
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sulfamides direct radical-mediated chlorination of aliphatic C–H bonds†
Melanie A.
Short
 ,
Mina F.
Shehata
,
Matthew A.
Sanders
and
Jennifer L.
Roizen
*
,
Mina F.
Shehata
,
Matthew A.
Sanders
and
Jennifer L.
Roizen
*
Duke University, Department of Chemistry, Box 90346, Durham, North Carolina 27709-0354, USA. E-mail: j.roizen@duke.edu
First published on 8th November 2019
Abstract
Given the prevalence of aliphatic amines in bioactive small molecules, amine derivatives are opportune as directing groups. Herein, sulfamides serve as amine surrogates to guide intermolecular chlorine-transfer at γ-C(sp3) centers. This unusual position-selectivity arises because accessed sulfamidyl radical intermediates engage preferentially in otherwise rare 1,6-hydrogen-atom transfer (HAT) processes through seven-membered transition states. The site-selectivity of C–H abstraction can be modulated by adjusting the steric and electronic properties of the sulfamide nitrogen substituents, an ability that has not been demonstrated with other substrate classes. The disclosed reaction relies on a light-initiated radical chain-propagation mechanism to oxidize C(sp3)–H bonds efficiently.
Introduction
Aliphatic amines are important structural motifs within organic molecules, making alkyl amine derivatives readily available. These derivatives can be used to guide position-selective C–H functionalization reactions1 to α-,2 β-, γ-, and δ-positions.3 Nevertheless, few strategies result in γ-C(sp3)–H functionalization. Amine derivatives template γ-selective cyclometallation processes4–6 (Scheme 1A), and can stabilize metallonitrenoid or metalloradical intermediates to facilitate C–H amination reactions (Scheme 1B).7–9 As a mechanistic complement to these approaches, herein disclosed is the first reaction in which an amine surrogate guides γ-C(sp3)–H functionalization by way of free radical intermediates,10 enabling a sulfamide-guided chlorine-transfer process (Scheme 1C).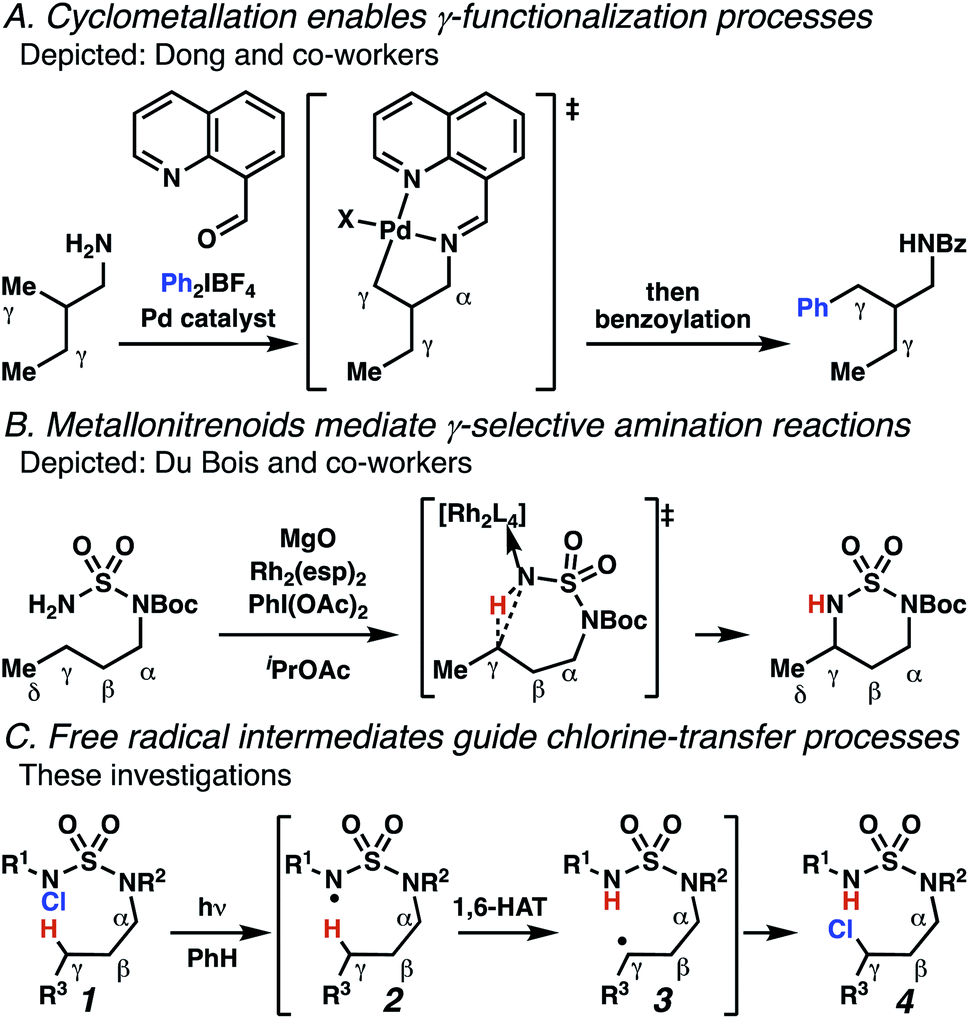 | ||
| Scheme 1 γ-C(sp3)–H reactions of amine derivatives. | ||
In these reactions, intermediate sulfamidyl radicals 2 engage in otherwise rare 1,6-HAT processes. With the exception of the recent discovery of sulfamate ester-templated reactions,11–13 transformations that employ cleavable linkers in 1,6-HAT processes lack generality. Fortunately, sulfamides appear to direct reactions based on 1,6-HAT processes,10 consistent with the geometrically originated prediction that elongated S–N bonds (∼1.58–1.69 Å)14 kinetically favor a seven-membered transition state15 for C–H abstraction.
This concept is developed to enable position-selective chlorine-transfer reactions (Scheme 1C). Alkyl chlorides are durable, yet versatile synthetic intermediates,16 and can be found in bioactive small molecules.17 Yet, directed C(sp3)–H chlorination reactions18 can be plagued by competitive off-site chlorine-installation arising from unguided C–H abstraction. The developed sulfamide-directed reactions offer high levels of position-selectivity with the unusual ability to predictably modulate site-selectivity based on variations in the steric and electronic properties of the substituents on the sulfamide nitrogen atoms. With appropriate substituents, the site of chlorine-transfer is complementary to that available based on other techniques,19 including sulfamate ester-guided processes,11 traditional Hoffman–Löffler–Freytag protocols,20 templated methods,21 and unguided processes that rely on innate selectivity.1a,b,22,23
Results and discussion
Sulfamide substrates present two chemically distinct nitrogen atoms that can support nitrogen-centered radicals as reaction intermediates. To simplify mechanistic investigations, we chose to access sulfamidyl radicals from N-chlorosulfamides 1 and 5via light-initiated nitrogen–chlorine bond homolysis. The requisite sulfamides are prepared from alcohols through Mitsunobu reactions,24 or from amines using a recently disclosed sulfamoylation strategy.25 The generated sulfamides react with an electrophilic chlorinating reagent to provide structurally diverse N-chlorosulfamides.As anticipated, N-chlorosulfamides prepared such that the chlorine atom initially resides on the “internal” sulfamide nitrogen (i.e., 5), engage in selective 1,5-HAT processes upon photoirradiation. These substrates provide δ-chlorinated alkanes 6 in good yield with exquisite selectivity (Scheme 2). As this selectivity mimics that observed in related HLF-type processes, our investigations primarily focus on reactions that target transformation of γ-C(sp3)–H bonds of amine derivatives, as technologies for γ-C(sp3)–H functionalization are limited (Scheme 1).
