
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular assembly of bent dinuclear amphiphilic alkynylplatinum(II) terpyridine complexes: diverse nanostructures through subtle tuning of the mode of molecular stacking†
Sam Ka-Ming
Leung
,
Alan Kwun-Wa
Chan
,
Sammual Yu-Lut
Leung
,
Ming-Yi
Leung
and
Vivian Wing-Wah
Yam
 *
*
Institute of Molecular Functional Materials (Areas of Excellence Scheme, University Grants Committee (Hong Kong)), Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong. E-mail: wwyam@hku.hk
First published on 27th November 2019
Abstract
A new class of bent amphiphilic alkynylplatinum(II) terpyridine complexes was found to adopt different modes of molecular stacking to give diverse nanostructures. In chlorinated solvents, the complexes aggregate in the presence of water droplets and assist in the formation of porous networks, while in DMSO solutions, they self-assemble to give fibrous nanostructures. The complexes would adopt a head-to-tail tetragonal stacking arrangement, as revealed by X-ray crystallographic studies, computational studies and powder X-ray diffraction (PXRD) studies. Their self-assembly follows a cooperative growth mechanism in DMSO and an isodesmic growth mechanism in DMSO–H2O mixture. The balance between hydrophobic and hydrophilic components of the complex system, in conjunction with the nuclearity and the positioning of the substituents, are found to govern the mode of molecular stacking and the fabrication of precise functional nanostructures. This class of complexes serve as versatile building blocks to construct orderly packed molecular materials and functional materials in a well-controlled manner.
Introduction
Investigations into d8 square-planar platinum(II) complexes with polydentate π-conjugated ligands have been receiving tremendous attention because of their unique photophysical and luminescence properties, along with their propensity to self-assemble intermolecularly with the assistance of Pt⋯Pt and π–π stacking interactions.1–18 In the literature, there exist a number of strategies for inducing self-assembly by noncovalent interactions of this type19–21 to give various nanostructures,22–30 providing potential applications in materials science.Among the nanostructures reported, ordered porous networks, which are considered as one of the most important structures in nanotechnology, have found their utility in catalysis,31 enantiomer sieving,31 molecular recognition,31,32 surface adsorption,32 membrane channels33 and lithium storage34 by rational molecular design.31 Therefore, exploring ways of producing ordered porous networks and manipulating pore sizes to perform a certain function are of paramount significance. One of the approaches to fabricating regular porous networks is by breath figure,35 in which water droplets condense onto a substrate containing a solution capable of stabilizing the water droplets and facilitate the formation of an array of pores before evaporating.36,37 For successful preparation of the porous network, the humidity should be high enough for the water droplets to condense onto the surface of the substrate, and volatile solvents, such as carbon disulfide, chloroform, dichloromethane, etc. should be utilized such that they evaporate easily and thereby providing a cold surface for condensation of the water droplets.36 As for the materials dissolved in the volatile solvent and cast onto the substrate, polymers are usually chosen, but it has been found recently that amphiphilic small molecules with the capability to aggregate are an equally viable candidate for the preparation of porous networks.36 Generally, with this method, porous networks of a greater pore size are developed.36
Another strategy is by molecular stacking, which makes use of molecules of rationally designed shapes and angles to form the desired porous network structure.31,32 For instance, rigid hexagonal macrocycles and bent molecules have been used to form porous network through dimeric stacks,31 or through electrostatic spreading of pre-assembled macrocycles.32 There have also been reports where metal coordination plays a role to assist formation of monomeric pores before aggregation to form a porous network by molecular stacking.38,39 With this method, the dimension of the pores is related to that of the molecules used,31,32,38,39 and specific side chains can be easily attached to the rigid backbone to give special functions to the porous network.31
