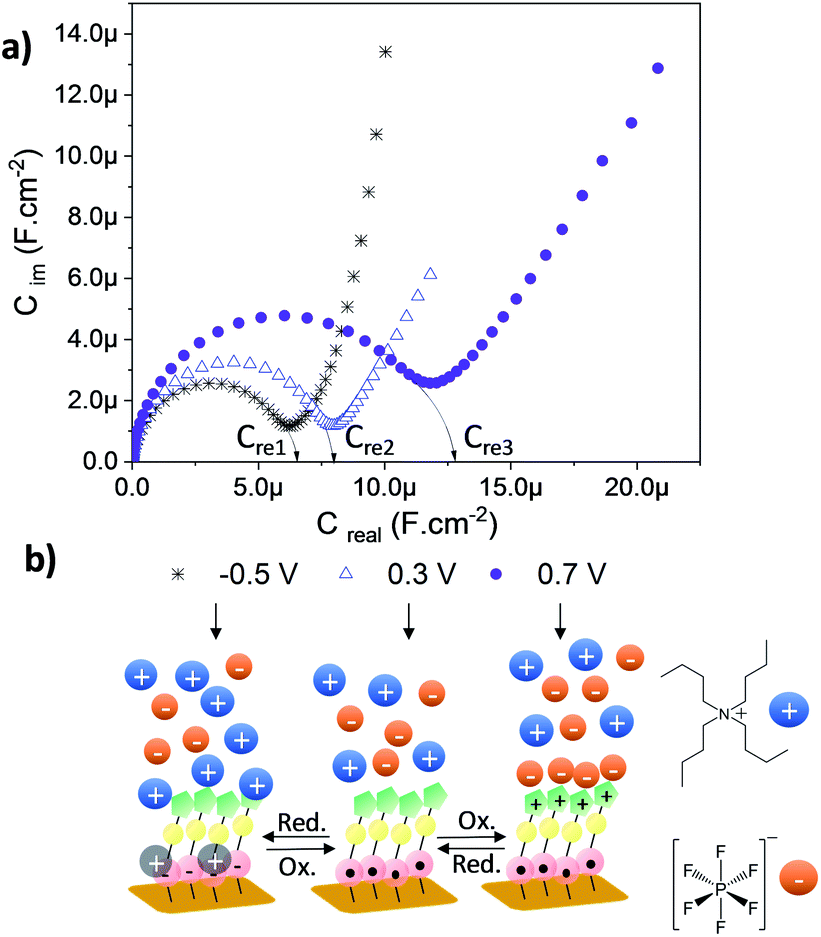 | ||
| Fig. 5 (a) Cole–Cole plots recorded for the SAM-3-rad at different applied potentials to obtain three different redox states. The electrolytic medium (CH2Cl2 + 0.2 M Bu4NPF6) was used and the frequency was varied from 100 kHz to 0.1 Hz with a 20 mV amplitude. (b) Scheme of the three different interfaces at the specific applied voltage. | ||
Clearly, the higher Cre value for the positively charged SAM (Cre3) is attributed to a well-defined charged double layer at the interface. The charged ferrocenium species are exposed to the top solid–electrolyte interface favoring the formation of a tight ion pair with the electrolyte anion (Fig. 5b, Cre3). In the case of the PTManion–Fc SAM, the negative charge of the PTM anion is located in the central carbon atom, which is shielded by the bulky chlorinated triphenyl rings. So, it forms a loose ion pair with the electrolyte cation.69 Therefore, this ill-defined double layer generated at the PTManion–Fc SAM/electrolyte interface is expected to be very similar to that of the neutral PTM–Fc SAM (Cre2).
Charge transport measurements across PTM monolayers bound to gold
Charge transport measurements across the AuTS/SAM-N-rad/liquid metal were performed in order to evaluate the SAMs as molecular wires, and to examine the influence of the D–A dyad. Moreover, the electrical output of the junction could be used to evaluate the second moiety introduced through the click chemistry on the PTM radicals (Fig. 6). The eutectic gallium indium alloy (EGaIn) was chosen as the top electrode,70,71 since it has been successfully used previously to investigate different PTM-based SAMs and films.72–74 The spontaneously formed oxide skin (mainly Ga2O3) allows the electrode to be shaped as a cone. In this work, the area of the GaOx/EGaIn tip that was brought into contact with the sample surfaces was around 1000–2500 μm2 in order to obtain stable measurements (see ESI† for details on the experimental setup).75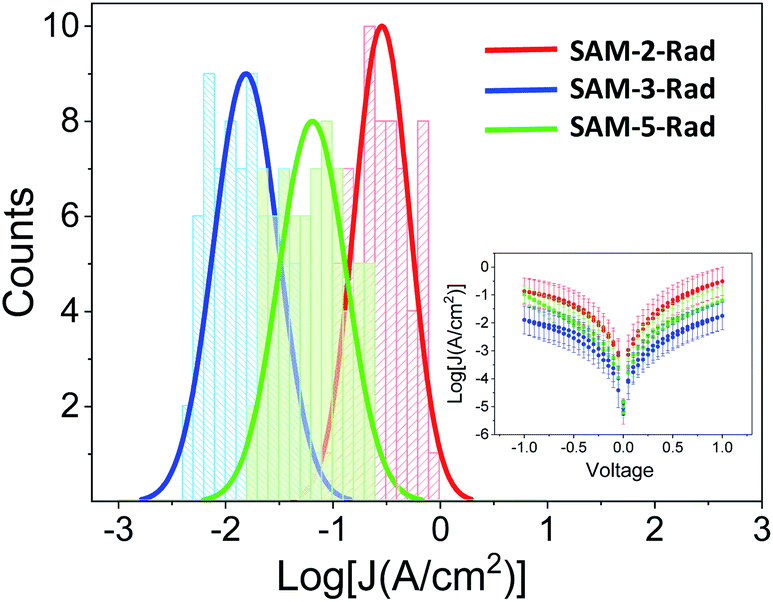 | ||
| Fig. 6 Histograms and Gaussian fits of log|J| at 1 V for junctions AuTS/SAM-2,3 and 5-Rad//GaOx/EGaIn. Inset: Plots of mean log|J| versus E(V). | ||
Fig. 6 shows that the three monolayers display a different molecular wire behaviour, in terms of the measured current through the junction, showing a current density (J) (SAM-2-Rad) > J (SAM-5-Rad) > J (SAM-3-Rad) (see ESI† for details on the statistical analysis). The differences between the three layers were mainly attributed to the different tunneling distances between the two electrodes arising from the thickness of the molecular layer. Taking into account that an approximately 50% of Fc incorporation was achieved to form SAM-5-Rad through the click reaction on SAM-2-Rad, the electrical response measured for this SAM was considered to be an average contribution of SAM-2-Rad and SAM-3-Rad. Current rectification was previously reported for SAMs incorporating Fc as the redox-active moiety.73,76 Here, in the ±1 V range, we did not observe such a behaviour (inset in Fig. 6). This could be attributed to a poor molecular order within the layer and hence a poor directionality of the Fc moiety towards the top electrode, compared to well-ordered Fc-thiolated SAMs presenting a well-defined interface with the liquid-metal electrode.76
