
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible RNA acylation for control of CRISPR–Cas9 gene editing†
Maryam
Habibian
 ,
Colin
McKinlay
,
Timothy R.
Blake
,
Anna M.
Kietrys
,
Robert M.
Waymouth
,
Paul A.
Wender
and
Eric T.
Kool
*
,
Colin
McKinlay
,
Timothy R.
Blake
,
Anna M.
Kietrys
,
Robert M.
Waymouth
,
Paul A.
Wender
and
Eric T.
Kool
*
Department of Chemistry, Stanford University, 450 Serra Mall, Stanford, CA 94305, USA
First published on 2nd December 2019
Abstract
We report the development of post-transcriptional chemical methods that enable control over CRISPR–Cas9 gene editing activity both in in vitro assays and in living cells. We show that an azide-substituted acyl imidazole reagent (NAI-N3) efficiently acylates CRISPR single guide RNAs (sgRNAs) in 20 minutes in buffer. Poly-acylated (“cloaked”) sgRNA was completely inactive in DNA cleavage with Cas9 in vitro, and activity was quantitatively restored after phosphine treatment. Delivery of cloaked sgRNA and Cas9 mRNA into HeLa cells was enabled by the use of charge-altering releasable transporters (CARTs), which outperformed commercial transfection reagents in transfecting sgRNA co-complexed with Cas9 encoding functional mRNA. Genomic DNA cleavage in the cells by CRISPR–Cas9 was efficiently restored after treatment with phosphine to remove the blocking acyl groups. Our results highlight the utility of reversible RNA acylation as a novel method for temporal control of genome-editing function.
Introduction
CRISPR–Cas9 gene editing has emerged as an immensely powerful technology with widespread applications ranging from accelerating biological research to the development of therapeutic agents and diagnostic tools. Co-opted from bacteria, CRISPR–Cas9 systems are particularly attractive for their ability to target and cleave arbitrary DNA sequences via complementary base pairing using a programmable CRISPR single-guide RNA (sgRNA).1 The sgRNA recruits the Cas9 nuclease to the target DNA to create a double-stranded break. This ability has been further exploited in editing genomes, activating2 or repressing3 gene expression, imaging DNA loci,4 generating targeted mutational diversity5 and modifying epigenetic markers.6Initiation of CRISPR–Cas9 function in cells is typically a slow multistep process, involving cellular transfection and uptake of components, transcription and translation of the Cas9 protein, assembly of the active complex, followed by editing and repair of the targeted genes. This long process – perhaps a day in duration – makes it difficult to study the activity of the editing complex and its downstream events in time. Ideally, one might trigger a pulse of CRISPR–Cas9 activity by an initiating signal after the complex is already introduced and assembled in cells, making it easier to follow the subsequent gene cleavage and alteration of gene expression. In addition, temporal control of CRISPR could enable pulse-chase experiments involving the complex (see for example our studies of sgRNA lifetime below). Further, regulation of CRISPR–Cas9 could potentially reduce off-target effects, and regulate cells in time during differentiation.
While a good deal of attention has been devoted to altering the activity and specificity of the Cas9 enzyme,7 as well as strategies for spatial and temporal control of gene targets,8 strategies to switch off and on CRISPR–Cas9 activity have not been extensively explored.7f,g Most of the efforts in CRISPR–Cas9 regulation have focused on chemical9 or optical10 control of the Cas9 protein. A less-explored alternative to this approach is to regulate the sgRNA instead. An advantage to sgRNA control is that it could support control of specific gene editing while other CRISPR–Cas9 enzymes remain operative. Moreover, as we show below, its control can be carried out more simply and with commercially available sgRNAs co-complexation with Cas9 mRNA using the CART delivery platform.11 Previous sgRNA controlling methods were focused on using RNA-binding ligands that recruit other ligand-dependent proteins or ribozymes to regulate or irreversibly terminate the function of an otherwise active sgRNA.9b,12 In addition, Doudna and coworkers reported ligand-controlled sgRNAs engineered to contain riboswitches, although the system was inactive in eukaryotic cells.13 In another report, an RNA strand complementary to the sgRNA was used to inhibit sgRNA function;14 the current work has the opposite goal, of turning on CRISPR function.
Here, we report a simple alternative chemical strategy for control of CRISPR RNA. We recently reported “RNA cloaking” as a mild and reversible chemical approach to controlling RNA hybridization, folding, and enzymatic interactions.15 In this method, an azide-substituted acyl imidazole reagent (NAI-N3) (Fig. 1a) is used to acylate the 2′-OH groups of RNAs post-synthetically, resulting in blocking of the RNAs' folding, hybridization, and function. Activity of such poly-acylated (“cloaked”) RNAs in vitro was efficiently recovered upon treatment with a water-soluble phosphine (Fig. 1b) that triggers Staudinger reduction of the azide, followed by spontaneous loss of acyl groups (“uncloaking”). Here, we test the reversible cloaking/uncloaking strategy with sgRNAs as a strategy to control the CRISPR–Cas9 gene editing efficiency in vitro and in living cells. We hypothesized that a one-step treatment of sgRNA might block DNA cleavage by the CRISPR–Cas9 complex, either by inhibiting RNA–DNA hybridization, or by blocking sgRNA folding, and/or by inhibiting the binding of sgRNA by Cas-9. Consequently, one could restore DNA cleaving activity by treatment with a phosphine to de-acylate the sgRNA (Fig. 2a).
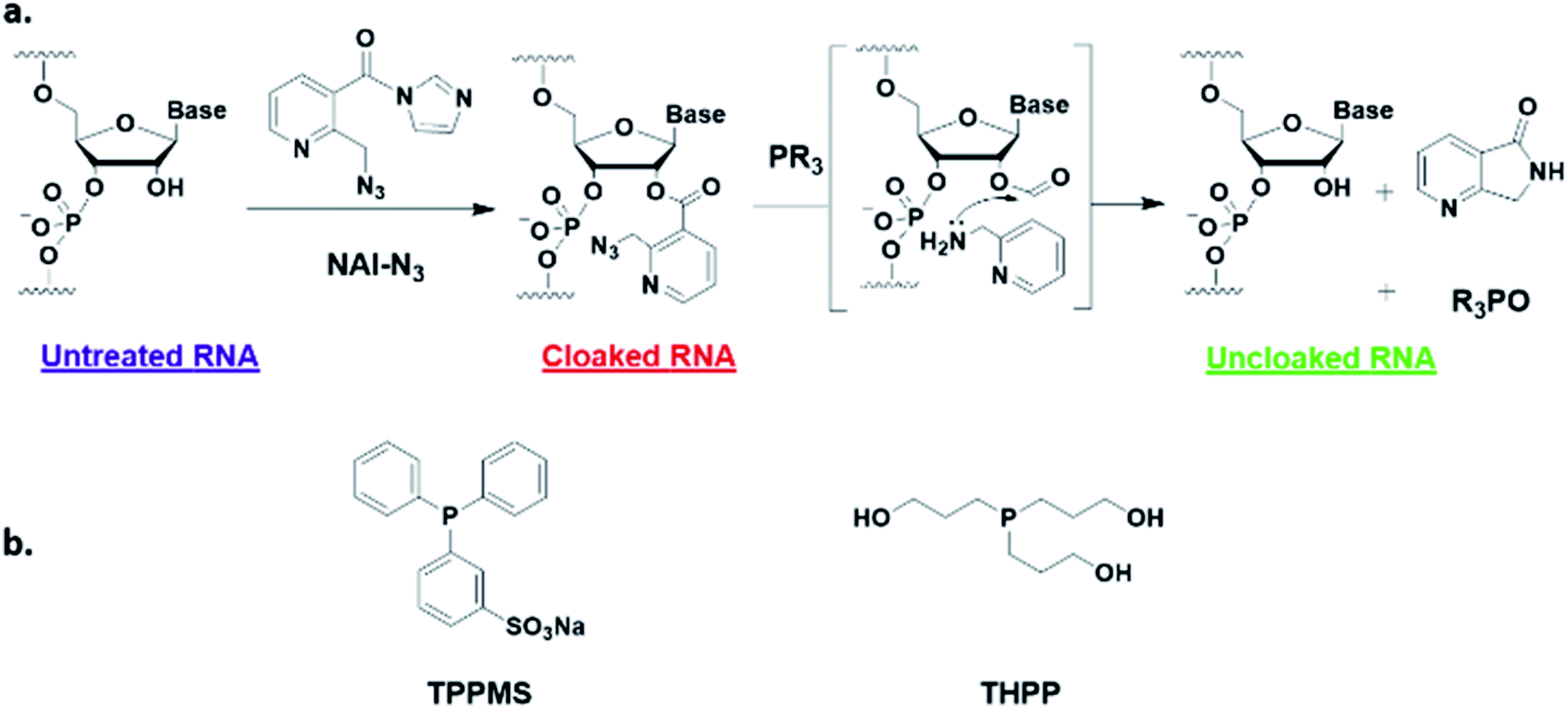 | ||
| Fig. 1 (a) Mechanism of RNA cloaking using NAI-N3 and Staudinger uncloaking by a soluble phosphine. (b) Structures of phosphines used in this study. | ||
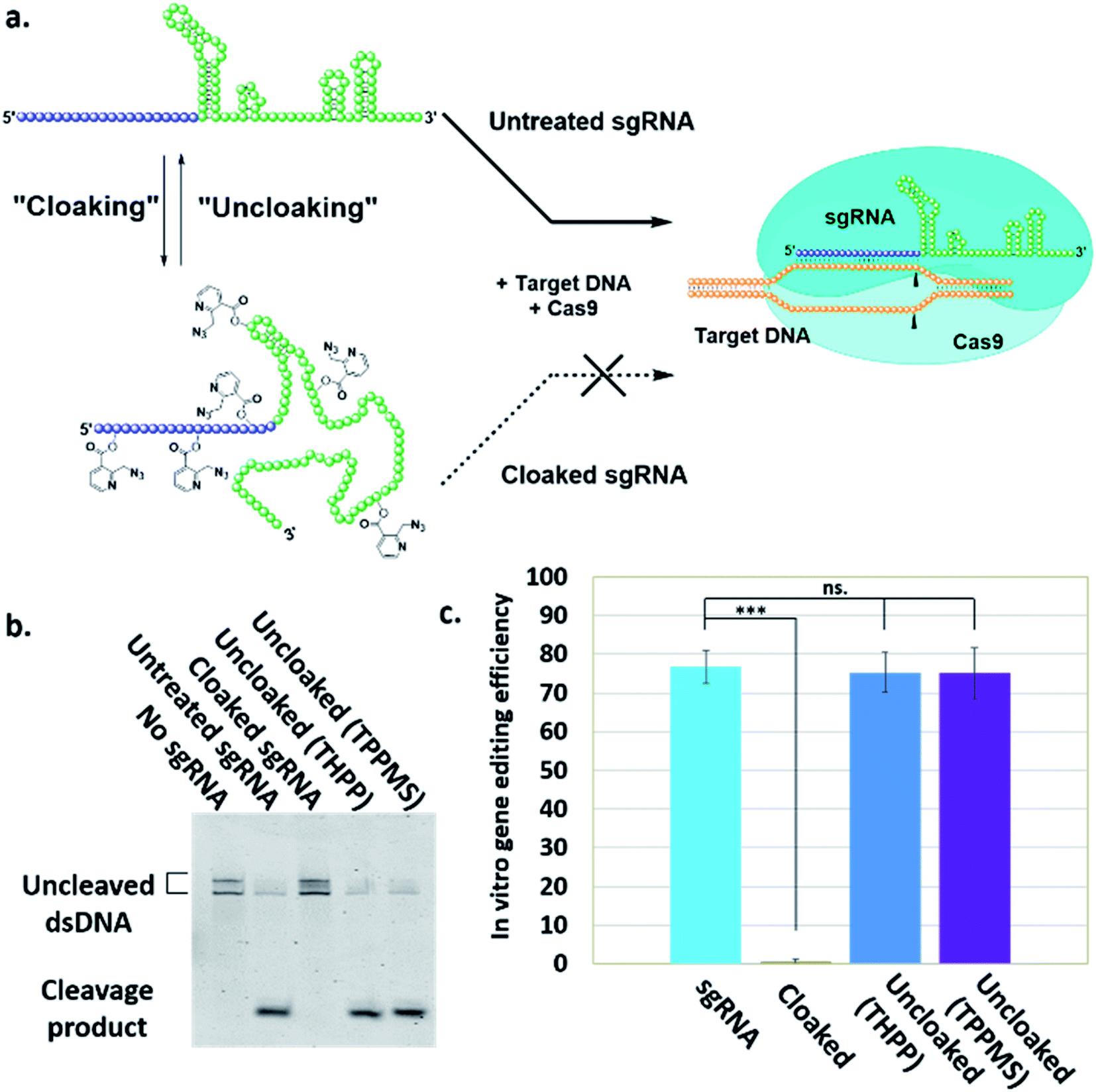 | ||
| Fig. 2 (a) Mechanism of NAI-N3-enabled inhibition of CRISPR–Cas9 gene editing. sgRNA cloaking inhibits the RNA-guided DNA double strand cleavage by Cas9 nuclease (b) PAGE analysis of Cas9 nuclease assay in vitro using Cy5-labelled dsDNA and untreated, cloaked, and uncloaked (phosphine-treated) sgRNA. (c) Bar chart represents the fraction of cleaved DNA after incubation of the sgRNAs with Cas9. Error bars represent ±s.d. and p-values: ***p < 0.001, ns = not significant. | ||
Results and discussion
In vitro studies