Although there have been examples of porous networks resulted from transition metal complexes38–42 and metal–organic frameworks (MOFs),43–50 along with those from covalent organic frameworks (COFs),51–54 fabrication of extensive porous structures driven by supramolecular assembly of organometallic compounds, together with synthesis of organometallic compounds which are capable of fabricating both porous structures and other nanostructures, has been relatively less explored.55 Herein, a series of bent alkynylplatinum(II) terpyridine complexes of rich luminescent properties 1–9 (Scheme 1) has been demonstrated to display diverse nanostructures, from porous networks to nanofibers, by a subtle change in solvent composition or a slight, rational structural modification. In terms of molecular design, among 1–4, which contain hydrophilic groups in the core meta-phenylene ethynylene (mPE) moiety and hydrophobic groups attached to the peripheral terpyridine units, the extents of hydrophilicity and hydrophobicity are varied to study their effects on the self-assembly of this class of complexes. Compared to 1–4, 5 and 6 are designed to have the positions of the hydrophilic and the hydrophobic groups exchanged to allow for the study of the effect of the positioning of different groups on the spectroscopic and supramolecular assembly properties. 7–9 serve as control complexes to highlight the importance of nuclearity, hydrophobicity and hydrophilicity in the process of molecular stacking and aggregation. Further guiding principles of the design and construction of supramolecular materials with functional properties, which are of high significance in molecular engineering, are expected to result from such investigations.
 | ||
| Scheme 1 Structures of a series of mono- and dinuclear alkynylplatinum(II) terpyridine complexes 1–9. | ||
Results and discussion
Synthesis and characterization
Complexes 1–9 were synthesized by treating chloroplatinum(II) terpyridine complexes with the corresponding alkynyl ligands through copper(I)-catalyzed dehydrohalogenation reactions in dichloromethane solutions. They were purified by column chromatography, followed by recrystallization by diffusion of diethyl ether into concentrated dichloromethane solutions of the respective complexes (see the ESI and Scheme S1 for the detailed procedures†). All the complexes were characterized by 1H NMR spectroscopy and high-resolution ESI mass spectrometry.X-Ray crystal structure and powder X-ray diffraction analysis
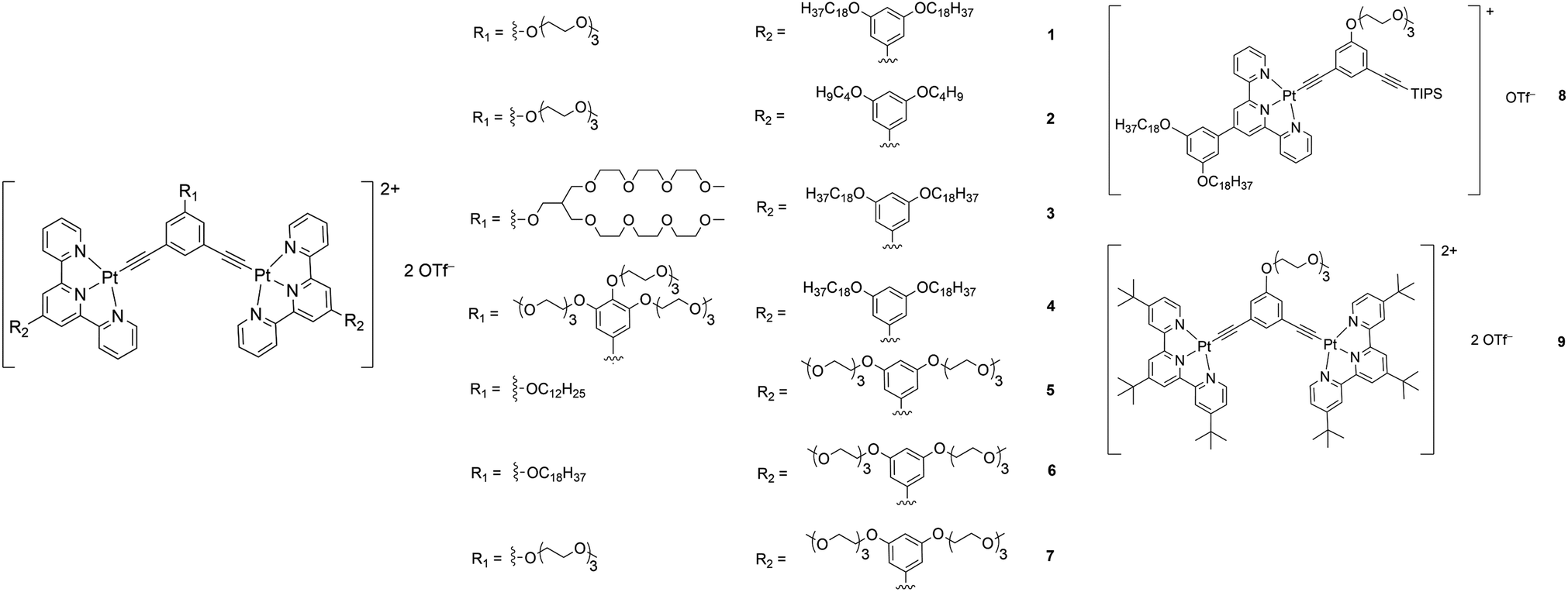
Recrystallization by diffusion of diethyl ether into a concentrated dichloromethane solution of 9 afforded yellow crystals suitable for X-ray crystallographic studies. The perspective drawing of 9 is depicted in Fig. S1a,† while the crystal packing diagrams are shown in Fig. S1b and c.† The crystal structure determination data are summarized in Table S1,† while selected bond lengths and angles are given in Table S2.† The platinum(II) metal center adopts a distorted square-planar geometry. Between pairs of complex molecules, a head-to-tail stacking arrangement is observed. The Pt⋯Pt distances were determined to be greater than 4 Å, which reveals the absence of Pt⋯Pt interactions. This can be justified by the steric hindrance brought about by the tert-butyl groups on the terpyridine moieties. It is expected that without the sterically bulky tert-butyl groups, Pt⋯Pt interactions could be observed. The interplanar separation between the phenyl ring of the core mPE unit and the plane of platinum terpyridine was determined to be 3.398 Å, indicating the presence of π–π interactions. It is noteworthy that the head-to-tail stacking arrangement of this class of bent complexes facilitates the formation of porous networks, as opposed to the head-to-head stacking arrangement, which is expected to form linear chains.Powder X-ray diffraction (PXRD) experiment was performed on 1, 3 and 6 (Fig. 1 and S2†). It was found that in the PXRD patterns of all the three complexes, a peak at d-spacing of 0.34 nm was observed, suggesting the existence of intermolecular π–π stacking and Pt⋯Pt interactions between the adjacent molecules in the solid state. More importantly, analysis of the X-ray diffraction pattern of 1 reveals d-spacings at 2.15, 1.75, 1.61, 0.93 and 0.63 nm with a ratio of 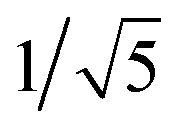
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: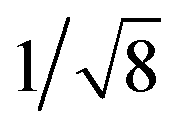 :1/3:
:1/3: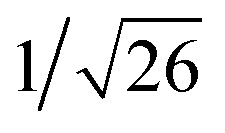 :
: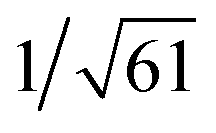 , which are characteristic of a tetragonal packing in the self-assembled architectures, whereas analysis from that of 3 affords d-spacings at 2.27, 1.96, 1.67, 0.83 and 0.58 nm, with a ratio of 1/3:
, which are characteristic of a tetragonal packing in the self-assembled architectures, whereas analysis from that of 3 affords d-spacings at 2.27, 1.96, 1.67, 0.83 and 0.58 nm, with a ratio of 1/3: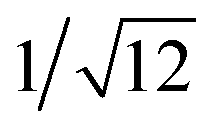 :
: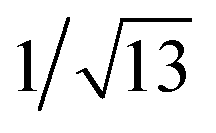 :
: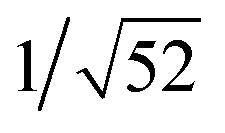 :
: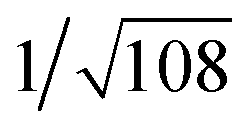 , which are characteristic of a hexagonal packing in the self-assembled architectures. The lattice packing parameters a were determined to be 4.81 and 7.86 nm for 1 and 3, respectively. The diffraction peaks from the X-ray diffraction patterns of 1 and 3 (Fig. 1) have also been indexed.
, which are characteristic of a hexagonal packing in the self-assembled architectures. The lattice packing parameters a were determined to be 4.81 and 7.86 nm for 1 and 3, respectively. The diffraction peaks from the X-ray diffraction patterns of 1 and 3 (Fig. 1) have also been indexed.
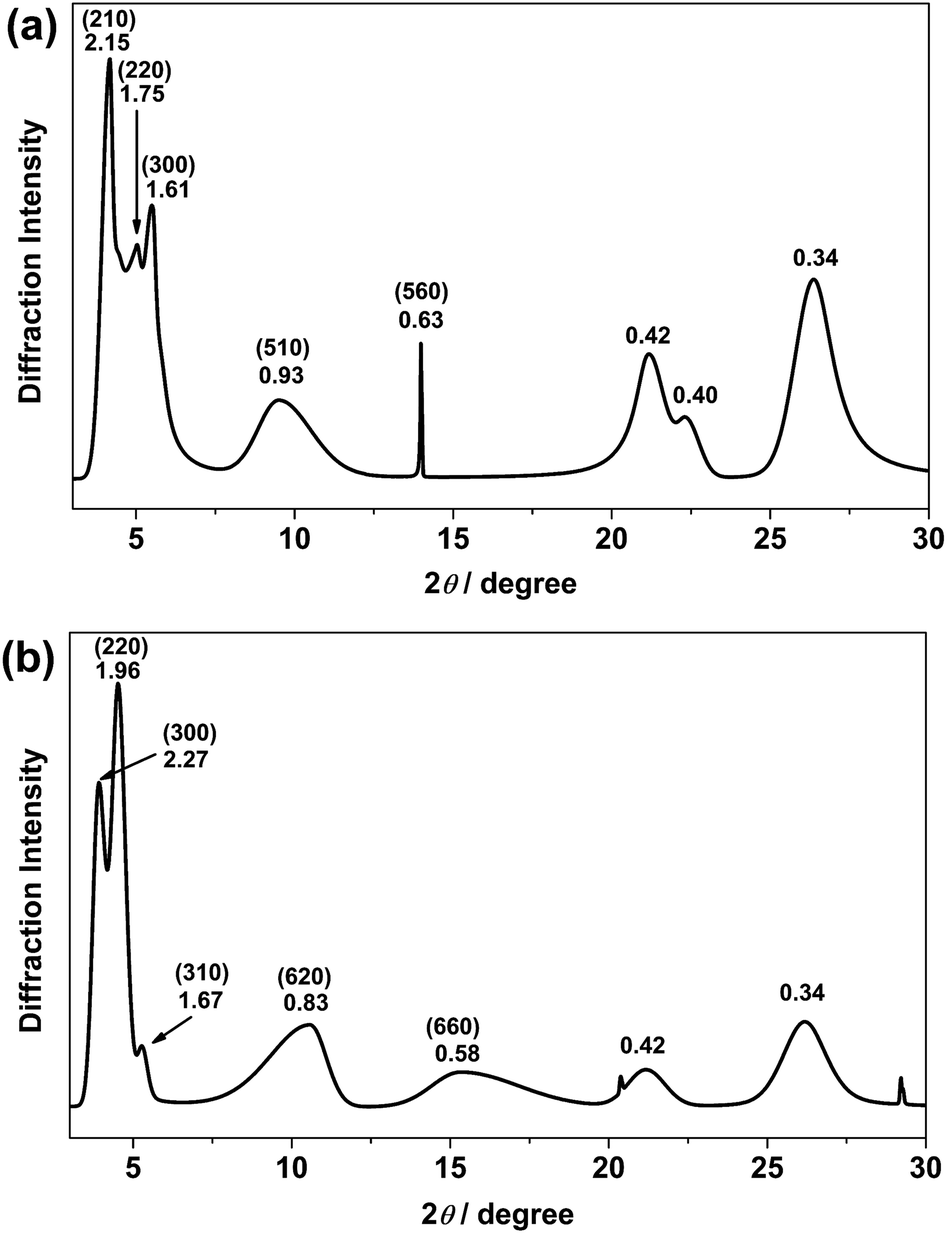 | ||
| Fig. 1 PXRD patterns on the bulk samples of (a) 1 and (b) 3. Numerical values indicate d-spacings (in nm). Values in parentheses are Miller indices. | ||
Self-assembly and morphological studies in chlorinated solvents
The yellow to orange solutions of 1–9 in dichloromethane were subjected to UV-vis absorption and emission studies. Their photophysical data are summarized in Table S3.† In general, at [Pt] = ∼10−5 M, low-energy absorption bands, assignable to a mixture of metal-to-ligand charge transfer (MLCT) and ligand-to-ligand charge transfer (LLCT) transitions, together with structureless 3MLCT/3LLCT emission bands, are observed (Fig. S3 and S4†).17,18 The nanosecond transient absorption spectrum of 1 (Fig. S5†), which shows an excited-stated absorption band corresponding to the terpyridine radical anion, further confirms the assignment of the 3MLCT/3LLCT excited state.56–61 Interestingly, at [Pt] = ∼10−3 M, upon increasing concentration from 10−5 to 10−3 M, the color of the complex changes from yellow to red, suggesting the aggregation of the complex. This observation contrasts with the mononuclear complex 8, which was observed to be yellow in color in dichloromethane at [Pt] = ∼10−3 M. In NMR studies, 1 gave very broad 1H NMR signals in CDCl3 solution, even at high temperature (Fig. S6†), contrary to 8, which gave well-resolved 1H NMR signals in CDCl3 solution suitable for characterization. In addition, 1 and 8 were subjected to concentration-dependent UV-vis absorption and emission studies in dichloromethane solutions. While the UV-vis absorption spectra of both complexes were observed to obey Beer's law in the concentration range from 10−5 to 10−3 M (Fig. S7 and S8†), the emission studies showed that upon increasing concentration, a red shift in the structureless emission band maximum from 640 nm to 728 nm was observed in 1 (Fig. S9†), which is absent in 8 (Fig. S10†). Furthermore, the excitation spectra of the two emission bands were found to be different (Fig. S11†), suggesting that its emission origin changes and that the lower-energy emission band originates from ground-state aggregation instead of formation of excimers. Therefore, significant ground-state aggregation of the dinuclear complex 1 exists in chlorinated solvents at high concentrations, with only a small association constant leading to the inability to observe deviation from Beer's law in the less sensitive UV-vis absorption studies. The aggregation may be contributed by Pt⋯Pt and π–π stacking interactions of the platinum(II) complexes upon increasing concentration, resulting in the triplet metal–metal-to-ligand charge transfer (MMLCT) emission in the near-infra-red region.To investigate the morphologies of this class of complexes in chlorinated solvents, TEM studies were conducted for 1 and 8 prepared form their dichloromethane solutions. The studies were performed by drop-casting the solution onto a pure carbon TEM grid and allowing the solvent to evaporate. Interestingly, a porous network structure was observed upon evaporating a dichloromethane solution of 1 ([Pt] = ∼10−4 M) (Fig. 2a). On the contrary, upon evaporating a dichloromethane solution of the mononuclear complex 8, no well-defined morphologies could be observed. The formation of a porous network was further confirmed by SEM (Fig. 2b) and AFM (Fig. 2c) studies. Based on the TEM and SEM images, the side length of each pore was estimated to be 1.6 ± 0.3 μm, while the pore size was estimated to be between 1 and 2 μm. Based on the height profile obtained from the AFM studies (Fig. 2d), the thickness of the layer was estimated to be 450 ± 50 nm. To study the effect of concentration on the development of the structure, TEM studies were conducted with dichloromethane solutions of 1 of [Pt] = ∼10−3 M and of [Pt] = ∼10−5 M. While no well-defined morphologies were observed for the solution of [Pt] = ∼10−5 M, some pores were observed for the solution of [Pt] = ∼10−3 M, but they were not regularly spread onto the surface of the grid (Fig. S12a†). Therefore, fabrication of porous networks by dichloromethane solutions of this class of complexes is dependent on the concentration of the solution prepared.
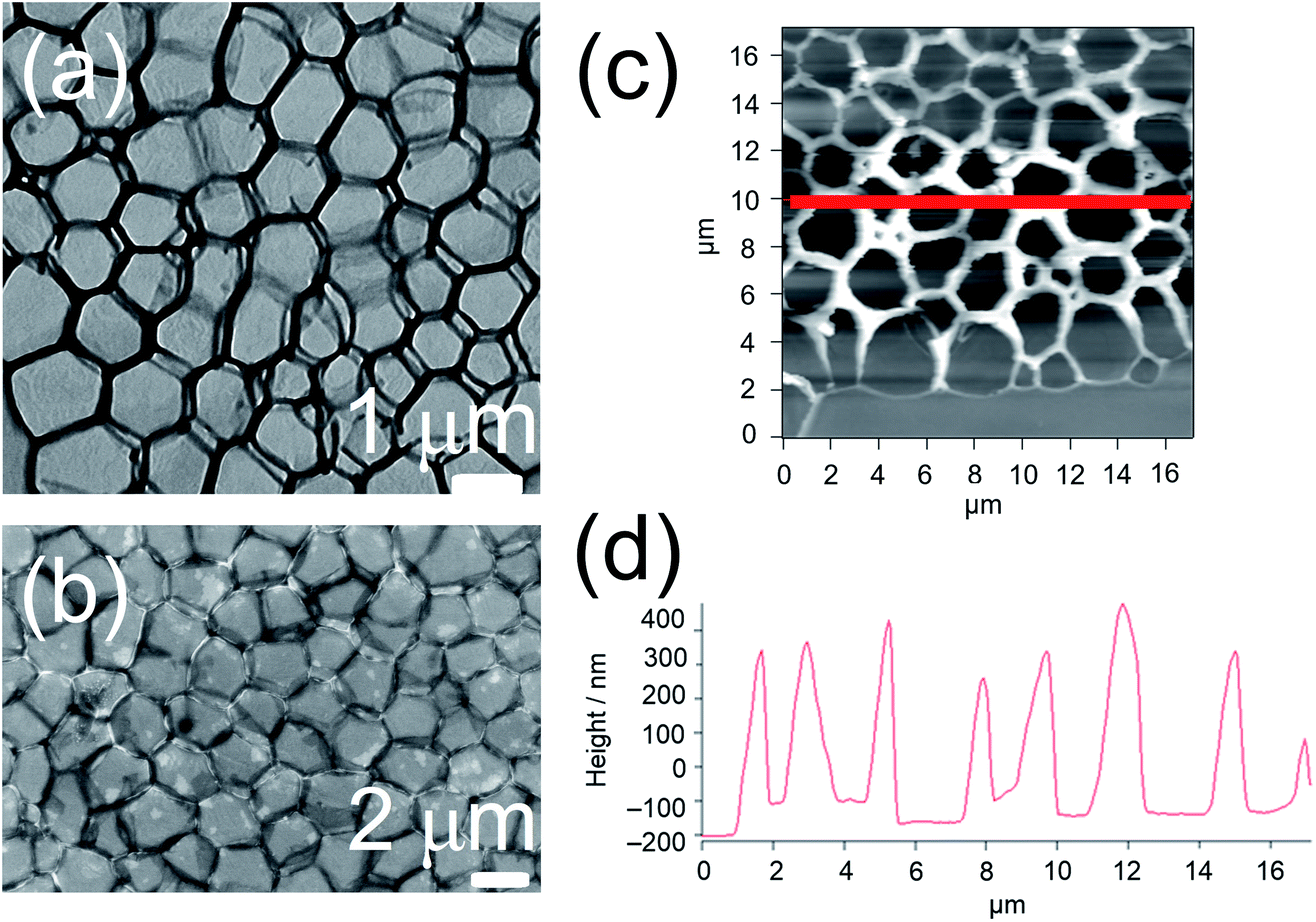 | ||
| Fig. 2 (a) TEM and (b) SEM images of an air-dried sample of a dichloromethane solution of 1 ([Pt] = 2.0 × 10−4 M). (c) Tapping mode AFM image of an air-dried sample of a dichloromethane solution of 1 ([Pt] = 2.0 × 10−4 M). (d) Height profile obtained along the red, solid line in the AFM image. | ||
With reference to the literature and our previous work, the porous network is likely formed by breath figure.35–37,42 In the dichloromethane solution of the amphiphilic dinuclear complex, aggregation exists with the aid of Pt⋯Pt and π–π stacking interactions. Upon drop-casting the dichloromethane solution of the already aggregated amphiphilic dinuclear complex onto the TEM grid, the volatile dichloromethane solvent provides a cold surface for the condensation of water droplets. With the water droplets, the hydrophobic chains attached to the alkynylplatinum(II) terpyridine moieties prefer to stay in the relatively less polar dichloromethane solution, while the hydrophilic chains attached to the core mPE unit of the complex prefer to dissolve in the water droplets. As a result, the water droplets are stabilized by the strengthened aggregation of the amphiphilic complex. Upon evaporation of the water droplets regularly oriented on the TEM grid, a regular porous network is formed. The significance of water during evaporation is supported by the lack of well-defined morphologies observed on the TEM grid prepared from drop-casting the dichloromethane solution of 1 ([Pt] = ∼10−4 M) under dry nitrogen atmosphere (Fig. S12b†). The proposed mechanism of formation of the structure is summarized in Fig. 3. Based on this mechanism, the inability to form a porous network from the mononuclear complex 8 or from a dilute solution of the dinuclear complex 1 may be attributed to the lack of sufficient hydrophobic, Pt⋯Pt and π–π stacking interactions for complex aggregation and thereby water droplet stabilization. The irregular pores formed from the concentrated solution of 1 might suggest that the crowded environment of the aggregates of the complex is unfavorable for the water droplets to evenly condense onto the surface. In other words, the dinuclear nature and the appropriate mix of hydrophobicity and hydrophilicity of this class of complexes play a crucial role in formation of the porous networks.
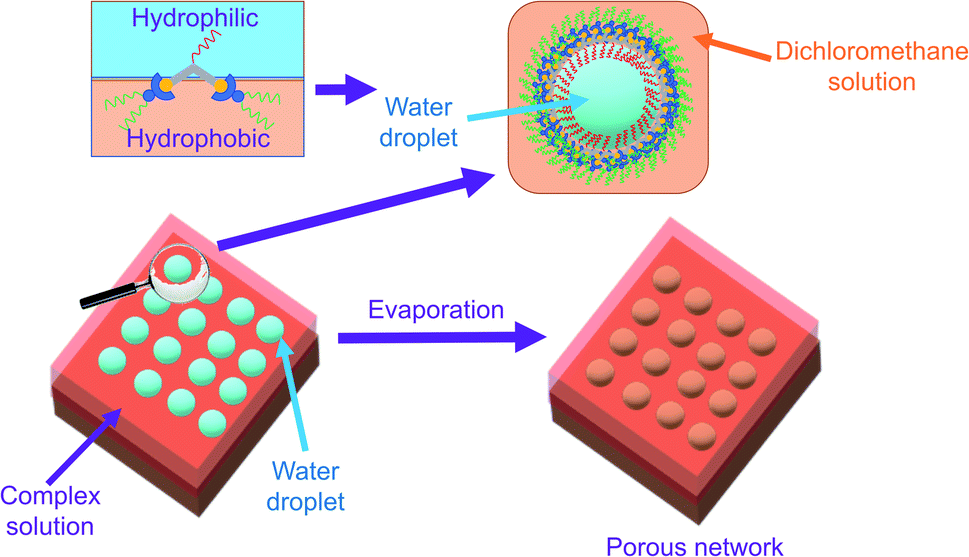 | ||
| Fig. 3 Graphical representation of formation of the porous networks assisted by 1. | ||
Self-assembly and morphological studies in DMSO
It was noted that only at elevated temperature could this series of dinuclear complexes give sharp 1H NMR signals in DMSO-d6 and be characterized, and that the solutions changed from orange to yellow on heating, indicative of the existence of ground-state aggregation at decreasing temperature. To understand their ground-state aggregation behavior in DMSO, 1–7 were studied at various temperatures in DMSO under UV-vis absorption spectroscopy. At >330 K, the UV-vis absorption spectra of 1–7 exhibit intense intraligand (IL) absorption bands at 310–385 nm, and moderately intense MLCT/LLCT absorption bands at 385–550 nm (Fig. 4 and S13–S16†).17,18 Among 1–7, the UV-vis absorption spectra of 1, 3 and 4 show a drastic change with temperature. Upon decreasing temperature, a growth in an even lower-energy absorption tail at ∼560 nm, accompanied by a drop in the MLCT/LLCT absorption and a growth in the high-energy IL absorption, was observed, with well-defined isosbestic points (Fig. 4). With reference to the literature,17,18 these even lower-energy absorption tails are assignable to MMLCT transitions, and can be attributed to the formation of Pt⋯Pt and π–π stacking interactions. It should be noted that compared to 1, 2 differs structurally only in the length of the hydrophobic chains attached to the peripheral terpyridine units. This highlights the importance of hydrophobic chain length in self-assembly in DMSO solutions. Compared to 1–4, the positions of the hydrophilic chains and the hydrophobic chains are swapped in 5 and 6, while 7 does not possess any hydrophobic chains. The lack of drastic spectral changes in variable-temperature UV-vis absorption studies in DMSO solutions reveals the importance of the presence of hydrophobic chains and the positioning of the hydrophobic chains in the self-assembly in DMSO solutions.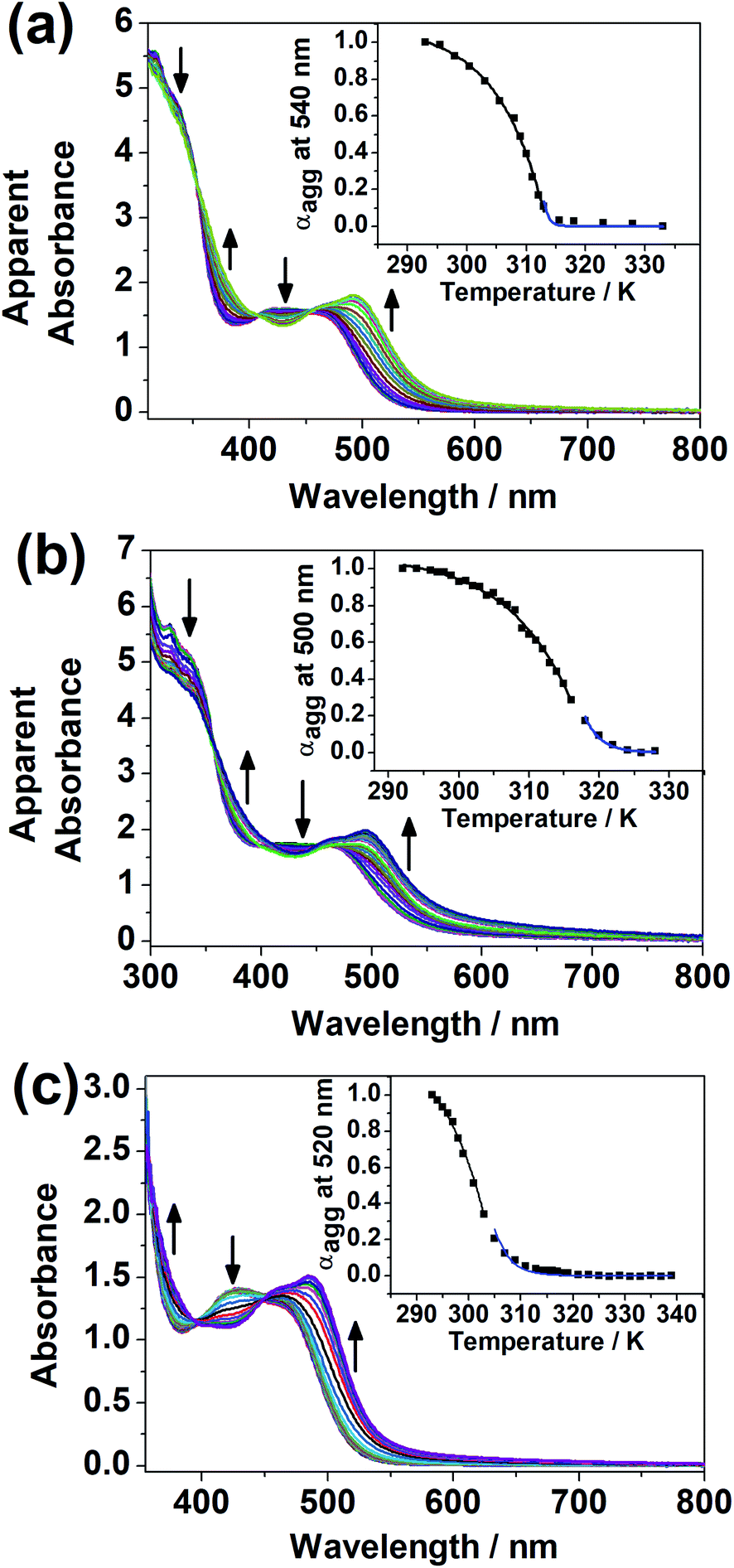 | ||
| Fig. 4 UV-Vis absorption spectra of (a) 1 ([Pt] = 2.8 × 10−4 M), (b) 3 ([Pt] = 2.8 × 10−4 M) and (c) 4 ([Pt] = 1.6 × 10−4 M) in DMSO upon decreasing temperature (a) from 333 to 293 K, (b) from 328 to 292 K and (c) from 339 to 293 K. The apparent absorbance values were obtained by correcting to a 1 cm path length equivalence. Insets: plots of degree of aggregation against temperature for the cooling of the DMSO solutions of the complexes. The solid lines show the fitted curves in the elongation (black) and the nucleation (blue) regimes. | ||
Based on the UV-vis absorption spectral traces monitored at the lowest-energy absorption bands, 1, 3 and 4 would undergo supramolecular assembly via the cooperative mechanism (Fig. 4). With the adoption of the nucleation–elongation model devised by Meijer and co-workers,62 the thermodynamic parameters were obtained (Table 1). The nucleus sizes of the complexes, 〈Nn(Te)〉, were found to be 2 or 3, implying that dimers or trimers had to be formed before elongation occurred below elongation temperature. The formation of dimers in 4 can even be observed at a low concentration, as revealed in the high-resolution ESI mass spectrum of 4, in which the isotopic pattern corresponding to its dimeric form was observed in addition to the monomeric pattern (Fig. S17†). Unlike other related systems,30,63 where the nucleus sizes of similar dinuclear platinum(II) systems obtained are usually around 10 and the equilibrium constants for the nucleation step lie in the range from 10−4 to 10−3, the nucleus sizes obtained in this system are relatively small, and the nucleation steps have a relatively large equilibrium constant and are thus more favorable. The difference might be explained by the structural difference in that the core mPE unit in this system is shorter than those in the previously reported systems, such that the platinum(II) moieties from the two ends are closer in proximity, which might reduce the size of the nucleus and thus the number of molecules required before elongation. Compared to the related platinum(II) systems,30,63 the enthalpy changes obtained in this system are more negative in value. From 1, 3 to 4, the enthalpy change of the cooperative self-assembly process becomes less negative, which can be attributed to the hindrance of intermolecular association arising from an increase in the steric bulk in the core mPE unit.
| Complex | ϕ SAT | ΔHe/kJ mol−1 | T e/K | K a | 〈Nn(Te)〉 |
|---|---|---|---|---|---|
| 1 | 1.086 | −102.1 | 313.6 | 0.0574 | 3 |
| 3 | 1.075 | −94.5 | 318.7 | 0.1340 | 2 |
| 4 | 1.363 | −84.5 | 305.5 | 0.1584 | 2 |
Furthermore, in the TEM and SEM studies conducted on 1, 3 and 4 in DMSO, fibers of different lengths were observed at different concentrations in the DMSO solutions of 1 (Fig. S18 and S19†), 3 (Fig. S20†) and 4 (Fig. S21†).
Self-assembly in mixed solvent compositions
The dinuclear complexes were subjected to UV-vis spectroscopic studies in mixed DMSO–H2O compositions in addition to studies in the single solvent. While 1–4 did not give any significant spectral changes, 5–7, with the hydrophilic triethylene glycol substituents positioned at the peripheral position of the complex, i.e. the terpyridine unit, instead of the central core, showed a growth in the absorption bands at ca. 510 nm, assignable to MMLCT transitions with Pt⋯Pt and π–π stacking interactions,17,18 upon increasing water content in the DMSO solutions of the complexes (Fig. 5). The difference in the spectral changes between 1–4 and 5–7 may be attributed to the higher sensitivity of the peripheral positions of the complex to changes in solvent compositions. Even though there were enough hydrophilic substituents on the complex, a change in the solvent polarity and hence the solvation did not give significant spectral changes when they were attached at the core of the complex.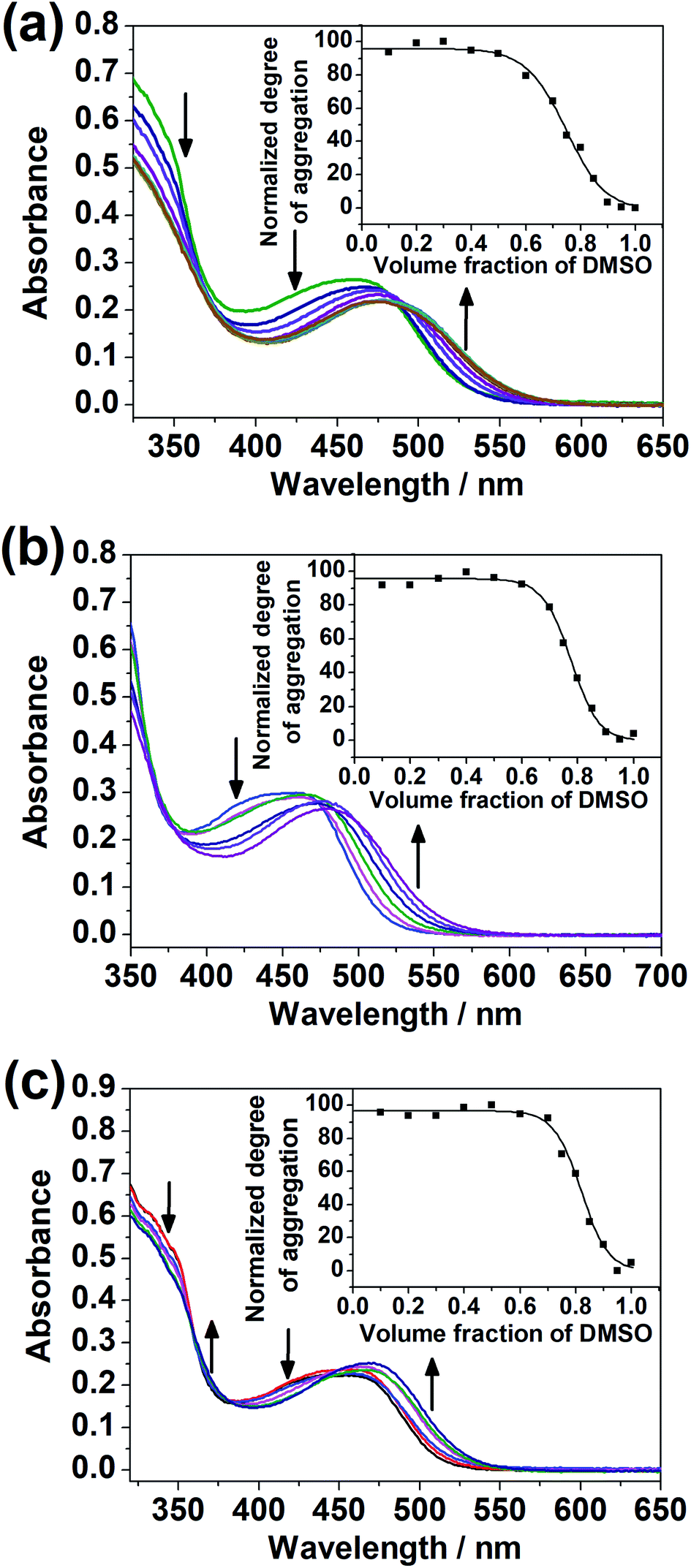 | ||
| Fig. 5 UV-Vis absorption spectra of (a) 5, (b) 6 and (c) 7 in DMSO ([Pt] = 4.0 × 10−5 M) with increasing water content (a) from 20 to 90%, (b) from 10 to 40% and (c) 5 to 30%. Insets: plots of normalized degree of aggregation of the complexes against volume fraction of DMSO. | ||
Variable-temperature UV-vis absorption studies were conducted for 5 and 6 in 30% H2O in DMSO (Fig. S22 and S23†). Upon decreasing temperature, a growth in the MMLCT absorption band, accompanied by a drop in the MLCT/LLCT absorption band, was observed, with well-defined isosbestic points, suggestive of ground-state aggregation of the complexes in DMSO–H2O mixtures by Pt⋯Pt and π–π stacking interactions.17,18 To quantify the self-assembly behavior of the complexes, the temperature-dependent nucleation–elongation model, developed by Meijer and co-workers,64 was adopted to perform curve-fitting (Fig. 5) and to obtain the thermodynamic parameters (Table 2). The more negative ΔG° value for 6 compared to 5 can be attributed to the additional hydrophobic interactions by the longer alkoxy chains.65–67 This observation, which highlights the importance of long alkoxy substituents in the self-assembly of transition-metal complexes, agrees with other works in the literature.65–67 The more negative ΔG° value for 7 compared to 5 and 6 might hint that the interactions between the hydrophilic substituents with water can constitute another dominating factor relative to hydrophobic interactions in promoting self-assembly in DMSO–H2O mixtures.
| Complex | ΔG°/kJ mol−1 | m/kJ mol−1 | σ |
|---|---|---|---|
| 5 | −53.8 ± 2.9 | 37.9 ± 3.7 | 1 |
| 6 | −67.8 ± 3.0 | 54.5 ± 3.8 | 1 |
| 7 | −71.7 ± 4.3 | 56.7 ± 5.3 | 1 |
In the emission studies on 5, a shift in the emission band maximum from 630 nm to 745 nm upon increasing water content (Fig. S24†) further confirms a clear conversion from the non-aggregated state to the aggregated state. Furthermore, in 1H NMR studies, upon increasing D2O content in the DMSO-d6 solution of the complex, the aromatic proton signals were upfield-shifted and broadened (Fig. 6), suggestive of the presence of ground-state aggregation of the complex, possibly promoted by π–π stacking interactions.
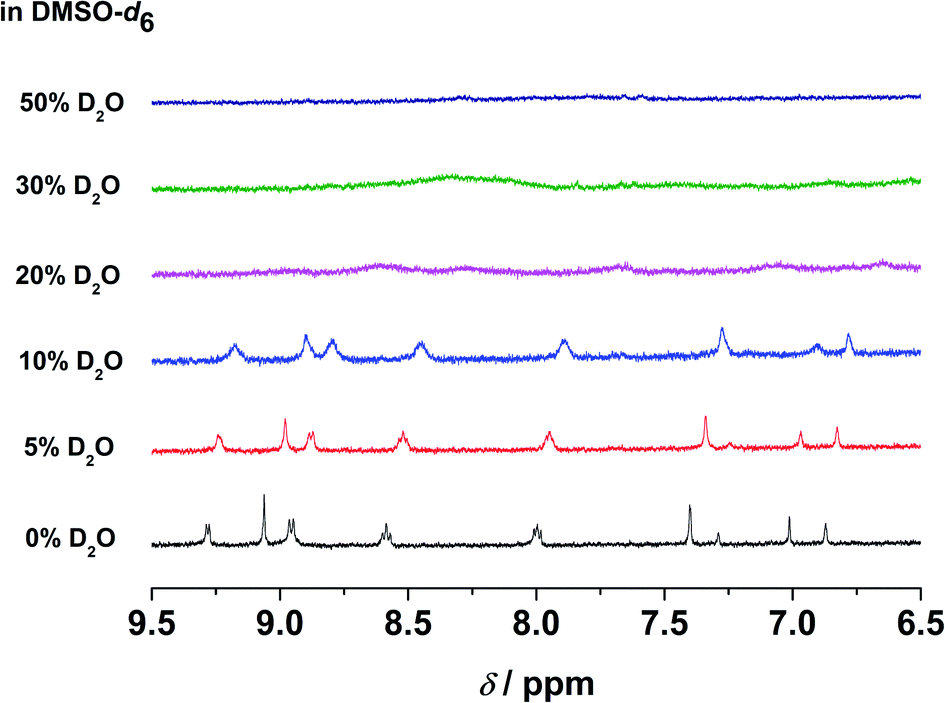 | ||
| Fig. 6 Partial 1H NMR spectra of 5 in DMSO-d6 upon increasing D2O content ([Pt] = 2.0 × 10−4 M). | ||
Computational studies
In view of the head-to-tail stacking observed in the X-ray crystal structure, geometry optimization was performed on the dimer of the model complex of 1 with head-to-tail stacking. The optimized structure of the model complex of 1, with some important structural parameters, is shown in Fig. S25.† The Pt–N and Pt–C distances, which range from 1.989–2.053 Å and 1.979–1.980 Å respectively, in conjunction with the N–Pt–N angles, are in good agreement with the experimental X-ray crystal structure. The interplanar distance between the pyridine rings on the terpyridine ligands within the dimer is around 3.5 Å, implying that π–π interactions exist within the dimer. The optimized ground-state structure of the dimer of the model complex displays Pt⋯Pt interactions in the head-to-tail stacking, with the Pt⋯Pt distance of 3.41 Å (Fig. S26†). Although Pt⋯Pt interactions are not observed in the X-ray crystal structure, it is believed that Pt⋯Pt interactions would exist if the sterically bulky tert-butyl groups are removed. The Cartesian coordinates of the structures are shown in Tables S4 and S5.†Plausible packing arrangement and factors affecting the self-assembly of this class of complexes
Based on the X-ray crystal structure of 9 and the computational studies of the backbone of this class of complexes, it is expected that it would adopt a head-to-tail stacking. The PXRD pattern on the bulk sample of 1 reveals that the complex would adopt tetragonal packing, with the lattice packing parameter of 4.81 nm. Taking into consideration the dimension of the complex, together with the nucleus size of 3 obtained from the nucleation–elongation model in the variable-temperature UV-vis absorption studies, the plausible packing arrangement of 1 is proposed as follows. In the stage of nucleation, the trimers of the complex, which adopt head-to-tail stacking, would first be formed. Afterwards, in the process of elongation, hydrophobic, Pt⋯Pt and π–π stacking interactions would allow the molecules to re-arrange to give a tetragonal packing, which would lead to formation of nanofibers macroscopically when the molecules continue to stack tetragonally, as observed in the TEM and SEM studies. The proposed mechanism of the self-assembly of 1, which establishes the relationship between the molecular stacking and the macroscopic nanostructures observed, is summarized in Fig. 7. It is noteworthy that the self-assembly of this series of complexes is an interplay of three factors, namely (1) nuclearity, in which only dinuclear complexes can self-assemble under suitable conditions; (2) hydrophobicity and hydrophilicity, in which only enough interactions between the hydrophobic and/or hydrophilic chains with solvents would lead to self-assembly; and (3) positioning of the hydrophobic and hydrophilic substituents, in which substituents sensitive to environmental changes should be attached to the peripheral positions of the molecule for the maximization of self-assembly.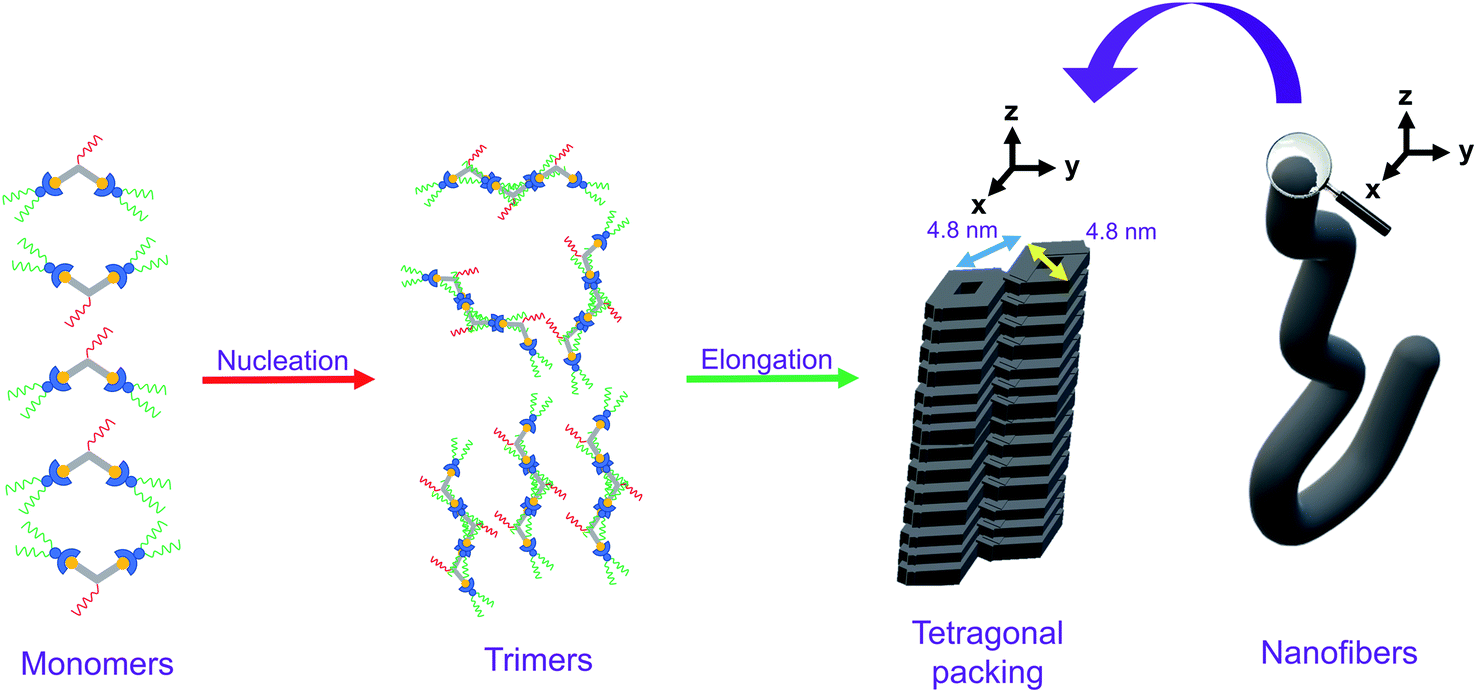 | ||
| Fig. 7 Schematic drawing of the self-assembly of 1. | ||
Conclusion
In conclusion, a series of luminescent amphiphilic alkynylplatinum(II) terpyridine complexes was designed and synthesized. In chlorinated solvents, this class of dinuclear complexes was demonstrated to aggregate and assist formation of porous networks, which was found to be concentration dependent. In DMSO solutions, it was shown to undergo self-assembly with a cooperative growth mechanism, and the steric hindrance of the core mPE unit was found to play an important role in the extent of intermolecular association. Complexes with higher hydrophilicity were demonstrated to undergo isodesmic self-assembly processes in DMSO–H2O mixtures, and the hydrophilicity and the hydrophobicity of the complexes were found to affect the thermodynamic parameters of the aggregation behavior. X-Ray crystallography, PXRD and computational studies were also performed to gain further insights into the molecular packing of this series of complexes. The present work has provided the understanding of the factors affecting self-assembly, in conjunction with the strategies for the programmable control of formation of nanostructures, which provide an alternative in building molecular framework. It is believed that with an understanding of the guiding principles of their self-assembly properties, this class of complexes serve as versatile building blocks to construct orderly packed molecular materials and functional materials in a well-controlled manner.Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. W.-W. Y. acknowledges the support from the Strategically Oriented Research Theme (SORT) on Functional Materials for Molecular Electronics of The University of Hong Kong. This work has been supported by the University Grants Committee Areas of Excellence (AoE) Scheme (AoE/P-03/08) and a General Research Fund (GRF) grant from the Research Grants Council of the Hong Kong Special Administrative Region, P. R. China (HKU 17302918). S. K.-M. L. acknowledges the receipt of a postgraduate studentship and a University Postgraduate Fellowship, both administered by The University of Hong Kong. We acknowledge UGC funding administered by The University of Hong Kong for supporting the Electrospray Ionization Quadrupole Time-of-Flight Mass Spectrometry Facilities under the Support for Interdisciplinary Research in Chemical Science, and The University Development Fund and the Dr Hui Wai Haan Fund of The University of Hong Kong for funding the Bruker D8 VENTURE X-Ray Diffractometer. Dr Kam-Hung Low is gratefully acknowledged for solving the X-ray crystal structure and collecting X-ray data, and Dr Fred Ka-Wai Kong is gratefully acknowledged for his assistance in transient absorption and emission lifetime measurements. We thank Mr Frankie Yu-Fee Chan at the Electron Microscope Unit of The University of Hong Kong for his helpful technical assistance, and the Information Technology Services (ITS) of The University of Hong Kong for providing computational resources.References
- G. Magnus, Ann. Phys., 1828, 14, 239–242 CrossRef.
- K. Krogmann, Angew. Chem., Int. Ed., 1969, 8, 35–42 CrossRef CAS.
- K. W. Jennette, S. J. Lippard, G. A. Vassiliades and W. R. Bauer, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3839–3843 CrossRef CAS PubMed.
- J. Van Houten and R. Watts, J. Am. Chem. Soc., 1976, 98, 4853–4858 CrossRef CAS.
- J. W. Schindler, R. C. Fukuda and A. W. Adamson, J. Am. Chem. Soc., 1982, 104, 3596–3600 CrossRef CAS.
- V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1989, 28, 1529–1533 CrossRef CAS.
- V. H. Houlding and V. M. Miskowski, Coord. Chem. Rev., 1991, 111, 145–152 CrossRef CAS.
- V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1991, 30, 4446–4452 CrossRef CAS.
- H.-K. Yip, L.-K. Cheng, K.-K. Cheung and C.-M. Che, J. Chem. Soc., Dalton Trans., 1993, 2933–2938 RSC.
- R. H. Herber, M. Croft, M. J. Coyer, B. Bilash and A. Sahiner, Inorg. Chem., 1994, 33, 2422–2426 CrossRef CAS.
- J. A. Bailey, M. G. Hill, R. E. Marsh, V. M. Miskowski, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1995, 34, 4591–4599 CrossRef CAS.
- W. B. Connick, R. E. Marsh, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1997, 36, 913–922 CrossRef CAS.
- G. Arena, G. Calogero, S. Campagna, L. M. Scolaro, V. Ricevuto and R. Romeo, Inorg. Chem., 1998, 37, 2763–2769 CrossRef CAS PubMed.
- R. Büchner, C. T. Cunningham, J. S. Field, R. J. Haines, D. R. McMillin and G. C. Summerton, J. Chem. Soc., Dalton Trans., 1999, 5, 711–718 RSC.
- V. W.-W. Yam, R. P.-L. Tang, K. M.-C. Wong and K.-K. Cheung, Organometallics, 2001, 20, 4476–4482 CrossRef CAS.
- V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506–6507 CrossRef CAS PubMed.
- K. M.-C. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 44, 424–434 CrossRef CAS PubMed.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- C. Yu, K. M.-C. Wong, K. H.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2005, 44, 791–794 CrossRef CAS PubMed.
- V. W.-W. Yam, K. H.-Y. Chan, K. M.-C. Wong and B. W.-K. Chu, Angew. Chem., Int. Ed., 2006, 45, 6169–6173 CrossRef CAS PubMed.
- Y. Tanaka, K. M.-C. Wong and V. W.-W. Yam, Chem. Sci., 2012, 3, 1185–1191 RSC.
- W. Lu, S. S.-Y. Chui, K.-M. Ng and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 4568–4572 CrossRef CAS PubMed.
- M.-Y. Yuen, V. A. L. Roy, W. Lu, S. C. F. Kui, G. S. M. Tong, M.-H. So, S. S.-Y. Chui, M. Muccini, J. Q. Ning, S. J. Xu and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 9895–9899 CrossRef CAS PubMed.
- Y. Chen, K. Li, W. Lu, S. S.-Y. Chui, C.-W. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 9909–9913 CrossRef CAS PubMed.
- C. Po, A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12136–12143 CrossRef CAS PubMed.
- H.-L. Au-Yeung, S. Y.-L. Leung, A. Y.-Y. Tam and V. W.-W. Yam, J. Am. Chem. Soc., 2014, 136, 17910–17913 CrossRef CAS PubMed.
- C. Po, A. Y.-Y. Tam and V. W.-W. Yam, Chem. Sci., 2014, 5, 2688–2695 RSC.
- A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2015, 8, 10–15 CrossRef PubMed.
- M. E. Robinson, D. J. Lunn, A. Nazemi, G. R. Whittell, L. De Cola and I. Manners, Chem. Commun., 2015, 51, 15921–15924 RSC.
- S. Y.-L. Leung, K. M.-C. Wong and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2845–2850 CrossRef CAS PubMed.
- B. Sun, Y. Kim, Y. Wang, H. Wang, J. Kim, X. Liu and M. Lee, Nat. Mater., 2018, 17, 599–604 CrossRef CAS PubMed.
- X. Liu, H. Li, Y. Kim and M. Lee, Chem. Commun., 2018, 54, 3102–3105 RSC.
- R. N. Das, Y. P. Kumar, S. A. Kumar, O. M. Schütte, C. Steinem and J. Dash, Chem.–Eur. J., 2018, 24, 4002–4005 CrossRef CAS PubMed.
- H. Wang, J. Wang, D. Cao, H. Gu, B. Li, X. Lu, X. Han, A. L. Rogach and C. Niu, J. Mater. Chem. A, 2017, 5, 6817–6824 RSC.
- G. Widawski, M. Rawiso and B. François, Nature, 1994, 369, 387–389 CrossRef CAS.
- H. Bai, C. Du, A. Zhang and L. Li, Angew. Chem., Int. Ed., 2013, 52, 12240–12255 CrossRef CAS PubMed.
- Y. Liu, H. Ma, Y. Tian, F. Xie and X. Wang, Macromol. Chem. Phys., 2014, 215, 1446–1455 CrossRef CAS.
- H. O. Lintang, K. Kinbara, K. Tanaka, T. Yamashita and T. Aida, Angew. Chem., Int. Ed., 2010, 49, 4241–4245 CrossRef CAS PubMed.
- H. O. Lintang, K. Kinbara, T. Yamashita and T. Aida, Chem.–Asian J., 2012, 7, 2068–2072 CrossRef CAS PubMed.
- X.-D. Xu, J. Zhang, L.-J. Chen, X.-L. Zhao, D.-X. Wang and H.-B. Yang, Chem.–Eur. J., 2012, 18, 1659–1667 CrossRef CAS PubMed.
- Y. Chen, C.-M. Che and W. Lu, Chem. Commun., 2015, 51, 5371–5374 RSC.
- K.-C. Yim, V. K.-M. Au, L.-L. Hung, K. M.-C. Wong and V. W.-W. Yam, Chem.–Eur. J., 2016, 22, 16258–16270 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- R. J. Kuppler, D. J. Timmons, Q.-R. Fang, J.-R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang and H.-C. Zhou, Coord. Chem. Rev., 2009, 253, 3042–3066 CrossRef CAS.
- O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43, 1166–1175 CrossRef CAS PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- Y.-Z. Chen, R. Zhang, L. Jiao and H.-L. Jiang, Coord. Chem. Rev., 2018, 362, 1–23 CrossRef CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, J. Am. Chem. Soc., 2008, 130, 11580–11581 CrossRef CAS PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed.
- Z. Chen, A. K.-W. Chan, V. C.-H. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2019, 141, 11204–11211 CrossRef CAS PubMed.
- D. Zhang, L.-Z. Wu, L. Zhou, X. Han, Q.-Z. Yang, L.-P. Zhang and C.-H. Tung, J. Am. Chem. Soc., 2004, 126, 3440–3441 CrossRef CAS PubMed.
- E. O. Danilov, I. E. Pomestchenko, S. Kinayyigit, P. L. Gentili, M. Hissler, R. Ziessel and F. N. Castellano, J. Phys. Chem. A, 2005, 109, 2465–2471 CrossRef CAS PubMed.
- F. N. Castellano, I. E. Pomestchenko, E. Shikhova, F. Hua, M. L. Muro and N. Rajapakse, Coord. Chem. Rev., 2006, 250, 1819–1828 CrossRef CAS.
- E. Shikhova, E. O. Danilov, S. Kinayyigit, I. E. Pomestchenko, A. D. Tregubov, F. Camerel, P. Retailleau, R. Ziessel and F. N. Castellano, Inorg. Chem., 2007, 46, 3038–3048 CrossRef CAS PubMed.
- P. Jarosz, P. Du, J. Schneider, S.-H. Lee, D. McCamant and R. Eisenberg, Inorg. Chem., 2009, 48, 9653–9663 CrossRef CAS PubMed.
- P. Jarosz, K. Lotito, J. Schneider, D. Kumaresan, R. Schmehl and R. Eisenberg, Inorg. Chem., 2009, 48, 2420–2428 CrossRef CAS PubMed.
- P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS PubMed.
- M. H.-Y. Chan, S. Y.-L. Leung and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 7637–7646 CrossRef CAS PubMed.
- P. A. Korevaar, C. Schaefer, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482–13491 CrossRef CAS PubMed.
- A. K.-W. Chan, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2015, 137, 6920–6931 CrossRef CAS PubMed.
- H. L.-K. Fu, S. Y.-L. Leung and V. W.-W. Yam, Chem. Commun., 2017, 53, 11349–11352 RSC.
- H. L.-K. Fu, C. Po, S. Y.-L. Leung and V. W.-W. Yam, ACS Appl. Mater. Interfaces, 2017, 9, 2786–2795 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Photophysical measurements and instrumentation, experimental details, crystallographic data, PXRD pattern, UV-vis absorption spectra, emission spectra, transient absorption difference spectra, 1H NMR spectra, TEM images and ESI mass spectra. CCDC 1945676. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04475b |
| This journal is © The Royal Society of Chemistry 2020 |