XPS and Near Edge X-ray Absorption Fine Structure (NEXAFS) measurements helped to confirm this interpretation. NEXAFS measurements of SAM-5-Rad show a clear Fe signal (Fig. 7d), however, the survey XPS and the Fe 2p core level spectra at 800 eV photon energy (Fi. S18 in ESI†) are characterized by a signal that is proportional to a lower concentration of iron atoms in comparison with the theoretical stoichiometry. These findings can be taken as an indication that SAM-5-Rad was obtained, however, the monolayer has a mixed nature: also molecules that do not carry the ferrocene unit are present, i.e., SAM-2-Rad, and they contribute to the signal. NEXAFS measurements also allow determining the average molecular arrangement of the molecules in the films.77 The C–K edge of SAM-5-Rad shows typical features due to transitions from the C 1s core levels to the unoccupied states, π* and σ* (Fig. 7b). In analogy with previous NEXAFS measurements of derivatives of the PTM radical, two main regions can be identified in the spectra in Fig. 7: the π* region up to around 290 eV and the σ* region in the photon energy range above 290 eV.73,78,79 We can assign the feature at around 285.4 eV to contributions due to transitions from the C 1s core levels located in the carbon atoms of the aromatic rings and of the perchlorinated benzene rings to π* orbitals. The feature at 287.2 eV (see Fig. 7b, grazing incidence) is typical of ferrocene C–K NEXAFS spectra and is due to the C 1s to Fe 3d/π* transitions.80–82 We observe a shoulder at 285.9 eV that is also assigned to contributions belonging to the ferrocene moiety.80–82 The intensity at around 282.2 eV corresponds to transitions from the C 1s core levels to the singly unoccupied molecular orbital (SUMO).60
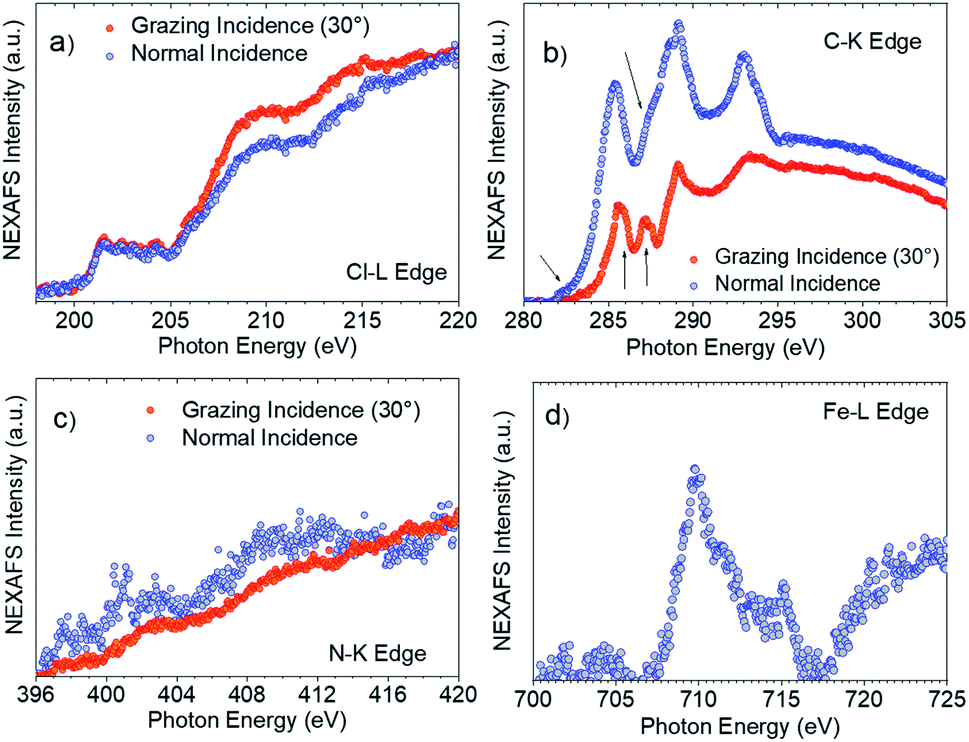 | ||
| Fig. 7 SAM-5-Rad (a) Cl–L edge, (b) C–K edge (the arrows indicate the transitions as discussed in the text) and (c) N–K edge in grazing (30° to the substrate, red circles) and normal incidences (blue circles). (d) Fe–L edge NEXAFS spectrum at grazing incidence. | ||
The determination of the average molecular orientation of SAM-5-Rad is very challenging, the Cl–L edge signal shows a very weak dichroism (Fig. 7a), and the resonances in the C–K edge spectra due to transitions from aromatic carbon atoms are all very close in energy (Fig. 7b). Thus, we have focused on the single building blocks. We have calculated an average molecular orientation of the ferrocene moiety of 22° with respect to the substrate using the intensities for the two polarizations at 285.9 eV and cross-checking with the intensities at 287.2 eV (21°).77,83,84 The relative torsion of the component blocks in SAM-5-Rad together with the presence of SAM-2-Rad might hinder a good interaction with the top contact and, thus, contribute to the absence of a current rectification behaviour.