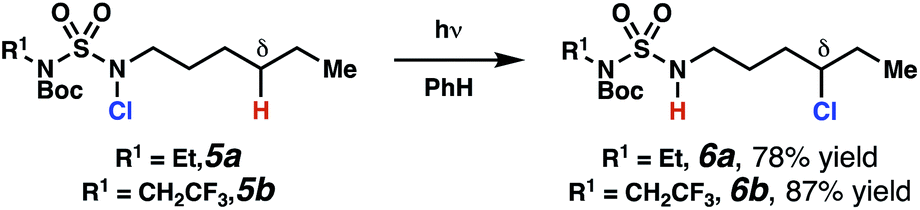 | ||
| Scheme 2 Chlorine-transfer from “internal” sulfamide nitrogen occurs through a 1,5-HAT process to provide δ-chlorinated alkanes. | ||
Building upon our laboratory's interest in transformations governed by 1,6-HAT processes,11,13a,d we sought to exploit N-chlorosulfamides in reactions to access γ-chlorinated alkanes as a complement to more traditional chlorination methods. To our delight, when employing N-chlorosulfamides where the radical is generated on the “external” sulfamide nitrogen (i.e., 1), chlorine-transfer proceeds in synthetically useful yields at primary, secondary, and tertiary C–H bonds (Table 1). These C–H bonds have bond dissociation energies (BDEs) that cover a broad range (BDE ≈ 96–101 kcal mol−1),26 demonstrating the generality of the transformation. In particular, this chlorine-transfer reaction provides access to primary alkyl chlorides (4a) in excellent yield, outperforming related sulfamate ester-11–13 and sulfamide-guided10 methods in transforming strong primary C–H bonds. This sulfamide-guided process oxidizes C–H bonds at acyclic or cyclic centers (entries 4–6), and is compatible with pendant ester (entry 7) and masked amine (entry 8) functionalities. Moreover, naturally abundant amines, such as leucine-derivatives, are appropriate substrate precursors (e.g.4f).
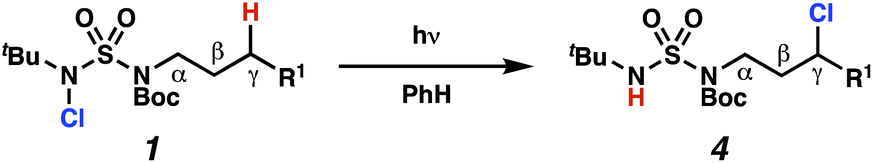
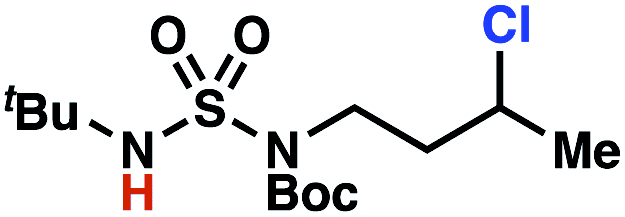
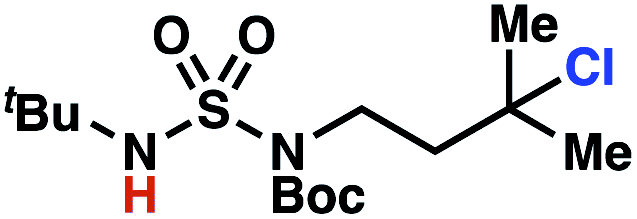
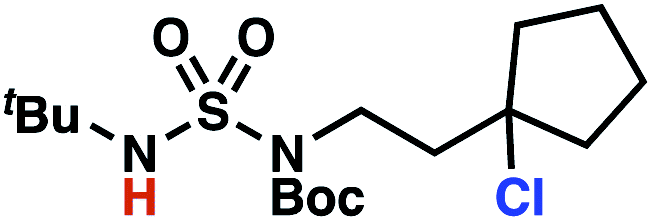
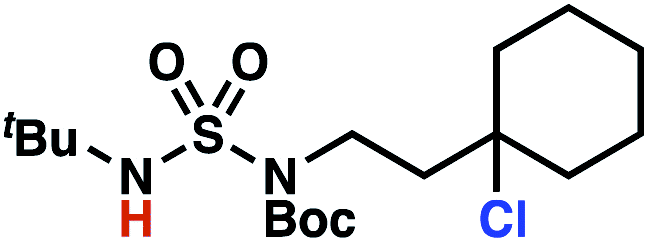
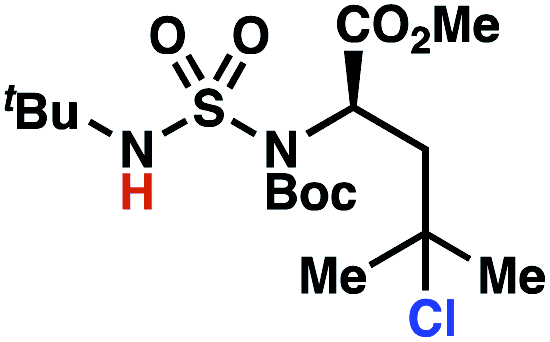
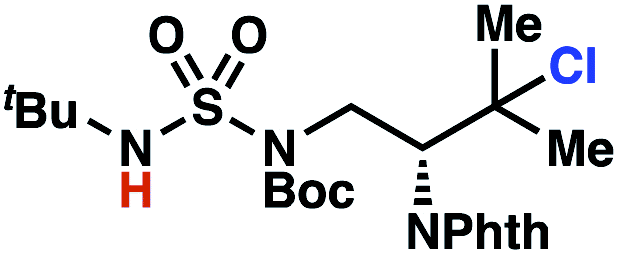
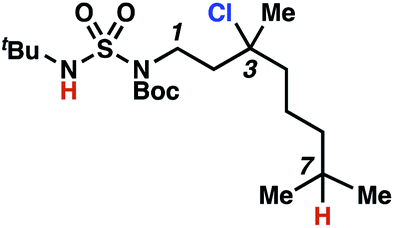
This guided chlorine-transfer process overcomes site-selectivity arising from inductive deactivation. Electron-withdrawing groups, such as sulfamides, inductively deactivate proximate C–H bonds to abstraction by electrophilic radicals. Consequently, unguided C–H functionalization reactions engage more distal C–H bonds preferentially. This effect is particularly evident when employing unguided, radical-mediated reaction protocols with 3,7-dimethyloctyl derivatives where the C(7)–H bond serves as the predominant site of oxidation in azidation,27 amination,28 fluorination,29 trifluoromethylthiolation,30 and hydroxylation31 processes. By contrast, sulfamide 1h undergoes templated chlorination at C(3)–H with exquisite site-selectivity (entries 9 and 10).
While the most consistently efficient protocol for chlorine-transfer relies on irradiation with UV light in benzene, some substrates react efficiently in iPrOAc upon photolysis with compact fluorescent lights (CFLs, entries 5 and 10).
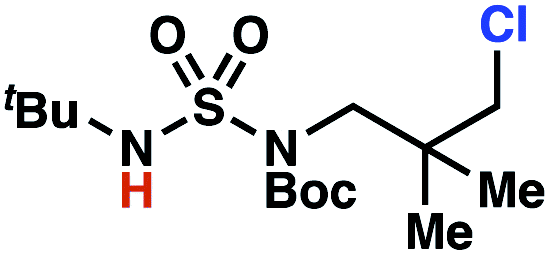
Surprisingly, sulfamide substrates undergo competitive γ- and δ-chlorination when they incorporate γ-C(sp3)–H bonds in proximity to weaker δ-C(sp3)–H bonds, a phenomenon not generally observed in related sulfamate ester-guided reactions.11–13 For example, N-pentyl sulfamide 1i yields a crude 8.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of γ- and δ-chlorinated 4i and 7i from which γ-chlorinated 4i can be isolated in 75% yield (Scheme 3A). In principle, δ-chlorinated minor product 7i could form via either a substrate-guided 1,7-HAT process that relies on an eight-membered transition state, or an intermolecular C–H abstraction process.
:1 mixture of γ- and δ-chlorinated 4i and 7i from which γ-chlorinated 4i can be isolated in 75% yield (Scheme 3A). In principle, δ-chlorinated minor product 7i could form via either a substrate-guided 1,7-HAT process that relies on an eight-membered transition state, or an intermolecular C–H abstraction process.
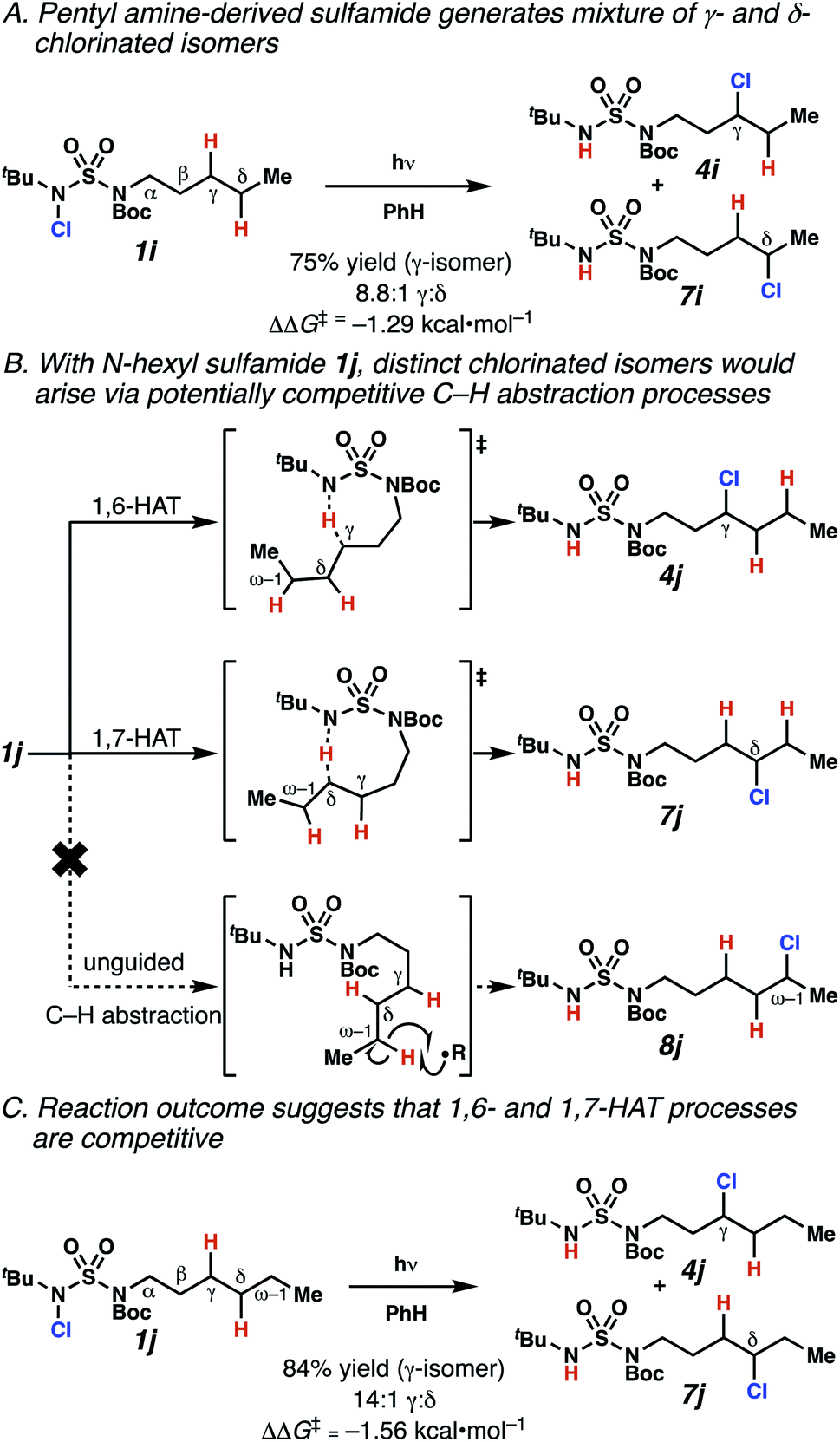 | ||
| Scheme 3 1,6- and 1,7-HAT processes appear competitive. | ||
To discriminate between these pathways, N-hexyl sulfamide 1j was employed (Scheme 3B). With this substrate, a 1,7-HAT process would generate a δ-chlorinated product, whereas, a reaction reliant on innate selectivity would engage the most distal, secondary C–H bond to form (ω−1)-chlorinated 8j. This reaction provides a crude 14:1 mixture of γ-chlorinated 4j and δ-chlorinated 7j (Scheme 3C). Fortuitously, (ω−1)-chlorinated 8j is not detected, suggesting that δ-chlorinated 7j may form through a guided 1,7-HAT process.
If a guided 1,7-HAT process provides the δ-chlorinated product, the ratio of γ-chlorinated 4 to δ-chlorinated 7 will quantitatively reflect the energetic difference between transition state barriers for competing 1,6- and 1,7-HAT processes (Scheme 3, Table 2). To relate the measured product ratios to the difference in the transition state barrier heights (Gibbs free energies), we employed the equation
| (ΔΔG‡ = −RTln(γ-chlorinated 4/δ-chlorinated 7)). |
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Product | γ:δb |
ΔΔG‡c (kcal mol−1) | Yieldd | |
|
a Conditions A.
b Determined by 1H or 19F NMR of crude mixture.
c Calculated from experimental product ratios.
d Isolated yield of depicted product.
e δ-Chlorinated-isomer not detected.
f Calculated assuming ≥20:1 ratio of 4:7.
g Isolated as a mixture of γ- and δ-chlorinated isomers.
h Conditions: 1.0 equiv. N-chlorosulfamate 9, PhH (0.07 M), 2 blue Kessil lamps.11
|
|||||
| 1 |
|
4k | —e | ≤−1.77f | 83 |
| 2 |
|
4l | >20:1 |
≤−1.77f | 87 |
| 3 |
|
4m | —e | ≤−1.77f | 97 |
| 4 |
|
4n | —e | ≤−1.77f | 63 |
| 5 |
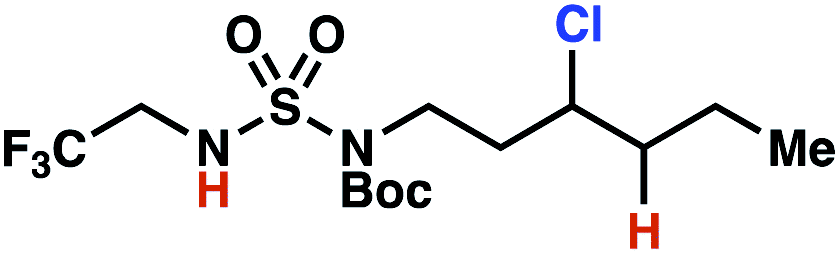
|
4o | 11:1 |
−1.39 | 79 |
| 6 |
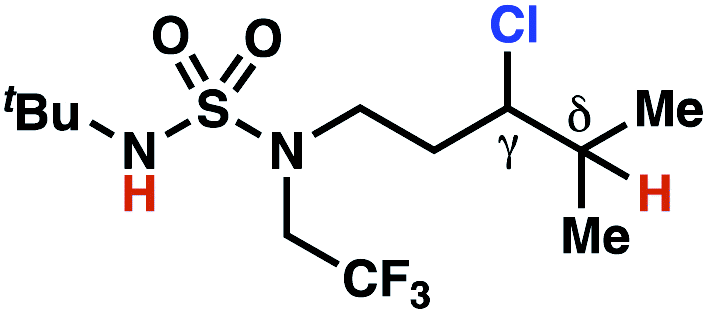
|
4p | 13:1 |
−1.52 | 70 |
| 7 |
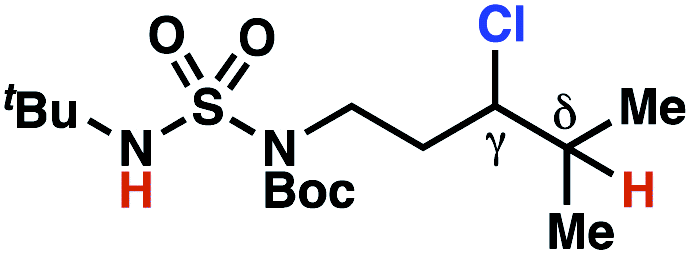
|
4q | 2:1 |
−0.41 | 98g |
| 8 |
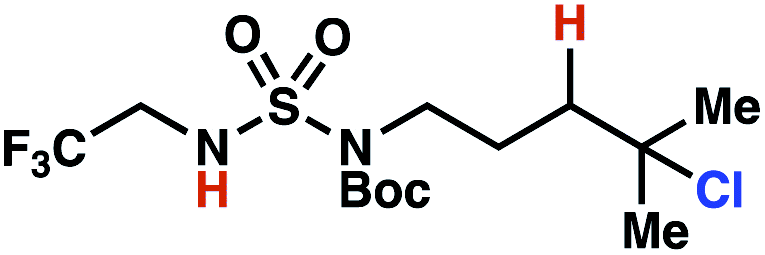
|
7r | 1:2 |
+0.38 | 84g |
| 9h |
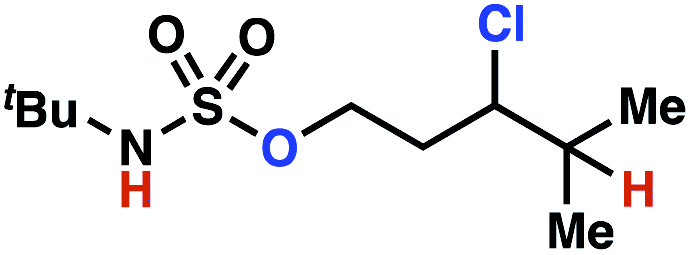
|
10a | 3:1 |
−0.69 | 57g |
| 10h |
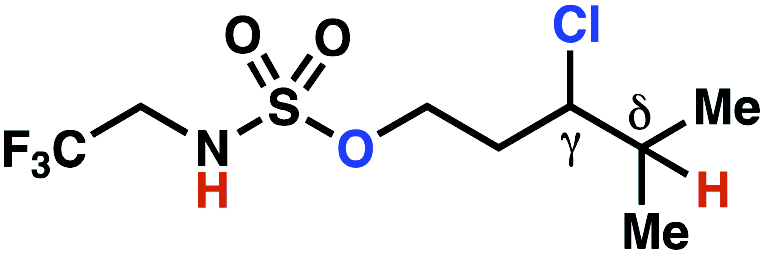
|
10b | 4:1 |
−0.82 | 67g |
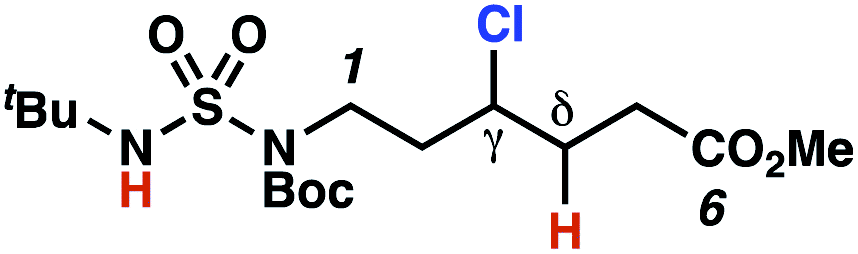
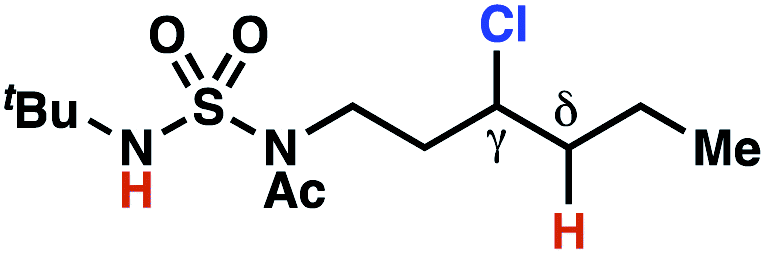
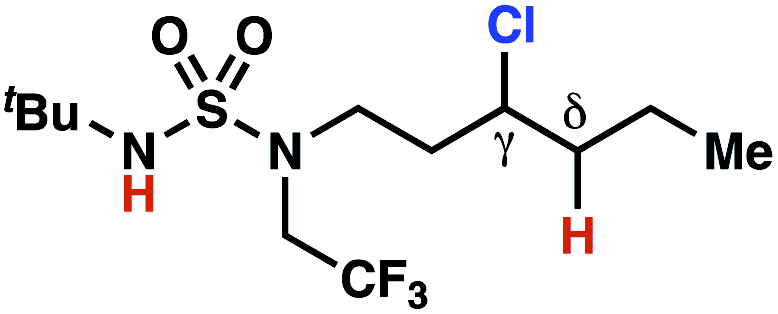
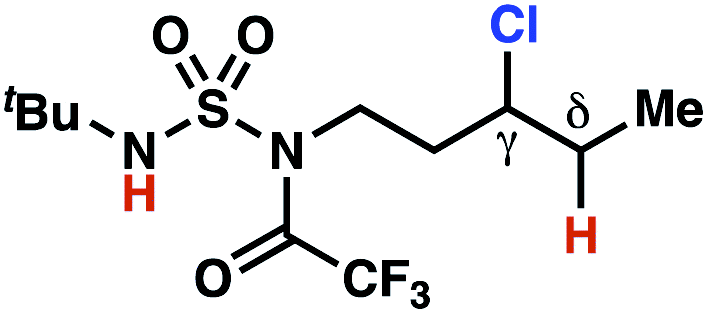
The ratio of γ- and δ-chlorinated 4 to 7 is sensitive to variations in substrate structure. Predictably, a C(6) ester inductively deactivates the δ-C–H bond to reaction, such that γ-chlorinated 4k forms exclusively (entry 1). Unexpectedly, the ratio of γ- to δ-chlorinated 4 to 7 increases as substituents on the tertiary nitrogen of the sulfamide become more electron withdrawing, from tert-butoxycarbonyl-, to acetyl-, to trifluoroacetyl-, and 2,2,2-trifluoroethyl-groups (Scheme 3B; Table 2, entries 2–4). This is the opposite of the trend that would be predicted based on BDEs. By contrast, the ratio of γ- to δ-chlorinated products decreases as substituents on the secondary nitrogen of the sulfamide decrease in electron density from tert-butyl- to 2,2,2-trifluoroethyl-groups (Scheme 3B; Table 2, entries 5 and 8).
These trends are more evident with substrates bearing γ-secondary C–H bonds and weaker, δ-tertiary C–H bonds (entries 6–8). Moreover, the effects of these trends are synergistic. Indeed, the combination of these trends can be used to favor the formation of δ-chlorinated 7r as the major product, possibly based on an eight-membered transition state (entry 8).
By contrast, with sulfamate esters 9, substitution has a less pronounced effect on product ratios, with an influence over the ratio of γ- to δ-chlorinated product isomers that is not statistically significant (Table 2, entries 9–10). Apparently, sulfamate ester- and sulfamide-directed processes differ substantively owing to the marked influence of nitrogen substituents on the site of sulfamide-directed HAT processes. This pronounced effect distinguishes this method from comparable sulfamate ester-templated reactions.
To the best of our knowledge, this is the first series of experimental data to provide evidence of the relative transition state barriers for competitive intramolecular radical-mediated processes. As such, we anticipate that the data published herein can serve as a benchmark that can be used to gauge the quality of computational transition state calculation methods.
In general, care should be taken when calculating transition state energies between radical intermediates. Few data sets highlight differences in barrier heights for competitive radical-mediated reaction pathways.32c Consequently, the quality of transition state calculations in radical pathways is often inferred based on agreement between computational methods. In such cases, extremely simple systems have been employed to provide limited experimental input regarding transition state energy measurements.32
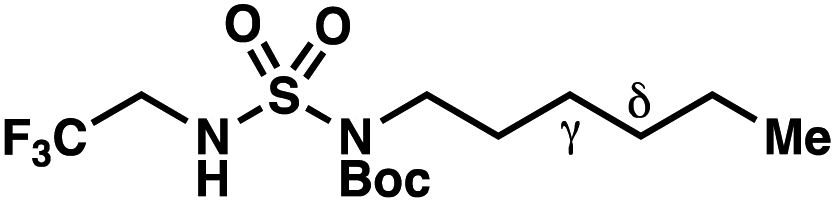
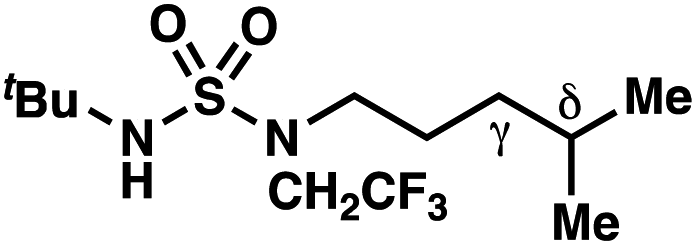
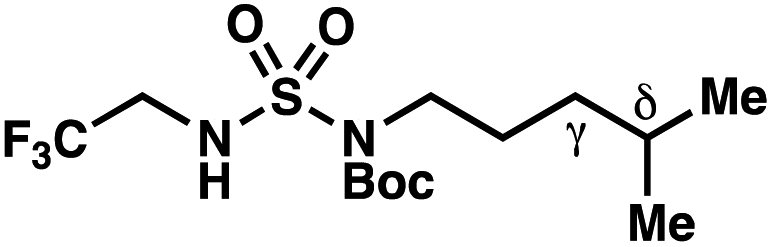
We set out to evaluate the ability of one of the more commonly recommended functional/basis set combinations to recapitulate qualitative trends in barrier heights associated with the disclosed radical-mediated transformations. To this end, we have modeled the product-determining intermediates (2, 3, 11) and transition states (3-TS, 11-TS) for a subset of sulfamide and sulfamate ester chlorine-transfer processes using density functional theory (DFT, Fig. 1, Table 3). Using the uB3LYP functional and the 6-31+G(d,p) basis set, we observe that the DFT method over-predicts the stability of the 7-membered ring transition states adopted for 1,6-HAT processes with both sulfamate ester substrates (entries 1 and 2) as well as the hexyl-derived sulfamide substrates (entries 3–5). Within these classes of compounds, calculations qualitatively correlate well with experimental results.
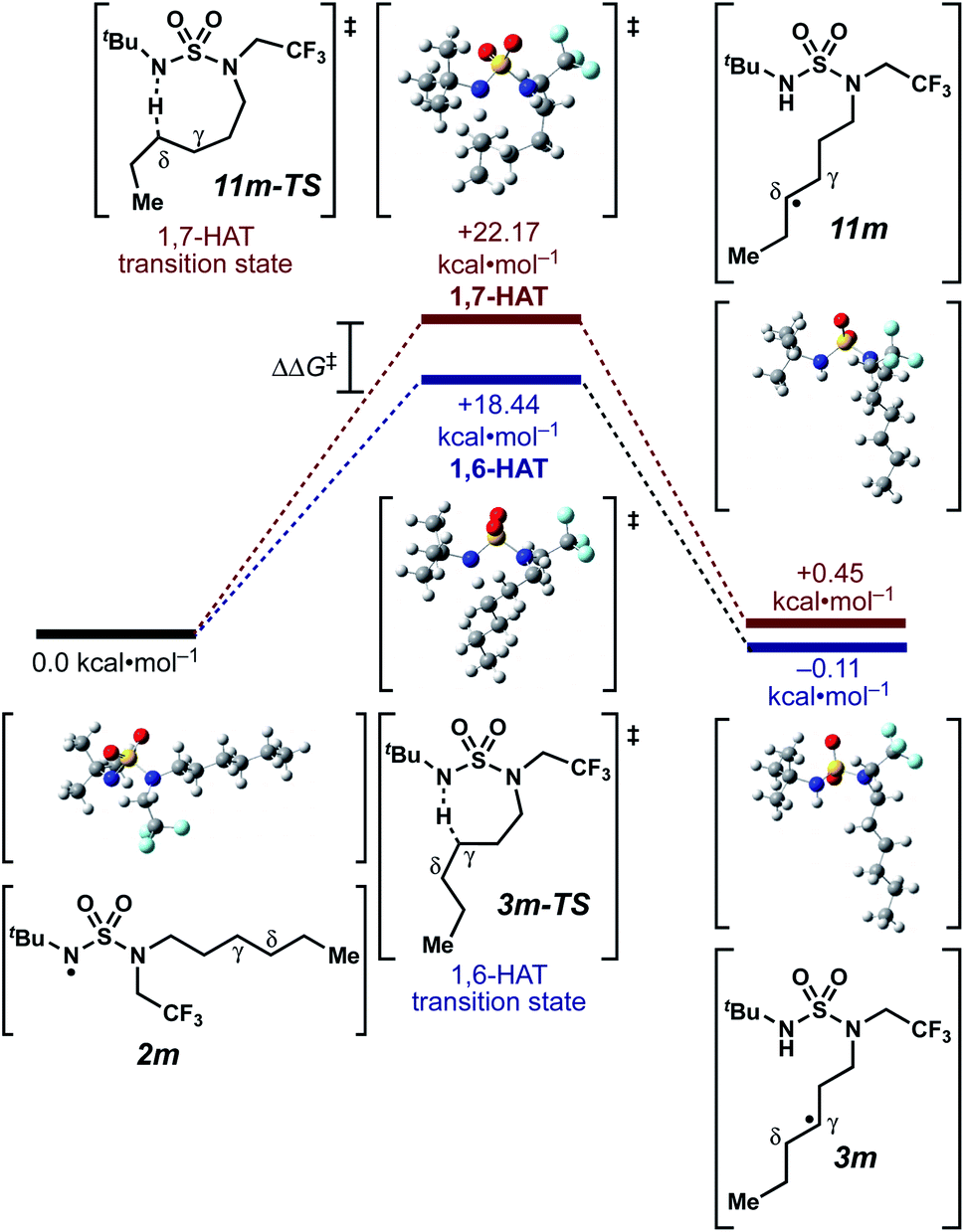 | ||
| Fig. 1 Representative example of calculated energies and structures for competing 1,6- and 1,7-HAT pathways. Density functional calculations were performed using Gaussian 09 (revision D.01) using the μB3LYP functional and the 6-31+G(d,p) basis set. See ESI† for further computational details. | ||
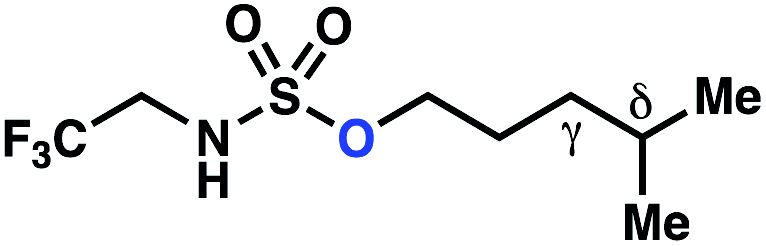
| Entry | Parent compound | Experimental ΔΔG‡a (kcal mol−1) | Calculated ΔΔG‡b (kcal mol−1) |
|---|---|---|---|
|
a ΔΔG‡ = −RTln(γ-chlorinated product/δ-chlorinated product) ad determined by 1H or 19F NMR of crude reaction mixture.
b ΔΔG‡ = (ΔG(1,6-HAT TS) − ΔG(1,7-HAT TS)) as determined from the calculated Gibbs free energies using uB3LYP/6-31+G(d,p).
|
|||
| 1 |
|
−0.82 | −3.33 |
| 2 |
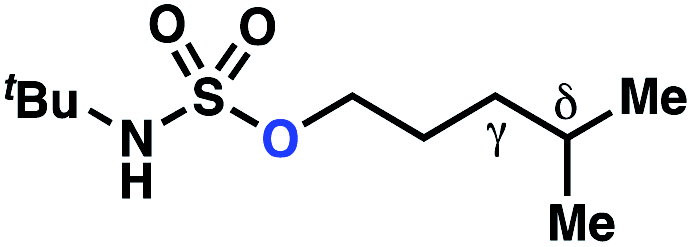
|
−0.69 | −1.09 |
| 3 |
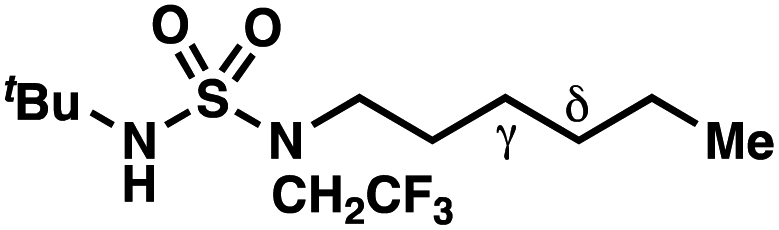
|
≤−1.77 | −3.74 |
| 4 |
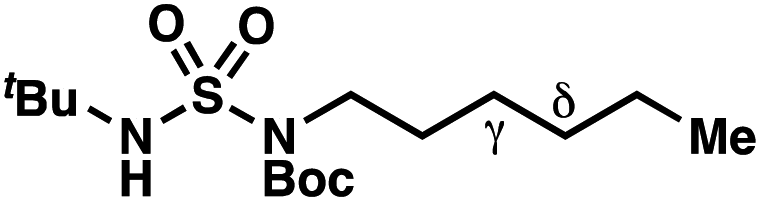
|
−1.56 | −2.98 |
| 5 |
|
−1.39 | −2.06 |
| 6 |
|
−1.52 | +0.35 |
| 7 |

|
−0.41 | +2.24 |
| 8 |
|
+0.38 | +1.04 |
By contrast, experimental and calculated differences in transition state barriers are poorly correlated when performed on 4-methylpentyl-derived sulfamides (entries 6–8), where 1,6-HAT results in abstraction of a hydrogen atom from a secondary center (BDE ≈ 98 kcal mol−1) and 1,7-HAT requires abstraction from a weaker tertiary center (BDE ≈ 96 kcal mol−1). The qualitative inconsistency between our experimental and computational results is evidence that our synthetically oriented community should exercise extreme caution when making claims based on calculated energies for transition state barriers between radical intermediates.
Rigorous experiments can provide insight into the mechanism of these chlorine-transfer reactions. In principle, chlorine-transfer could involve a radical chain propagation mechanism or a closed reaction pathway. To initiate either of these processes, light-promoted N–Cl bond homolysis would convert N-chlorosulfamide 1h to chlorine radical and nitrogen-centered radical 2h. Sulfamidyl radical 2h then performs a site-selective hydrogen-atom abstraction through a seven-membered transition state to generate carbon-centered radical 3h.
The two feasible reaction paths differ in terms of the carbon–chlorine bond forming events. In a radical-chain propagation process, carbon-centered radical 3h engages another equivalent of N-chlorosulfamide substrate 1h in chlorine-atom abstraction (Scheme 4). This sequence would produce desired chlorinated 4h along with another equivalent of nitrogen-centered radical 2h, which would propagate this chain reaction. Alternatively, in a closed reaction mechanism, intermediate carbon-centered radical 3h would recombine with the initially generated chlorine radical to terminate the reaction and afford chlorinated 4h (not depicted).
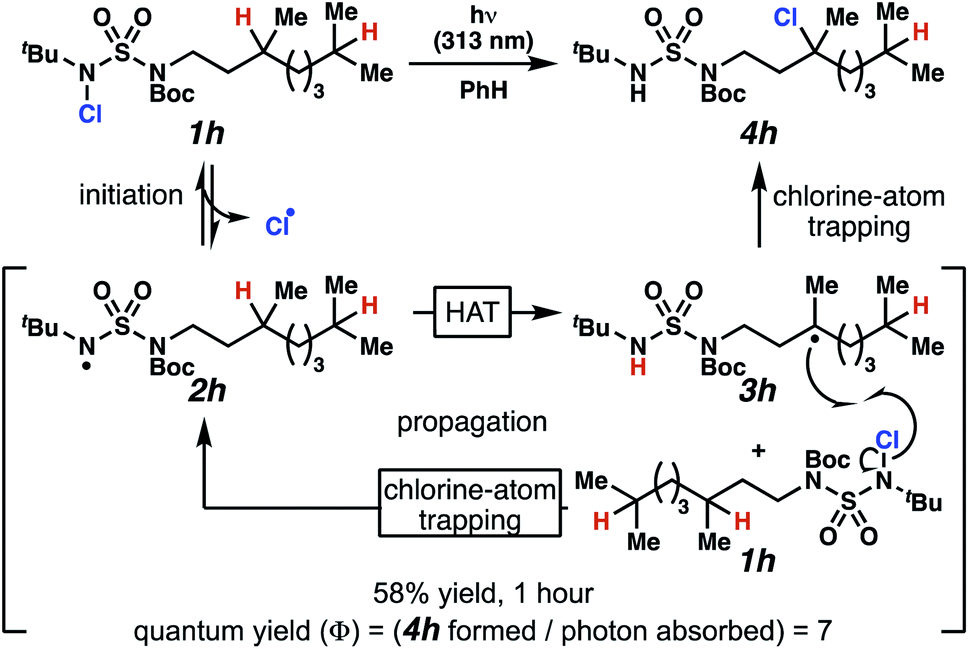 | ||
| Scheme 4 Chlorination proceeds through a light-initiated chain propagation. | ||
These reaction pathways would differ in terms of the equivalents of product formed per absorbed photon, defined as the quantum yield (Φ). In a radical chain propagation process, each absorbed photon could initiate the formation of multiple equivalents of product (Φ > 1). By contrast, in a closed process, each absorbed photon could initiate the preparation of a maximum of one product molecule (Φ ≤ 1).
Quantum yield measurements suggest that the reaction engages a light-initiated chain propagation mechanism. To determine the number of photons available to a sample in a fluorimeter, we rely on standard chemical actinometry using potassium ferrioxalate at 313 nm.33,34 In the calibrated fluorimeter, 1 hour of irradiation of N-chlorinated 1h in benzene furnishes chloroalkane 4h in 58% isolated yield. This yield indicates that at least 7 equivalents of product have formed for each absorbed photon (Φ = 7), a value that is consistent with chlorination via a radical chain propagation process.
In spite of the rapid speed of radical chain propagation, the generated radical has a long enough lifetime to promote ring-opening of an appropriately positioned cyclopropane (Scheme 5). Upon photoirradiation, N-chlorosulfamide 1s reacts to furnish ring-opened isomer 12s in 80% yield, with exclusive detection of ring-opened products. This cascade sequence provides position-selective access to a more distally ζ-chlorinated product with an intervening olefin. The presence of an intact olefins is interesting, as alkenes are not tolerated under typical N-chlorination conditions.
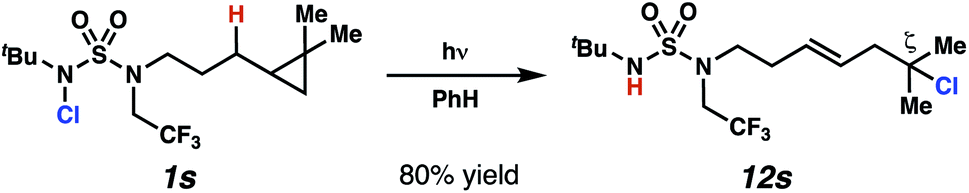 | ||
| Scheme 5 Cyclopropyl-containing substrate provides evidence for radical reaction pathway. | ||
Conclusions
These investigations demonstrate that sulfamides guide 1,6-HAT processes. This mechanistic manifold has been employed to access alkyl chlorides, which are high-value synthetic intermediates. Consequently, this sulfamide-directed process establishes the premise for a broadly translatable γ-C(sp3)–H functionalization approach that complements known alkane functionalization technologies.Furthermore, these investigations establish that sulfamide substitution can be used to predictably vary the site-selectivity of C–H abstraction processes. Initial calculations have not qualitatively recapitulated experimental trends. Fortunately, the mere ability to experimentally quantify the relative transition state barriers for two competing radical-mediated reaction steps is of benefit as a benchmark for computational methods, where quantitative data relating to barrier heights is scarce.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Within this disclosure, the National Institutes of Health (R35GM128741-01) funded research documenting sulfamate ester reactivity. The remainder of this project was funded by Duke University, and through Predoctoral Fellowships to partially support MAS (NSF DCG 1106401, and Burroughs Wellcome), and MFS (U.S. Department of Education GAANN Fellowship, Award No. P200A150114). Computational data has been acquired using the Extreme Science and Engineering Discovery Environment (XSEDE) resource Comet at the San Diego Supercomputer Center through a startup allocation (MAS, TG-CHE19001). We would like to thank Mikey Kwon, Erin Viere, Martina Zafferani, and Dr Peng Zhang of the Duke Chemistry Department for initial assistance in performing computational calculations. Characterization data were obtained on instrumentation secured by funding from the NSF (CHE-0923097, ESI-MS, G. Dubay, Duke Dept. of Chemistry Instrument Center), or the NSF, NIH, HHMI, North Carolina Biotechnology Center and Duke (Duke Magnetic Resonance Spectroscopy Center).Notes and references
- (a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (c) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed.
- K. Nakajima, Y. Miyake and Y. Nishibayashi, Acc. Chem. Res., 2016, 49, 1946 CrossRef CAS PubMed.
- J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62 ( Angew. Chem. , 2018 , 130 , 64 ) CrossRef CAS PubMed.
- Y. Xu and G. Dong, Chem. Sci., 2018, 9, 1424 RSC.
- (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed; (b) Y. Xu, M. C. Young, C. Wang, D. M. Magness and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 9084 ( Angew. Chem. , 2016 , 128 , 9230 ) CrossRef CAS PubMed; (c) Y. Liu and H. Ge, Nat. Chem., 2017, 9, 26 CrossRef CAS; (d) Y. Wu, Y. Q. Chen, T. Liu, M. D. Eastgate and J. Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554 CrossRef CAS PubMed; (e) A. Yada, W. Liao, Y. Sato and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 1073 ( Angew. Chem. , 2017 , 129 , 1093 ) CrossRef CAS PubMed.
- J. Calleja, D. Pla, T. W. Gorman, V. Domingo, B. Haffemayer and M. J. Gaunt, Nat. Chem., 2015, 7, 1009 CrossRef CAS PubMed.
- (a) C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378 CrossRef CAS PubMed; (b) T. Kurokawa, M. Kim and J. Du Bois, Angew. Chem., Int. Ed., 2009, 48, 2777 ( Angew. Chem. , 2009 , 121 , 2815 ) CrossRef CAS PubMed.
- (a) H. Lu, H. Jiang, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2010, 49, 10192 ( Angew. Chem. , 2010 , 122 , 10390 ) CrossRef CAS PubMed; (b) H. Lu, Y. Hu, H. Jiang, L. Wojtas and X. P. Zhang, Org. Lett., 2012, 14, 5158 CrossRef CAS PubMed; (c) H. Lu, K. Lang, H. Jiang, L. Wojtas and X. P. Zhang, Chem. Sci., 2016, 7, 6934 RSC.
- X. Xiao, C. Hou, Z. Zhang, Z. Ke, J. Lan, H. Jiang and W. Zeng, Angew. Chem., Int. Ed., 2016, 55, 11897 ( Angew. Chem. , 2016 , 128 , 12076 ) CrossRef CAS PubMed.
- Concurrently, another PI and co-workers have advanced a sulfamide-guided radical-mediated group-transfer process: T. Duhamel, M. D. Matínez, I. K. Sideri and K. Muñiz, ACS Catal., 2019, 9, 7741 CrossRef CAS.
- M. A. Short, J. M. Blackburn and J. L. Roizen, Angew. Chem., Int. Ed., 2018, 57, 296–299 ( Angew. Chem. , 2018 , 130 , 302 ) CrossRef CAS PubMed.
- S. Sathyamoorthi, S. Banerjee, J. Du Bois, N. Z. Burns and R. N. Zare, Chem. Sci., 2018, 9, 100 RSC.
- (a) K. Muñiz, E. Del Castillo, M. D. Martínez, A. E. Bosnidou, T. Duhamel, C. Q. O'Broin, H. Zhang, E. C. Escudero-Adán and M. Martínez-Belmonte, Chem.–Eur. J., 2018, 24, 17225 CrossRef PubMed; (b) S. K. Ayer and J. L. Roizen, J. Org. Chem., 2019, 84, 3508 CrossRef CAS PubMed; (c) Z.-Y. Ma, L.-N. Guo, Y. You, F. Yang, M. Hu and X.-H. Duan, Org. Lett., 2019, 21, 5500 CrossRef CAS PubMed; (d) A. L. G. Kanegusuku, T. Castanheiro, S. K. Ayer and J. L. Roizen, Org. Lett., 2019, 21, 6089 CrossRef CAS PubMed; (e) W. Shu, H. Zhang and Y. Huang, Org. Lett., 2019, 21, 6107 CrossRef CAS PubMed.
- See ESI† for S–N bond lengths in related sulfamides.
- M. Nechab, S. Mondal and M. P. Bertrand, Chem.–Eur. J., 2014, 20, 16034 CrossRef CAS PubMed.
- W. R. Gutenkunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC.
- G. W. Gribble, Naturally Occurring Organohalogen Compounds: A Comprehensive Update, Springer-Verlag, Weinheim, Germany, 2010 Search PubMed.
- P. S. Skell and H. N. Baxter, J. Am. Chem. Soc., 1985, 107, 2823 CrossRef CAS.
- J. Y. Su, D. C. Grünenfelder, K. Takeuchi and S. Reisman, Org. Lett., 2018, 20, 4912 CrossRef CAS PubMed.
- (a) Q. Qin and S. Yu, Org. Lett., 2015, 17, 1894 CrossRef CAS PubMed; (b) J. Ozawa and M. Kanai, Org. Lett., 2017, 19, 1430 CrossRef CAS PubMed; (c) S. P. Morcillo, E. M. Dauncey, J. H. Kim, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 12945 ( Angew. Chem. , 2018 , 130 , 13127 ) CrossRef CAS PubMed; (d) E. A. Wappes, A. Vanitcha and D. A. Nagib, Chem. Sci., 2018, 9, 4500 RSC.
- (a) R. Breslow, R. J. Corcoran, B. B. Snider, R. J. Doll, P. L. Khanna and R. Kaleya, J. Am. Chem. Soc., 1977, 99, 905 CrossRef CAS PubMed; (b) R. Breslow and D. Heyer, J. Am. Chem. Soc., 1982, 104, 2045 CrossRef CAS; (c) H. Guan, S. Sun, Y. Mao, L. Chen, R. Lu, J. Huang and L. Liu, Angew. Chem., Int. Ed., 2018, 57, 11413 CrossRef PubMed.
- (a) T. Brückl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826 CrossRef PubMed; (b) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC.
- (a) R. A. Quinn, Z. A. Könst, S. E. Michalak, Y. Schmidt, A. R. Szklarski, A. R. Flores, S. Nam, D. A. Horne, C. D. Vanderwal and E. Alexanian, J. Am. Chem. Soc., 2016, 138, 696 CrossRef CAS PubMed; (b) W. Liu and J. T. Groves, J. Am. Chem. Soc., 2010, 132, 12847 CrossRef CAS PubMed; (c) W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727 CrossRef CAS PubMed; (d) G. Li, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, Angew. Chem., Int. Ed., 2018, 57, 1251 CrossRef CAS PubMed; (e) S. Liu, Q. Zhang, X. Tian, S. Fan, J. Huang and A. Whiting, Green Chem., 2018, 20, 4729 RSC; (f) F. Minisci, R. Galli, A. Galli and R. Bernardi, Tetrahedron Lett., 1967, 8, 2207 CrossRef; (g) N. C. Deno, R. Fishbein and J. C. Wyckoff, J. Am. Chem. Soc., 1971, 93, 2065 CrossRef CAS.
- Y. Iso, T. Irie, T. Iwaki, M. Ku, Y. Sendo, K. Motokawa and Y. Nishitani, J. Antibiot., 1996, 49, 478 CrossRef CAS PubMed.
- (a) J. M. Blackburn, M. A. Short, T. Castanheiro, S. K. Ayer, T. D. Muellers and J. L. Roizen, Org. Lett., 2017, 19, 6012 CrossRef CAS PubMed; (b) M. F. Shehata, M. A. Short, M. A. Sanders and J. L. Roizen, Tetrahedron, 2019, 75, 3186 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. John R. Rumble, 98th edn, (Internet Version 2018), CRC Press/Taylor & Francis, Boca Raton, FL, 2018 Search PubMed.
- (a) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600 CrossRef CAS PubMed; (b) K. A. Margrey, W. L. Czaplyski, D. A. Nicewicz and E. J. Alexanian, J. Am. Chem. Soc., 2018, 140, 4213 CrossRef CAS PubMed; (c) X. Li and Z.-J. Shi, Org. Chem. Front., 2016, 3, 1326 RSC.
- F. Collet, C. Lescot, C. Liang and P. Dauban, Dalton Trans., 2010, 39, 10401 RSC.
- (a) C. Gal, G. Ben-Shoshan and S. Rozen, Tetrahedron Lett., 1980, 21, 5067 CrossRef CAS; (b) S. Rozen and C. Gal, J. Org. Chem., 1987, 52, 4928 CrossRef CAS.
- S. Mukherjee, B. Maji, A. Tlahuext-Aca and F. Glorius, J. Am. Chem. Soc., 2016, 138, 16200 CrossRef CAS PubMed.
- J. M. Howell, K. Feng, J. R. Clark, L. J. Trzepkowski and M. C. White, J. Am. Chem. Soc., 2015, 137, 14590 CrossRef CAS PubMed.
- (a) N. Mardirossian and M. Head-Gordon, Mol. Phys., 2017, 115, 2315 CrossRef CAS; (b) A. J. Cohen, P. Mori-Sánchez and W. Yang, Chem. Rev., 2012, 112, 289 CrossRef CAS PubMed; (c) X. Xu, I. M. Alecu and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 1667 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
- C. G. Hatchard and C. A. Parker, Proc. R. Soc. London, Ser. A, 1956, 235, 518 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03428e |
| This journal is © The Royal Society of Chemistry 2020 |