To test our hypothesis, we first designed an in vitro gene editing model platform with a 103 nucleotide (nt) long synthetic sgRNA targeting a Cy5-labeled double-stranded DNA (dsDNA) target encoding a portion of the green fluorescent protein (GFP) (Table S1†). Using this system, we tested the acylation of sgRNA in varied buffer conditions and at different NAI-N3 concentrations and reaction times (ESI Note 2†). The cloaked sgRNAs were then isolated by precipitation and employed in a gel-based assay to measure DNA cleavage (ESI Note 3†). Prior RNA cloaking conditions reported by our group required a high level of acylation to block RNA hybridization.15 However, in the present case we aimed to obtain the lowest level of sgRNA cloaking required to inhibit the CRISPR–Cas9 DNA cleavage, in order to maximize recovery upon phosphine treatment.We found that the minimum level of cloaking required to strongly inhibit the Cas9-mediated DNA cleavage (<1% DNA cleavage) was obtained by treating the sgRNA with 0.2 M NAI-N3 for 20 min in 100 mM MOPS buffer with 6 mM MgCl2 and 100 mM NaCl at pH 7.5 (Fig. S1† and 2b).
Next, we tested whether our proposed Staudinger reduction strategy could efficiently de-acylate the polyacylated sgRNA and restore its function in our Cas9-mediated DNA cleavage assay. Initial uncloaking experiments were performed using a range of phosphines at varying reaction conditions (phosphine concentration, incubation times and temperatures; ESI Note 4†). The results showed that the highest levels of Cas9-mediated DNA cleavage – quantitatively complete restoration of activity to the level of untreated sgRNA – were obtained when the cloaked sgRNA was incubated with tris(hydroxypropyl)phosphine (THPP) or diphenylphosphinobenzene-3-sulfonate (TPPMS) in phosphate-buffered saline (PBS) buffer at 37 °C for 1 hour at 1–5 mM concentrations (Fig. 2b, c and S2†). This strikingly robust performance in vitro suggests possible utility of this method in control of timing and initiation in diagnostic applications of CRISPR–Cas9.
Cellular control of gene editing
Our success in controlling the CRISPR–Cas9 complex in vitro suggested the intriguing possibility of regulating CRISPR–Cas9 function using the same approach in living systems. Such a cellular experiment faces the additional challenges of the delivery of the sgRNA, the Cas9 protein, and the phosphine into the nucleus of cells. To report on DNA-cleavage activity, we performed our experiments using engineered GFP-positive HeLa cells (ESI Note 5†).To implement this, GFP-positive HeLa cells were initially transfected with GFP-targeting sgRNA and Cas9 protein using nucleofection or commercial cationic lipids (Lipofectamine CRISPRMAX). However, these experiments showed relatively poor effectiveness of the ensuing DNA-cleaving activity. We then tested CART delivery vehicles,11,16 a novel platform recently developed for mRNA delivery in cells and living animals, and observed much more effective biological activity (Fig. S3 and S4†). In our experiments, after introduction of sgRNA and Cas9 mRNA, cells were cultured for five days to allow for depletion of existing GFP protein, before their residual GFP expression level was analyzed by flow cytometry and fluorescence microscopy. These initial experiments using the CART delivery system showed excellent gene editing efficiency, yielding a 95% decrease in the number of GFP-positive cells (ESI Note 6; Fig. S4†). Further studies were carried out using this delivery strategy.
Next, we tested whether GFP-targeting sgRNAs that were cloaked based on the same procedure as in the in vitro experiments (0.2 M NAI-N3 for 20 min in MOPS buffer, pH 7.5) and delivered by CART were able to block CRISPR–Cas9 genome editing in the GFP-positive HeLa cells. We observed that the GFP-positive cells that were transfected with cloaked sgRNA/Cas9 mRNA expressed the same level of GFP fluorescence as untreated cells, confirming that acylation of sgRNA blocks essentially all sgRNA activity in live cells (Fig. 3).
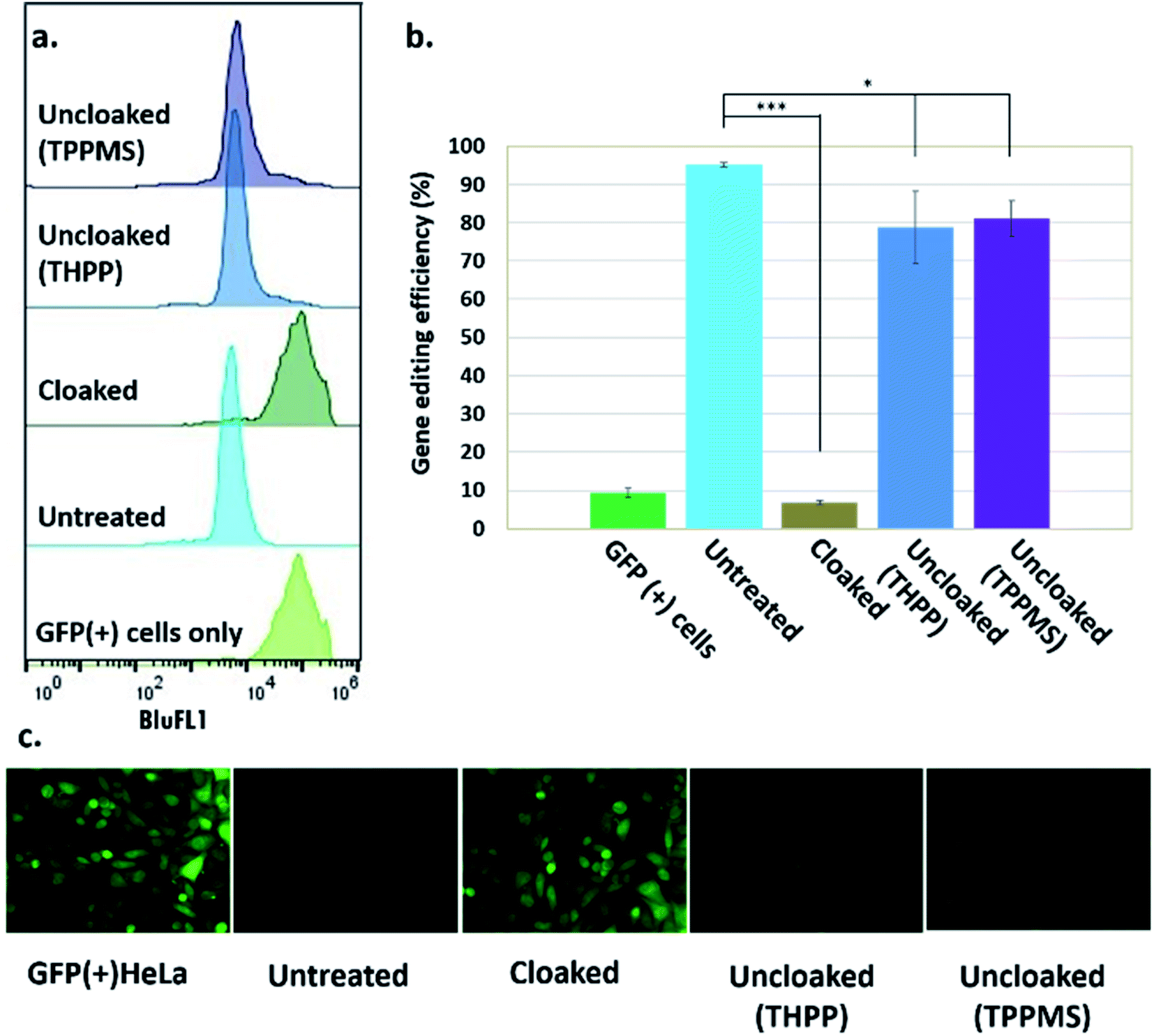 | ||
| Fig. 3 Phosphine control of cloaked CRISPR–Cas9 activity in human cells. (a) Flow cytometric analysis of GFP(+) HeLa cells showing loss of activity of cloaked sgRNA, and restoration of editing with phosphine treatment; (b) chart showing gene editing efficiencies (from flow cytometry data) of untreated, cloaked, and phosphine-uncloaked sgRNA (error bars represent ±s.d., n = 3 and p-values: ***p < 0.001, *p < 0.05.); (c) epifluorescence microscope images showing GFP knockout upon transfection with untreated sgRNA and Cas9 mRNA, lack of editing activity with cloaked sgRNA leaving GFP fluorescence unaffected, and restoration of editing with phosphine treatment of the cells. | ||
Before testing sgRNA uncloaking in HeLa cells using phosphines, we confirmed the tolerance of the cells for the phosphines THPP and TPPMS at 1–5 mM concentrations (ESI Note 7; Fig. S5†). We then proceeded to use cloaked CART-delivered sgRNA along with Cas9 mRNA and investigated the effect of THPP and TPPMS phosphine additions at 1 and 5 mM concentrations 6 hours post-transfection, potentially to recover gene-editing activity. Phosphine-triggered recovery of activity was indeed observed, with the highest levels of gene editing efficiency obtained when cells were incubated for 17 hours at 37 °C with 5 mM THPP and 1 mM TPPMS, although recovery was largely complete after 6 h (Fig. S6†). A maximal level of 81% recovery of gene editing activity (relative to untreated sgRNA) was observed using 1 mM TPPMS as quantified by flow cytometry (Fig. 3b). These results confirmed that under these conditions, polyacylated sgRNAs delivered by CARTs were efficiently uncloaked in GFP-positive HeLa cells with little or no toxicity and could elicit the CRISPR–Cas9 system to knock out the GFP gene effectively. In an initial test of generality, we also tested the effects of polyacylation and deacylation of a different sgRNA targeted to an endogenous gene (RUNX1) in HeLa cells, evaluating gene editing by a quantitative endonuclease assay. The results showed successful acylation-based suppression and significant phosphine-triggered recovery of activity in this case as well (ESI Note 9, Fig. S10†).
A time course study investigating the gene editing efficiency when cloaked sgRNA was uncloaked (using 5 mM THPP for 17 hours) from 4 hours up to 3 days after the transfection revealed a gradual reduction in the gene editing efficiency as the phosphine was added at later time points post-transfection (Fig. S7†). This was anticipated, as the sgRNA is expected to be degraded in the cells over time. To be effective, it is of course necessary that the uncloaking be carried out within the stability lifetime of the cloaked sgRNA. As expected, we observed the highest recovered gene editing efficiency (82%) at 4 hours and the lowest efficiency (6%) after 3 days from transfection, suggesting that the cloaked sgRNA/mRNA combination has a similar half-life as the unmodified combination, and that CART release of the RNAs is relatively rapid on this time scale.
To test possible mechanisms by which cloaking inhibits CRISPR–Cas9 function, we explored the possibility that acylation of the DNA hybridization domain at the 5′ end of the sgRNA might inhibit recognition of the target DNA. We tested this hypothesis with a molecular beacon (MB) fluorescence probe complementary to the hybridization domain of the GFP-targeting sgRNA (ESI Note 8 and Fig. S9†). To quantify the hybridization, we measured the emitted fluorescence upon addition of the MB to the RNA. As expected, a strong increase in MB fluorescence was observed upon addition of target DNA to the untreated sgRNA. However, sgRNA cloaking largely blocked this hybridization signal (>70% reduced; Fig. S9†). These results indicate that cloaking by NAI-N3 strongly impairs the ability of CRISPR sgRNA to hybridize to a complementary DNA target. It is noteworthy that uncloaking the sgRNA with THPP and TPPMS (5 mM or 1 mM, 37 °C, PBS buffer, 1 hour) restored the molecular beacon fluorescence up to 60% (THPP) and 70% (TPPMS). To test a second possible mechanism of inhibition, in which acylation might disrupt the Cas9 protein–sgRNA complex, we measured protein–RNA binding by native gel electrophoresis (ESI Note 10, Fig. S9†). The results showed that sgRNA acylation did not prevent RNA–protein binding, but it did appear to disrupt conformation, as judged by altered gel mobility of the complex. These preliminary results establish two mechanisms by which cloaking with NAI-N3 may control CRISPR–Cas9 function. The ability to block hybridization of the sgRNA to the DNA target suggests future uses of this approach to control non-editing functions of CRISPR systems, such as in CRISPR interference and in diagnostic applications involving RNA or DNA detection.
Conclusions
Taken together, our results establish reversible sgRNA acylation with delivery by CARTs as a novel strategy for temporal control of genome-editing function in vitro and in living cells. We have demonstrated optimized conditions for cloaking and uncloaking of sgRNAs in vitro and in HeLa cells and identified a CART delivery platform as being particularly effective for intracellular delivery of active CRISPR–Cas9. Our study provides proof of principle for establishing temporal control over any RNA-guided CRISPR–Cas9 system, or potentially any other RNA-guided enzymatic reaction, using methods similar to those described here. Our cloaking method is exceptionally simple, requiring a single step and 20 minutes, and is potentially universal if applied to any of the thousands of sgRNAs that are commercially available. The method utilizes inexpensive reagents, functions both with chemically synthesized or transcribed sgRNAs, and requires no sgRNA or Cas9 engineering. Such systems can provide a platform to conduct large-scale screens to probe a desired biological activity through dose- and/or time-dependent release of the RNA function in the system. Moreover, CART delivery can offer activity in distinct cell populations11b or localized to specific organs.11c Finally, the new methodology offers promise in studying the biological roles of specific genes by making it possible to knock-in or knock-out different sets of genes in the same system, in a time- and dose-dependent manner.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the U.S. National Institutes of Health (GM127295 to ETK; CA031845 to PAW), the National Science Foundation (CHE848280 to PAW, CHE1607092 to RMW), the Stanford Cancer Translational Nanotechnology Training T32 Training Grant CA196585 funded by the National Cancer Institute (to T. R. B.), the Child Health Research Institute at Stanford University for support.References
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816 CrossRef CAS PubMed.
- (a) L. A. Gilbert, M. H. Larson, L. Morsut, Z. Liu, G. A. Brar, S. E. Torres, N. Stern-Ginossar, O. Brandman, E. H. Whitehead, J. A. Doudna, W. A. Lim, J. S. Weissman and L. S. Qi, Cell, 2013, 154, 442 CrossRef CAS PubMed; (b) P. Perez-Pinera, D. D. Kocak, C. M. Vockley, A. F. Adler, A. M. Kabadi, L. R. Polstein, P. I. Thakore, K. A. Glass, D. G. Ousterout, K. W. Leong, F. Guilak, G. E. Crawford, T. E. Reddy and C. A. Gersbach, Nat. Methods, 2013, 10, 973 CrossRef CAS PubMed.
- L. S. Qi, M. H. Larson, L. A. Gilbert, J. A. Doudna, J. S. Weissman, A. P. Arkin and W. A. Lim, Cell, 2013, 152, 1173 CrossRef CAS PubMed.
- B. Chen, L. A. Gilbert, B. A. Cimini, J. Schnitzbauer, W. Zhang, G.-W. Li, J. Park, E. H. Blackburn, J. S. Weissman, L. S. Qi and B. Huang, Cell, 2013, 155, 1479 CrossRef CAS PubMed.
- G. M. Findlay, E. A. Boyle, R. J. Hause, J. C. Klein and J. Shendure, Nature, 2014, 513, 120 CrossRef CAS PubMed.
- I. B. Hilton, A. M. D'Ippolito, C. M. Vockley, P. I. Thakore, G. E. Crawford, T. E. Reddy and C. A. Gersbach, Nat. Biotechnol., 2015, 33, 510 CrossRef CAS PubMed.
- (a) J. P. Guilinger, D. B. Thompson and D. R. Liu, Nat. Biotechnol., 2014, 32, 577 CrossRef CAS PubMed; (b) F. A. Ran, P. D. Hsu, C. Y. Lin, J. S. Gootenberg, S. Konermann, A. E. Trevino, D. A. Scott, A. Inoue, S. Matoba, Y. Zhang and F. Zhang, Cell, 2013, 154, 1380 CrossRef CAS PubMed; (c) B. Shen, W. Zhang, J. Zhang, J. Zhou, J. Wang, L. Chen, L. Wang, A. Hodgkins, V. Iyer, X. Huang and W. C. Skarnes, Nat. Methods, 2014, 11, 399 CrossRef CAS PubMed; (d) I. M. Slaymaker, L. Gao, B. Zetsche, D. A. Scott, W. X. Yan and F. Zhang, Science, 2016, 351, 84 CrossRef CAS PubMed; (e) J. A. Zuris, D. B. Thompson, Y. Shu, J. P. Guilinger, J. L. Bessen, J. H. Hu, M. L. Maeder, J. K. Joung, Z. Y. Chen and D. R. Liu, Nat. Biotechnol., 2015, 33, 73 CrossRef CAS PubMed; (f) J. K. Nuñez, L. B. Harrington and J. A. Doudna, ACS Chem. Biol., 2016, 11, 681 CrossRef PubMed; (g) F. Richter, I. Fonfara, R. Gelfert, J. Nack, E. Charpentier and A. Moglich, Curr. Opin. Biotechnol., 2017, 48, 119 CrossRef CAS PubMed.
- K. M. Davis, V. Pattanayak, D. B. Thompson, J. A. Zuris and D. R. Liu, Nat. Chem. Biol., 2015, 11, 316 CrossRef CAS PubMed.
- (a) K. I. Liu, M. N. B. Ramli, C. W. A. Woo, Y. Wang, T. Zhao, X. Zhang, G. R. D. Yim, B. Y. Chong, A. Gowher, M. Z. H. Chua, J. Jung, J. H. J. Lee and M. H. Tan, Nat. Chem. Biol., 2016, 12, 980 CrossRef CAS PubMed; (b) B. Maji, C. L. Moore, B. Zetsche, S. E. Volz, F. Zhang, M. D. Shoulders and A. Choudhary, Nat. Chem. Biol., 2017, 13, 9 CrossRef CAS PubMed; (c) D. P. Nguyen, Y. Miyaoka, L. A. Gilbert, S. J. Mayerl, B. H. Lee, J. S. Weissman, B. R. Conklin and J. A. Wells, Nat. Commun., 2016, 7, 12009 CrossRef CAS PubMed; (d) B. Zetsche, S. E. Volz and F. Zhang, Nat. Biotechnol., 2015, 33, 139 CrossRef CAS PubMed; (e) Y. Gao, X. Xiong, S. Wong, E. J. Charles, W. A. Lim and L. S. Qi, Nat. Methods, 2016, 13, 1043 CrossRef CAS PubMed.
- (a) J. Hemphill, E. K. Borchardt, K. Brown, A. Asokan and A. Deiters, J. Am. Chem. Soc., 2015, 137, 5642 CrossRef CAS PubMed; (b) Y. Nihongaki, F. Kawano, T. Nakajima and M. Sato, Nat. Biotechnol., 2015, 33, 755 CrossRef CAS PubMed; (c) L. R. Polstein and C. A. Gersbach, Nat. Chem. Biol., 2015, 11, 198 CrossRef CAS PubMed.
- (a) C. J. McKinlay, J. R. Vargas, T. R. Blake, J. W. Hardy, M. Kanada, C. H. Contag, P. A. Wender and R. M. Waymouth, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E448 CrossRef CAS PubMed; (b) C. J. McKinlay, N. L. Benner, O. A. Haabeth, R. M. Waymouth and P. A. Wender, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5859 CrossRef PubMed; (c) O. A. W. Haabeth, T. R. Blake, C. J. McKinlay, R. M. Waymouth, P. A. Wender and R. Levy, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E9153 CrossRef PubMed.
- (a) Y. Liu, Y. Zhan, Z. Chen, A. He, J. Li, H. Wu, L. Liu, C. Zhuang, J. Lin, X. Guo, Q. Zhang, W. Huang and Z. Cai, Nat. Methods, 2016, 13, 938 CrossRef CAS PubMed; (b) W. Tang, J. H. Hu and D. R. Liu, Nat. Commun., 2017, 8, 15939 CrossRef CAS PubMed; (c) Q. R. V. Ferry, R. Lyutova and T. A. Fulga, Nat. Commun., 2017, 8, 14633 CrossRef PubMed.
- K. Kundert, J. E. Lucas, K. E. Watters, C. Fellmann, A. H. Ng, B. M. Heineike, C. M. Fitzsimmons, B. L. Oakes, J. Qu, N. Prasad, O. S. Rosenberg, D. F. Savage, H. El-Samad, J. A. Doudna and T. Kortemme, Nat. Commun., 2019, 10, 2127 CrossRef PubMed.
- Y. J. Lee, A. Hoynes-O'Connor, M. C. Leong and T. S. Moon, Nucleic Acids Res., 2016, 44, 2462 CrossRef CAS PubMed.
- A. Kadina, A. M. Kietrys and E. T. Kool, Angew. Chem., Int. Ed., 2018, 57, 3059 CrossRef CAS PubMed.
- (a) N. Benner, K. Near, M. Bachmann, C. Contag, R. Waymouth and P. Wender, Biomacromolecules, 2018, 19, 2812 CrossRef CAS PubMed; (b) N. L. Benner, R. L. McClellan, C. R. Turlington, O. A. W. Haabeth, R. M. Waymouth and P. A. Wender, J. Am. Chem. Soc., 2019, 141, 8416 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03639c |
| This journal is © The Royal Society of Chemistry 2020 |