Conclusions
In summary, we have demonstrated that the functionalization of electro- and magnetically-active perchlorotriphenylmethyl radicals with terminal alkynes clearly permits us to expand their applicability. It has been shown that using the alkyne group as the grafting unit, chemically bonded self-assembled monolayers can be successfully prepared both on hydrogenated silicon and on gold in one step and preserving the radical character. The electronic properties of Si have allowed us to fabricate a light-triggered capacitance switch exploiting the redox properties of the grafted radicals. In the case of gold, not only the formation of the SAM but also its post-modification with other functional molecules through click chemistry has demonstrated the potential of such a versatile platform for achieving multifunctional layers displaying the fascinating properties of the attached organic radicals. In particular, here a radical donor–acceptor dyad has been on-surface synthesized following optimized conditions that enable the spin to be unaltered. For all this, we believe that the findings reported in this manuscript are a significant step forward in the implementation of organic radicals in molecular based devices with different properties and applications in fields such as energy storage and conversion, sensing, imaging, memory devices, and spintronics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. Ababou-Girard and C. Meriadec (Institut de Physique de Rennes, UMR6251, France) are fully acknowledged for XPS measurements on the Si–H surfaces. Dr Vega Lloveras and Amable Bernabé are acknowledged for the EPR and MALDI-ToF measurements, respectively. We also would like to thank Thomas Chassé for allowing us to access the photoelectron laboratory at the University of Tübingen, Hilmar Adler for technical support, and Helmholtz-Zentrum Berlin (HZB) for providing beamtime at BESSY II. This work was funded by the Spanish Ministry project FANCYCTQ2016-80030-R, the Generalitat de Catalunya (2017SGR918) and the Spanish Ministry of Economy and Competitiveness, through the “Severo Ochoa” Programme for Centers of Excellence in R&D (SEV-2015-0496), the CSIC with the i-Link+ 2018 (Ref. LINKA20128) and CIBER-BBN. J. A. de S. is enrolled in the Materials Science PhD program of UAB. J.A. de S. thanks the Spanish Ministry for the FPI fellowship. Financial support from the German Research Foundation (DFG) under the contract CA852/11-1 and from Helmholtz-Zentrum Berlin is gratefully acknowledged.Notes and references
- K. T. Butler, G. Sai Gautam and P. Canepa, npj Comput. Mater., 2019, 19 DOI:10.1038/s41524-019-0160-9.
- M. S. Maglione, S. Casalini, S. Georgakopoulos, M. Barbalinardo, V. Parkula, N. Crivillers, C. Rovira, P. Greco and M. Mas-Torrent, Small, 2018, 14, 1703344–1703351 CrossRef PubMed.
- X. Su and T. A. Hatton, Phys. Chem. Chem. Phys., 2017, 19, 23570–23584 RSC.
- C. Jia and X. Guo, Chem. Soc. Rev., 2013, 42, 5642–5660 RSC.
- R. M. Arnold, N. E. Huddleston and J. Locklin, J. Mater. Chem., 2012, 22, 19357–19365 RSC.
- G. A. Hudalla and W. L. Murphy, Soft Matter, 2011, 7, 9561–9571 RSC.
- S. Hosseinpour, M. Forslund, C. M. Johnson, J. Pan and C. Leygraf, Surf. Sci., 2016, 648, 170–176 CrossRef CAS.
- D. Xiang, X. Wang, C. Jia, T. Lee and X. Guo, Chem. Rev., 2016, 116, 4318–4440 CrossRef CAS PubMed.
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Nature, 2018, 563, 536–540 CrossRef CAS PubMed.
- H. Guo, Q. Peng, X.-K. Chen, Q. Gu, S. Dong, E. W. Evans, A. J. Gillett, X. Ai, M. Zhang, D. Credgington, V. Coropceanu, R. H. Friend, J.-L. Brédas and F. Li, Nat. Mater., 2019, 18, 977–984 CrossRef CAS PubMed.
- D. A. Wilcox, V. Agarkar, S. Mukherjee and B. W. Boudouris, Annu. Rev. Chem. Biomol. Eng., 2018, 9, 83–103 CrossRef PubMed.
- Y. Morita, S. Nishida, T. Murata, M. Moriguchi, A. Ueda, M. Satoh, K. Arifuku, K. Sato and T. Takui, Nat. Mater., 2011, 10, 947–951 CrossRef CAS PubMed.
- M. Mas-Torrent, N. Crivillers, V. Mugnaini, I. Ratera, C. Rovira and J. Veciana, J. Mater. Chem., 2009, 19, 1691–1695 RSC.
- M. A. Sowers, J. R. Mccombs, Y. Wang, J. T. Paletta, S. W. Morton, E. C. Dreaden, M. D. Boska, M. Francesca Ottaviani, P. T. Hammond, A. Rajca and J. A. Johnson, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- M. Hou, X. Lu, Z. Zhang, Q. Xia, C. Yan, Z. Yu, Y. Xu and R. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 44316–44323 CrossRef CAS PubMed.
- G. Seber, J. Muñoz, S. Sandoval, C. Rovira, G. Tobias, M. Mas-Torrent and N. Crivillers, Adv. Mater. Interfaces, 2018, 5, 1701072 CrossRef.
- C. Simão, M. Mas-Torrent, N. Crivillers, V. Lloveras, J. M. Artés, P. Gorostiza, J. Veciana and C. Rovira, Nat. Chem., 2011, 3, 359–364 CrossRef PubMed.
- C. Simão, M. Mas-Torrent, J. Veciana and C. Rovira, Nano Lett., 2011, 11, 4382–4385 CrossRef PubMed.
- J. Lee, E. Lee, S. Kim, G. S. Bang, D. A. Shultz, R. D. Schmidt, M. D. E. Forbes and H. Lee, Angew. Chem., Int. Ed., 2011, 50, 4414–4418 CrossRef CAS PubMed.
- M. Mas-Torrent, J. Veciana, N. Crivillers, C. Rovira and J. Veciana, Chem. Rev., 2012, 112, 2506–2527 CrossRef CAS PubMed.
- M. Halik and A. Hirsch, Adv. Mater., 2011, 23, 2689–2695 CrossRef CAS PubMed.
- M. Mas-Torrent, C. Rovira and J. Veciana, Adv. Mater., 2013, 25, 462–468 CrossRef CAS PubMed.
- T. Zaba, A. Noworolska, C. M. Bowers, B. Breiten, G. M. Whitesides and P. Cyganik, J. Am. Chem. Soc., 2014, 136, 11918–11921 CrossRef CAS PubMed.
- S. Zhang, K. L. Chandra and C. B. Gorman, J. Am. Chem. Soc., 2007, 129, 4876–4877 CrossRef CAS PubMed.
- F. Bejarano, I. J. Olavarria-Contreras, A. Droghetti, I. Rungger, A. Rudnev, D. Gutiérrez, M. Mas-Torrent, J. Veciana, H. S. J. Van Der Zant, C. Rovira, E. Burzurĺ and N. Crivillers, J. Am. Chem. Soc., 2018, 140, 1691–1696 CrossRef CAS PubMed.
- J. M. Buriak, Chem. Rev., 2002, 102, 1271–1308 CrossRef CAS PubMed.
- S. Ciampi, J. B. Harper and J. J. Gooding, Chem. Soc. Rev., 2010, 39, 2158–2183 RSC.
- B. Fabre, Chem. Rev., 2016, 116, 4808–4849 CrossRef PubMed.
- J. Li, P. S. Thiara and M. Mrksich, Langmuir, 2007, 23, 11826–11835 CrossRef CAS PubMed.
- J. P. Collman, N. K. Devaraj and C. E. D. Chidsey, Langmuir, 2004, 20, 1051–1053 CrossRef CAS PubMed.
- T. Klasen, T. Winkler, R. Chelmowski, A. Terfort, S. D. Köster, C. Wöll, N. Metzler-Nolte and D. Käfer, Langmuir, 2009, 25, 11480–11485 CrossRef PubMed.
- T. Lummerstorfer and H. Hoffmann, J. Phys. Chem. B, 2004, 108, 3963–3966 CrossRef CAS.
- J. Mehlich and B. J. Ravoo, Org. Biomol. Chem., 2011, 9, 4108–4115 RSC.
- M. Ballester, J. Riera, J. Castafier, C. Badía and J. M. Monsó, J. Am. Chem. Soc., 1971, 93, 2215–2225 CrossRef CAS.
- B. Fabre, Y. Li, L. Scheres, S. P. Pujari and H. Zuilhof, Angew. Chem., Int. Ed., 2013, 52, 12024–12027 CrossRef CAS PubMed.
- M. H. Choudhury, S. Ciampi, Y. Yang, R. Tavallaie, Y. Zhu, L. Zarei, V. R. Gonçales and J. J. Gooding, Chem. Sci., 2015, 6, 6769–6776 RSC.
- L. Zhang, Y. B. Vogel, B. B. Noble, V. R. Gonçales, N. Darwish, A. Le Brun, J. J. Gooding, G. G. Wallace, M. L. Coote and S. Ciampi, J. Am. Chem. Soc., 2016, 138, 9611–9619 CrossRef CAS PubMed.
- D. Zigah, C. Herrier, L. Scheres, M. Giesbers, B. Fabre, P. Hapiot and H. Zuilhof, Angew. Chem., Int. Ed., 2010, 49, 3157–3160 CrossRef CAS PubMed.
- L. Scheres, J. Ter Maat, M. Giesbers and H. Zuilhof, Small, 2010, 6, 642–650 CrossRef CAS PubMed.
- J. M. Buriak, Chem. Mater., 2014, 26, 763–772 CrossRef CAS.
- W. Peng, W. J. I. Debenedetti, S. Kim, M. A. Hines and Y. J. Chabal, Appl. Phys. Lett., 2014, 104, 241601 CrossRef.
- K. Uno, A. Namiki, S. Zaima, T. Nakamura and N. Ohtake, Surf. Sci., 1988, 193, 321–335 CrossRef CAS.
- X. G. Zhang, Electrochemistry of Silicon and Its Oxide, 2004, Kluwer Acad., New York Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods; Fundamentals and Applications, John Wiley & Sons, Ltd, 2002 Search PubMed.
- Y. B. Vogel, A. Molina, J. Gonzalez and S. Ciampi, Anal. Chem., 2019, 91, 5929–5937 CrossRef CAS PubMed.
- A. W. Bott and D. Ph, Curr. Sep., 1998, 17, 87–91 CAS.
- P. R. Bueno, F. Fabregat-Santiago and J. J. Davis, Anal. Chem., 2013, 85, 411–417 CrossRef CAS PubMed.
- J. Gonzalez and A. Molina, J. Electroanal. Chem., 2003, 557, 157–165 CrossRef CAS.
- B. Fabre, Acc. Chem. Res., 2010, 43, 1509–1518 CrossRef CAS PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572–2590 CrossRef CAS.
- L. Zhu, C. J. Brassard, X. Zhang, P. M. Guha and R. J. Clark, Chem. Rec., 2016, 16, 1501–1517 CrossRef CAS PubMed.
- D. N. Barsoum, N. Okashah, X. Zhang and L. Zhu, J. Org. Chem., 2015, 80, 9542–9551 CrossRef CAS PubMed.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- M. Meldal and C. W. Tomøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- J. Castañer, M. Ballester and J. Riera, Tetrahedron Lett., 1978, 20, 643–644 Search PubMed.
- E. A. Weiss, G. K. Kaufman, J. K. Kriebel, Z. Li, R. Schalek and G. M. Whitesides, Langmuir, 2007, 23, 9686–9694 CrossRef CAS PubMed.
- L. Yuan, L. Jiang, B. Zhang and C. A. Nijhuis, Angew. Chem., Int. Ed., 2014, 53, 3377–3381 CrossRef CAS PubMed.
- A. V Rudnev, C. Franco, N. Crivillers, G. Seber, A. Droghetti, I. Rungger, I. V Pobelov, J. Veciana, M. Mas-Torrent and C. Rovira, Phys. Chem. Chem. Phys., 2016, 18, 27733–27737 RSC.
- P. G. Santangelo, G. M. Miskelly and N. S. Lewis, J. Phys. Chem., 1988, 92, 6359–6367 CrossRef CAS.
- K. Siegbahn, C. Nordling, A. Fahlman, R. Nordberg, K. Hamrin, J. Hedman, G. Johansson, T. Bergmark, S.-E. Karlsson, I. Lindgren and B. Lindberg, ESCA: Atomic, Molecular, and Solid State Structured Studied by Means Electron Spectroscopy, Almqvist and WiksellsBoktryckeri AB, Uppsala, 1967 Search PubMed.
- M. B. Casu, Acc. Chem. Res., 2018, 51, 753–760 CrossRef CAS PubMed.
- S. A. Savu, I. Biswas, L. Sorace, M. Mannini, D. Rovai, A. Caneschi, T. Chassé and M. B. Casu, Chem.–Eur. J., 2013, 19, 3445–3450 CrossRef CAS PubMed.
- S. A. Savu, M. B. Casu, S. Schundelmeier, S. Abb, C. Tönshoff, H. F. Bettinger and T. Chassé, RSC Adv., 2012, 2, 5112–5118 RSC.
- E. Marchante, M. S. Maglione, N. Crivillers, C. Rovira and M. Mas-Torrent, RSC Adv., 2017, 7, 5636–5641 RSC.
- E. Marchante, N. Crivillers, M. Buhl, J. Veciana and M. Mas-Torrent, Angew. Chem., Int. Ed., 2016, 55, 368–372 CrossRef CAS PubMed.
- J. Casado-Montenegro, E. Marchante, N. Crivillers, C. Rovira and M. Mas-Torrent, ChemPhysChem, 2016, 17, 1810–1814 CrossRef CAS PubMed.
- J. Veciana, J. Riera, J. Castañer and N. Ferrer, J. Organomet. Chem., 1985, 297, 131–141 CrossRef CAS.
- W. F. Reus, M. M. Thuo, N. D. Shapiro, C. A. Nijhuis and G. M. Whitesides, ACS Nano, 2012, 6, 4806–4822 CrossRef CAS PubMed.
- C. A. Nijhuis, W. F. Reus and G. M. Whitesides, J. Am. Chem. Soc., 2009, 131, 17814–17827 CrossRef CAS PubMed.
- L. Yuan, C. Franco, N. Crivillers, M. Mas-Torrent, L. Cao, C. S. S. Sangeeth, C. Rovira, J. Veciana and C. A. Nijhuis, Nat. Commun., 2016, 7, 12066–12075 CrossRef CAS PubMed.
- M. Souto, V. Díez-Cabanes, L. Yuan, A. R. Kyvik, I. Ratera, C. A. Nijhuis, J. Cornil and J. Veciana, Phys. Chem. Chem. Phys., 2018, 20, 25638–25647 RSC.
- D. Gutiérrez, S. Riera-Galindo, M. R. Ajayakumar, J. Veciana, C. Rovira, M. Mas-Torrent and N. Crivillers, J. Phys. Chem. C, 2018, 122, 17784–17791 CrossRef.
- F. C. Simeone, H. J. Yoon, M. M. Thuo, J. R. Barber, B. Smith and G. M. Whitesides, J. Am. Chem. Soc., 2013, 135, 18131–18144 CrossRef CAS PubMed.
- L. Yuan, N. Nerngchamnong, L. Cao, H. Hamoudi, E. Del Barco, M. Roemer, R. K. Sriramula, D. Thompson and C. A. Nijhuis, Nat. Commun., 2015, 6, 6324–6334 CrossRef CAS PubMed.
- J. Stohr, NEXAFS Spectroscopy, Springer, 2003 Search PubMed.
- O. Shekhah, N. Roques, V. Mugnaini, C. Munuera, C. Ocal, J. Veciana and C. Wöll, Langmuir, 2008, 24, 6640–6648 CrossRef CAS PubMed.
- V. Mugnaini, A. Calzolari, R. Ovsyannikov, A. Vollmer, M. Gonidec, I. Alcon, J. Veciana and M. Pedio, J. Phys. Chem. Lett., 2015, 6, 2101–2106 CrossRef CAS PubMed.
- E. Otero, R. G. Wilks, T. Regier, R. I. R. Blyth, A. Moewes and S. G. Urquhart, J. Phys. Chem. A, 2008, 112, 624–634 CrossRef CAS PubMed.
- A. Shaporenko, K. Rössler, H. Lang and M. Zharnikov, J. Phys. Chem. B, 2006, 110, 24621–24628 CrossRef CAS PubMed.
- S. Watcharinyanon, E. Moons and L. S. O. Johansson, J. Phys. Chem. C, 2009, 113, 1972–1979 CrossRef CAS.
- M. B. Casu, A. Schöll, K. R. Bauchspiess, D. Hübner, T. Schmidt, C. Heske and E. Umbach, J. Phys. Chem. C, 2009, 113, 10990–10996 CrossRef CAS.
- M. B. Casu, P. Cosseddu, D. Batchelor, A. Bonfiglio and E. Umbach, J. Chem. Phys., 2008, 128, 74702 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details and characterization of the prepared compounds, details of the preparation of SAMs on Si–H and on Au, additional electrochemical characterization of SAMs, survey and XPS spectra of SAM-3-Rad and SAM-2-Rad together with their stoichiometric and experimental elemental ratios and instrumentation. See DOI: 10.1039/c9sc04499j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |