
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe rise of continuous flow biocatalysis – fundamentals, very recent developments and future perspectives
Piera
De Santis†
 ,
Lars-Erik
Meyer†
and
Selin
Kara
*
,
Lars-Erik
Meyer†
and
Selin
Kara
*
Aarhus University, Department of Engineering, Biological and Chemical Engineering Section, Biocatalysis and Bioprocessing Group, Gustav Wieds Vej 10, DK 8000 Aarhus, Denmark. E-mail: selin.kara@eng.au.dk
First published on 15th October 2020
Abstract
Biocatalysis community has witnessed a drastic increase in the number of studies for the use of enzymes in continuously operated flow reactors. This significant interest arose from the possibility of combining the strengths of the two worlds: enhanced mass transfer and resource efficient synthesis achieved in flow chemistry at micro-scales and excellent selectivities obtained in biocatalysis. Within this review, we present very recent (from 2018 to September 2020) developments in the field of biocatalysis in continuously operated systems. Briefly, we describe the fundamentals of continuously operated reactors with a special focus on enzyme-catalyzed reactions. We devoted special attention on future perspectives in this key emerging technological area ranging from process analytical technologies to digitalization.
 Piera De Santis | Piera De Santis studied chemistry at the University of Naples Federico II (Italy). Her master thesis has been partly conducted at the Molecular and Industrial Biotechnology Laboratory under the supervision of Prof. Giovanni Sannia and Prof. Georg Guebitz (University of Natural Resources and Life Sciences, Vienna). The project focused on the opportunity of developing and optimizing a nanoparticles production's protocol suitable for the synthesis of polyhydroxyalkanoates (PHAs) and their characterization. In February 2020, she started her Ph.D. in the Biocatalysis and Bioprocessing Group headed by Assoc. Prof. Selin Kara and she is currently working on enzyme immobilization, photocatalysis and continuous flow processes. |
 Lars-Erik Meyer | Lars-Erik Meyer studied chemistry and received his Ph.D. from the University of Rostock (Germany) under the supervision of Dr. Jan von Langermann in 2020. In his dissertation, he investigated thermal separation processes for integrated downstream processing in biocatalytic reactions including adsorption techniques and the use of biocatalysis in non-conventional thermomorphic solvent media. In April 2020, he joined the Biocatalysis and Bioprocessing Group headed by Assoc. Prof. Selin Kara and he is currently working on flow biocatalysis, novel downstream processing approaches for enzyme-catalyzed reactions and photobiocatalytic reactions in continuously operated reactors. |
 Selin Kara | Selin Kara studied Chemical Engineering and Food Engineering at the Middle East Technical University (Turkey). After her M.Sc. study in Biotechnology and Ph.D. at the Hamburg University of Technology (TUHH, Germany), she moved to the TU Delft for her Postdoctoral research with Prof. F. Hollmann. In September 2013, she started her Habilitation at the TU Dresden (Germany) in the group of Prof. M. Ansorge-Schumacher. In May 2018, she finalized her Habilitation at TUHH in the group of Prof. A. Liese. Since July 2018 she is leading Biocatalysis and Bioprocessing Group within Department of Engineering at the Aarhus University (AU, Denmark). |
1. Introduction
Chemical reactions being carried out in continuously operated stirred tank reactors (CSTRs) have developed into an emerging research focus in all sub-disciplines of chemistry and it will receive further and increasing attention in the near future.1 More precisely, the concept of ‘flow chemistry’ defines a very general range of chemical processes that take place in a continuously flowing stream and is not limited to the reactor zone itself, but must be considered as a flow reactor environment that includes e.g. pumps, mixers and downstream units.2Especially, miniaturized flow reactors were studied intensively in the last decade.3 On the other hand, the use of biocatalysts to run chemical transformations has grown to an important and influencing key element in research and industry for the selective synthesis of essential organic compounds.4–9 Merging the knowledge and expertise of chemical reaction engineering10,11 and biocatalysis to a so-called ‘continuous flow biocatalysis’ has recently gained great attention in the scientific community.
Miniaturized flow reactors are applied for continuous processing, whereby reactions take place under rigorously controlled conditions in a confined space. Reduced environmental impact, an improved heat and mass transfer and high-energy efficiencies are only some of the advantages to be mentioned here for continuous flow technology running in miniaturized flow reactors.12,13 Some of their other advantages are: (i) ease of increasing capacity by prolonging reaction time or building series- and/or parallel reactors, (ii) reduced risk associated with accumulation and storage of hazardous intermediates since their transient amounts are below the safety limits, (iii) reduced attrition of enzyme activity compared to using immobilized enzymes under stirring conditions, (iv) easy reaction parameter (temperature, pressure, flow rate) set-up and monitor resulting in more reliable and reproducible processes. Being both a green and sustainable technology,14 academia and industry are now concentrating on continuous production.15
Remarkable well-written reviews16–20 focusing on biocatalysis in continuous flow are available and can complete the current picture of this research area, as this review article in hand cannot claim to provide a complete overview. What should not be forgotten in this context are the recently published excellent review articles dealing with ‘flow chemistry’ in general.21–29 In addition, to make it clearer, we have briefly listed recent reviews and their core interests: a review from Yu et al. is about continuous flow enantioselective catalysis,30 a publication by Noël and co-workers describes the field of continuous flow photochemistry in organic synthesis, material science, and water treatment photo flux catalysis,31 and a review written by Power et al.32 is available about organolithium bases in flow chemistry.
In their recent review, Zhu et al. outstandingly focus on microfluidic immobilized enzyme reactors for continuous biocatalysis.33 Fernandes et al. present a perspective on the field of microfluidics when applied to chemical engineering and biotechnology,34 and Šalić et al. focus on microreactors as an effective tool for biotransformations.35 We would also like to refer to the recently published review article from Žnidaršič-Plazl, which gives a detailed overview of current examples of the implementation of microfluidic devices in biocatalytic process development.36 Worth to mention is also the recently published special issues devoted to microreactors in biotechnology.37–42
Additionally, a very recent review article was published by Hughes dealing with applications of flow chemistry in the pharmaceutical industry focusing in the patent literature,43 whereas Lee et al. emphasized on the translation of the pharmaceutical manufacturing from batch to flow,44 and Baumann et al. concentrate on the perspectives of continuous flow chemistry in the pharmaceutical industry.45 Monbaliu and co-workers summarize the impact of continuous flow on platform-chemicals derived from biomass.46 Also the continuous synthesis of polymers have been reviewed by Zaquen et al. very recently.47
Different from the above-mentioned literature data, in this review, we aim at presenting only very recent developments in the field of continuous flow biocatalysis being published from 2018 to September 2020 with a special focus on potential future developments in this key emerging research area. After a short outline of the fundamentals of continuous flow biocatalysis in chapter 2, the most recent and significant examples appeared in the common literature will be presented in particular in chapter 3. In chapter 4, we focus on enzyme (co)immobilization strategies for continuous processing. Finally, in chapter 5, future perspectives in the research area are outlined in detail. With this review article we would also like to motivate other colleagues in the research field to establish consistency in reporting on biocatalytic reactions and hereby refer to the brilliantly written guide by Halling and co-workers.48
2. Fundamentals of continuous flow and biocatalysis
2.1 General aspects
It is assumed that continuous flow systems have the potential to improve ca. 50% of all chemical processes based mainly on microreactor technology.49 Flow chemistry in particular has developed rapidly at both industrial and academic levels.16,50–54 It is a modular technique providing a toolbox for synthetic chemists and it has also been comprehensively reviewed in the last years.55–61 Novel developments in the field of magnetic microreactors with immobilized enzymes have been recently reviewed by Gkantzou et al. discussing how different magnetic particles can be combined with the appropriate biocatalysts.62 Those systems may constitute a powerful microsystem and provide a highly explorable scope.Continuous flow reactors are generally smaller than batch reactors, but in an ideal arrangement and under optimized conditions, these bench-scale reactors can produce more product in a given time than an analog batch reactor. Furthermore, another advantage is the direct transfer of flow systems to large-scale production without significant further optimization, which is in stark contrast to the upscaling of batch processes.25,63 The traditional ‘upscaling’ of (bio)catalysis in large batch reactors has significantly changed during the last decades where ‘miniaturization’ and ‘numbering-up’ of catalytic reactions being performed in continuous flow has become a trendsetter.
A typical continuous flow system for synthetic applications is composed of different modules opening also the possibility for a quick substitution of each module (Fig. 1i). Therefore, the whole setup can be designed to the desired reaction system (cf.Fig. 2 for further details). The system's centerpiece, indeed, is the type of reactor and here, three basic concepts are well established: i) the chip type, ii) the coil type and iii) the packed-bed type reactors. Depending on the bulk density of the particles, the latter can be referred to as packed, fluidized or mixed-bed reactor (Fig. 1ii). If gas–liquid reactions are carried out under continuous conditions, basically three flow regimes are usually observed at a constant liquid flow rate (Fig. 1iii). Additionally, tube-in-tube flow reactor concepts were also studied and described.64,65 Within immiscible liquid–liquid systems, slug flow is observed (Fig. 1iv), whereby the flow regime depends on the Reynolds number (Re), see below.
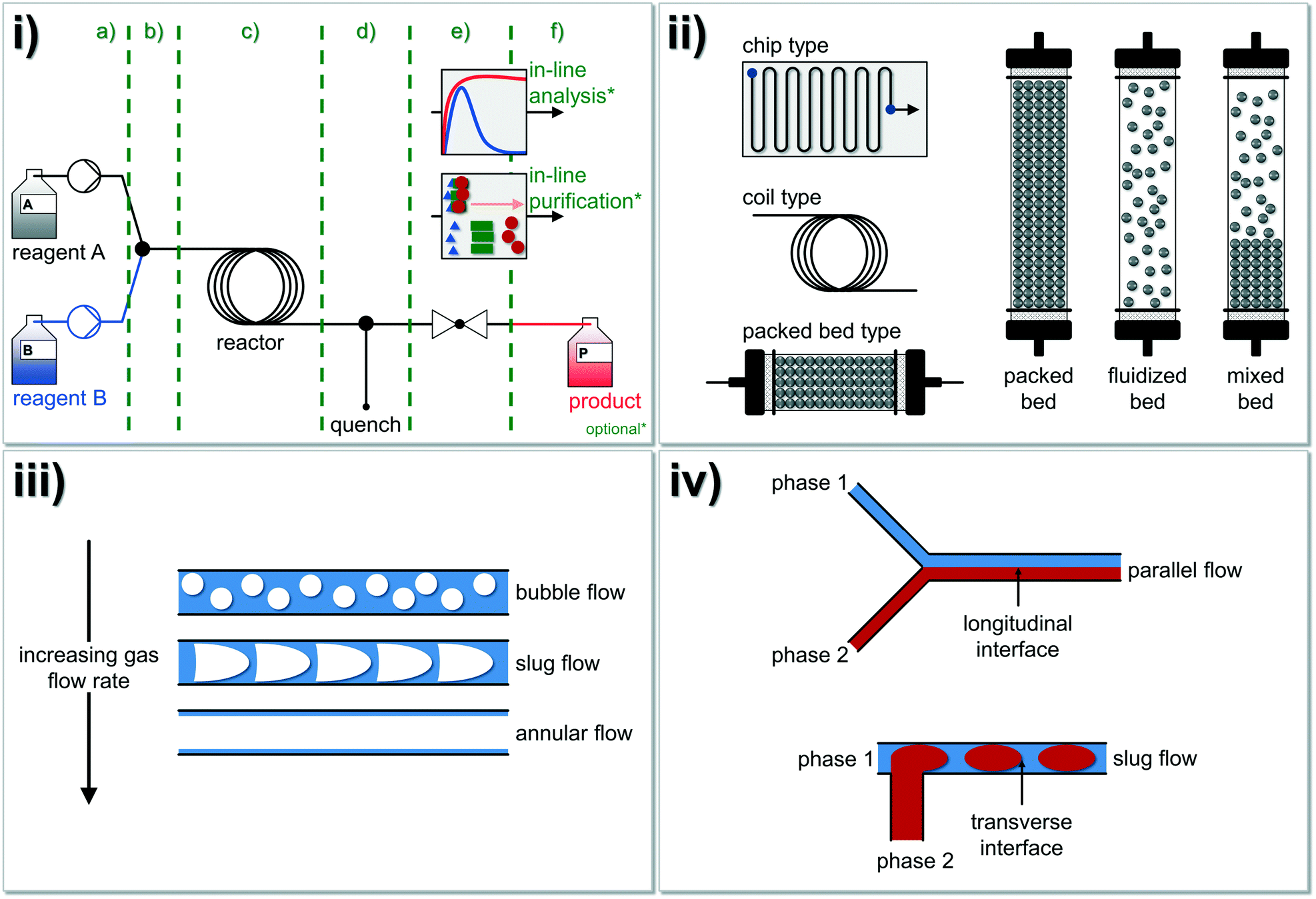 | ||
| Fig. 1 Overview of the fundamental principles of chemistry in continuously operated reactor systems (‘flow chemistry’): i) a typical continuous flow system is composed of different modules: a) reagent delivery pump-zone, b) mixing, c) reactor, d) quenching, e) back pressure regulation (BPR), f) collection, analysis, and purification. Optionally, inline analysis and/or inline purification techniques can be additionally coupled to the system (see also Fig. 2 for further details). ii) In general, three different reactor types (chip type, coil type and packed-bed reactor type) for continuous flow chemistry were developed and intensively studied in the past decades. Please note, that many more types of microreactors are also available.27 Different bed types (packed, fluidized and mixed bed) can be described for solid–liquid reactors where typically slug flow or turbulent flow occur. iii) If gas–liquid mixtures in microfluidic devices are investigated, three flow regimes are usually observed at a constant liquid flow rate. iv) Immiscible liquid–liquid fluids running either in parallel flow or in slug flow.73 Regularly, slug flow occurs under common conditions in tubular reactors with a diameter of >0.25 mm. Figure compiled and adapted from Plutschack and et al.21 | ||
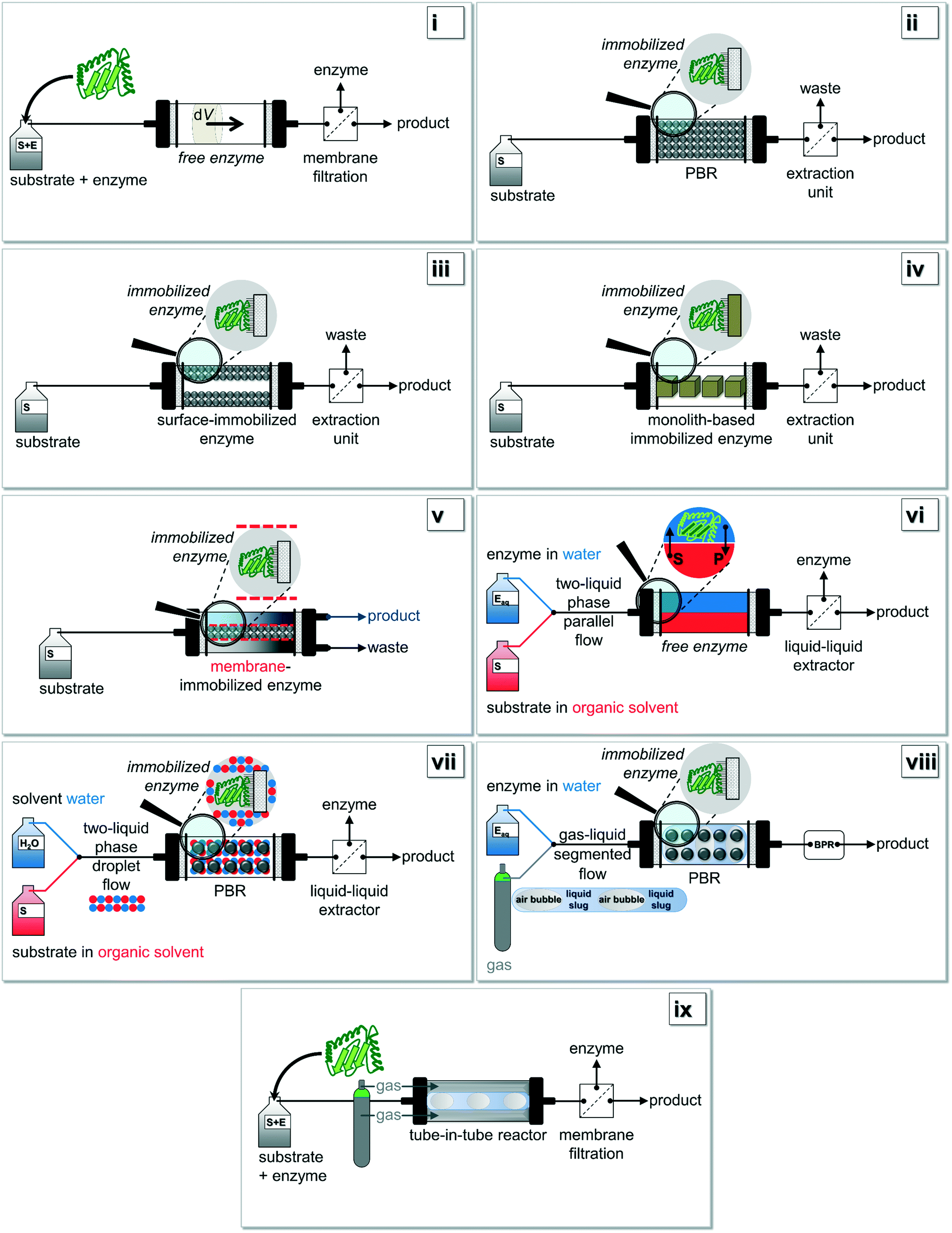 | ||
| Fig. 2 Based on Fig. 1i): various setup-configurations are possible for biocatalytic reactions being carried out under continuous operation. Figure adapted from Tamborini and co-workers.16 Further details for (i)–(ix) can be found in chapter 2.2. | ||
Growing out of the beaker, biocatalysis in continuously operated systems can be more productive, controlled and sustainable.27,66 In order to reproduce and compare the results of biotransformations in flow reactors, certain key parameters are available, but unfortunately not used in the scientific community in a standardized way. The situation is further complicated by the fact that a comparison between biocatalytic reactions in batch and continuous-flow is rather difficult because the basic physical conditions are too different. Fortunately, some key parameters can be compared: the reaction time t in a batch reactor is referred to as the time a possible reaction requires to achieve a certain (previously defined) conversion. In continuous operated systems, the residence time τ can be used and is defined as the time required for the reagents to flow through the reactor. The residence time τ can be determined by tracer experiments (pulse/step experiments).67 An important dimensionless number in fluid mechanics is the Reynolds number (Re) helping to predict flow patterns in different fluid flow situations. At low Re numbers, flows tend to be dominated by laminar flow whereas turbulent flow occurs within higher Re numbers. It is defined as the ratio of inertial forces to viscous forces within a fluid: Re = (ρ·v·d)·μ−1 with the density of the fluid (ρ), the flow speed (v), the diameter of the tube (d), and the dynamic viscosity (μ) of the fluid.68
Another very important parameter is the space time yield (STY) describing the amount of product (mP) formed within the residence time τ, which is present in the applied reaction volume (VR): STY = mP·(τ·VR)−1. Usually, this value is reported in g L−1 h−1 or g L−1 d−1 and depending on the product costs, values for the STY in the range of >100 g L−1 d−1 (for low-amount and high-value products) up to values >500 g L−1 d−1 (for high-amount and low-value products) are essential.69–71 Further parameters like the biocatalyst loading (amount of enzyme used, e.g. mg or g of immobilized biocatalysts and/or the enzyme activity U), the reactor productivity (STY normalized by the reactor volume), the process stability (conversion at different operation-times) and the biocatalyst productivity are additional important metrics.16 The latter can be expressed as total turnover number (TTN) as the quotient of the apparent turnover frequency kcat and the first-order deactivation rate constant kd.72
The precise and uniform description of the flow reactor's volume is certainly one of the biggest challenges reviewing the recent literature. The data summarized in Table 2 depend on the literature information without further calculations. According to different literature sources, various information for the ‘reactor volume’ are available: e.g. (i) total volume of the reactor, (ii) effective volume of the reactor, (iii) packed volume, (iv) column volume or even (v) total bed volume minus the space filled by the catalyst, as calculated by the difference in column mass before and after filling with water. Often, further explanations are missing how the authors calculated or measured the volume. Herein, we strongly suggest to use exact descriptions of the reactors being used. For a correct description of the PBR at least the definition of the reactor (total, column) and bed (packed) volume or alternatively the void (effective) volume is required. We hope, that this issue will be fixed in the next years and we would like to recommend authors to precisely explain and describe the methodology how the reactor volumes were determined.
In general, the advantages of micro flow-reactors (at least one characteristic dimension below 1 mm inside diameter [i.d.]74–77) are evident: they show (i) high heat transfer surface to product volume ratios, (ii) good heat transfer capabilities ideally suited for optimizing reaction conditions, (iii) efficient mixings and (iv) improved flow capacities, and (v) lower flow capacities, (vi) lower pressure drops, (vii) no blocking of channels and preparation of multikilogram quantities if mesoreactors are taken into account. Please note, that slightly different definitions for micro- and mesoreactors are also available in the literature.12,16,25
Nevertheless, micro- and mesoreactors still show some challenges and a careful balance of pros and cons must be taken into consideration when a reaction should be operated in flow (Table 1). When searching the general technical literature, we noticed that the terms are not used uniformly – or not at all. In order to change this, we consider a standardized use of technical terminology to be necessary for further development in the research field.
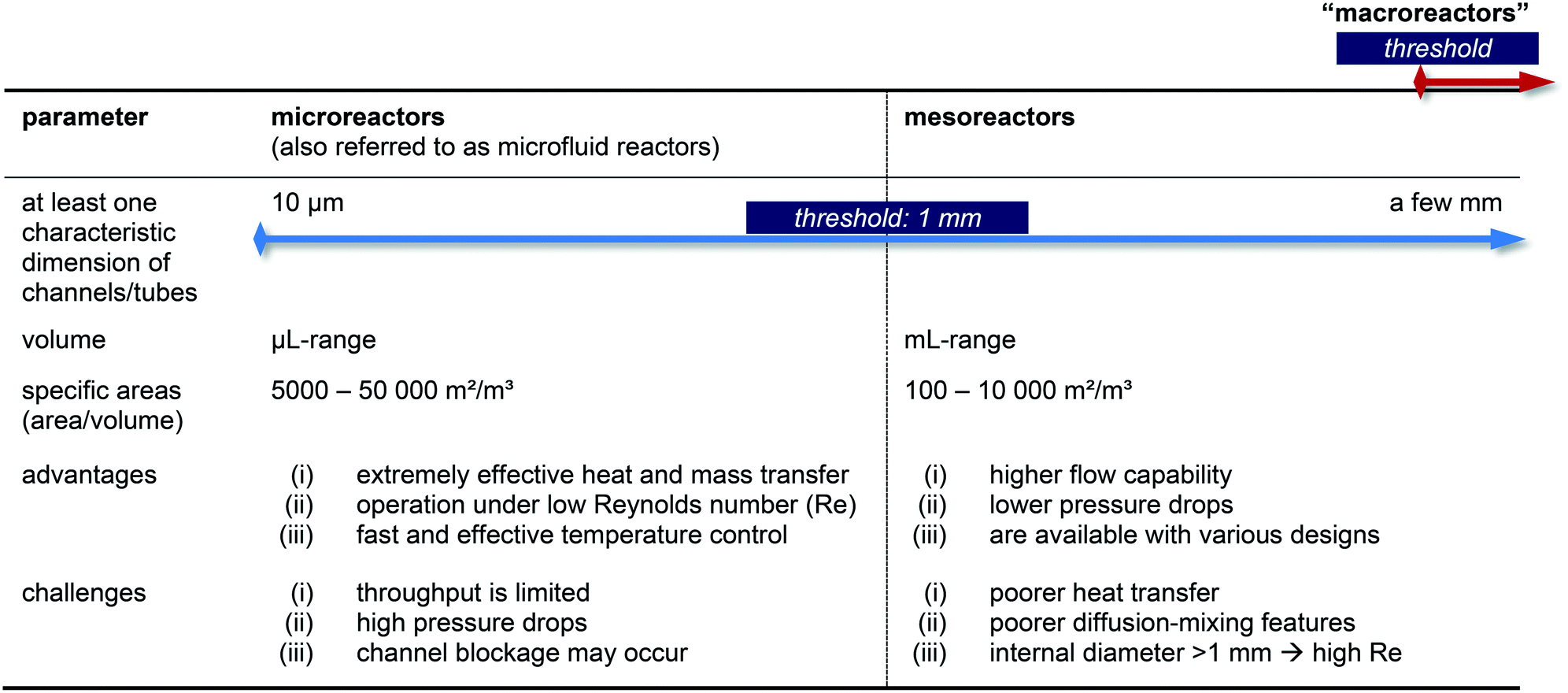
|
2.2 General overview of different reactor set-ups for flow biocatalysis
In Fig. 2, the most common configurations for a continuous biocatalysis setup are shown in detail. Two main groups of flow reactors for enzymatic reactions can be defined according to the form of the biocatalyst: (i) setups using free enzymes and (ii) systems containing immobilized enzymes. This review paper specifically focusses on recent developments in the second group of immobilized enzymes, but in the interests of clarity and completeness of contents, the first group should be mentioned briefly. If setups with free enzymes are used, in the easiest possible scenario the substrate and the enzyme are simultaneously pumped through the reactor (Fig. 2i). Within the volume element dV, the time-independent but location-dependent enzymatic reaction takes place. The residence time τ is the key parameter to achieve full conversion and a possible work-up of the outflow could be performed via membrane filtration. To overcome the problematic obstacle to run reactions with an immiscible hydrophobic substrate, a more elaborate procedure using e.g. a liquid–liquid parallel laminar flow system could be applied (Fig. 2vi).29,78 Here, the enzyme is mainly present in the aqueous phase whereas the substrate is diluted in an organic medium. Especially surface-active lipases might benefit from such a setup design. If gas–liquid reaction systems are investigated, the aforementioned ‘tube-in-tube’ reactor configuration is most commonly used (Fig. 2ix).64,65The simplest reactor concept in the group of immobilized enzymes is the use of a fixed-bed reactor in which the enzyme is directly bound to the packaging material (Fig. 2ii).79 High pressure drops and channeling effects occurring in this setup might be prevented by using a reactor with surface-immobilized enzymes (Fig. 2iii).80 Additionally, the enzymes can be immobilized onto monolith materials (Fig. 2iv),12 or they can be retained by e.g. size-exclusion membranes (Fig. 2v).20,61 Finally, immobilized enzymes can also be used for a configuration within a liquid–liquid system composed of immiscible solvents (Fig. 2vii) or within a gas–liquid system (Fig. 2viii). In the first case, most commonly the two liquids are mixed together so that a droplet flow occurs. In the second case, a segmented air–liquid flow is mainly applied.81
In addition to the established reactor concepts shown in Fig. 2, new developments have recently been discussed by Wirth and co-workers.82 The authors studied among others a biphasic biocatalytic sesquiterpene syntheses in a high-performance counter current chromatography (HPCCC) system. Here, the most important innovation is the extremely rapid mass transfer between two immiscible phases and therefore, a 70-times faster reaction rate was observed in HPCCC than in batch. Consequently, denaturation of the enzymes is avoided reducing emulsion formation, which was detected in the batch and segmented flow systems of the study.82
3. Selected examples of continuously operated meso-scale reactors for flow biocatalysis
The following section will review the latest developments and publications in the field of biocatalysis in continuous flow since 2018. Publications that can be categorized with the help of Fig. 2 are presented in Table 2. Selected examples from the very recent literature showing a significant progress in the research field will be otherwise discussed in detail.| Entry | Reaction system | Biocatalyst | Reactor configuratione | Reactor volume | Flow rate(-range) | Residence time(-range) τ | STYa | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| [mL] | [mL min−1] | [min] | [g L−1 h−1] | ||||||
| n.a. = not available. a If several STYs were reported, the maximum number was always given in the table. b Effective volume. c Reactor volume. d Bed volume. e The information is directly taken from the literature for naming of the reactors used. f E-Factor = amount of waste generated per amount of target product. | |||||||||
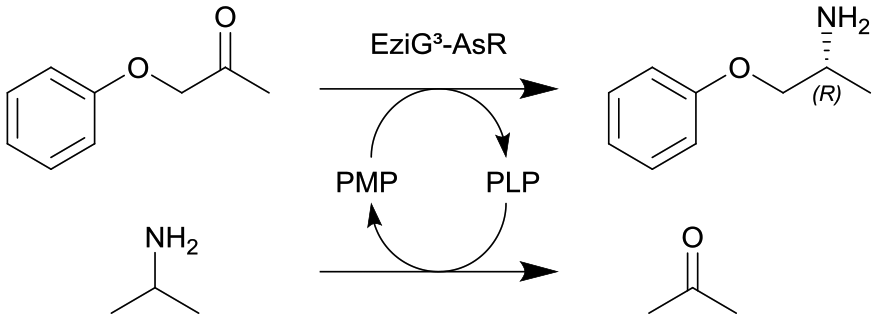
| 1 |
|
ω-Transaminase from Arthrobacter sp. (AsR-ωTA) immobilized on EziG3 support (EziG3-AsR) | PBR | 1.82b | 0.2 | 9.1 | 1.99 | Loop operation mode, stainless steel pre-column for adjusting the water activity via salt hydrate pairs, neat organic solvent toluene (aw = 0.7), 70% conversion in 72 h, 90% conversion in 120 h | 108 |
| 2 |
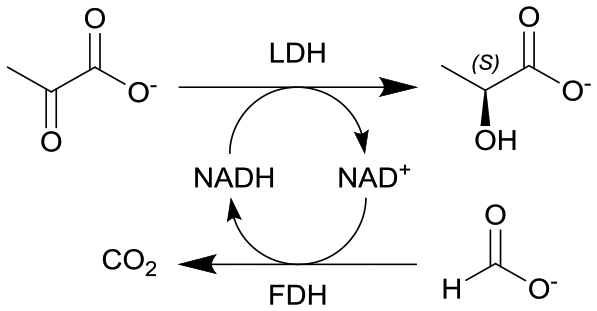
|
Lactate dehydrogenase (LDH) and formate dehydrogenase (FDH) immobilized on carbon particles (Black Pearls 2000) | PBR | 0.48b | (17.2–51.6) × 10−3 | 10–30 | 22.9 | Biocatalyst particles were mixed with glass beads, 100% conversion in 0.5 h residence time, low E-factorsf between 2.5–11, long-term stability of the biocatalytic reactor was investigated as maintained over more than 30 h | 109 |
| 3 | Transesterification of crude coconut oil | Lipase from Burkholderia cepacia (BCL) immobilized on protic ionic liquid (PIL)-modified silica | Either one PBR or two PBRs in series | 7.85c | 0.5 | n.a. | n.a. | Ca. 65% conversion after 96 h at 40 °C | 110 |
| 4 |
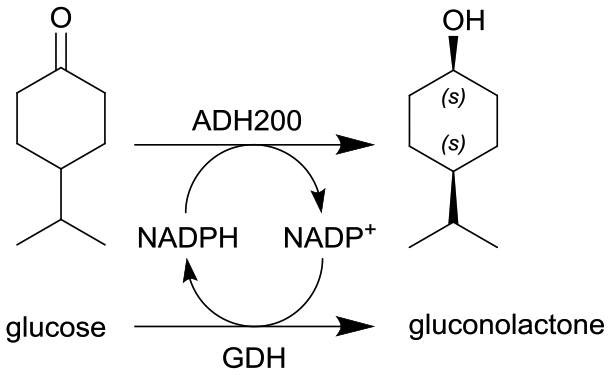
|
Alcohol dehydrogenase (ADH200) and glucose dehydrogenase (GDH) for cofactor regeneration | Stirred membrane reactor as CSTR model with an ultrafiltration membrane (regenerated cellulose, 5 kDa cut-off) | 12b | 0.2 | 60 | n.a. | 80% conversion after 24 h at 30 °C, for a DSP at the membrane reactor outlet, solution was mixed to n-hexane and the biphasic segmented flow was separated via a commercial inline liquid–liquid separator from Zaiput® (hydrophobic membrane OP-900) | 111 |
| 5 |
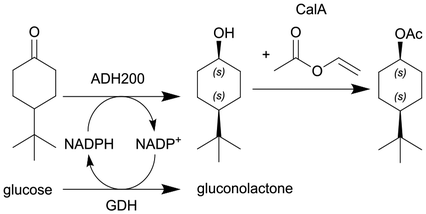
|
ADH200 and GDH (see entry 4) and subsequent enzymatic acetylation with lipase A from Candida antarctica (CalA) | See entry 4 for reduction, PBR for acetylation | 12b | 0.2 | 60 min for reduction and 11 min for acetylation | n.a. | 85% conversion after 24 h, for the DSP see entry 4 | 111 |
| 6 |
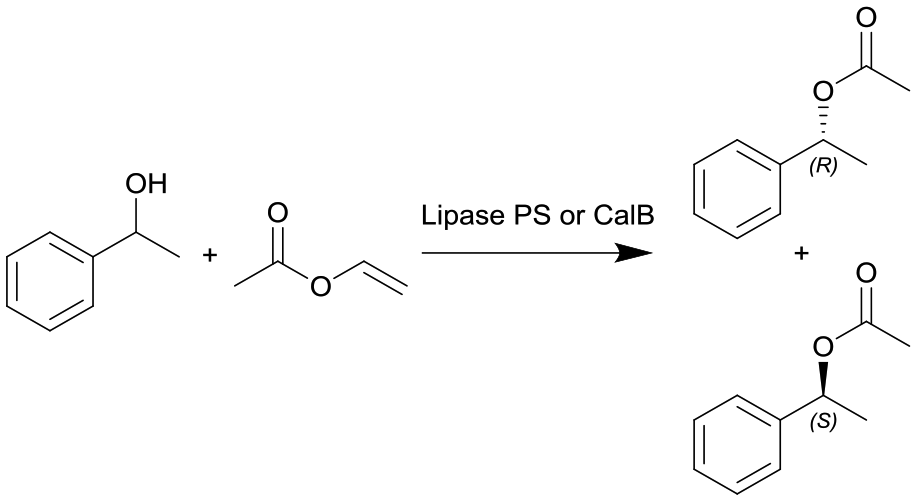
|
Lipase from Burkholderia cepacia (lipase PS) or lipase B from Candida antarctica (CalB) immobilized on hollow silica microspheres (M540) by bisepoxide activation | PBR | 0.816c | 0.3 (lipase PS) | n.a. | n.a. | In dry toluene, various temperatures (30–100 °C), compared to batch reaction, ee(R) = 99% | 112 |
| 7 | 0.1–0.2 (CalB) | n.a. | n.a. | In hexane/MTBE (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), various temperatures (0–90 °C), 2.6 equiv. of vinyl acetate :1), various temperatures (0–90 °C), 2.6 equiv. of vinyl acetate |
113 | ||||
| 8 |
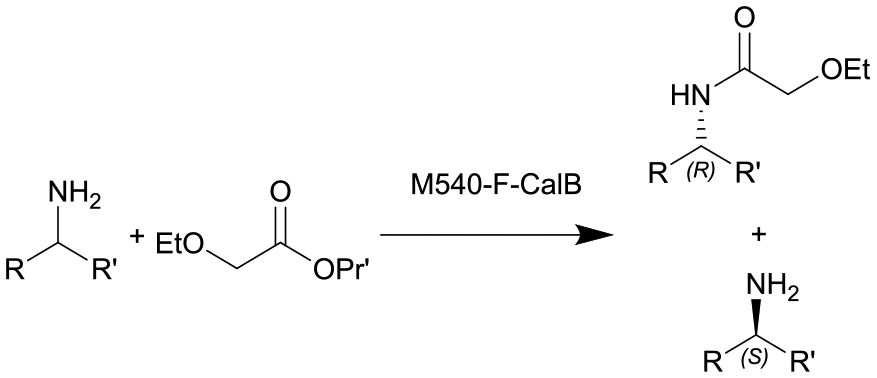
|
See entry 7 | PBR | 0.816c | 0.1 | n.a. | n.a. | In dry toluene, 60 °C, 0.6 equiv. of isopropyl 2-ethoxyacetate | 113 |
| 9 | Aldehyde tag conversion | Formylglycine generating enzyme (TcFGEC187A,Y273F) from Thermomonospora curvata immobilized on epoxy-activated Sepharose beads | PBR | 1c | (50–500) × 10−3 | 2–20 | 21.6 | 10 times higher productivity compared to batch reactions | 114 |
| 10 |
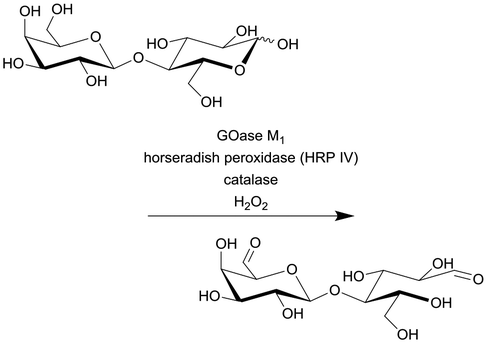
|
Galactose oxidase (GOase) | PFR | 2.6c | 0.108 | 24 | 56.7 | 224-Fold increase in productivity (STY) and 7-fold improvement in efficiency (gproduct genzyme−1) compared with the batch reaction | 115 |
| 11 |
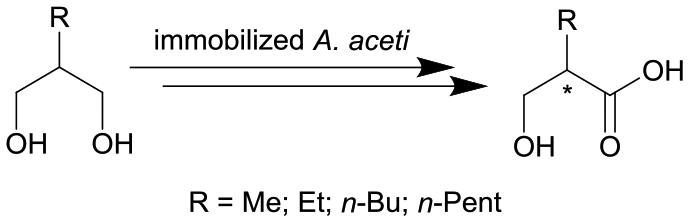
|
Cells of Acetobacter aceti MIM 2000/28, cells immobilized in alginate beads | PBR, see #viii in Fig. 2 | 5.1 | (15–60) × 10−3 | 10 | n.a. | Air/water segmented flow, ion exchange resin column for inline purification step | 116 |
| 12 |
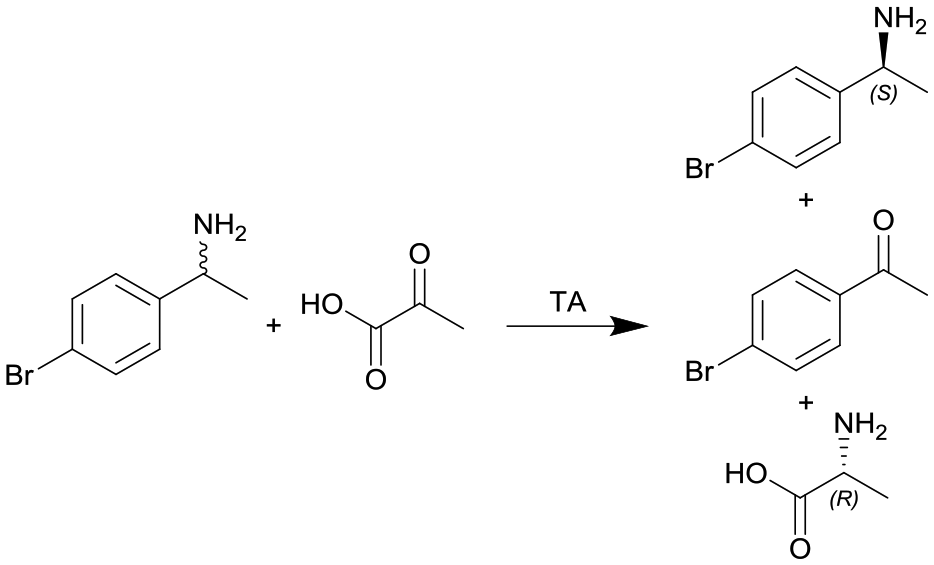
|
Transaminase (TA) ATA-117 immobilized onto macrocellular silica monoliths | PTFE tube, see #iv in Fig. 2 | n.a. | 0.11 | n.a. | n.a. | n.a. | 117 |
| 13 |
|
E. coli cells containing overexpressed transaminases (TAs) and hollow silica microspheres as supporting agent were immobilized by a sol–gel process | PBR | 0.816c | (R)-Isomer: (40) × 10−3 | n.a. | (R)-Isomer: 4.8 | Total three (S)-selective TAs and three (R)-selective TAs were investigated for kinetic resolution of the rac-amine leaving the unconverted enantiomer, ee(R) = 99.1, yield(R) = 45%, ee(S) = 99.2, yield(S) = 44%. Additionally, three more amine substrates were investigated | 118 |
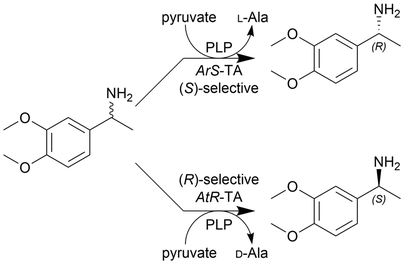
| (S)-isomer: (60) × 10−3 | (S)-isomer: 1.8 | ||||||||
| 14 |
|
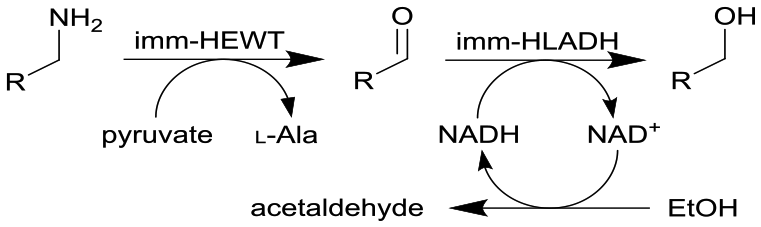
Combined ω-transaminase from Halomonas elongata (HEWT) with horse liver alcohol dehydrogenase (HLADH) and immobilization (imm-) onto epoxy resin | Two PBRs in series with work up column, see #ii in Fig. 2 | 1.7 | n.a. | 30–45 | n.a. | An inline purification step was added trapping any trace of unreacted aldehydes, simplified work-up procedure, use of pyruvate as the amino acceptor strongly favours the equilibrium reaction of the first step, the flow rate was varied and optimized for each individual reaction and the resulting organic phase was purified using a QuadraPure™ BZA (benzyl amine) scavenger | 119 |
| 15 |
|
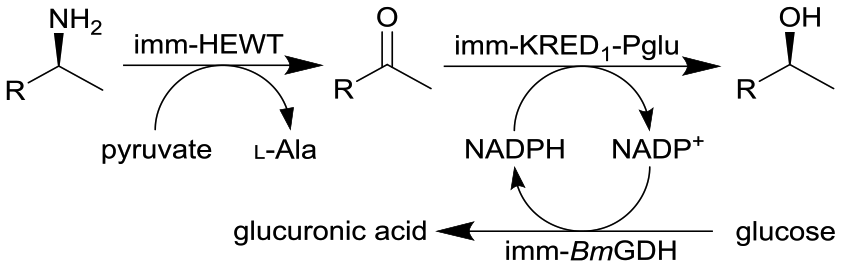
Combined ω-transaminase from Halomonas elongate (HEWT) with ketoreductase from Pichia glucozyma (KRED1-Pglu) and a glucose dehydrogenase from Bacillus megaterium (BmGDH) immobilized (imm-) onto epoxy resin | See entry 14 | 1.7 | n.a. | 180 | n.a. | The flow rate was varied and optimized for each individual reaction, the flow stream was extracted inline using EtOAc and the resulting organic phase was purified using a QuadraPure™ BZA (benzyl amine) scavenger | 119 |
| 16 |
|
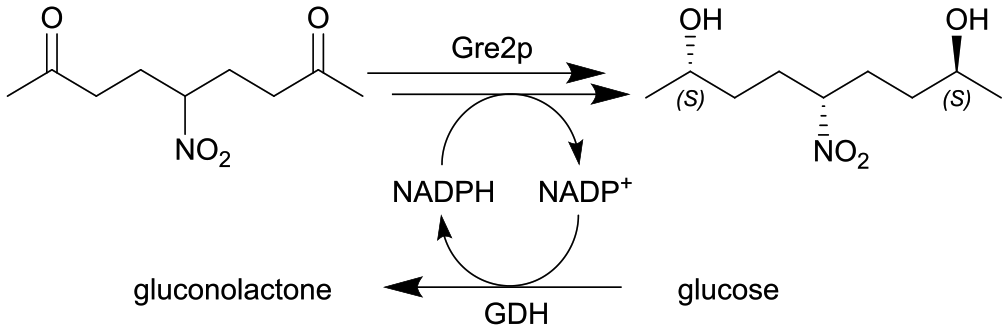
(S)-Stereoselective ketoreductase Gre2p from Saccharomyces cerevisiae and cofactor-regenerating glucose 1-dehydrogenase GDH from Bacillus subtilis in stable, active hydrogels | Self-prepared hydrogel flow reactor120 | 0.150c | (5–100) × 10−3 | 30 | 13.3 | Hydrogel microreactors showed activity loss first however a stable conversion rate plateau was reached during the course of the process, sequential use as well as parallelization by ‘numbering up’ of the flow reactor modules was investigated (up to a stack of six microreactors), also acetophenone and 4′-chloroacetophenone were reduced to their corresponding (S)-configured alcohols | 121 |
| Due to the size of the reactor (0.150 mL) this particular example represents a micro-scale application | |||||||||
| 17 |
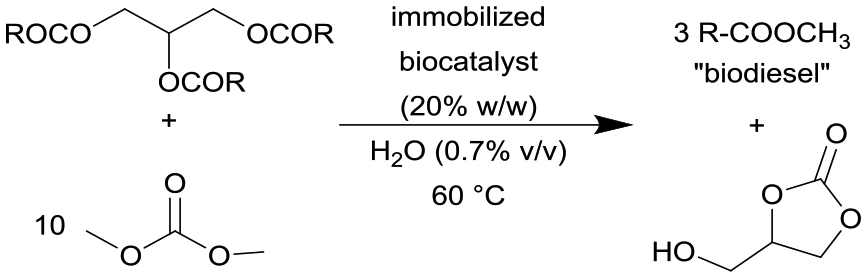
|
Immobilized lipases from porcine pancreas (PPL) and Candida antarctica (CalB) on epoxy resins | PBR | 8.83 | 0.1–0.05 | 88–176 | n.a. | Compared against commercially available Novozym 435 | 122 |
| 18 |
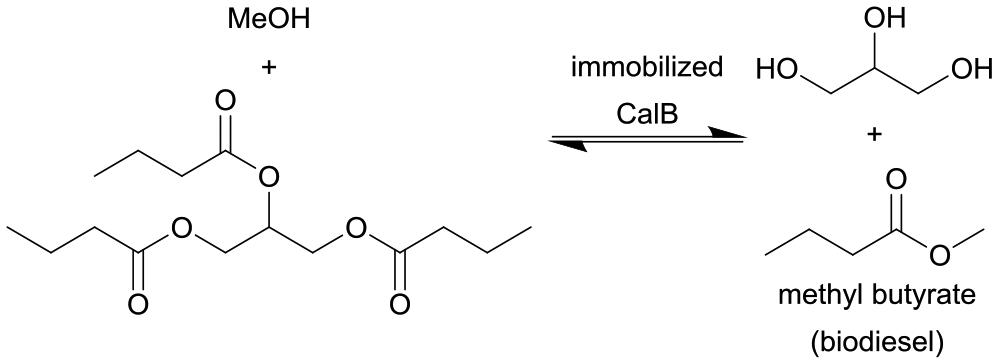
|
Candida antarctica lipase (CalB) immobilized on amine-free silica monoliths | Isothermal monolith microreactor | n.a. | (0.8–12.8) × 10−3 | 400–25 | n.a. | Solvent-mixture of 2:1 (methanol:tributyrin) at 30 °C, the optimum immobilized lipase microreactor exhibited almost quantitative ester production for >100 h without deactivation |
123 |
| 19 |
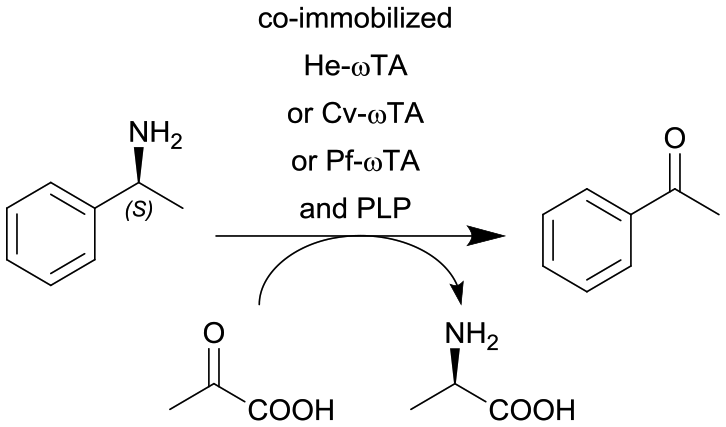
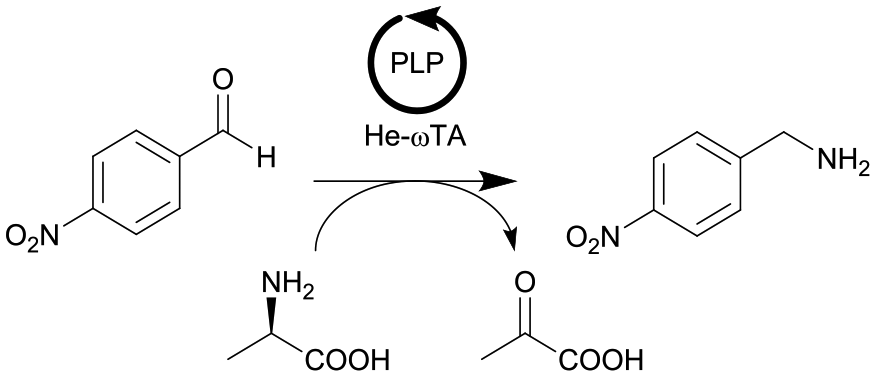
|
ω-Transaminase from Halomonas elongata (He-ωTA), Chromobacterium violaceum (Cv-ωTA) or Pseudomonas fluorescens (Pf-ωTA), coimmobilized with pyridoxal 5′-phosphate (PLP) onto porous methacrylate-based Purolite or Sepabeads carriers | PBR | 1.3–1.45d | 1.45 | 1 | n.a. | A R2+/R4 flow reactor commercially available from Vaportec equipped with an Omnifit glass column was used | 124 |
| 20 |
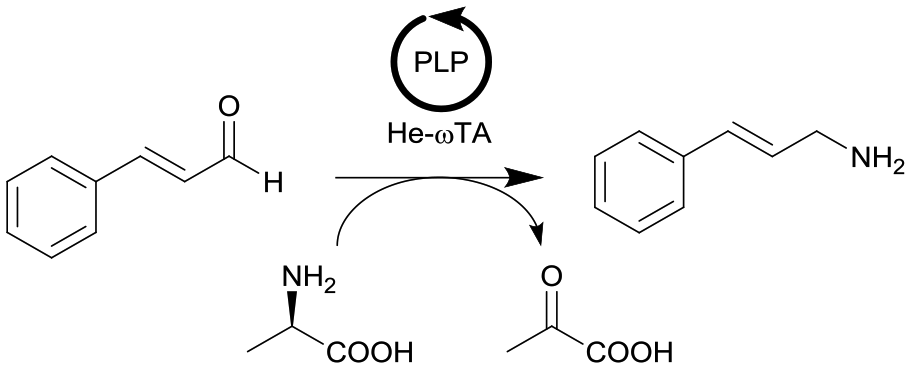
|
ω-Transaminase from Halomonas elongata (He-ωTA), coimmobilized with pyridoxal 5′-phosphate (PLP) onto porous methacrylate-based Purolite or Sepabeads carriers | PBR | 1.3–1.45d | 0.725 | 2 | n.a. | Reaction were performed using the equipment described in entry 19, E-factor values were calculated and discussed in detail | 124 |
| 21 |
|
See entry 20 | PBR | 1.3–1.45d | 0.725 | 2 | n.a. | See entry 20 | 124 |
| 22 |
|
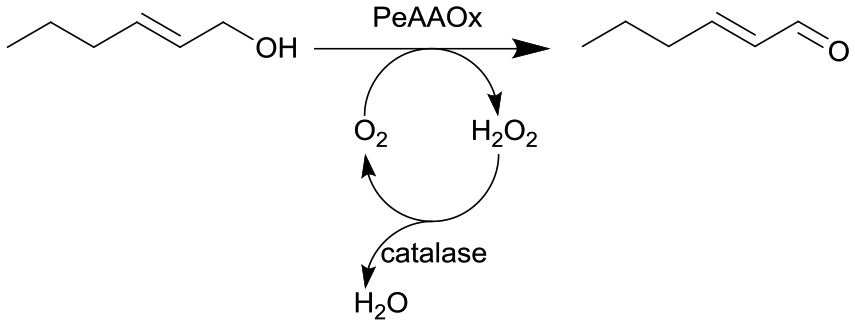
Novel O2-dependent enzyme aryl alcohol oxidase from Pleurotus eryngii (PeAAOx) | PFA slug-flow microreactor coils | 6 | 0.067–0.2 | 80 | n.a. | Different residence times and flow rates were investigated, turnover frequency up to 38 s−1 and turnover numbers more than 300000 could be achieved, semi-preparative scale reaction yielded 90% conversion after 18 h of total reaction |
50 |
| 23 |
|
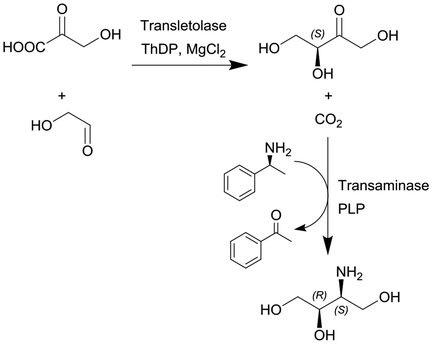
Transketolase (TK) and transaminase (TAm) | TK microreactor coupled with a micromixer followed by TAm coil reactor for the two-step translocase-transaminase catalyzed synthesis | TK: 0.24 | TK: (2–40) × 10−3 | TK: 6–120 | n.a. | Conversion into L-erythrulose in less than 10 min using a TK enzyme activity of 3.25 U ml−1 and when coupled with the TAm reaction, the volumetric activity of the TAm was increased up to 10.8 U ml−1 and a final yield of 100% of the product | 125 |
| TAm: 3 | TAm: (5–100) × 10−3 | TAm: 30–600 | Due to the size of the reactor (0.24 mL) this particular example represents a micro-scale application | ||||||
| 24 |
|
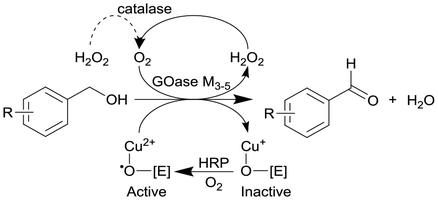
GOase M3–5, catalase and horseradish peroxidase (HRP) as GOase activator enzyme | Multipoint-injection flow reactor (MPIR) with a pressure capacity of 5.2 bar that is capable of sequentially dosing H2O2 across the full reactor manifold | 2.6c | (80) × 10−3 | 8–20 | 14.3 | A 2 mL CSTR and CSTR cascade also investigated with STY up to 168 g L−1 d−1 and 13 min of residence time within four CSTRs | 126 |
| 25 |
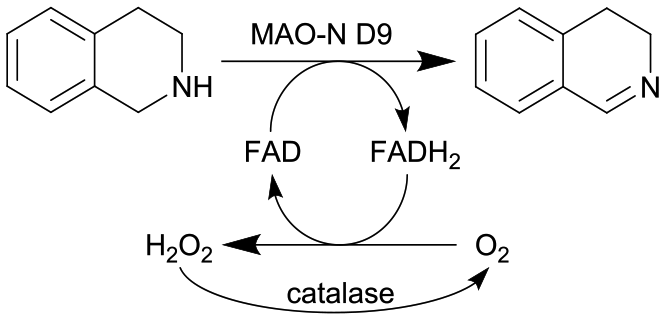
|
Oxygen-dependent biocatalyst monoamine oxidases (MAO-N) from Aspergillus niger | See entry 24, glass beads were packed into the flow channel to facilitate mixing | 1.6 | n.a. | 8–12 | n.a. | 97% conversion at steady state | 126 |
| 26 |
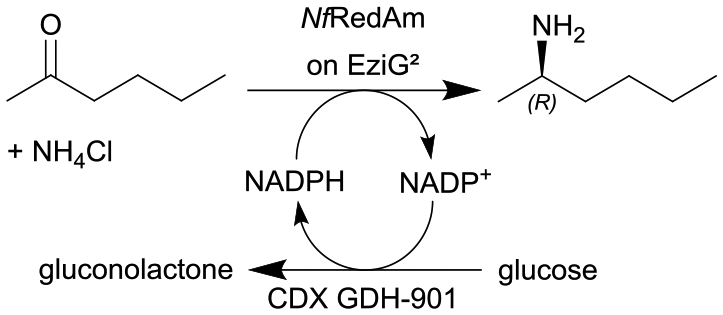
|
Two fungal reductive aminases (RedAms) from Neosartorya fumigatus (NfRedAm) on EziG2 support | PBR | 1 | (33) × 10−3 | 30 | 8.1 | Continuous production for up to 12 h, the biocatalyst productivity was 4.86 gproduct genzyme−1 with TTN up to 1400, also different EziG (EziG2 and EziG3) supports were tested and other flow conditions were investigated and also another RedAm from Neosartorya fischeri (NfisRedAm) | 127 |
| 27 |
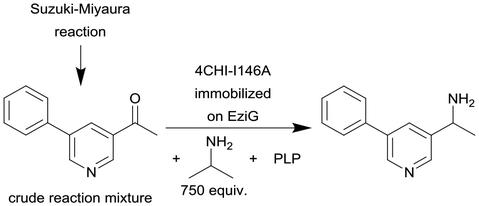
|
Single alanine mutations of an amine transaminases from Aspergillus fumigatus (4CHI-TA) | PBR | 0.3c | (1.7) × 10–3–3 | 210 | n.a. | The crude Suzuki–Miyaura reaction product was added in flow to an IPA and PLP containing solution and subsequently pumped through the column, in silico studies were used to identify possible amino acid residues around the active site of 4CHI-TA | 128 |
| Due to the size of the reactor (0.3 mL) this particular example represents a micro-scale application | |||||||||
| 28 |
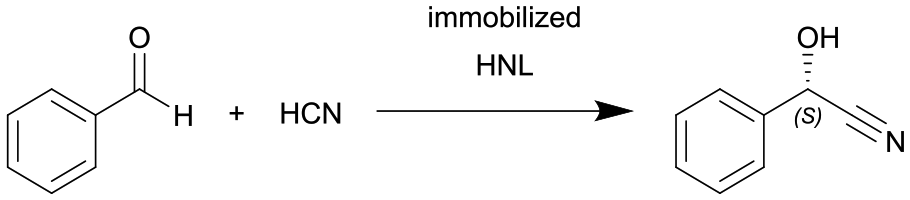
|
Hydroxynitrile lyases from Hevea brasiliensis (HbHNL) and Manihot esculenta (MeHNL) covalently immobilized in a siliceous monolithic microreactor | HNL-amino functionalized monolithic microreactor, see #iv in Fig. 2 | 0.96c | 0.045–08 | 1.2–21.3 | MeHNL: 1229 | Fast production of chiral cyanohydrins within 3.2 min with high conversion of 97% and high ee-values of 98% | 129 |
| HbHNL: 613 | |||||||||
Quite often, other aspects of a biocatalytic reaction play an enormous role when considering the overall performance of the process under consideration.83–85 Above all – and of course also in the case of continuous flow systems – cofactors and co-substrates play a decisive role. The use of stoichiometric amount of the expensive redox equivalent nicotinamide adenine dinucleotide [phosphate] (NAD[P](H)) is already a challenge when it is used in a batch system and even more so when used in a continuous mode of operation. Herein, reusability is particularly difficult, since the cofactors are often washed out (if not immobilized) thus driving the costs of the reaction to an economically unacceptable level.
Pietruszka and co-workers addressed this issue and presented a quasi-stationary recycling system for NADP(H) utilizing immobilized Halo-tagged alcohol dehydrogenase from L. brevis (HaloTag-LbADH).86 The authors set up a ‘simple flow-through system’ (system 1) and a ‘closed-loop cofactor regeneration system’ (system 2) and deeply compared the two devises (Fig. 3). System 2 enabled continuous production as well as quick substrate changes. According to the authors, the developed system could serve as a blueprint for a general cofactor regeneration unit for continuous biocatalytic devices using (co-)substrates being miscible in organic solvent. The asymmetrically reduction of acetophenone yielded (R)-phenylethan-1-ol as the product with a conversion of 92% using 50 mmol L−1 of substrate and 10% (v/v) 2-propanol with flow rates up to 65 μL min−1 in system 1. The comparison between both systems showed that system 2 produced comparable results to system 1 (e.g. for a combined flow rate of approx. 30 μL min−1, system 2 achieved a conversion of 96%). This closed-loop set up enabled a decrease of NADP(H) to catalytic amounts (0.1 mol% relative to the substrate). The reaction system 2 was broadened to the reduction of three further substrates (cf.Fig. 3, downright) yielding moderate to high STYs and high to very high TTNs with excellent enantioselectivities (>99%). The flexibility of the system was additionally proven by the successful use of three different extraction solvents. Moreover, the system confirmed its reliability by continuous runs over 32 h without loss in performance yielding 1.36 g of ethyl (S)-4-chloro-3-hydroxybutanoate with a STY of 121 g L−1 h−1 (96%). This conversion was also upheld for over 123 h without a loss on performance. Furthermore, the concept of system 2 was expanded to a consecutive experiment reducing two of the ketones shown in Fig. 3 with the very same setup, enzyme-batch and cofactor. Between the two reductions a rinsing step was carried out with acetone in potassium phosphate buffer (KPi) to prevent cross-contamination. This set-up shows the potential of NADP(H) recycling for continuous operated systems within biocatalytic processes. Recently, also Döbber et al. used the HaloTag-immobilization strategy for the continuous biocatalytic production of chiral alcohols and epoxides,87 and for a continuous enzymatic cascade towards a vicinal chiral diol.88
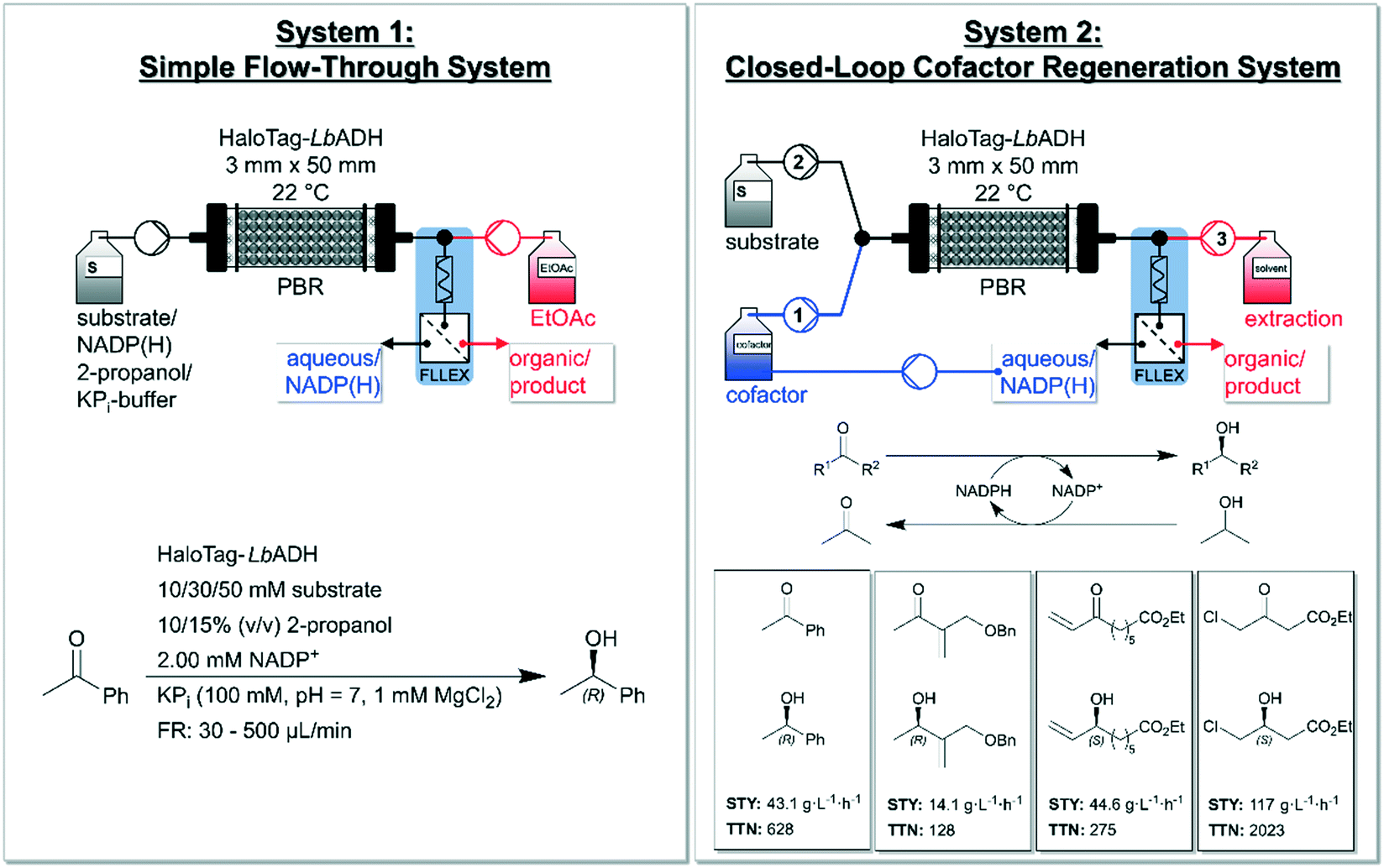 | ||
| Fig. 3 Overview of the developed ‘simple flow-through system’ (left) and the ‘closed-loop cofactor regeneration system’ (right) by Baumer and co-workers. The simple flow-through system (left) consists of a pump for the reaction mixture (substrate, NADP+, 2-propanol and KPi-buffer) and one for the organic extraction solvent (EtOAc = ethyl acetate). The packed-bed-reactor (PBR) is filled with Halo-tagged immobilized LbADH and a FLLEX system is applied for separating the phases. The applied reduction within this reactor concept is shown in the left-bottom (reducing acetophenone to (R)-phenylethan-1-ol together with substrate-coupled regeneration of NADPH). The closed-loop cofactor regeneration system (right) consists of two pumps containing four separated channels in total. Channel 1 is used for the aqueous/cofactor stream and channel 2 for the substrate and co-substrate (2-propanol) and both channels are connected via a y-piece. Channel 3 delivers the extraction solvent (dichloromethane or ethyl acetate or diisopropyl ether). Again, a column with immobilized HaloTag-LbADH as catalyst and a FLLEX system is applied for separating the layers. ‘FLLEX’ = Syrris Asia FLLEX (flow liquid–liquid extraction) system. The total turnover number (TTN) refers to NADP+ consumption. Figure adapted from Baumer et al.86 | ||
A recent publication correspondingly dealing with the cofactor regeneration in continuously operated biocatalytic reactions was published by Hartley et al. in late 2019.89 The authors cover the topics of cofactor supply and cofactor regeneration in continuous mode and enzyme immobilization without loss of activity. A complex multistep continuous flow reactor with three PBRs was designed for the cofactor-dependent continuous-flow biocatalysis of the antidiabetic drug D-fagomine (Fig. 4). Each biocatalyst consists of a genetically encoded multi-enzyme fusion protein together with a bounded cofactor being retained and regenerated. Additionally, these designed biocatalysts were immobilized on TFK-activated agarose and packed into glass columns to produce three PBR-columns: a phosphorylation reactor (23.1 mL packed volume), an oxidation reactor (25.7 mL packed volume) and a carboligation reactor (17.7 mL packed volume). Each step of the total synthesis was optimized and evaluated in detail (see green boxes in Fig. 4). The three-step continuous-flow reactor cascade maintained continuous product yields between 85% and 90% conversion of glycerol to N-carboxybenzyl-3S,4R-amino-3,4-dihydroxy-2-oxyhexyl phosphate (N-Cbz-3S,4R-ADHOP) at a temperature of 23 °C for more than 7 h. The TTNs for the cofactors exceeded 10000 (ca. 11000 for the NAD+-dependent oxidation reactor and ca. 17000 for the ATP-dependent phosphorylation reactor). In our opinion, fusion systems linking enzymes to cofactors and immobilization modules with the help of suitable synthetic spacers will develop to an important research field in the area of continuously operated biocatalysis in the near future.90
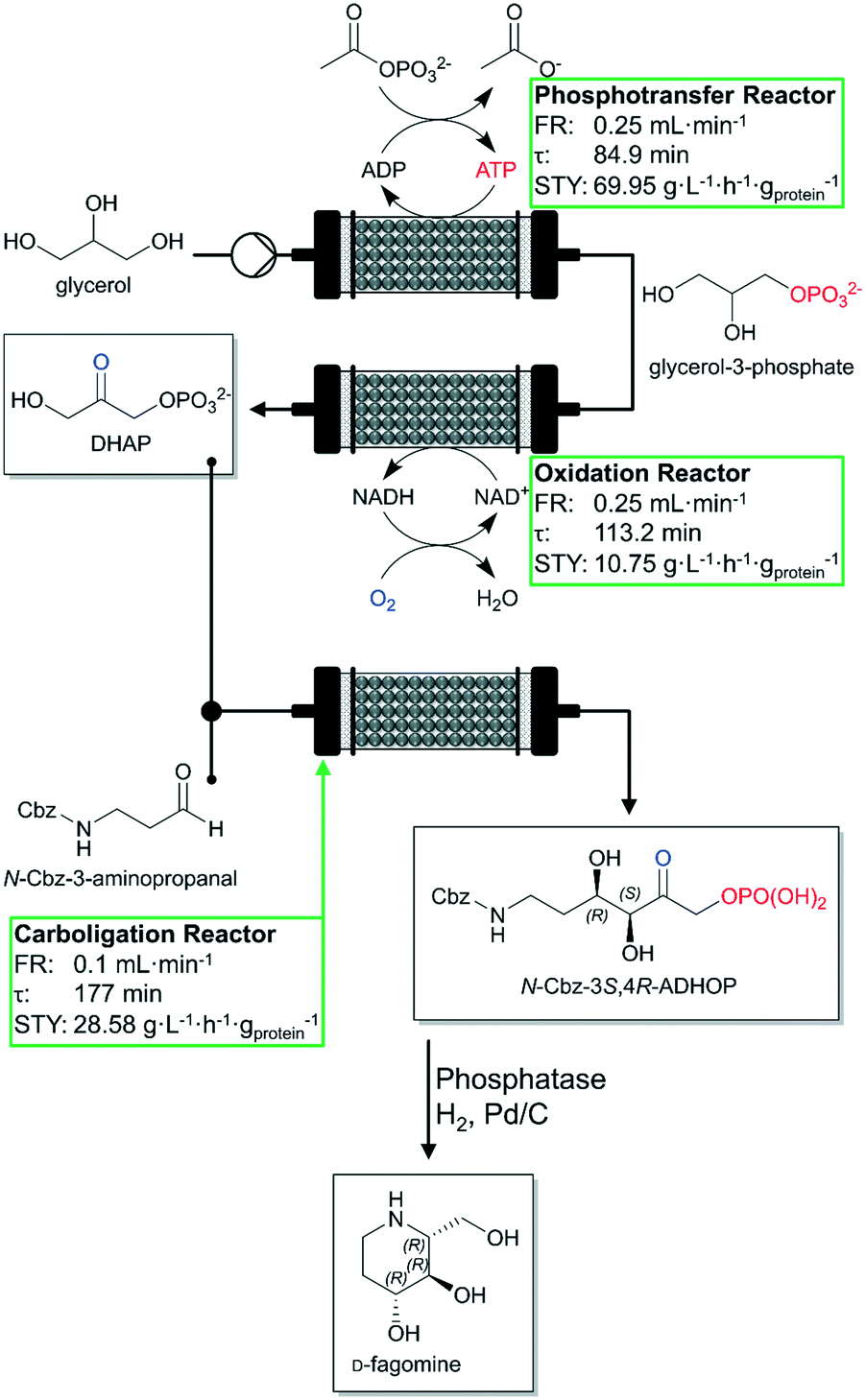 | ||
| Fig. 4 Schematic representation of the three-step continuous-flow reactor and associated biotransformations (phosphotransfer, oxidation and aldol addition) for the synthesis of D-fagomine developed by Hartley et al. Glycerol is phosphorylated forming glycerol-3-phosphate, then oxidized to form dihydroxyacetone phosphate (DHAP). Afterwards, it is then used in an aldolase-dependent reaction to produce the intermediate N-Cbz-3S,4R-ADHOP of the desired final product. Individual performance of each reactor is shown in the green box. FR: flow rate. Figure adapted from Hartley et al.89 The STYs indicated are mass specific (based of mg of proteins). | ||
As described earlier, the STY is a reasonable and important quantity for the comparability of continuously operated flow processes. Niemeyer and co-workers maximized the STY in flow reactors by self-immobilizing biocatalysts. The authors investigated and compared five different biocatalytic flow reactors with different immobilization concepts with the same (R)-selective alcohol dehydrogenase from Lactobacillus brevis ATCC 14869 (LbADH) for the stereoselective reduction of 5-nitrononane-2,8-dione (Scheme 1, top).91 To regenerate the cofactor NADPH, the authors used three different strategies within their study (cf.Scheme 1i)–iii)). The enzyme itself contained a genetically encoded streptavidin (STV)-binding peptide enabling one-step purification and self-immobilization on STV-coated surfaces. The authors describe different immobilization-methodologies like physisorption or chemisorption as monolayers on the flow channel walls or on magnetic microbeads in a packed-bed format as well as self-assembled all-enzyme hydrogels. Coatings of the reactor surface with the biocatalyst in mono- and multilayers lead to STY <10 g L−1 h−1 whereas this value was increased tenfold by using packed-bed reactors. The highest observed STY of >450 g L−1 h−1 in this study was achieved with room-filling packed hydrogels. Also, even without detailed optimization of the process parameters, a continuous production for more than six days can be achieved.
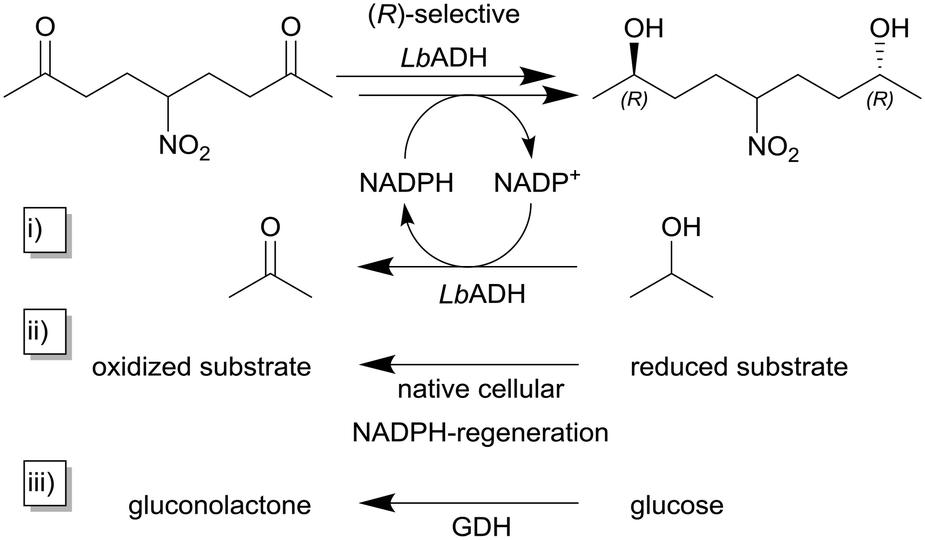 | ||
| Scheme 1 Reduction of 5-nitrononane-2,8-dione with (R)-selective alcohol dehydrogenase from Lactobacillus brevis (LbADH) and different methods for NADPH-cofactor regeneration.91 The NADPH cofactor regeneration can be implemented (i) as substrate coupled cofactor regeneration utilizing the same enzyme by the oxidation of isopropanol to acetone, (ii) within the host's native cellular metabolisms in the case of whole-cell biocatalysis or (iii) as enzyme coupled cofactor regeneration using a second enzyme e.g. in this case glucose dehydrogenase (GDH). | ||
In connection with hydrogels Menegatti et al. described a copolymeric immobilization of yeast cells on hydrogel basis for continuous biotransformation of fumaric acid in a microreactor.92 The authors developed an efficient microreactor with permeabilized Saccharomyces cerevisiae cells and used it for continuous biotransformation of fumaric acid into industrially relevant L-malic acid. Immobilization of the permeabilized cells resulted in up to 72% retention of fumarase activity, and the continuous biotransformation process using two layers of hydrogels integrated in a two plate microreactor resulted in a high space–time yield of 2.86 g L−1 h−1, with no loss of activity during seven days of continuous operation.
A major factor in biocatalytic flow processes that should not be underestimated are downstream processing (DSP) strategies to obtain the desired product directly and preferably in a high purity. In the best-case scenario, the DSP approach can be carried out inline and thus considerably improve the biocatalytic process (see also chapter 5.3.1). Semproli et al. successfully developed a new procedure for the synthesis of (S)-1-(5-fluoropyrimidin-2-yl)-ethanamine via flow biocatalysis with a subsequent inline DSP (Fig. 5).93 (S)-Selective amine transaminase from Vibrio fluvialis (Vf-ATA) was immobilized on glyoxyl-agarose or Sepabeads EC-EP/S and the (co)-substrate solution was pumped through the PBR at a flow rate of 0.2 mL min−1 (τ = 10 min). The exiting stream was fed into a second column packed with ion-exchange resin Dowex Marathon C and the resin trapped the desired amine. Afterwards, the resin was washed by H2O (flow rate of 0.25 mL min−1 for 20 min) and the desired product (S)-1-(5-fluoropyrimidin-2-yl)-ethanamine was subsequently released using a 1 mol L−1 solution of NH4OH (flow rate of 0.25 mL min−1 for 30 min). The pure amine was recovered with 35% isolated yield after evaporation of the solvent.
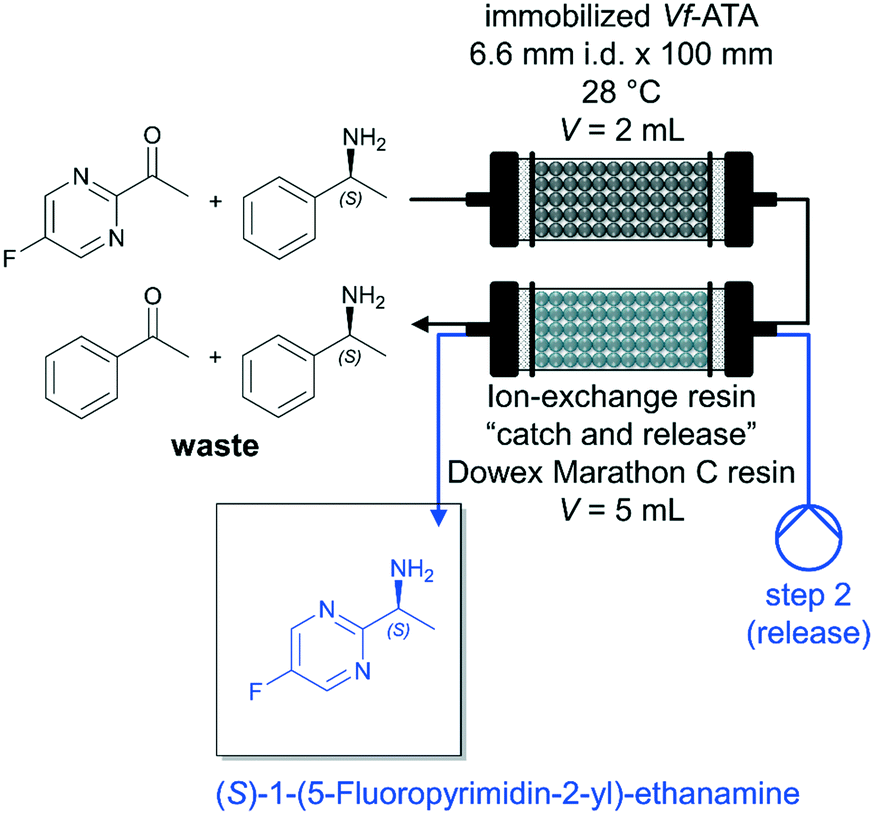 | ||
| Fig. 5 Synthesis of (S)-1-(5-fluoropyrimidin-2-yl)-ethanamine catalyzed by immobilized Vf-ATA in continuous flow followed by a continuous inline DSP approach by Semproli et al.93 | ||
A comparable approach was used by Jördening and co-workers for the continuous enzymatic production and adsorption of laminaribiose from sucrose and glucose in packed-bed reactors.94 The authors used a cylindrical glass column packed-bed reactor (36 mL bed volume) with a flow rate of 0.1 mL min−1 to produce laminaribiose continuously at 35 °C over 10 days. A subsequent adsorption on zeolite H-BEA 150 was carried out to remove the product inline. After successfully washing and desorption steps, a productivity of 5.6 mglaminaribiose Lenzyme bed−1 h−1 was achieved (Fig. 6).
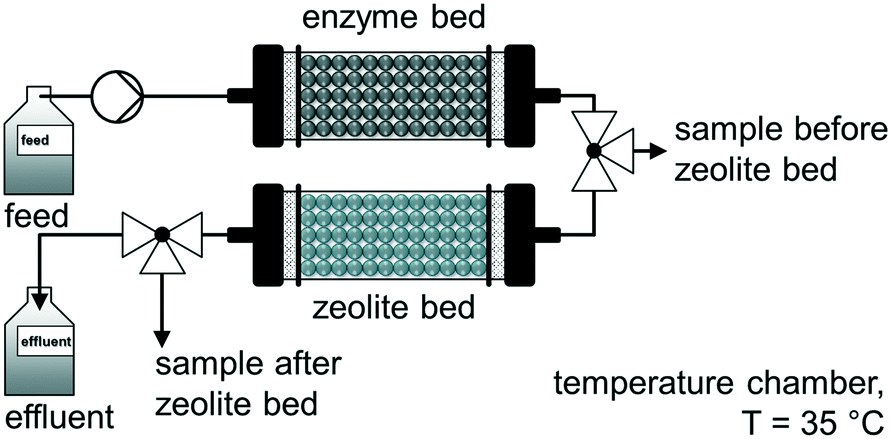 | ||
| Fig. 6 Continuous enzymatic production and adsorption of laminaribiose from sucrose and glucose in packed-bed reactors.94 | ||
Biocatalytic reactions do not always take place in an aqueous environment, and therefore aqueous-organic biphasic solvent systems for flow processes are also being researched.
Thus, Gröger and co-workers used a liquid–liquid segmented flow process to overcome work-up limitations of biphasic biocatalytic reaction mixtures.95 The authors used buffer/tert-butyl ether (MTBE) as biphasic medium for the enzymatic reduction of acetophenone and trifluoroacetophenone. The applicability of this flow system was demonstrated with two different enzymes and substrates (see Fig. 7). Additionally, in another study Gröger and colleagues used the same reaction system shown in Fig. 7 (ADH from Lactobacillus brevis for the reduction of acetophenone) to investigate a superabsorber-based immobilization strategy for application in a PBR and applicability in flow processes and observed initial conversions of up to 67% in such a continuously running process.96
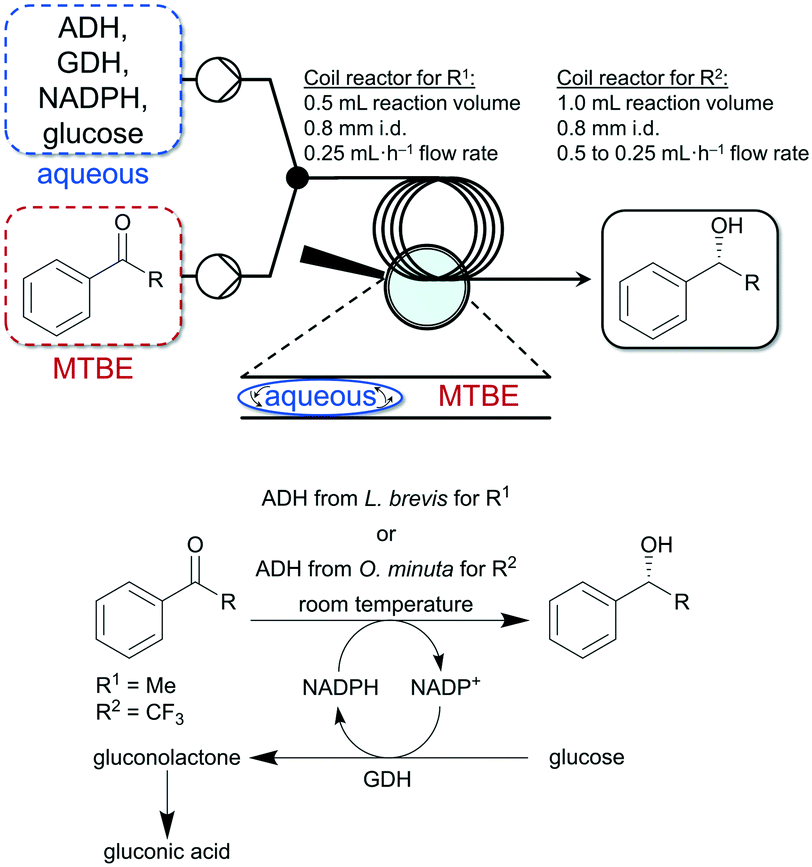 | ||
| Fig. 7 Enzymatic reduction of acetophenone (R1) and trifluoroacetophenone (R2) to the corresponding alcohols in a liquid–liquid segmented flow approach developed by Adebar et al.95 | ||
Hanefeld and co-workers used manganese dependent Granulicella tundricola hydroxynitrile lyase (GtHNL) in batch and continuous flow reactions in organic solvents for the synthesis of enantiopure cyanohydrin (Scheme 2).97 The continuous reaction was performed in acetate buffered methyl MTBE with flow rates from 0.1 to 1.0 mL min−1 (τ = 240 s to 24 s, respectively) and at optimal flow conditions conversions of 97% in 4 min at 0.1 mL min−1 were achieved.
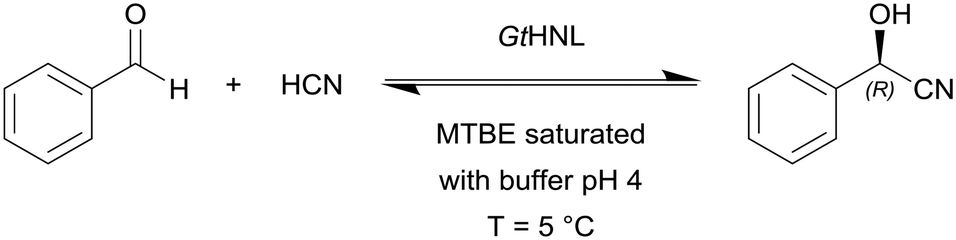 | ||
| Scheme 2 Granulicella tundricola hydroxynitrile lyase (GtHNL) catalysed hydrocyanation of benzaldehyde yielding (R)-mandelonitrile. | ||
Recently, also some pioneering developments have taken place at the interface between academia and industry: Wang and co-workers showed in their study that the implementation and use of biocatalysis in a plug-flow microreactor for the production of sitagliptin can lower the environmental factor (E-factor) about 74% compared to traditional processes.98 Goundry et al. reported a scale-up route to synthesize ATR inhibitor AZD6738, a medicament for the treatment of cancer. Within this multi-step industrial process, a biocatalytic step was developed installing the sulfoxide moiety with high enantioselectivity. Finally, a process on a plant scale with a CSTR cascade with four reactors was established and operated on a 46 kg scale with a total yield of 18%.99
Very recently, Tamborini and co-workers investigated the chemo-enzymatic flow synthesis of selected APIs butacaine, procaine and procainamide.100 The amide and ester intermediates were prepared in gram scale using a PBR with acyltransferase from Mycobacterium smegmatis immobilized on glyoxyl-agarose and inline purification with polymer-bound sulphonyl chloride. After washing the columns with toluene to recover the products, the solvent was evaporated under reduced pressure isolating the products in excellent (Fig. 8, compound A) to poorly (Fig. 8, compound C) yields. In a second flow synthesis step, the nitrogen group of the obtained intermediates was reduced to aniline using 10% Pd/C yielding the APIs mentioned above.
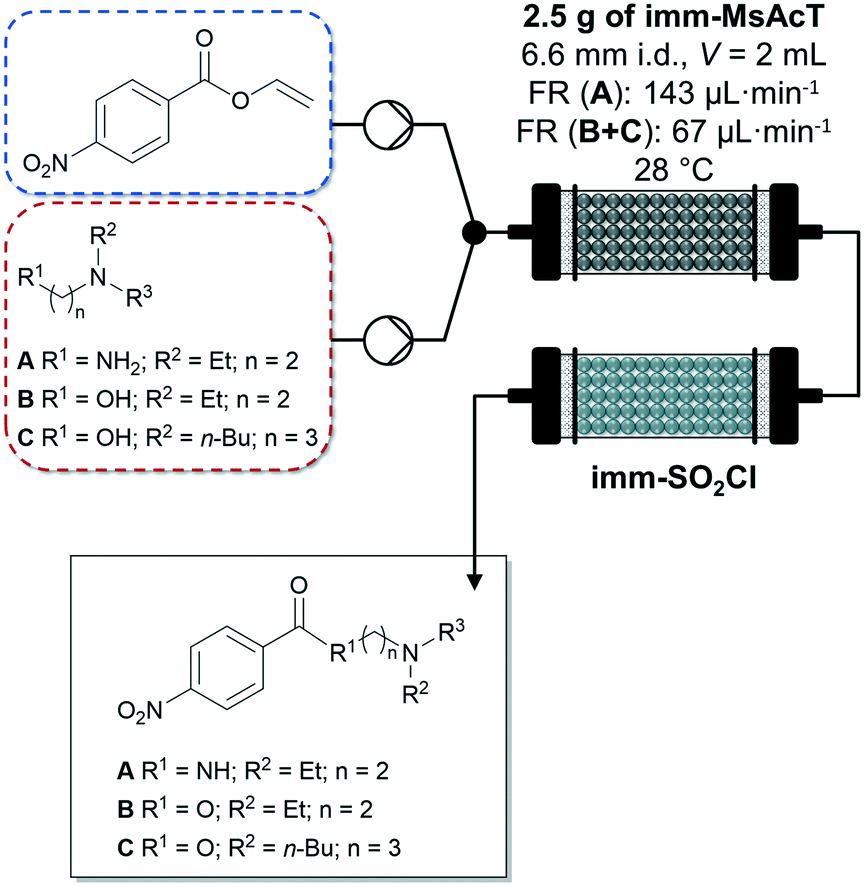 | ||
| Fig. 8 Enzymatic synthesis of amide and ester intermediates by immobilized acyltransferase from Mycobacterium smegmatis (imm-MsAcT) in continuous flow followed by a continuous inline DSP approach (imm-SO2Cl) by Tamborini and co-workers.100 Flow stream and solvent: toluene. | ||
Focusing specifically on microreactors, the recent publication of Huang et al. is worth mentioning, which presents the development of a new microfluidic biocatalysis–organocatalysis combination strategy for ring-opening copolymerizations of lactone, lactide and cyclic carbonate.101 Among other significant improvements, the authors state a shortened overall polymerization time (<40 min) and high monomer conversions (>95%). Žnidaršič-Plazl and co-workers theoretically and experimentally characterized a micro packed-bed reactor (μPBR) with immobilized Candida antarctica lipase B (Novozym 435) in their well-presented study.102 In addition, the authors performed a transesterification reaction in the μPBR (vinyl butyrate and 1-butanol into butyl butyrate with n-heptane as a solvent) and evaluated different operating conditions, e.g. flow rates and thus residence times and temperatures. Semenova et al. used a model-based analysis of biocatalytic processes and the performance of microbioreactors with integrated optical sensors. The model predictions (identification of the reaction mechanism, kinetics and limiting factors) were independently confirmed for μL- and mL-scale experiments.103 Vobecká et al. have recently presented an interesting work in which they present an enzymatic synthesis of cephalexin in a continuous-flow microfluidic device in aqueous two-phase system (ATPS) forming two-phase slug flow in a microfluidic capillary as the reaction-separation environment.104 ATPS consisted of 15 wt% of PEG 4000, 12 wt% of phosphates, and 73 wt% of water.
A canonical microreactor design for heterogeneously catalyzed continuous biotransformations has been studied in detail by Nidetzky and co-workers in their 2019 publication. The authors used a wall-coated, immobilized enzyme microreactor and demonstrated the important interaction of reaction/enzyme properties, microchannel geometry and reactor operation.105 Further work of the group includes among others process intensification for O2-dependent enzymatic transformations in continuous single-phase pressurized flow,106 and the continuous synthesis of lacto-N-triose II by engineered β-hexosaminidase immobilized on solid support.107
4. Enzyme immobilization for continuous processing
4.1 Fundamentals of enzyme immobilization with respect to continuously operated flow systems
In this section, we will highlight recent improvements in enzyme immobilization methods with respect to flow biocatalysis. Both carrier-free and carrier-bound strategies are well known in the literature,130–134 and will be briefly discussed and summarized below. In this context, the recently published review article by Bolivar et al. is worth mentioning describing the characterization and evaluation of immobilized enzymes for applications in continuous flow reactors in an excellent way.135 In general, two main enzyme immobilization techniques have been proven to be successful for continuous biocatalytic synthesis. Firstly, immobilization of the enzyme onto the reactor wall thereby creating a wall-coated reactor, and secondly, the use of a wide variety of support materials binding the enzyme onto particles or monolithic structures,136,137 thereby creating e.g. a fixed-bed reactor.15The strategies of enzyme immobilization are roughly divided into three categories: (i) encapsulation/entrapment, (ii) cross-linking and (iii) binding to a (porous) carrier, based on the respective physical or chemical interactions (Fig. 9).19,139,140 Each typology has its own advantages and disadvantages depending on the process/reaction conditions required. In cross-linking, as an example of covalent bonding, several enzymes are linked together with the help of a difunctional agent, usually glutaraldehyde.141 Conversely, encapsulation methods trap enzymes (both soluble and aggregated) within a bulk matrix such as a polymer network.19 In this context, Castiglione and co-workers recently used crosslinked polymersomes as nanoreactors in different biphasic reaction setups.142 Here, the solvent-sensitive enzyme mandelate racemase was protected from the organic phase and the enzyme remained its activity for more than 24 h whereas the free enzyme got completely inactivated after 1 h.
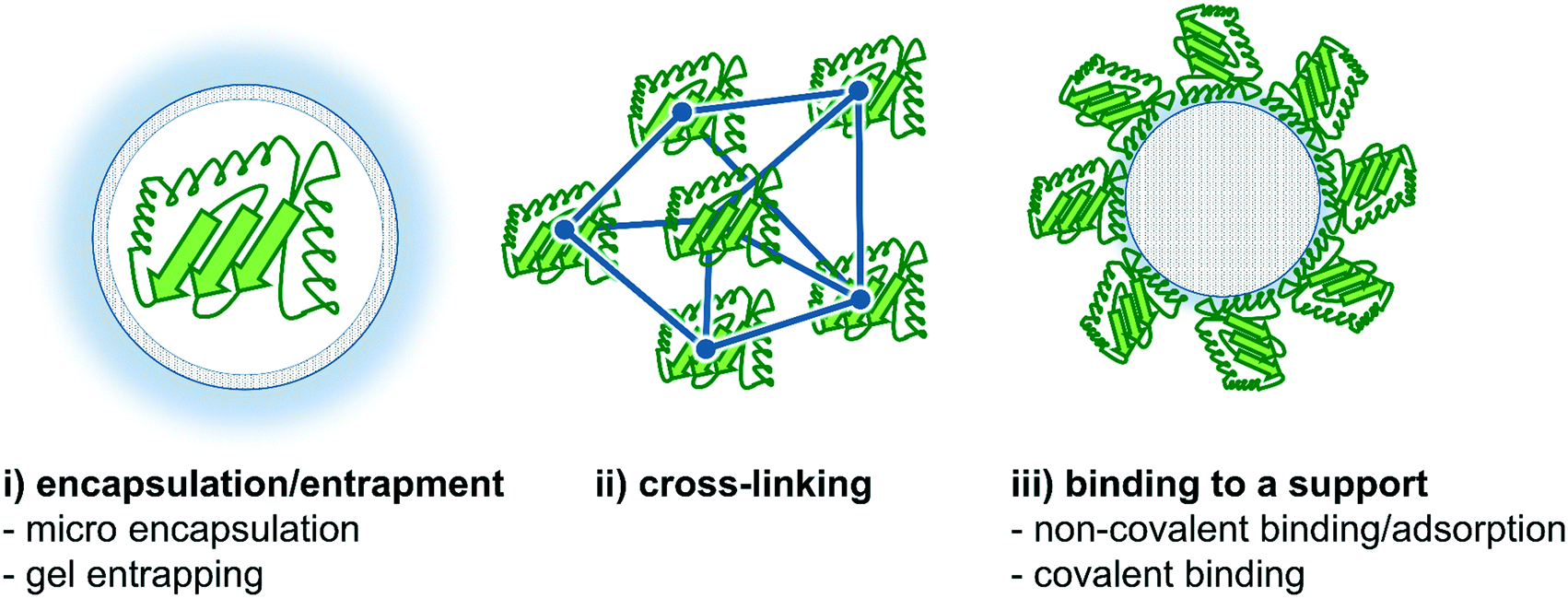 | ||
| Fig. 9 Different strategies for enzyme immobilization. Three main options are commonly applied: (i) encapsulation/entrapment (e.g. micro encapsulation or gel entrapping), (ii) cross-linking and (iii) binding to a carrier (e.g. non-covalent binding/adsorption or covalent binding).138 | ||
The widely applied carrier-based immobilization strategy is subdivided into a covalent and a noncovalent category. For covalent attachments, specific reactive groups (e.g. aldehydes, amino, epoxides, etc.) are required to anchor the enzyme to the support. Noncovalent approaches are instead based on physiochemical interactions like hydrophobic, charge–charge, van der Waals, hydrogen or affinity-tag binding.143 However, it is not always sufficient to select only the most suitable immobilization method, since other parameters must be taken into account when transferring immobilization methods to flow processes. In detail, several parameters have to be carefully selected to design an optimal flow process, especially if carrier-bound methods are applied. The most important parameters are (i) the pore size and (ii) the particle size of the respected carrier material. The pore size defines the specific area being available for the enzyme to anchor to the support. It directly affects the mass transfer, e.g. a bigger pore allows an easy entrance of substrates and cofactors as well as for (co)products' release. Conversely, the particle size plays a decisive role for the length of the diffusion path and thus the pressure drop in continuous-flow reactors.144 Additionally, all particles should have the same shape and size to avoid a high back-pressure in packed-bed column reactors.
4.2 Carrier-free methods
Carrier-free immobilization strategies benefit from the fact that no additional inactive mass is present as a support. The enzymes are bound together to form a solid phase dispersed in the bulk solution. In most cases, these systems can be produced by cross-linking different enzyme preparations such as dissolved enzymes, crystalline enzymes, spray-dried enzymes and physically aggregated enzymes.145 Often this immobilization technique requires the use of a chemical crosslinker such as glutaraldehyde to form the desired crosslinked structures. In addition, several factors such as the amount of the above mentioned crosslinker, temperature, pH and ionic strength must be carefully and finely balanced within the immobilization process. Carrier-free techniques offer clear advantages, including high enzyme stability and activity and a simple preparation procedure. Compared to carrier-bound methods, this method is fast and inexpensive because no additional carrier material is used.In conclusion, carrier-free methods prevent diffusion limitations caused by the blocking of the surface by a high amount of enzymes. Problems such as small effective surface areas of commonly available carrier materials are also advantageously avoided. Some of the work discussed below also refers to Table 2 of chapter 3.
Two major challenges need to be overcome for a possible industrial application of all-enzyme hydrogels: firstly, this approach is limited to multimeric enzymes (as a recent survey of the PDB database pointed out that only ∼10% of the enzymes are homotetramers),121 and secondly, a slight decrease in the enzyme activity is observed (probably due to mass transport limitations).120 Fortunately, this challenge could be solved by enzyme engineering. However, the aforementioned advantages and the constant improvement in continuous flow chemistry are the decisive factors for the transition to self-assembled all-enzyme hydrogels. In addition, the described approach proves to be suitable for semi-preparative scales thanks to the possibility of a sequential use as well as a parallelization by ‘numbering-up’ of the flow reactor modules as a further advantage (see also Table 2, entry 16 and Table 3, entry 1and 2).120,121
| Entry | Biocatalyst | Immobilization technique | Carrier material | Functional group | Particle size | Pore size | Ref. |
|---|---|---|---|---|---|---|---|
| [μm] | [Å] | ||||||
| n.a. = not available. | |||||||
| 1 | (S)-Stereoselective ketoreductase from Saccharomyces cerevisiae and | Self-assembling all-enzyme hydrogel | n.a. | n.a. | n.a. | n.a. | 121 |
| 2 | Cofactor-regenerating glucose 1 dehydrogenase from Bacillus subtilis | Self-assembling all-enzyme hydrogel | n.a. | n.a. | n.a. | n.a. | 121 |
| 3 | Lipase PS from Burkholderia cepacia | Covalent immobilization | Hollow silica microspheres | Bisepoxide | 10–30 | 150–300 | 112 |
| 4 | Lipase B from Candida antarctica | Covalent immobilization | Hollow silica microspheres | Bisepoxide | 10–30 | 150–300 | 113 |
| 5 | Lipase from Burkholderia cepacian | Ionic immobilization | Ionic liquid modified silica | Amine | n.a. | 42 | 110 |
| 6 | ω-Transaminase from Halomonas elongata | Covalent immobilization | Functionalized methacrylate-based carrier | Hydroxylamine/ethanolamine | 150–300 | 120–180 | 124 |
| 7 | ω-Transaminase from Halomonas elongata | Covalent immobilization | Functionalized methacrylate-based carrier | Ethanolamine/polyethylenimine (60 kDa) | 100–300 | 10–20 | 124 |
| 8 | Crude naringinase from Penicillium decumbens and a purified naringinase with high α-L-rhamnosidase activity | Covalent immobilization | Two-dimensional zeolite ITQ-2 after surface modification, surface area of 236 m2 g−1 | 3-(Aminopropyl) triethoxysilane (= NITQ-2), and subsequent treatment with glutaraldehyde, yielding aldehyde groups on the surface (= GITQ-2) able to react with amino groups of the enzyme | Thin zeolite sheets, 2.5 nm thick | n.a. | 174 |
| 9 | Alcohol dehydrogenase (ADH) | Covalent immobilization | Two-dimensional zeolite ITQ-2 after surface modification, surface area of 387 m2 g−1 | 3-(Aminopropyl) triethoxysilane (= NITQ-2) | Thin zeolite sheets, 2.5 nm thick | n.a. | 175 |
4.3 Carrier-bound methods
Anchoring an enzyme to a solid carrier is a well-known technique that is widely applied in industry. Beneficially, better enzyme stabilities and an enhanced eco-efficient downstream processing can be achieved resulting in greatly reduced process costs. Here, for the latter, an easy recovery from the reaction mixture and the reusability of the biocatalyst play a decisive role. A broad variety of materials are used as carriers and they are classified into organic, inorganic, hybrid or composite substances depending on their chemical composition.149 As aforementioned, covalent attachment is the most commonly applied procedure for attaching an enzyme to a solid support. However, an activation either of the enzyme or the support (or also both) is needed to achieve the required bound formation. This immobilization procedure is not only limited to isolated enzymes, since recently microorganisms like Penicillium funiculosum have also been immobilized on polyether based polyurethane foams and the overgrown foams have been successfully used for biological and continuous conversion of racemic mixture of 1-amino-1-(3′-pyridyl)methylphosphonic acid.150 Some of the work discussed below also refers to Table 2 of chapter 3.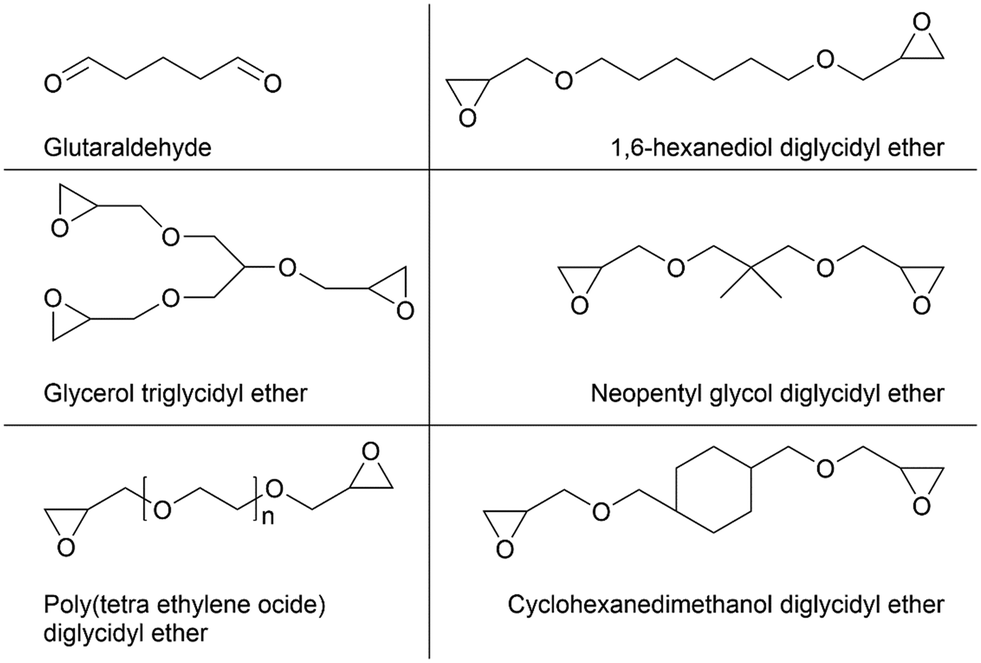 | ||
| Fig. 10 Overview of commonly used bisepoxides for covalent binding in enzyme immobilization. | ||
Several bisepoxides with different lengths, stiffnesses and hydrophobicities were selected and evaluated for the surface modification of hollow silica microspheres.149,152 The epoxy functions of the activator-agent bind covalently to the enzymes instead of a simple adsorption on the silica surface. The introduced bisepoxide moieties can covalently bind amine-, thiol- and carboxylate-functional groups under mild conditions.149 First results with bisepoxide-functionalized silica microspheres showed a strong tendency to link the enzyme compared to other glutaraldehyde-based support-activated functionalization strategies.149 Bisepoxides with long and flexible chains can also provide multi-point binding of the enzyme to the carrier to promote an even more robust anchoring. Moreover, the immobilized enzymes preserve their activity and selectivity in contrast to other covalent immobilization strategies because the enzyme's catalytically active site is not sterically hindered, especially if a longer and more flexible bifunctional linker is used.149,152 Thanks to their properties and numerous typologies of these activating agents, bisepoxides with different lengths and characteristics of the linker chain can be designed. This represents a cost-efficient and easy to perform technique for fine-tuned surface carriers.153 Recently it was shown, that polyethylene glycol diglycidyl ether (PDE) bisepoxide as the activating agent together with hollow silica microspheres M540 as the carrier and lipase PS as the catalyst obtained very high catalytic properties. PDE consists of a long and flexible chain attaching the enzyme via multipoint bindings together with an optimal enzyme orientation allowing the best entrance of the substrate into the active site.112,149,154
ILs are organic salts with a melting point below 100 °C and unique properties (if compared to inorganic salts) with broad liquid state ranges, high ionic conductivities, negligible vapor pressures, high chemical and thermal stabilities as well as good abilities to dissolve several organic, inorganic and polymer compounds.155
In contrast to organic solvents, ILs can be considered eco-friendly solvents thanks to their non-volatility and non-flammability.156–159 Overrating should nevertheless be avoided: the synthesis of some ILs can be very tedious and resource intensive, yet generalization is not possible and their potential should be further explored.160
The main advantage is that all these above-mentioned properties of ILs can be fine-tuned by carefully selecting the most appropriate combination of cations and anions according to the desired purpose.161 Thanks to these characteristics, the use of ILs as a pure solvent or as a cosolvent have gained high attention as a well-established strategy improving the enzyme activity and stability for the last two decades. Early works of Kragl, Welton, Seddon, Sheldon and other co-workers paved the way for this scientific field.162–167
Unfortunately, their liquid nature leads also to many disadvantages like e.g. a delicate catalyst recovery, a difficult product isolation and a possible increase in cost due to the high amount of solvent needed. An elegant solution to tackle these drawbacks is to combine ILs with solid supports, coating just the surface of the carrier. This new class of composites takes advantage of both ILs and the carrier-bound strategy. Several IL-carrier-fusions have been evaluated in the last years and, among them, the most promising ones are based on protonic ILs with either silica supports (see also Table 2, entry 3 and Table 3, entry 5),110 or magnetic carboxymethyl cellulose nanoparticles.156 The results obtained being applied to both lipases and laccases showed a higher protein loading on the support and a higher activity than for both free and immobilized enzymes in the absence of ILs. It has been shown that ILs lead to an increase in the material surface area and pore volume size facilitating the covalent immobilization of the enzyme.155 In addition, an interface activation promoted by ILs have been observed.110,156,168 Finally, it should be emphasized that ILs enable the creation of a microenvironment protecting the biocatalysts from denaturation by chemicals such as urea.156
This presented approach has already been evaluated in continuous flow processes in which redox cofactors and enzymes have been proven to remain bound to the support for up to 100 hours of operations. Consequently, the self-sufficient heterogeneous system proved to be highly stable over several operational cycles without requiring any exogenous cofactor (see also Table 2, entry 19, 20 and 21 and Table 3, entry 6 and 7).124
4.4 Evaluation of enzyme immobilization from the continuous flow perspective
The advantages of enzyme immobilization are well known. Among others, an increased stability, a recyclability and a simplification of the downstream processing are beneficial futures. However, from the continuous flow viewpoint, the predominant advantage of immobilization is that the catalyst remains in the reactor core, while reagents and products are easily and gently added by the solution being pumped through the reactor.171,172As highlighted by Bolivar and López Gallego, enzyme immobilization is a conditio sine qua non for the use of a green catalyst in continuous processes. Therefore, the evaluation of immobilization effects on the biocatalyst's activity is mandatory and parameters such as the activity of the immobilized enzyme (U mgcarrier−1), the enzyme loading on the carrier (mgenzyme mgcarrier−1), the space–yield time (g L−1 h−1), the specific productivity (gproduct h−1 mgenzyme−1) and the turnover frequency (s−1) are crucial for the evaluation.135 A systematic investigation of these parameters is very important and in addition we would like to highlight the publication of Aguillón et al. where the authors systematically screened 16 immobilized lipases for the continuous-flow kinetic resolution of (±)-1,2-propanediol.173
5. Future perspectives
5.1 Additive manufacturing/3D printing
Additive manufacturing is a method used to build up three-dimensional (3D) structures by adding a certain material stepwise onto a support. Nowadays, a wide variety of materials can be 3D-printed and this field of research has grown considerably in the last two decades. 3D printing has already been used in various industries such as medicine, fashion and chemical industry. With the help of computer-aided design (CAD) program codes, additive manufacturing has become one of the most promising technologies currently available.176–185Its secret weapons? The rapidly decreasing costs and almost infinite design possibilities: it allows to easily produce complex (up to 0.01 mm resolution) and highly elastic structures out of a wide variety of materials from polymers to metal alloys. Additionally, the waste of material is minimal.186 These advantages reduce the time needed for process development showing an easy and well-planned route to ‘rapid prototyping’.187
The fusion between 3D printing and continuous flow biocatalysis took place few years ago when researchers started to ask themselves how additive manufacturing's pros enhance continuous flow chemistry.188,189 Since then, additive manufacturing successfully overcome deficits in the field of continuous flow chemistry with respect to manufacturing of solid supports with immobilized enzymes. Some promising developments have been achieved recently and many studies focus on the development of 3D-printed carriers. The design of a tailor-made carrier material with a finely tuned porosity and pore diameter, and a defined particle size is highly beneficial here.
The entrapment of enzymes is widely applied together with 3D-printed carrier materials, and in addition, this physical entrapment not only allows the enzyme to be retained in the reactor, but also provides its sufficient purification.190 The broad applicability of 3D-printed carriers has already been investigated with different enzymes and under different conditions, indicating the universal applicability of those carrier materials and processes.186,191,192
Entrapping enzymes in 3D-printed hydrogel beads creates an optimal aqueous environment restricting the movement of macromolecules while substrates and products can pass through the hydrogel network. This hydrogel network can be specifically designed by a careful selection of monomers and cross-linkers.190,193 Next to others, this methodology benefits from high flexibility, easy automatization and fast implementation, especially compared to covalent immobilization techniques. Another positive aspect is the fact that hydrogels are stable at high temperatures and can operate in the presence of harsh reaction conditions.194 Owing to their enormous potential, we believe that hydrogels will be further explored for their use in flow biocatalysis. Nowadays, first types of bio-inks are investigated, such as biodegradable agarose-based hydrogels,187 which are also involved in the enzymatic reaction. Also, these include 3D-printed graphene carriers that can be applied in reactions where electron transfers are required.194
The most common strategy for the synthesis of enzyme carriers is to use an extrusion-based 3D printer building up the regarded material layer by layer.185,188 Various types of inks have already been evaluated for enzyme immobilization strategies, including the most promising ones based on poly(ethylene glycol) diacrylate,190 and nylon.186
Recently, Liese and co-workers investigated polyethylene terephthalate (PET) as an additively manufactured carrier material for the immobilization of phenolic acid decarboxylase (PAD) from Mycobacterium colombiense for the decarboxylation of ferulic acid. PAD was genetically fused with an anchor peptide and non-covalently immobilized on the support PET. Together with an in situ product removal (ISPR), a conversion of ca. 88% after 2 h was achieved.195,196
In addition to additively manufacturing of novel carrier materials, reactors, and other devices can also be 3D-printed. With its cost-efficiency it is indeed possible to assemble reactors with diverse and complex structures, channels and porosities.189,197–199 Moreover, each 3D-printed component can be modified easily (completely or partially) compared to the first design draft, if necessary, favoring a simple trial-and-error development process.192 This principle can be implemented when CAD modeling is combined with computational fluid dynamics (CFD), where it simplifies the development and optimization of the reactor design and allows digital visualization and understanding of the entire process.200
The most widespread technology for the production of reactors is also based on extrusion processes, whereby in particular fused deposition modelling (FDM) and fusion filament fabrication (FFF) are now enjoying great popularity.199 Compared to other 3D printing processes, FDM is advantageously simple and inexpensive: it does not require a support and thermoplastic materials such as acrylonitrile–butadiene–styrene (ABS) and polylactic acid (PLA) are used. They are also easily recyclable and recently, polyether ether ketone (PEEK) for example, has shown remarkably high chemical resistance.199 The application of the FFF techniques makes it possible to create cavities in the main structure without the need for subsequent modifications. Among others, this ensures a better mixing of the reactants in the 3D-printed reactors.
3D printing can also provide an alternative to traditional micro-technologies, which are mainly used for the construction of miniaturized reactors and other microfluidic devices. Some of the current limitations associated with their manufacture can be overcome by 3D printing, such as the long time required for their construction and the use of a narrow range of chip materials. From this perspective, laser additive manufacturing (LAM) is of particular interest as a 3D printing technique for microfluidic devices using metals or alloy powders that are melted to form specifically designed 3D structures.201 Yet, meso-scale reactors cannot be printed with the powder-based fusion (PBF) methods because their resolution is limited by the powder's diameter. However, the most suitable technology for meso-scale reactors with 3D printing is electrochemical additive manufacturing (ECAM), in which metal ions are deposited as metal atoms by electrochemical processes.202
Many steps have already been taken with regard to 3D printing, but some have yet to be taken, such as the chemical refinement of printed devices, which has received much less attention. Various research groups are now focusing to fill this gap by developing initial proof-of-concept studies for modifying and functionalizing 3D printing surfaces and devices, paving the way for a new strategy that combines enzyme immobilization, its recyclability and continuous flow processes.203
5.2 Enzyme cascades
Continuous flow chemistry has had an impressive effect on enzyme-catalyzed cascade reactions opening the doors to new potential pathways for biosynthetic and bioanalytical applications. Biocatalytic cascade reactions are atom-economic and energy efficient processes mimicking natural activities and synthesis pathways.204–208 The performance of cascade reactions in continuously operated flow-through systems includes further positive properties such as an improved mass and heat transfer and a better mixing of the reactants, e.g. increasing the reaction rate and having least distortion of the biocatalyst due to the lack of high shear-stress.209,210 Furthermore, the activity of the biocatalyst is increased, e.g. by avoiding substrate and product inhibition through a precise control of the residence time in relation to the reactor volume. By combining this strategy with real-time monitoring and control (see also 5.3.1), further decisive advantages can be achieved leading to a better understanding not only of the overall process, but also of the individual steps that make up the cascade.Multi-enzyme cascades in continuously operated flow systems can dramatically boost the efficiency and productivity of the biocatalytic process under consideration. Recently, Lauterbach and co-workers combined an imine reductase with a diamine oxidase and immobilized the enzymes onto polymer coated glass porous carriers.211 The enzymatic cascade was performed in a continuous flow reactor and both H2 and O2 were produced by electrolysis and transferred through a gas-permeable membrane into the flow system.211 Modeling of enzymatic cascades allows us to understand the effect of parameters and optimize thereof while running the cascades for continuous synthesis. Finnigan et al. combined both mechanistic and empirical modeling to optimize a two-enzyme system for continuous reductive amination.212 The multi-dimensional optimization shows a clear advantage compared to traditional time consuming one-factor at a time (OFAT) approaches. In addition, the availability of these models can be extended by modern public platforms such as github, and they can be operated with cloud-based Python notebooks. In our opinion, these in silico methods are an important groundbreaking development in flow-through biotechnology. Especially the time savings can contribute to the breakthrough of enzymatic reactions on an industrial scale due to the lower costs in Research and Development (R&D) departments. Despite all the euphoria, it should not be forgotten that the computers working with these models, require basic experimentally validated kinetic data (Km, kcat and Ki, etc.). The practical chemist working in the laboratory will therefore remain an absolute necessity for a long time to come.
5.3 Online analytics
Tracking a chemical reaction being performed in a continuous operated mode is a challenge due to a higher technical effort compared to classical batch reactions. In the last decades, several techniques have been successfully developed and implemented. Depending on the technical configuration (primarily with respect to the position of the sensor), different methods of continuous measurement can be distinguished: (i) inline, (ii) online, (iii) offline and (iv) atline analysis. ‘Inline analysis’ refers to a setup in which the sensor is in direct and continuous contact with the analytes. Whereas, if a bypass line is installed within the system, it can be described as ‘online analysis’. Conversely, a procedure in which a sample is taken from the bulk solution in order to subsequently carry out the analysis is referred to as ‘offline analysis’. However, if the analysis (e.g. gas or liquid chromatography) is performed within a narrow time frame and in close proximity to the reactor vessel together with automatic sampling, the method is defined as ‘atline analysis’.213 We think that the definitions mentioned above are important to have a common and standardized communication on the type of analysis. Below the chapter emphasizes the future perspectives of online analysis for continuous synthesis.Recently, a new device was developed by the ALMAC group (one of the leader company in development of innovative technologies in pharmaceutical and biotech sectors). The device offers a new platform that is able to facilitate substitutions of functional groups. Here, chemical reactions using high energy, high pressure, oxidation, and photochemical transformations can be performed in a safe and scalable manner. This flow system allows to obtain better results to those reached under batch conditions in terms of both productivity and minimization of genotoxic impurity intermediates formation. First results obtained with the above-mentioned FAST system have been presented by Rahman et al. The authors successfully used a FAST hydrogenation continuous platform for greener aromatic nitroreductions in aqueous solution at low pressures of H2.224 We believe that the (bio)catalysis community will further witness developments at the interface of academia and industry to speed up the research and technical implementation phases for the production of chemicals.
5.4 Downstream processing
Often, the main focus in continuous biocatalysis concentrate on the development and improvement of the flow system itself. In our opinion, not the same level of attention is paid so far to the downstream processing (DSP), even though, simultaneously to the development of continuous flow chemistry, continuous DSP has gained some attention. The development goals here are primarily to increase process efficiency, improve product yields and quality and reduce space requirements and the cost of goods.230 This new approach has found application in biopharmaceuticals purifying the final product. The continuous approach can be performed using various techniques and tools, such as continuous centrifugation, depth filtration or tangential flow filtration (TFF) representing primary clarification techniques at a manufacturing scale. In connection with the separation and purification of biomacromolecules we would like to refer to the recently published tutorial review by Vicente et al.231However, by far the most commonly used continuous DSP technique is continuous chromatography.232 Continuous chromatography benefits from two main advantages: firstly, a reduction in processing volume and secondly, greater tolerance to unstable compounds. Therefore, a continuous flow operation improves a greater number of purification cycles within a smaller column while utilizing shorter process times, which is a major advantage when purifying less stable proteins.233
Generally, in situ solid phase adsorption can be used for continuous flow biocatalysis. Although various adsorption materials are now moderately used in the literature (see entry 1, 5 and 7 in Table 2), a systematic screening of the large number of commercially available adsorber materials (and ion exchange resins) is still lacking in the research field. Recently, von Langermann and co-workers presented two studies with promising results for the DSP of products from biocatalytic reactions in shaking flask experiments, which could be transferred to the use in enzymatic continuous processes.234,235 Additionally, in situ product recovery techniques (ISPR) like in situ product crystallization (ISPC),236 and in situ product adsorption (ISPA) can help to implement continuous DSP.237
5.5 Non-conventional media
Biocatalysis and green chemistry changed our way to approach chemical synthesis, especially on an industrial scale. On the one hand, they brought several advantages to the research field, but on the other hand they also caused many new problems and doubts. In enzymatic processes, most biocatalysts operate best under aqueous reaction conditions where they reach their maximum activity. In contrast, hydrophobic substrates are often hardly soluble in aqueous environments and the applied chemist becomes severely restricted in his freedom of action.To overcome these issues, the subfield of enzyme catalysis in organic solvents was constituted early and eventually.238–242 It was 1980s–90s when the use of enzymes in non-aqueous media was documented by pioneers of the field: Klibanov,243 Halling,244 Mattiasson and Adlercreutz.245 Furthermore, it was at the beginning of the 2000s, when the first papers were published on biocatalytic reactions in ionic liquids (ILs) (see also above, chapter 4.2.2).
Recently, Wang and co-workers studied 16 different ILs as a cosolvent for microfluidic biocatalysis.246 The authors used a novel recombinant RhaB1 enzyme (a bacterial α-L-rhamnosidase belonging to glycoside hydrolase family 78 (GH78)). Without any further purification steps, rutin was hydrolyzed yielding isoquercitrin. Using a continuous flow glass-PDMS microchannel reactor together with an aqueous solution of the colloid enzyme, the recombinant RhaB1 had agglomerated in the microchannel after 4 h and blocked the channels. When IL [Toma][Tf2N] was used as a cosolvent in the same time, the solubility and liquidity of RhaB1 was increased, no agglomeration occurred and the microchannel was not blocked. Additionally, the authors confirmed a positive effect on RhaB1 activity resulting from the use of the IL. The main channel dimension was 200 μm wide, 100 μm deep and 1 m long. The flow regime was stable and laminar. The reaction substrate at 0.01 g L−1 (rutin at pH 5.0) and the recombinant RhaB1 solution containing 0.02 g mL−1 [Toma][Tf2N] were both pumped into the microchannel by a two-channel syringe pump. The flow rates of the two phases were the same at a range from 1 to 10 μL min−1 and different temperatures were used. Compared with a batch reactor, the reaction time was reduced by ca. 98%, the Km was decreased to ca. 1/3, productivity was improved ca. 61 times and the reaction time was reduced by ca. 75%. Under optimum conditions, an isoquercitrin yield of ca. 99% was achieved in 10 min using the microchannel reactor. With this study, the authors were able to show that the disadvantages of microreactors can be overcome through the systematic and intelligent use of cosolvents like ILs.
The work of Grollmisch et al., which recently demonstrated the immobilization of lipase CalB in polymerized ionic liquids (PILs), should be mentioned here even if the material is not yet used in continuous systems, but it is certainly pointing the way forward.247 In addition, Villa et al. discuss non-conventional media in biocatalysis and ISPR with microflow systems.248
The topic of ‘biocatalysis in non-conventional media’ (BNCM) was recently augmented by the field of using deep eutectic solvents (DES) as efficient solvents and reaction (co)media in biocatalysis.240,249–254 DESs gained much attention in the last years being neoteric solvents which special benefits, such as an easy and fast preparation procedure and several tunable properties. Eutectic mixtures are obtained mixing Lewis or Brønsted acids and bases containing a variety of anionic and/or cationic species.255–257 However, the final solvent has a lower intrinsic toxicity compared to ionic liquids, making them to promising substitutes.258 Its main disadvantage is the high viscosity (which depends on the starting materials) being a problem for the scale-up of the process, but it can be easily overcome by mixing them with other (co)solvents. In this context, adding water/buffer (e.g. up to 20% v/v) has shown a significant decrease in viscosity.256,259–261 All these properties make DESs particularly suitable for use in continuous flow chemistry, and their tailor-made properties make multi-stage continuous flow processes and cascade reactions with them ideal.
5.6 Photobiocatalysis in continuous flow
The use of light for catalysis is an elegant approach, that has been taking great attention within the scientific community. Nowadays, photocatalysis has been proven to extend the substrate scope by using (transition) metal or organic catalysts under milder conditions compared to light-independent alternatives. Herein, photocatalytic reactions take advantages for the enhanced reactivity of the photocatalyst in its excited state, e.g. allowing single-electron transfer (SET) processes with organic substrates, producing radicals playing a key role in organic synthesis.262Increasing number of studies focus on photobiocatalytic reactions covering enzymes that are coupled with a photocatalyst263–265 as well as photo-enzymes that are strictly light-dependent being able to catalyze a reaction only upon illumination.266 Recently, Schmermund et al.267 and Park and co-workers268 have reviewed this constantly growing research field of photobiocatalysis. Indeed, nowadays light-driven enzymatic biocatalysis are extensively investigated,269–296 and – in our opinion – the integration and consolidation of photobiocatalysis and continuous flow chemistry seems to be the next logical step broadening the knowledge in this interesting field of research. For example, low light penetration depth and inhomogeneity of light distribution inside the reaction medium can be overcome by continuous flow technology. Combining the pros (and cons) of both research parts can lead to a more efficient catalysis era and we hypothesize that proof-of-concept examples will be published in the near future.
Conclusions and outlook
The fusion of biocatalysis and flow chemistry is in progress and is increasing. It stands to reason: high surface-to-volume ratios, improved mixing and mass transfer, a superior temperature control and small volumes requiring significantly reduced amounts of reagents and a shorter time from idea to application. All these advantageous parameters will boost and inspire research. Compared to classical ‘beaker-biocatalysis’, flow chemistry can be more productive, resource-efficient, controlled and environmentally friendly.In the near future, we are expecting an intense increase of the use of flow biocatalysis in academia and industry for the synthesis of chemicals of our daily need. Especially the opportunity of downscaling of a biocatalytic reaction enables a sustainable approach for screening of process parameters with high degree of freedom in a more resource efficient way. In addition, the modularization of reaction cascades in flow rather than one-pot synthesis is a particularly attractive route. Particularly in pharmaceutical production, we expect an increasing interest of the industry for flow biocatalysis as flow biocatalysis can play a significant role to reduce the time-to-market. Perspectively seen, we think that a model-based scale-up of flow biocatalysis and the use of commercial modular systems for industrial implementation will contribute to the success of continuous flow biocatalysis.
Within our review, we specifically focused on future perspectives like 3D printing techniques, enzyme cascades in continuously operated reactors and online analytics. Additionally, future developments in continuous downstream processing and the use of nonconventional media in flow systems will make their contribution to the successful and ongoing progress. Finally, we also hope that the scientific community will be stimulated to report consistently about biocatalysis in continuously operated reactors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank to Danmarks Frie Forskningsfond (DFF), Independent Research Fund Denmark (IRFD), PHOTOX-f project, grant agreement No 9063-00031B. The authors also thank video telecommunications for the possibility to stay in contact with colleagues and friends during the 2019–2020 COVID-19 pandemic. An ongoing discussion is crucial developing such a review paper.References
- N. G. Anderson, Org. Process Res. Dev., 2012, 16, 852–869 CrossRef CAS.
- F. Darvas, V. Hessel and G. Dorman, Flow Chemistry, Volume 1: Fundamentals, De Gruyter, Berlin/Boston, 2014 Search PubMed.
- F. E. Valera, M. Quaranta, A. Moran, J. Blacker, A. Armstrong, J. T. Cabral and D. G. Blackmond, Angew. Chem., Int. Ed., 2010, 49, 2478–2485 CrossRef CAS.
- M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef CAS.
- K. Faber, W.-D. Fessner and N. J. Turner, Adv. Synth. Catal., 2019, 361, 2373–2376 CrossRef CAS.
- J. M. Woodley, Appl. Microbiol. Biotechnol., 2019, 103, 4733–4739 CrossRef CAS.
- U. T. Bornscheuer, B. Hauer, K. E. Jaeger and U. Schwaneberg, Angew. Chem., Int. Ed., 2019, 58, 36–40 CrossRef CAS.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202006648.
- J. P. Adams, M. J. B. Brown, A. Diaz-Rodriguez, R. C. Lloyd and G. D. Roiban, Adv. Synth. Catal., 2019, 361, 2421–2432 CAS.
- U. Kragl, Chimia, 2020, 74, 378–381 CrossRef CAS.
- A. Petrenz, P. D. D. María, A. Ramanathan, U. Hanefeld, M. B. Ansorge-Schumacher and S. Kara, J. Mol. Catal. B: Enzym., 2015, 114, 42–49 CrossRef CAS.
- R. Wohlgemuth, I. Plazl, P. Žnidaršič-Plazl, K. V. Gernaey and J. M. Woodley, Trends Biotechnol., 2015, 33, 302–314 CrossRef CAS.
- V. Hessel, D. Kralisch, N. Kockmann, T. Noël and Q. Wang, ChemSusChem, 2013, 6, 746–789 CrossRef CAS.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS.
- R. M. Lindeque and J. M. Woodley, Catalysts, 2019, 9, 262 CrossRef CAS.
- L. Tamborini, P. Fernandes, F. Paradisi and F. Molinari, Trends Biotechnol., 2018, 36, 73–88 CrossRef CAS.
- J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891–5918 RSC.
- J. M. Woodley, React. Chem. Eng., 2020, 5, 632–640 RSC.
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2018, 23, 9–18 CrossRef.
- L. Hajba and A. Guttman, J. Flow Chem., 2016, 6, 8–12 CrossRef CAS.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2015, 20, 2–25 CrossRef.
- S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC.
- X. Y. Mak, P. Laurino and P. H. Seeberger, Beilstein J. Org. Chem., 2009, 5, 19 Search PubMed.
- J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583–4592 RSC.
- S. V. Ley, Chem. Rec., 2012, 12, 378–390 CrossRef CAS.
- K. F. Jensen, AIChE J., 2017, 63, 858–869 CrossRef CAS.
- P. Gruber, M. P. C. Marques, B. O'Sullivan, F. Baganz, R. Wohlgemuth and N. Szita, Biotechnol. J., 2017, 12, 1700030 CrossRef.
- J. M. Bolivar and B. Nidetzky, Green Process. Synth., 2013, 2, 541–559 CAS.
- T. Yu, Z. Ding, W. Nie, J. Jiao, H. Zhang, Q. Zhang, C. Xue, X. Duan, Y. M. A. Yamada and P. Li, Chem. – Eur. J., 2020, 26, 5729–5747 CrossRef CAS.
- D. Cambie, C. Bottecchia, N. J. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef CAS.
- M. Power, E. Alcock and G. P. McGlacken, Org. Process Res. Dev., 2020, 24, 1814–1838 CrossRef CAS.
- Y. Zhu, Q. Chen, L. Shao, Y. Jia and X. Zhang, React. Chem. Eng., 2020, 5, 9–32 RSC.
- A. C. Fernandes, K. V. Gernaey and U. Kruhne, Biotechnol. Adv., 2018, 36, 1341–1366 CrossRef CAS.
- A. Šalić and B. Zelić, Food Technol. Biotechnol., 2018, 56, 464–479 Search PubMed.
- P. Žnidaršič-Plazl, Biotechnol. J., 2019, 14, e1800580 CrossRef.
- A. C. Fernandes, B. Petersen, L. Møller, K. V. Gernaey and U. Krühne, New Biotechnol., 2018, 47, 39–49 CrossRef CAS.
- N. Miložič, G. Stojkovič, A. Vogel, D. Bouwes and P. Žnidaršič-Plazl, New Biotechnol., 2018, 47, 18–24 CrossRef.
- D. Grajales, J. C. Mateos, D. Padro, P. Ramos-Cabrer and F. López-Gallego, New Biotechnol., 2018, 47, 25–30 CrossRef CAS.
- G. Kulsharova, N. Dimov, M. P. C. Marques, N. Szita and F. Baganz, New Biotechnol., 2018, 47, 31–38 CrossRef CAS.
- A. Šalić, A. J. Tušekb, A. Sander and B. Zelić, New Biotechnol., 2018, 47, 80–88 CrossRef.
- L. Vobecká, A. Romanov, Z. Slouka, P. Hasal and M. Přibyl, New Biotechnol., 2018, 47, 73–79 CrossRef.
- D. L. Hughes, Org. Process Res. Dev., 2020, 24, 1850–1860 CrossRef CAS.
- S. L. Lee, T. F. O'Connor, X. Yang, C. N. Cruz, S. Chatterjee, R. D. Madurawe, C. M. V. Moore, L. X. Yu and J. Woodcock, J. Pharm. Innov., 2015, 10, 191–199 CrossRef.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802–1813 CrossRef CAS.
- R. Gerardy, D. P. Debecker, J. Estager, P. Luis and J. M. Monbaliu, Chem. Rev., 2020, 120, 7219–7347 CrossRef CAS.
- N. Zaquen, M. Rubens, N. Corrigan, J. T. Xu, P. B. Zetterlund, C. Boyer and T. Junkers, Prog. Polym. Sci., 2020, 107, 101256 CrossRef CAS.
- L. Gardossi, P. B. Poulsen, A. Ballesteros, K. Hult, V. K. Svedas, D. Vasic-Racki, G. Carrea, A. Magnusson, A. Schmid, R. Wohlgemuth and P. J. Halling, Trends Biotechnol., 2010, 28, 171–180 CrossRef CAS.
- D. M. Roberge, L. Ducry, N. Bieler, P. Cretton and B. Zimmermann, Chem. Eng. Technol., 2005, 28, 318–323 CrossRef CAS.
- M. van Schie, T. Pedroso de Almeida, G. Laudadio, F. Tieves, E. Fernandez-Fueyo, T. Noël, I. Arends and F. Hollmann, Beilstein J. Org. Chem., 2018, 14, 697–703 CrossRef CAS.
- L. Vaccaro, Sustainable Flow Chemistry : Methods and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017 Search PubMed.
- C. J. Mallia and I. R. Baxendale, Org. Process Res. Dev., 2015, 20, 327–360 CrossRef.
- M. Movsisyan, E. I. Delbeke, J. K. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, ACS Cent. Sci., 2016, 2, 131–138 CrossRef CAS.
- T. Wirth, Microreactors in Organic Chemistry and Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013 Search PubMed.
- K. S. Elvira, X. Casadevall i Solvas, R. C. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef CAS.
- S. K. R. Cherlo, K. Sreenath and S. Pushpavanam, Ind. Eng. Chem. Res., 2009, 48, 8678–8684 CrossRef CAS.
- X. Yao, Y. Zhang, L. Du, J. Liu and J. Yao, Renewable Sustainable Energy Rev., 2015, 47, 519–539 CrossRef CAS.
- R. C. Wheeler, O. Benali, M. Deal, E. Farrant, S. J. F. MacDonald and B. H. Warrington, Org. Process Res. Dev., 2007, 11, 704–710 CrossRef CAS.
- K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406–446 CrossRef.
- E. Laurenti and A. dos Santos Vianna Jr, Biocatalysis, 2015, 1, 148–165 Search PubMed.
- E. Gkantzou, M. Patila and H. Stamatis, Catalysts, 2018, 8, 282 CrossRef.
- K. Geyer, J. D. Codee and P. H. Seeberger, Chemistry, 2006, 12, 8434–8442 CrossRef CAS.
- R. H. Ringborg, A. Toftgaard Pedersen and J. M. Woodley, ChemCatChem, 2017, 9, 3285–3288 CrossRef CAS.
- B. Tomaszewski, A. Schmid and K. Buehler, Org. Process Res. Dev., 2014, 18, 1516–1526 CrossRef CAS.
- C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC.
- O. Levenspiel, Chemical reaction engineering, John Wiley & Sons, New York, USA, 3rd edn, 1999 Search PubMed.
- O. Reynolds, Philos. Trans. R. Soc. London, 1883, 174, 935–982, DOI:10.1098/rstl.1883.0029.
- R. Yuryev, S. Strompen and A. Liese, Beilstein J. Org. Chem., 2011, 7, 1449–1467 CrossRef CAS.
- A. Liese, C. Wandrey and K. Seelbach, Industrial biotransformations, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2nd edn, 2006 Search PubMed.
- K.-E. Jaeger, A. Liese and C. Syldatk, Einführung in die Enzymtechnologie, Springer Spektrum, Berlin, Germany, 2018 Search PubMed.
- T. A. Rogers and A. S. Bommarius, Chem. Eng. Sci., 2010, 65, 2118–2124 CrossRef CAS.
- J. Jovanović, E. V. Rebrov, T. A. Nijhuis, M. T. Kreutzer, V. Hessel and J. C. Schouten, Ind. Eng. Chem. Res., 2011, 51, 1015–1026 CrossRef.
- B. Sun, H. Zhu, Y. Jin, K. Qiao, W. Xu and J. Jiang, Chem. Eng. Technol., 2019, 42, 252–256 CrossRef CAS.
- J. Yue, Catal. Today, 2018, 308, 3–19 CrossRef CAS.
- T. Hardwick and N. Ahmed, RSC Adv., 2018, 8, 22233–22249 RSC.
- P. Watts and C. Wiles, Chem. Commun., 2007, 443–467 RSC.
- P. Žnidaršič-Plazl, J. Flow Chem., 2017, 7, 111–117 CrossRef.
- I. I. Junior, M. C. Flores, F. K. Sutili, S. G. F. Leite, L. S. M. de Miranda, I. C. R. Leal and R. O. M. A. de Souza, Org. Process Res. Dev., 2011, 16, 1098–1101 CrossRef.
- J. M. Bolivar and B. Nidetzky, Chim. Oggi, 2013, 31, 50–54 CAS.
- K. Robertson, Chem. Cent. J., 2017, 11, 4 CrossRef CAS.
- F. Huynh, M. Tailby, A. Finniear, K. Stephens, R. K. Allemann and T. Wirth, Angew. Chem., Int. Ed., 2020, 59, 16490–16495 CrossRef CAS.
- S. Kara, J. H. Schrittwieser, F. Hollmann and M. B. Ansorge-Schumacher, Appl. Microbiol. Biotechnol., 2014, 98, 1517–1529 CrossRef CAS.
- W. Hummel and H. Groger, J. Biotechnol., 2014, 191, 22–31 CrossRef CAS.
- S. Kara, J. H. Schrittwieser and F. Hollmann, in Synthetic Methods for Biologically Active Molecules, ed. E. Brenna, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013, pp. 209–238 Search PubMed.
- B. Baumer, T. Classen, M. Pohl and J. Pietruszka, Adv. Synth. Catal., 2020, 362, 2894–2901 CrossRef CAS.
- J. Döbber, M. Pohl, S. V. Ley and B. Musio, React. Chem. Eng., 2018, 3, 8–12 RSC.
- J. Döbber, T. Gerlach, H. Offermann, D. Rother and M. Pohl, Green Chem., 2018, 20, 544–552 RSC.
- C. J. Hartley, C. C. Williams, J. A. Scoble, Q. I. Churches, A. North, N. G. French, T. Nebl, G. Coia, A. C. Warden, G. Simpson, A. R. Frazer, C. N. Jensen, N. J. Turner and C. Scott, Nat. Catal., 2019, 2, 1006–1015 CrossRef CAS.
- M. L. Contente and F. Molinari, Nat. Catal., 2019, 2, 951–952 CrossRef CAS.
- T. Peschke, P. Bitterwolf, S. Hansen, J. Gasmi, K. Rabe and C. Niemeyer, Catalysts, 2019, 9, 164 CrossRef.
- T. Menegatti and P. Žnidaršič-Plazl, Micromachines, 2019, 10, 867 CrossRef.
- R. Semproli, G. Vaccaro, E. E. Ferrandi, M. Vanoni, T. Bavaro, G. Marrubini, F. Annunziata, P. Conti, G. Speranza, D. Monti, L. Tamborini and D. Ubiali, ChemCatChem, 2020, 12, 1359–1367 CrossRef CAS.
- A. Abi, D. Hartig, K. Vorländer, A. Wang, S. Scholl and H.-J. Jördening, Eng. Life Sci., 2019, 19, 4–12 CrossRef CAS.
- N. Adebar, J. E. Choi, L. Schober, R. Miyake, T. Iura, H. Kawabata and H. Gröger, ChemCatChem, 2019, 11, 5788–5793 CrossRef CAS.
- N. Adebar and H. Gröger, Bioengineering, 2019, 6, 99 CrossRef CAS.
- J. Coloma, Y. Guiavarc'h, P.-L. Hagedoorn and U. Hanefeld, Catal. Sci. Technol., 2020, 10, 3613–3621 RSC.
- C.-H. Ho, J. Yi and X. Wang, ACS Sustainable Chem. Eng., 2018, 7, 1038–1051 CrossRef.
- W. R. F. Goundry, K. Dai, M. Gonzalez, D. Legg, A. O'Kearney-McMullan, J. Morrison, A. Stark, P. Siedlecki, P. Tomlin and J. Yang, Org. Process Res. Dev., 2019, 23, 1333–1342 CrossRef CAS.
- F. Annunziata, M. Letizia Contente, D. Betti, C. Pinna, F. Molinari, L. Tamborini and A. Pinto, Catalysts, 2020, 10, 939 CrossRef CAS.
- W. Huang, N. Zhu, Y. Liu, J. Wang, J. Zhong, Q. Sun, T. Sun, X. Hu, Z. Fang and K. Guo, Chem. Eng. J., 2019, 356, 592–597 CrossRef CAS.
- F. Strniša, M. Bajić, P. Panjan, I. Plazl, A. M. Sesay and P. Žnidaršič-Plazl, Chem. Eng. J., 2018, 350, 541–550 CrossRef.
- D. Semenova, A. C. Fernandes, J. M. Bolivar, I. P. Rosinha Grundtvig, B. Vadot, S. Galvanin, T. Mayr, B. Nidetzky, A. Zubov and K. V. Gernaey, New Biotechnol., 2020, 56, 27–37 CrossRef CAS.
- L. Vobecká, L. Tichá, A. Atanasova, Z. Slouka, P. Hasal and M. Přibyl, Chem. Eng. J., 2020, 396, 125236 CrossRef.
- J. M. Bolivar, D. Valikhani and B. Nidetzky, Biotechnol. J., 2019, 14, e1800244 CrossRef.
- J. M. Bolivar, A. Mannsberger, M. S. Thomsen, G. Tekautz and B. Nidetzky, Biotechnol. Bioeng., 2019, 116, 503–514 CrossRef CAS.
- L. Ruzic, J. M. Bolivar and B. Nidetzky, Biotechnol. Bioeng., 2020, 117, 1597–1602 CrossRef CAS.
- W. Böhmer, A. Volkov, K. Engelmark Cassimjee and F. G. Mutti, Adv. Synth. Catal., 2020, 362, 1858–1867 CrossRef.
- B. Poznansky, L. A. Thompson, S. A. Warren, H. A. Reeve and K. A. Vincent, Org. Process Res. Dev., 2020, 24, 2281–2287 CrossRef CAS.
- J. L. Santana, J. M. Oliveira, J. S. Nascimento, S. Mattedi, L. C. Krause, L. S. Freitas, E. B. Cavalcanti, M. M. Pereira, A. S. Lima and C. M. F. Soares, Biotechnol. Appl. Biochem., 2020, 67, 404–413 CAS.
- F. Tentori, E. Brenna, M. Crotti, G. Pedrocchi-Fantoni, M. C. Ghezzi and D. Tessaro, Catalysts, 2020, 10, 102 CrossRef CAS.
- G. Hornyanszky, F. Nagy, K. Szabo and P. Bugovics, Period. Polytech., Chem. Eng., 2019, 63, 414–424 Search PubMed.
- G. Hornyánszky, C. Gaudreault, M. Gosselin, P. Kovács, Z. Boros, S. Suba and M. Oláh, Period. Polytech., Chem. Eng., 2018, 62, 519–532 Search PubMed.
- Q. Peng, B. Zang, W. Zhao, D. Li, J. Ren, F. Ji and L. Jia, Catal. Sci. Technol., 2020, 10, 484–492 RSC.
- S. C. Cosgrove, A. P. Mattey, M. Riese, M. R. Chapman, W. R. Birmingham, A. J. Blacker, N. Kapur, N. J. Turner and S. L. Flitsch, ACS Catal., 2019, 9, 11658–11662 CrossRef CAS.
- V. De Vitis, F. Dall'Oglio, F. Tentori, M. Contente, D. Romano, E. Brenna, L. Tamborini and F. Molinari, Catalysts, 2019, 9, 208 CrossRef.
- L. van den Biggelaar, P. Soumillion and D. P. Debecker, RSC Adv., 2019, 9, 18538–18546 RSC.
- Z. Molnár, E. Farkas, Á. Lakó, B. Erdélyi, W. Kroutil, B. G. Vértessy, C. Paizs and L. Poppe, Catalysts, 2019, 9, 438 CrossRef.
- M. L. Contente and F. Paradisi, Nat. Catal., 2018, 1, 452–459 CrossRef CAS.
- T. Peschke, P. Bitterwolf, S. Gallus, Y. Hu, C. Oelschlaeger, N. Willenbacher, K. S. Rabe and C. M. Niemeyer, Angew. Chem., Int. Ed., 2018, 57, 17028–17032 CrossRef CAS.
- P. Bitterwolf, S. Gallus, T. Peschke, E. Mittmann, C. Oelschlaeger, N. Willenbacher, K. S. Rabe and C. M. Niemeyer, Chem. Sci., 2019, 10, 9752–9757 RSC.
- M. A. do Nascimento, L. E. Gotardo, R. A. C. Leão, A. M. de Castro, R. O. M. A. de Souza and I. Itabaiana, ACS Omega, 2019, 4, 860–869 CrossRef CAS.
- M. Alotaibi, J. C. Manayil, G. M. Greenway, S. J. Haswell, S. M. Kelly, A. F. Lee, K. Wilson and G. Kyriakou, React. Chem. Eng., 2018, 3, 68–74 RSC.
- A. I. Benítez-Mateos, M. L. Contente, S. Velasco-Lozano, F. Paradisi and F. López-Gallego, ACS Sustainable Chem. Eng., 2018, 6, 13151–13159 CrossRef.
- P. Gruber, F. Carvalho, M. P. C. Marques, B. O'Sullivan, F. Subrizi, D. Dobrijevic, J. Ward, H. C. Hailes, P. Fernandes, R. Wohlgemuth, F. Baganz and N. Szita, Biotechnol. Bioeng., 2018, 115, 586–596 CrossRef CAS.
- M. R. Chapman, S. C. Cosgrove, N. J. Turner, N. Kapur and A. J. Blacker, Angew. Chem., Int. Ed., 2018, 57, 10535–10539 CrossRef CAS.
- J. Mangas-Sanchez, M. Sharma, S. C. Cosgrove, J. I. Ramsden, J. R. Marshall, T. W. Thorpe, R. B. Palmer, G. Grogan and N. J. Turner, Chem. Sci., 2020, 11, 5052–5057 RSC.
- A. W. H. Dawood, J. Bassut, R. de Souza and U. T. Bornscheuer, Chem. – Eur. J., 2018, 24, 16009–16013 CrossRef CAS.
- M. P. van der Helm, P. Bracco, H. Busch, K. Szymańska, A. B. Jarzębski and U. Hanefeld, Catal. Sci. Technol., 2019, 9, 1189–1200 RSC.
- E. Magner, Chem. Soc. Rev., 2013, 42, 6213–6222 RSC.
- A. Liese and L. Hilterhaus, Chem. Soc. Rev., 2013, 42, 6236–6249 RSC.
- S. Cantone, V. Ferrario, L. Corici, C. Ebert, D. Fattor, P. Spizzo and L. Gardossi, Chem. Soc. Rev., 2013, 42, 6262–6276 RSC.
- H. J. Federsel, J. Pesti and M. P. Thompson, in Catalyst Immobilization, ed. M. Benaglia and A. Puglisi, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2020, pp. 437–463 Search PubMed.
- K. Xu, X. Chen, R. Zheng and Y. Zheng, Front. Bioeng. Biotechnol., 2020, 8, 660 CrossRef.
- J. M. Bolivar and F. López-Gallego, Curr. Opin. Green Sustain. Chem., 2020, 25, 100349 CrossRef.
- F. Rinaldi, J. Fernandez-Lucas, D. de la Fuente, C. Zheng, T. Bavaro, B. Peters, G. Massolini, F. Annunziata, P. Conti, I. de la Mata, M. Terreni and E. Calleri, Bioresour. Technol., 2020, 307, 123258 CrossRef CAS.
- C. Hou, N. Gheczy, D. Messmer, K. Szymanska, J. Adamcik, R. Mezzenga, A. B. Jarzebski and P. Walde, ACS Omega, 2019, 4, 7795–7806 CrossRef CAS.
- A. Vogel and O. May, Industrial enzyme applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2019 Search PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- F. Golombek and K. Castiglione, Biotechnol. J., 2020, e1900561, DOI:10.1002/biot.201900561.
- S. Bormann, B. O. Burek, R. Ulber and D. Holtmann, Mol. Catal., 2020, 492, 110999 CrossRef CAS.
- Z. Boros, P. Falus, M. Márkus, D. Weiser, M. Oláh, G. Hornyánszky, J. Nagy and L. Poppe, J. Mol. Catal. B: Enzym., 2013, 85-86, 119–125 CrossRef CAS.
- L. Cao, L. V. Langen and R. A. Sheldon, Curr. Opin. Biotechnol., 2003, 14, 387–394 CrossRef CAS.
- Y. Wang, J. Tian, Y. Xiao, Y. Wang, H. Sun, Y. Chang and H. Luo, Biotechnol. Lett., 2019, 41, 987–994 CrossRef CAS.
- P. Bitterwolf, F. Ott, K. S. Rabe and C. M. Niemeyer, Micromachines, 2019, 10, 783 CrossRef.
- E. Mittmann, S. Gallus, P. Bitterwolf, C. Oelschlaeger, N. Willenbacher, C. M. Niemeyer and K. S. Rabe, Micromachines, 2019, 10, 795 CrossRef.
- F. Nagy, K. Szabó, P. Bugovics and G. Hornyánszky, Period. Polytech., Chem. Eng., 2019, 63, 414–424 CAS.
- E. Zymanczyk-Duda, N. Dunal, M. Brzezinska-Rodak, A. Osiewala, T. K. Olszewski, M. Klimek-Ochab and M. Serafin-Lewanczuk, Bioorg. Chem., 2019, 93, 102751 CrossRef CAS.
- S. P. de Souza, R. A. D. de Almeida, G. G. Garcia, R. A. C. Leão, J. Bassut, R. O. M. A. de Souza and I. Itabaiana, J. Chem. Technol. Biotechnol., 2018, 93, 105–111 CrossRef CAS.
- B. Szokol, G. Hornyanszky and J. Nagy, Stud. Univ. Babes-Bolyai, Chem., 2019, 64, 69–78 CAS.
- E. Abaházi, P. Sátorhelyi, B. Erdélyi, B. G. Vértessy, H. Land, C. Paizs, P. Berglund and L. Poppe, Biochem. Eng. J., 2018, 132, 270–278 CrossRef.
- E. Krisch, D. Balogh-Weiser, J. Klimko, B. Gyarmati, K. Laszlo, L. Poppe and A. Szilagyi, eXPRESS Polym. Lett., 2019, 13, 512–523 CrossRef CAS.
- M. S. Barbosa, A. J. Santos, N. B. Carvalho, R. T. Figueiredo, M. M. Pereira, Á. S. Lima, M. G. Freire, R. Y. Cabrera-Padilla and C. M. F. Soares, ACS Sustainable Chem. Eng., 2019, 7, 15648–15659 CrossRef CAS.
- H. Suo, L. Xu, Y. Xue, X. Qiu, H. Huang and Y. Hu, Carbohydr. Polym., 2020, 234, 115914 CrossRef CAS.
- J. Claus, F. O. Sommer and U. Kragl, Solid State Ionics, 2018, 314, 119–128 CrossRef CAS.
- L. E. Meyer, J. von Langermann and U. Kragl, Biophys. Rev., 2018, 10, 901–910 CrossRef CAS.
- L. E. Meyer, A. Gummesson, U. Kragl and J. von Langermann, Biotechnol. J., 2019, 14, e1900215 CrossRef.
- M. Deetlefs and K. R. Seddon, Green Chem., 2010, 12, 17–30 RSC.
- S. HajKacem, S. Galai, F. J. Hernandez Fernandez, A. Perez de Los Rios, I. Smaali and J. Quesada Medina, Appl. Biochem. Biotechnol., 2020, 190, 1–17 CrossRef CAS.
- S. H. Schöfer, N. Kaftzik, P. Wasserscheid and U. Kragl, Chem. Commun., 2001, 425–426 RSC.
- R. Madeira Lau, F. van Rantwijk, K. R. Seddon and R. A. Sheldon, Org. Lett., 2000, 2, 4189–4191 CrossRef CAS.
- M. Erbeldinger, A. J. Mesiano and A. J. Russell, Biotechnol. Prog., 2000, 16, 1129–1131 CrossRef CAS.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS.
- K. R. Seddon, J. Chem. Technol. Biotechnol., 1997, 68, 351–356 CrossRef CAS.
- U. Kragl, M. Eckstein and N. Kaftzik, Curr. Opin. Biotechnol., 2002, 13, 565–571 CrossRef CAS.
- H. Suo, L. Xu, C. Xu, X. Qiu, H. Huang and Y. Hu, J. Colloid Interface Sci., 2019, 553, 494–502 CrossRef CAS.
- A. I. Benitez-Mateos, E. San Sebastian, N. Rios-Lombardia, F. Moris, J. Gonzalez-Sabin and F. Lopez-Gallego, Chem. – Eur. J., 2017, 23, 16843–16852 CrossRef CAS.
- S. Velasco-Lozano, A. I. Benitez-Mateos and F. Lopez-Gallego, Angew. Chem., Int. Ed., 2017, 56, 771–775 CrossRef CAS.
- M. Romero-Fernandez and F. Paradisi, Curr. Opin. Chem. Biol., 2020, 55, 1–8 CrossRef CAS.
- M. T. De Martino, F. Tonin, N. A. Yewdall, M. Abdelghani, D. S. Williams, U. Hanefeld, F. P. J. T. Rutjes, L. K. E. A. Abdelmohsen and J. C. M. van Hest, Chem. Sci., 2020, 11, 2765–2769 RSC.
- A. R. Aguillón, M. N. Avelar, L. E. Gotardo, S. P. de Souza, R. A. C. Leão, I. Itabaiana, L. S. M. Miranda and R. O. M. A. de Souza, Mol. Catal., 2019, 467, 128–134 CrossRef.
- J. M. Carceller, J. P. M. Galan, R. Monti, J. C. Bassan, M. Filice, J. H. Yu, M. J. Climent, S. Iborra and A. Corma, ChemCatChem, 2020, 12, 4502–4511 CrossRef CAS.
- J. M. Carceller, M. Mifsud, M. J. Climent, S. Iborra and A. Corma, Green Chem., 2020, 22, 2767–2777 RSC.
- X. Wang, M. Jiang, Z. Zhou, J. Gou and D. Hui, Composites, Part B, 2017, 110, 442–458 CrossRef CAS.
- T. D. Ngo, A. Kashani, G. Imbalzano, K. T. Q. Nguyen and D. Hui, Composites, Part B, 2018, 143, 172–196 CrossRef CAS.
- J. Kietzmann, L. Pitt and P. Berthon, Bus. Horiz., 2015, 58, 209–215 CrossRef.
- I. Karakurt and L. Lin, Curr. Opin. Chem. Eng., 2020, 28, 134–143 CrossRef.
- B. C. Gross, J. L. Erkal, S. Y. Lockwood, C. Chen and D. M. Spence, Anal. Chem., 2014, 86, 3240–3253 CrossRef CAS.
- S. El-Sayegh, L. Romdhane and S. Manjikian, Arch. Civ. Mech. Eng., 2020, 20, 34 CrossRef.
- D. Chimene, K. K. Lennox, R. R. Kaunas and A. K. Gaharwar, Ann. Biomed. Eng., 2016, 44, 2090–2102 CrossRef.
- A. K. Au, W. Huynh, L. F. Horowitz and A. Folch, Angew. Chem., Int. Ed., 2016, 55, 3862–3881 CrossRef CAS.
- I. Lignos, H. Ow, J. P. Lopez, D. McCollum, H. Zhang, J. Imbrogno, Y. Shen, S. Chang, W. Wang and K. F. Jensen, ACS Appl. Mater. Interfaces, 2020, 12, 6699–6706 CrossRef CAS.
- Y. Qiao, Y. Yao, Y. Liu, C. Chen, X. Wang, G. Zhong, D. Liu and L. Hu, Small, 2020, 16, e2000509 CrossRef.
- F. D. S. Belgrano, O. Diegel, N. Pereira, Jr. and R. Hatti-Kaul, Bioresour. Technol., 2018, 249, 777–782 CrossRef CAS.
- M. Peng, E. Mittmann, L. Wenger, J. Hubbuch, M. K. M. Engqvist, C. M. Niemeyer and K. S. Rabe, Chem. – Eur. J., 2019, 25, 15998–16001 CrossRef CAS.
- N. Bhattacharjee, A. Urrios, S. Kang and A. Folch, Lab Chip, 2016, 16, 1720–1742 RSC.
- S. Danaci, L. Protasova, V. Middelkoop, N. Ray, M. Jouve, A. Bengaouer and P. Marty, React. Chem. Eng., 2019, 4, 1318–1330 RSC.
- B. Schmieg, A. Schimek and M. Franzreb, Eng. Life Sci., 2018, 18, 659–667 CrossRef CAS.
- X. Shen, M. Yang, C. Cui and H. Cao, Colloids Surf., A, 2019, 568, 411–418 CrossRef CAS.
- J. Ye, T. Chu, J. Chu, B. Gao and B. He, ACS Sustainable Chem. Eng., 2019, 7, 18048–18054 CrossRef CAS.
- A. Steier, B. Schmieg, Y. Irtel von Brenndorff, M. Meier, H. Nirschl, M. Franzreb and J. Lahann, Macromol. Biosci., 2020, e2000154, DOI:10.1002/mabi.202000154.
- A. M. Lopez Marzo, C. C. Mayorga-Martinez and M. Pumera, Biosens. Bioelectron., 2020, 151, 111980 CrossRef CAS.
- N. Büscher, G. V. Sayoga, K. Rübsam, F. Jakob, U. Schwaneberg, S. Kara and A. Liese, Org. Process Res. Dev., 2019, 23, 1852–1859 CrossRef.
- A. Liese, M. Schlüter, D. Herzog, M. Maiwald, A. Dawood, G. Sayogo, J. Kracht, C. Spille and N. Büscher, Counter-currently operated reactive extractor with additively manufactured enzyme carrier structure, 2020, DOI:10.26434/chemrxiv.12152679.v1.
- E. Fornells, E. Murray, S. Waheed, A. Morrin, D. Diamond, B. Paull and M. Breadmore, Anal. Chim. Acta, 2020, 1098, 94–101 CrossRef.
- C. Li, B. Ding, L. Zhang, K. Song and S. Tao, J. Mater. Chem. C, 2019, 7, 9167–9174 RSC.
- M. J. Harding, S. Brady, H. O'Connor, R. Lopez-Rodriguez, M. D. Edwards, S. Tracy, D. Dowling, G. Gibson, K. P. Girard and S. Ferguson, React. Chem. Eng., 2020, 5, 728–735 RSC.
- S. Bettermann, F. Kandelhard, H.-U. Moritz and W. Pauer, Chem. Eng. Res. Des., 2019, 152, 71–84 CrossRef CAS.
- G. Scotti, S. M. E. Nilsson, V. P. Matilainen, M. Haapala, G. Boije Af Gennas, J. Yli-Kauhaluoma, A. Salminen and T. Kotiaho, Heliyon, 2019, 5, e02002 CrossRef.
- P. Liu, Y. Guo, Y. Wu, J. Chen and Y. Yang, Crystals, 2020, 10, 257 CrossRef CAS.
- E. Peris, O. Okafor, E. Kulcinskaja, R. Goodridge, S. V. Luis, E. Garcia-Verdugo, E. O'Reilly and V. Sans, Green Chem., 2017, 19, 5345–5349 RSC.
- L. Huang, F. S. Aalbers, W. Tang, R. Röllig, M. W. Fraaije and S. Kara, ChemBioChem, 2019, 20, 1653–1658 CrossRef CAS.
- J. Engel, K. S. Mthethwa, D. J. Opperman and S. Kara, Mol. Catal., 2019, 468, 44–51 CrossRef CAS.
- C. Scherkus, S. Schmidt, U. T. Bornscheuer, H. Gröger, S. Kara and A. Liese, Biotechnol. Bioeng., 2017, 114, 1215–1221 CrossRef CAS.
- A. Bornadel, R. Hatti-Kaul, F. Hollmann and S. Kara, Tetrahedron, 2016, 72, 7222–7228 CrossRef CAS.
- A. Bornadel, R. Hatti-Kaul, F. Hollmann and S. Kara, ChemCatChem, 2015, 7, 2442–2445 CrossRef CAS.
- L. J. Durndell, M. A. Isaacs, C. E. Li, C. M. A. Parlett, K. Wilson and A. F. Lee, ACS Catal., 2019, 9, 5345–5352 CrossRef CAS.
- C. Altuğ, M. J. Muñoz-Batista, D. Rodríguez-Padrón, A. M. Balu, A. A. Romero and R. Luque, Green Chem., 2019, 21, 300–306 RSC.
- A. Al-Shameri, M. C. Petrich, K. Junge Puring, U. P. Apfel, B. M. Nestl and L. Lauterbach, Angew. Chem., Int. Ed., 2020, 59, 10929–10933 CrossRef CAS.
- W. Finnigan, J. Citoler, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2020, 24, 1969–1977 CrossRef CAS.
- C. Minnich, S. Hardy and S. Krämer, Chem. Ing. Tech., 2016, 88, 694–697 CrossRef CAS.
- A. Gioiello, A. Piccinno, A. M. Lozza and B. Cerra, J. Med. Chem., 2020, 63, 6624–6647 CrossRef.
- C. Mateos, M. J. Nieves-Remacha and J. A. Rincón, React. Chem. Eng., 2019, 4, 1536–1544 RSC.
- M. Hosoya, S. Nishijima and N. Kurose, Org. Process Res. Dev., 2020, 24, 1095–1103 CrossRef CAS.
- K. S. Rao, F. St-Jean and A. Kumar, Org. Process Res. Dev., 2019, 23, 945–951 CrossRef CAS.
- S. D. Riegel and N. G. A. Bell, Magn. Reson. Chem., 2020 DOI:10.1002/mrc.5057.
- R. W. Morrison and M. Zhang, Magn. Reson. Chem., 2020 DOI:10.1002/mrc.5031.
- W. G. Lee, M. T. Zell, T. Ouchi and M. J. Milton, Magn. Reson. Chem., 2020 DOI:10.1002/mrc.5035.
- C. Claassen, K. Mack and D. Rother, ChemCatChem, 2020, 12, 1190–1199 CrossRef CAS.
- K. E. Anderssen and E. R. McCarney, Food Control, 2020, 112, 107053 CrossRef CAS.
- A. Soyler, D. Bouillaud, J. Farjon, P. Giraudeau and M. H. Oztop, LWT--Food Sci. Technol., 2020, 118, 108832 CrossRef CAS.
- M. D. T. Rahman, S. Wharry, M. Smyth, H. Manyar and T. S. Moody, Synlett, 2020, 31, 581–586 CrossRef CAS.
- J. Hirvonen and O. Ventä, IMETI 2009 - 2nd International Multi-Conference on Engineering and Technological Innovation, Proceedings, 2009, vol. 1.
- P. P. Plehiers, C. W. Coley, H. Gao, F. H. Vermeire, M. R. Dobbelaere, C. V. Stevens, K. M. Van Geem and W. H. Green, Front. Chem. Eng., 2020, 2, 5 CrossRef.
- N. Garg, J. M. Woodley, R. Gani and G. M. Kontogeorgis, Comput. Chem. Eng., 2019, 126, 499–519 CrossRef CAS.
- D. K. Babi, J. Holtbruegge, P. Lutze, A. Gorak, J. M. Woodley and R. Gani, Comput. Chem. Eng., 2015, 81, 218–244 CrossRef CAS.
- P. Lutze, R. Gani and J. M. Woodley, Chem. Eng. Process., 2010, 49, 547–558 CrossRef CAS.
- B. Somasundaram, K. Pleitt, E. Shave, K. Baker and L. H. L. Lua, Biotechnol. Bioeng., 2018, 115, 2893–2907 CrossRef CAS.
- F. A. Vicente, I. Plazl, S. P. M. Ventura and P. Žnidaršič-Plazl, Green Chem., 2020, 22, 4391–4410 RSC.
- H. Schmidt-Traub, M. Schulte and A. Seidel-Morgenstern, Preparative Chromatography, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2020 Search PubMed.
- C. Goussen, L. Goldstein, C. Breque, B. You, S. Boyer, D. Bataille and L. Burlot, J. Chromatogr., B, 2020, 1145, 122056 CrossRef CAS.
- L.-E. Meyer, K. Plasch, U. Kragl and J. von Langermann, Org. Process Res. Dev., 2018, 22, 963–970 CrossRef CAS.
- L. E. Meyer, H. Brundiek and J. von Langermann, Biotechnol. Prog., 2020, 36, e3024 CrossRef CAS.
- D. Hülsewede, E. Temmel, P. Kumm and J. V. Langermann, Crystals, 2020, 10, 345 CrossRef.
- D. Hülsewede, L. E. Meyer and J. von Langermann, Chem. – Eur. J., 2019, 25, 4871–4884 CrossRef.
- S. Kara, D. Spickermann, A. Weckbecker, C. Leggewie, I. W. C. E. Arends and F. Hollmann, ChemCatChem, 2014, 6, 973–976 CrossRef CAS.
- D. Spickermann, S. Kara, I. Barackov, F. Hollmann, U. Schwaneberg, P. Duenkelmann and C. Leggewie, J. Mol. Catal. B: Enzym., 2014, 103, 24–28 CrossRef CAS.
- L. Huang, P. Domínguez de María and S. Kara, Chim. Oggi, 2018, 36, 48–56 CAS.
- G. de Gonzalo, A. R. Alcantara and P. Dominguez de Maria, ChemSusChem, 2019, 12, 2083–2097 CrossRef CAS.
- P. Žnidaršič-Plazl, Chim. Oggi, 2014, 32, 54–60 Search PubMed.
- R. Z. Kazandjian, J. S. Dordick and A. M. Klibanov, Biotechnol. Bioeng., 1986, 28, 417–421 CrossRef CAS.
- P. J. Halling, Enzyme Microb. Technol., 1994, 16, 178–206 CrossRef CAS.
- P. Aldercreutz and B. Mattiasson, Biocatalysis, 1987, 1, 99–108 CrossRef CAS.
- F. Wang, S. He, C. Zhu, U. Rabausch, W. Streit and J. Wang, J. Chem. Technol. Biotechnol., 2018, 93, 2671–2680 CrossRef CAS.
- A. Grollmisch, U. Kragl and J. Großeheilmann, Synopen, 2018, 2, 192–199 CrossRef.
- R. Villa, E. Alvarez, R. Porcar, E. Garcia-Verdugo, S. V. Luis and P. Lozano, Green Chem., 2019, 21, 6527–6544 RSC.
- M. Patzold, S. Siebenhaller, S. Kara, A. Liese, C. Syldatk and D. Holtmann, Trends Biotechnol., 2019, 37, 943–959 CrossRef.
- Y. Ma, P. Li, Y. Li, S. J. Willot, W. Zhang, D. Ribitsch, Y. H. Choi, R. Verpoorte, T. Zhang, F. Hollmann and Y. Wang, ChemSusChem, 2019, 12, 1310–1315 CrossRef CAS.
- R. Hollenbach, K. Ochsenreither and C. Syldatk, Int. J. Mol. Sci., 2020, 21, 4342 CrossRef CAS.
- M. Hümmer, S. Kara, A. Liese, I. Huth, J. Schrader and D. Holtmann, Mol. Catal., 2018, 458, 67–72 CrossRef.
- N. Guajardo, C. R. Müller, R. Schrebler, C. Carlesi and P. Domínguez de María, ChemCatChem, 2016, 8, 1020–1027 CrossRef CAS.
- G. de Gonzalo, C. Martin and M. W. Fraaije, Catalysts, 2020, 10, 447 CrossRef CAS.
- S. Kara and J. von Langermann, BIOspektrum, 2020, 26, 215–217 CrossRef.
- L. Huang, J. P. Bittner, P. Dominguez de Maria, S. Jakobtorweihen and S. Kara, ChemBioChem, 2020, 21, 811–817 CrossRef CAS.
- P. Domínguez de María, N. Guajardo and S. Kara, in Deep Eutectic Solvents, ed. D. J. Ramón and G. Guillena, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2019, pp. 257–271 Search PubMed.
- B. Grabner, A. K. Schweiger, K. Gavric, R. Kourist and H. Gruber-Woelfler, React. Chem. Eng., 2020, 5, 263–269 RSC.
- N. Guajardo, R. A. Schrebler and P. Dominguez de Maria, Bioresour. Technol., 2019, 273, 320–325 CrossRef CAS.
- N. Guajardo and P. Domínguez de María, ChemCatChem, 2019, 11, 3128–3137 CrossRef CAS.
- N. Guajardo, P. Domínguez de María, K. Ahumada, R. A. Schrebler, R. Ramírez-Tagle, F. A. Crespo and C. Carlesi, ChemCatChem, 2017, 9, 1393–1396 CrossRef CAS.
- C. Stephenson, T. Yoon and D. W. C. MacMillan, Visible Light Photocatalysis in Organic Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA Weinheim, Germany, 2018 Search PubMed.
- S. Kochius, Y. Ni, S. Kara, S. Gargiulo, J. Schrader, D. Holtmann and F. Hollmann, ChemPlusChem, 2014, 79, 1554–1557 CrossRef CAS.
- F. Feyza Özgen, M. E. Runda, B. O. Burek, P. Wied, J. Z. Bloh, R. Kourist and S. Schmidt, Angew. Chem., Int. Ed., 2020, 59, 3982–3987 CrossRef.
- W. Zhang, B. O. Burek, E. Fernandez-Fueyo, M. Alcalde, J. Z. Bloh and F. Hollmann, Angew. Chem., Int. Ed., 2017, 56, 15451–15455 CrossRef CAS.
- D. Sorigue, B. Legeret, S. Cuine, S. Blangy, S. Moulin, E. Billon, P. Richaud, S. Brugiere, Y. Coute, D. Nurizzo, P. Muller, K. Brettel, D. Pignol, P. Arnoux, Y. Li-Beisson, G. Peltier and F. Beisson, Science, 2017, 357, 903–907 CrossRef CAS.
- L. Schmermund, V. Jurkaš, F. F. Özgen, G. D. Barone, H. C. Büchsenschütz, C. K. Winkler, S. Schmidt, R. Kourist and W. Kroutil, ACS Catal., 2019, 9, 4115–4144 CrossRef CAS.
- S. H. Lee, D. S. Choi, S. K. Kuk and C. B. Park, Angew. Chem., Int. Ed., 2018, 57, 7958–7985 CrossRef CAS.
- K. F. Biegasiewicz, S. J. Cooper, M. A. Emmanuel, D. C. Miller and T. K. Hyster, Nat. Chem., 2018, 10, 770–775 CrossRef CAS.
- X. Guo, Y. Okamoto, M. R. Schreier, T. R. Ward and O. S. Wenger, Chem. Sci., 2018, 9, 5052–5056 RSC.
- J. W. Hindley, Y. Elani, C. M. McGilvery, S. Ali, C. L. Bevan, R. V. Law and O. Ces, Nat. Commun., 2018, 9, 1093 CrossRef.
- G. T. Hofler, E. Fernandez-Fueyo, M. Pesic, S. H. Younes, E. G. Choi, Y. H. Kim, V. B. Urlacher, I. Arends and F. Hollmann, ChemBioChem, 2018, 19, 2344–2347 CrossRef.
- M. M. E. Huijbers, W. Zhang, F. Tonin and F. Hollmann, Angew. Chem., Int. Ed., 2018, 57, 13648–13651 CrossRef CAS.
- J. Kim, S. H. Lee, F. Tieves, D. S. Choi, F. Hollmann, C. E. Paul and C. B. Park, Angew. Chem., Int. Ed., 2018, 57, 13825–13828 CrossRef CAS.
- K. Lauder, A. Toscani, Y. Qi, J. Lim, S. J. Charnock, K. Korah and D. Castagnolo, Angew. Chem., Int. Ed., 2018, 57, 5803–5807 CrossRef CAS.
- C. J. Seel, A. Králík, M. Hacker, A. Frank, B. König and T. Gulder, ChemCatChem, 2018, 10, 3960–3963 CrossRef CAS.
- Q. Yang, F. Zhao, N. Zhang, M. Liu, H. Hu, J. Zhang and S. Zhou, Chem. Commun., 2018, 54, 14065–14068 RSC.
- W. Zhang, E. Fernandez-Fueyo, Y. Ni, M. van Schie, J. Gacs, R. Renirie, R. Wever, F. G. Mutti, D. Rother, M. Alcalde and F. Hollmann, Nat. Catal., 2018, 1, 55–62 CrossRef CAS.
- B. O. Burek, S. R. de Boer, F. Tieves, W. Zhang, M. van Schie, S. Bormann, M. Alcalde, D. Holtmann, F. Hollmann, D. W. Bahnemann and J. Z. Bloh, ChemCatChem, 2019, 11, 3093–3100 CrossRef CAS.
- X. Ding, C. L. Dong, Z. Guan and Y. H. He, Angew. Chem., Int. Ed., 2019, 58, 118–124 CrossRef CAS.
- A. Hoschek, B. Buhler and A. Schmid, Biotechnol. Bioeng., 2019, 116, 1887–1900 CrossRef CAS.
- M. Kato, M. Melkie, J. Li, B. Foley, H. T. Nguyen, L. Leti and L. Cheruzel, Arch. Biochem. Biophys., 2019, 672, 108077 CrossRef CAS.
- J. Kim, S. H. Lee, F. Tieves, C. E. Paul, F. Hollmann and C. B. Park, Sci. Adv., 2019, 5, eaax0501 CrossRef CAS.
- K. Kinastowska, J. Liu, J. M. Tobin, Y. Rakovich, F. Vilela, Z. Xu, W. Bartkowiak and M. Grzelczak, Appl. Catal., B, 2019, 243, 686–692 CrossRef CAS.
- T.-K. Le, J. H. Park, D. S. Choi, G.-Y. Lee, W. S. Choi, K. J. Jeong, C. B. Park and C.-H. Yun, Green Chem., 2019, 21, 515–525 RSC.
- Y. Ma, X. Zhang, W. Zhang, P. Li, Y. Li, F. Hollmann and Y. Wang, ChemPhotoChem, 2019, 4, 39–44 Search PubMed.
- B. A. Sandoval, S. I. Kurtoic, M. M. Chung, K. F. Biegasiewicz and T. K. Hyster, Angew. Chem., Int. Ed., 2019, 58, 8714–8718 CrossRef CAS.
- M. van Schie, C. E. Paul, I. Arends and F. Hollmann, Chem. Commun., 2019, 55, 1790–1792 RSC.
- S. J. Willot, E. Fernandez-Fueyo, F. Tieves, M. Pesic, M. Alcalde, I. Arends, C. B. Park and F. Hollmann, ACS Catal., 2019, 9, 890–894 CrossRef CAS.
- J. Xu, Y. Hu, J. Fan, M. Arkin, D. Li, Y. Peng, W. Xu, X. Lin and Q. Wu, Angew. Chem., Int. Ed., 2019, 58, 8474–8478 CrossRef CAS.
- J. Yoon, S. H. Lee, F. Tieves, M. Rauch, F. Hollmann and C. B. Park, ACS Sustainable Chem. Eng., 2019, 7, 5632–5637 CrossRef CAS.
- W. Zhang, E. F. Fueyo, F. Hollmann, L. L. Martin, M. Pesic, R. Wardenga, M. Hohne and S. Schmidt, Eur. J. Org. Chem., 2019, 2019, 80–84 CrossRef CAS.
- W. Zhang, M. Ma, M. M. E. Huijbers, G. A. Filonenko, E. A. Pidko, M. van Schie, S. de Boer, B. O. Burek, J. Z. Bloh, W. J. H. van Berkel, W. A. Smith and F. Hollmann, J. Am. Chem. Soc., 2019, 141, 3116–3120 CrossRef CAS.
- H. J. Cha, S. Y. Hwang, D. S. Lee, A. R. Kumar, Y. U. Kwon, M. Voss, E. Schuiten, U. T. Bornscheuer, F. Hollmann, D. K. Oh and J. B. Park, Angew. Chem., Int. Ed., 2020, 59, 7024–7028 CrossRef CAS.
- W. Zhang, J. H. Lee, S. H. H. Younes, F. Tonin, P. L. Hagedoorn, H. Pichler, Y. Baeg, J. B. Park, R. Kourist and F. Hollmann, Nat. Commun., 2020, 11, 2258 CAS.
- D. Zheng, Y. Zhang, X. Liu and J. Wang, Photosynth. Res., 2020, 143, 221–231 CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |