
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTackling elemental mercury removal from the wet-gas phase by enhancing the performance of redox-active copper-based adsorbents utilising an operando pre-heating system†
Syamzari
Rafeen
*a,
Rafin
Ramli
a and
Geetha
Srinivasan
 *ab
*ab
aPETRONAS Research Sdn. Bhd, Lot 3288 & 3289, Off Jalan Ayer Itam, Kawasan Institusi Bangi, 43000 Kajang, Selangor, Malaysia. E-mail: syamzari_rafeen@petronas.com.my; geetha.srinivasan@petronas.com
bQUILL, The Queen's University of Belfast, Belfast, BT9 5AG, UK
First published on 5th August 2020
Abstract
A simple and facile technology to capture the elemental mercury selectively from moisture-containing natural gas (wet-gas) streams of petrochemical industries is presented. We aimed at exploiting the established redox chemistry of copper(II) towards mercury(0), by utilising simple copper(II) chloride impregnated adsorbents. Tuning the copper coordination sphere with temperature as a control by taking advantage of moisture in the feed, has been successful in enhancing the mercury adsorption through a viable process design. Mercury removal was achieved through chemisorption when the CuCl2 (Cu(II)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Hg(II) mol ratio = 1.35:1) impregnated on solid supports, such as silica, γ-alumina and activated carbon via the wet incipient method and have been used as adsorbents. Supported CuCl2-based adsorbents were characterised using UV-visible, scanning electron microscopy, energy dispersive X-ray spectroscopy and powder X-ray diffraction, wherein copper complexation was not observed on freshly prepared adsorbents. An in-house experimental setup (wet-gas rig) was designed to perform mercury breakthrough experiments through mercury online monitoring, wherein an add-on moisture saturator was included to allow water vapour in the feed and mimic a wet-gas scenario. In the wet-gas testing rig, two modes of testing were implemented, i.e., with and without pre-heating the incoming gas feed in front of the adsorbent column. The gas pre-heating method introduced for the test under wet-gas conditions resulted in an enhancement of mercury adsorption performance as compared to the dry conditions for selected CuCl2-containing adsorbents. This enhancement of mercury removal was particularly predominant for CuCl2 supported on silica, in which the performance increased by four-fold, accounting for a slight pre-heating by 10 °C for the wet-gas in comparison to the test under dry-gas conditions. The redox centre on copper at the hydration sphere, was influenced particularly on the silica support, which consequently resulted in superior reactivity with elemental mercury in the vapour state. Moreover, the gas pre-heating avoided capillary condensation, thereby contributing to the enhancement on mercury adsorbent's life-time. The mercury capture on the adsorbent has been expected to be due to complexation on the silica support that led to a better fixation.
:Hg(II) mol ratio = 1.35:1) impregnated on solid supports, such as silica, γ-alumina and activated carbon via the wet incipient method and have been used as adsorbents. Supported CuCl2-based adsorbents were characterised using UV-visible, scanning electron microscopy, energy dispersive X-ray spectroscopy and powder X-ray diffraction, wherein copper complexation was not observed on freshly prepared adsorbents. An in-house experimental setup (wet-gas rig) was designed to perform mercury breakthrough experiments through mercury online monitoring, wherein an add-on moisture saturator was included to allow water vapour in the feed and mimic a wet-gas scenario. In the wet-gas testing rig, two modes of testing were implemented, i.e., with and without pre-heating the incoming gas feed in front of the adsorbent column. The gas pre-heating method introduced for the test under wet-gas conditions resulted in an enhancement of mercury adsorption performance as compared to the dry conditions for selected CuCl2-containing adsorbents. This enhancement of mercury removal was particularly predominant for CuCl2 supported on silica, in which the performance increased by four-fold, accounting for a slight pre-heating by 10 °C for the wet-gas in comparison to the test under dry-gas conditions. The redox centre on copper at the hydration sphere, was influenced particularly on the silica support, which consequently resulted in superior reactivity with elemental mercury in the vapour state. Moreover, the gas pre-heating avoided capillary condensation, thereby contributing to the enhancement on mercury adsorbent's life-time. The mercury capture on the adsorbent has been expected to be due to complexation on the silica support that led to a better fixation.
Introduction
Mercury occurs naturally but is a toxic contaminant in the petroleum stream that results in detrimental effects, causing equipment degradation and catalyst poisoning, and is also a health hazard to workers of the petroleum industry.1 In the fossil fuel combustion and metallurgical industries, the mercury release to atmosphere has always been a major environmental threat2 and the risk from the petrochemical industry is not any less. Mercury can be categorized as elemental, inorganic and organomercury and all forms of mercury are toxic depending on dosage. With the latter being fatal.1Mercury exposure causes neurological dysfunction, wherein the exposure to mercury may be in the form of inhalation, ingestion and dermal absorption.1,3 The notorious example of human mercury poisoning is the Minamata tragedy, where a neurological syndrome developed due to organomercury(II) poisoning that originated from a chemical factory release, which used mercury(II) sulfate as a catalyst.4 The release of mercury was estimated to be 81.3 tons due to inefficient plant operation between 1932–1968, and the methylation process induced by microorganisms converted the released inorganic mercury to highly toxic organomercury(II).4,5 On the other hand, trace amounts of mercury are known to severely affect the industrial plant integrity due to mercury-induced corrosion and degradation.6,7 Mercury can cause degradation by four basic mechanisms, namely, amalgam corrosion, amalgamation, liquid metal embrittlement (LME) and galvanic corrosion.7 Amalgam corrosion was believed to be the cause of the catastrophic explosion in Skikda LNG, Algeria in 1973.6
The incidents related to mercury showed that it is very critical that the mercury release and exposure be reduced to a minimal level, if not zero, and hence the need for effective mercury removal technologies. The United States Environmental Protection Agency (EPA) issued the first national standards for mercury pollution emitted from power plants, namely, the Mercury and Air Toxic Standards (MATS) in 2011, and amended in 2012 to set the limit for emission from boilers alone to be 2.0–3.0 tons per year.8,9 In terms of exposure, the US Occupational Safety and Health Administration (OSHA) imposed a permissible exposure limit (PEL) for mercury vapour of 0.1 mg m−3 of air as a ceiling limit. The National (US) Institute of Occupational Safety and Health (NIOSH) has also established the recommended exposure limit (REL) for mercury vapour to be 0.05 mg m−3 as a time-weighted average (TWA) for up to a 10 hour workday and a 40 hour work-week.3 Following the Skikda incident, the mercury tolerance level for the petroleum natural gas stream has become more stringent over time and the typical specification is <0.01 mg Nm−3.10 The ultimate goal for mercury removal in natural gas processing is to tackle it at its source, which is at the front end of the process, i.e. on an offshore platform. This is the most challenging situation with an additional limitation of concomitant space constraints.3,11 A solution to this problem in terms of either chemistry and/or chemical engineering will be a boon to the wealth and health of the petrochemical industry.
Fixed bed adsorption, either regenerative or non-regenerative, has been the preferred commercial approach to mercury removal from natural gas, where a solid adsorbent is used to capture mercury through adsorption, amalgamation, or oxidation followed by adsorption.12 The solid adsorbent typically consists of a substrate support (viz. zeolite, activated carbon, alumina, silica), onto which reactive components (e.g. silver nano particles, potassium iodide, sulfur, metal sulfide) are chemically or physically bonded/impregnated.1 Metal sulfide (e.g. CuS)-based adsorbents rely on the high reactivity of mercury with the metal centre of variable oxidation states and can operate in multiple streams such as dry and wet natural gas, and liquid hydrocarbons.13 Copper(II) sulfide-based adsorbent has been a commercial product by companies like UOP, Johnson Matthey, and Axens.14–20
The application of solid-supported CuCl2 has also been reported for elemental mercury removal from gas streams, especially for combustion flue gas.21–32 For the test mimicking the combustion flue gas conditions, CuCl2 supported on activated carbon (AC) was found to give a relatively better mercury removal, via elemental mercury, Hg(0) oxidation and adsorption, in comparison with CuCl2 supported on other types of solid supports such as clay, zeolite and alumina.28,29,31,33,34 Although the redox centre at CuCl2 was reported as a powerful Hg(0) oxidant, when supported on clay was not efficient in the subsequent adsorption of the oxidized mercury when compared to supported on AC. The carbon support was reported to be good in both the oxidation and adsorption of mercury. This may be due to the availability of functional groups on carbon sites for re-adsorption on the surface and within the pores.35 Tests using other non-carbon solid supports, i.e. zeolite and alumina also resulted in removal efficiencies that were relatively lower than AC.33 CuCl2 supported on ceria, zirconia and titania were also reported, which showed good Hg(0) oxidation with potential application in mercury removal from combustion flue gas.36,37
In a previous report, we used chlorocuprate(II) ionic liquid38 with CuCl2 as the precursor for application in mercury removal from a dry natural gas stream, which operated at a much lower temperature (∼30 °C) than the combustion flue gas. The applications of these solid-supported ionic liquid phase (SILP) materials have shown superior performance for mercury removal and have been successfully operating in our commercial gas plants since 2011.38–41
We report herein a solution for Hg removal from the natural gas phase that contains moisture in the lower temperature regions of the plant (25–40 °C) and more specifically, for application in wet-gas conditions, using CuCl2 as the reactive chemical supported on selected solid supports. Wet-gas is defined as the gas that does not meet the pipeline quality for natural gas due to the presence of undesirable components such as free water, water vapour and/or higher hydrocarbons that might result in condensation under pipeline conditions.42 Therefore, the development of mercury adsorbents for wet-gas needs to overcome the challenge of being robust towards increased amounts of contaminants in the gas feed. More specifically, moisture in the gas stream reacts with most chemicals, compromising their reactivity and selectivity to mercury(0) and/or results in capillary condensation, which leads to the poor performance of the physical adsorbents. On the contrary, dry-gas is defined as the natural gas that contains mole fractions of water less than 0.005 mol% [50 p.p.m. (mol)] in the vapour phase.42 In the current research, we utilized the established redox chemistry of copper towards mercury in a commercially viable chemical, i.e. copper(II) chloride, by designing an add-on feature (a gas pre-heating system) to the removal process that can smartly tune the copper coordination sphere by using the moisture in the stream as an advantage and thereby enhance the reactivity to the incoming mercury, this simple tweaking of the process design using temperature as a control also simultaneously avoided the capillary condensation, which is a physical property challenge to address in the wet-gas streams. Initial studies using this process enhancement for mercury removal from wet-gas showed that elemental mercury in the vapour state was successfully removed with a longer adsorbent lifetime, i.e. almost four times enhancement in mercury breakthrough times when compared to the same adsorbent without the gas pre-heating system using a common redox active material.
Materials and synthesis
Silica, γ-alumina and activated carbon were the solid supports used in this study. Silica and γ-alumina extrudates were obtained from Johnson Matthey, while activated carbon was obtained from Calgon Carbon for testing purposes. Chemically active ingredients impregnated on the solid support were anhydrous copper(II) chloride, sodium chloride, their mixtures and 1-butyl-3-methylimidazolium chloride ([C4mim]Cl) ionic liquid. Anhydrous copper(II) chloride (CuCl2), with purity of 97%, and sodium chloride (NaCl) with purity of 99.98% were used as purchased from Sigma-Aldrich. The ionic liquid 1-butyl-3-methylimidazolium chloride ([C4mim]Cl) was synthesized in-house.The preparation of the adsorbents was performed via a wet incipient method. For each type of adsorbent, a standard weight of 2 g of the solid support was used, and the loading of copper content on the solid support was restricted to 2 wt% of Cu(II) within the complex. In the case of salt and ionic liquids mixtures with CuCl2, the mole fractions of CuCl2, χCuCl2, were 0, 0.5, 0.67 or 1. An example for the synthesis of NaCl–CuCl2 on silica, χCuCl2, is described. NaCl (0.0368 g, 0.6295 mmol) was weighed in a sample vial (15 cm3). Placing this vial on the balance, anhydrous CuCl2 (0.0846 g, 0.6295 mmol) was added slowly to obtain the desired amount. Deionized water (2.8 cm3, this is the right amount of water to wet the adsorbent) was added and the mixture was stirred until dissolved to obtain a clear homogeneous solution. Silica (2.0 g) was then added to the salt solution and shaken vigorously for 10 min using a mechanical shaker to ensure a good coating of the reactive ingredients throughout the support. Subsequently, water was removed from the system by drying in the oven at 90 °C for 15 h (overnight).
These adsorbents were characterised in terms of the Brunauer–Emmett–Teller (BET) surface area and Barrett–Joyner–Halenda (BJH) porosity, thermogravimetric analysis (TGA), chemical speciation by UV-visible-near IR spectroscopy, morphology by scanning electron microscopy and elemental analysis by energy dispersive X-ray spectroscopy.
Experimental
The experimental set-up for mercury removal was an in-house design comprised of the commercially available 10.534 PSA Mercury Generator associated with the PSA Sir Galahad II Mercury Analyzer. Two different test rigs were designed, which were able to interchange between dry-gas and wet-gas testing. The adsorbents for testing were crushed and sieved to 300–500 μm in size for packing in the adsorbent column.Dry gas testing rig
The design of the dry-gas testing rig setup was to enable mercury extraction studies to be conducted under moisture-free conditions at a set temperature, and the schematic for the dry-gas testing rig is shown in Fig. 1.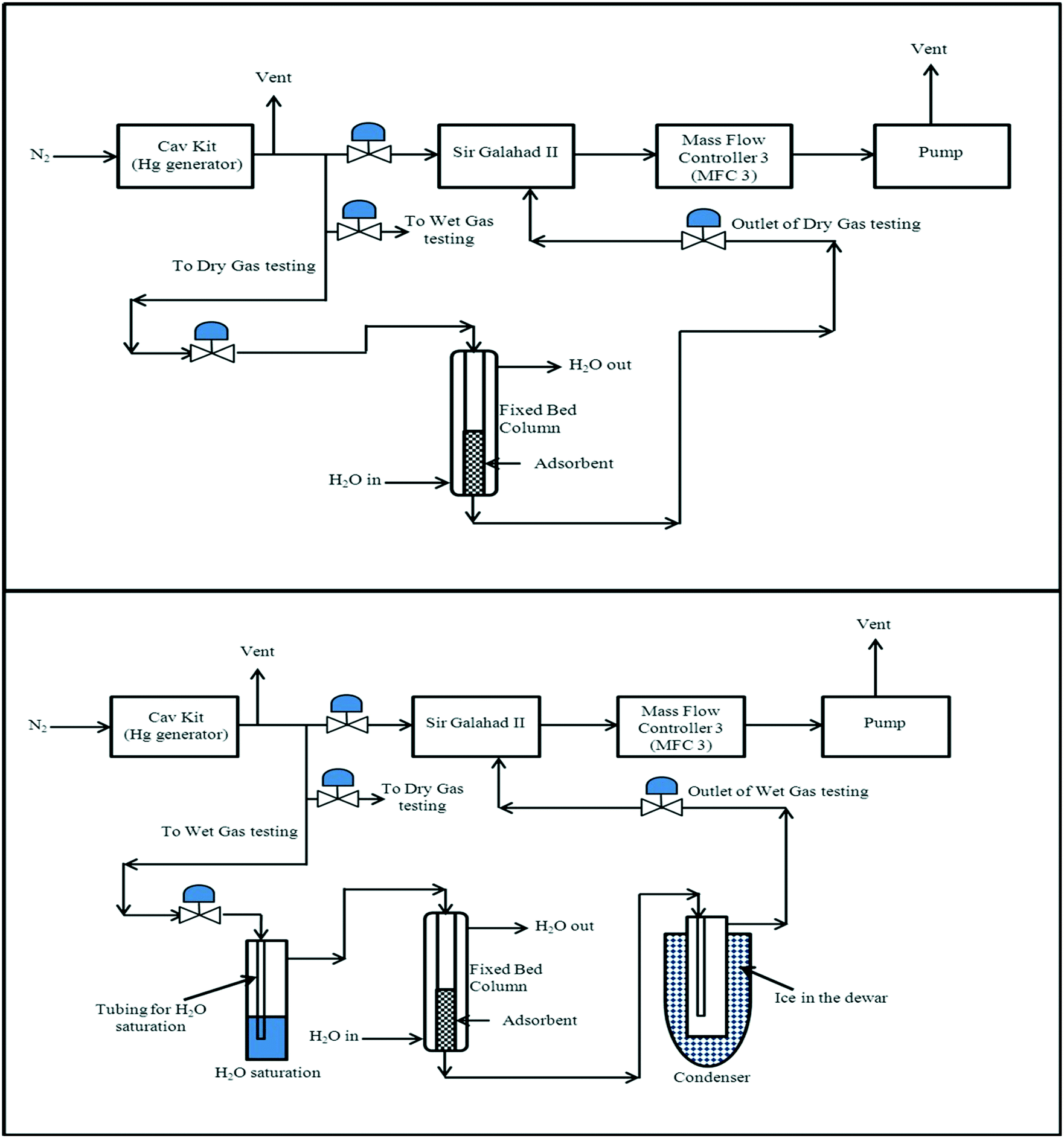 | ||
| Fig. 1 Schematic of the mercury extraction studies under dry and wet-gas conditions to validate and compare the extraction performance of the adsorbents prepared in this study. Typical experimental conditions for mercury extraction studies in the fixed bed column were as follows: (i) adsorbent weight, 30 mg; (ii) adsorbent size, 300–500 μm; (iii) column internal diameter, 2 mm; (iv) gas flow rate, 600 cm3 min−1; gas linear velocity, 31.8 m s−1; (v) contact time, ca. 0.005 s; (vi) inlet Hg(0) concentration, 2000 ng L−1; (vii) carrier gas, N2 (O2 free, 99.998% purity). | ||
Firstly, the carrier gas, nitrogen, was introduced through the Hg(0) vapour generator-10.534, where a wide range of mercury concentrations was generated (1.5–4400 ng L−1) by manipulating the reservoir temperature (30–57 °C), saturation gas flow (1–20 cm3 min−1) and dilution gas flow (1–20 L min−1). Mercury-containing nitrogen gas was then flowed into the jacketed fixed-bed column, where the gas flow rate was controlled by a mass flow controller (MFC 3) with a flow range of 0–1000 cm3 min−1, while the rest of the gas flow from the mercury generator was vented. The temperature of the fixed bed column was normally maintained at 20 °C, with the jacketed fixed-bed column allowed the mercury extraction studies to be carried out at elevated temperatures, if required, using a thermal fluid circulator. The concentration of mercury at the outlet of the fixed-bed column loaded with mercury adsorbent was analyzed by using the PSA Sir Galahad II Mercury Analyzer at specific time intervals of approximately 5 min. As recommended by the PSA, a vacuum pump was used to create the pressure drop to drive the flow through the test sections of the experimental set-up, and the outlet gas was finally vented to the fume hood exhaust after passing through a mercury trap (packed with HycaPure Hg™). All experimental procedures were carried out in the fume hood under ambient conditions unless specified.
Wet-gas testing rig
The aim of the design of the wet-gas testing setup was to enable mercury extraction studies to be conducted under wet-gas conditions (moisture containing) in two modes of testing, with and without gas-preheating; the schematic for the wet-gas testing setup can be seen in Fig. 1 and 2.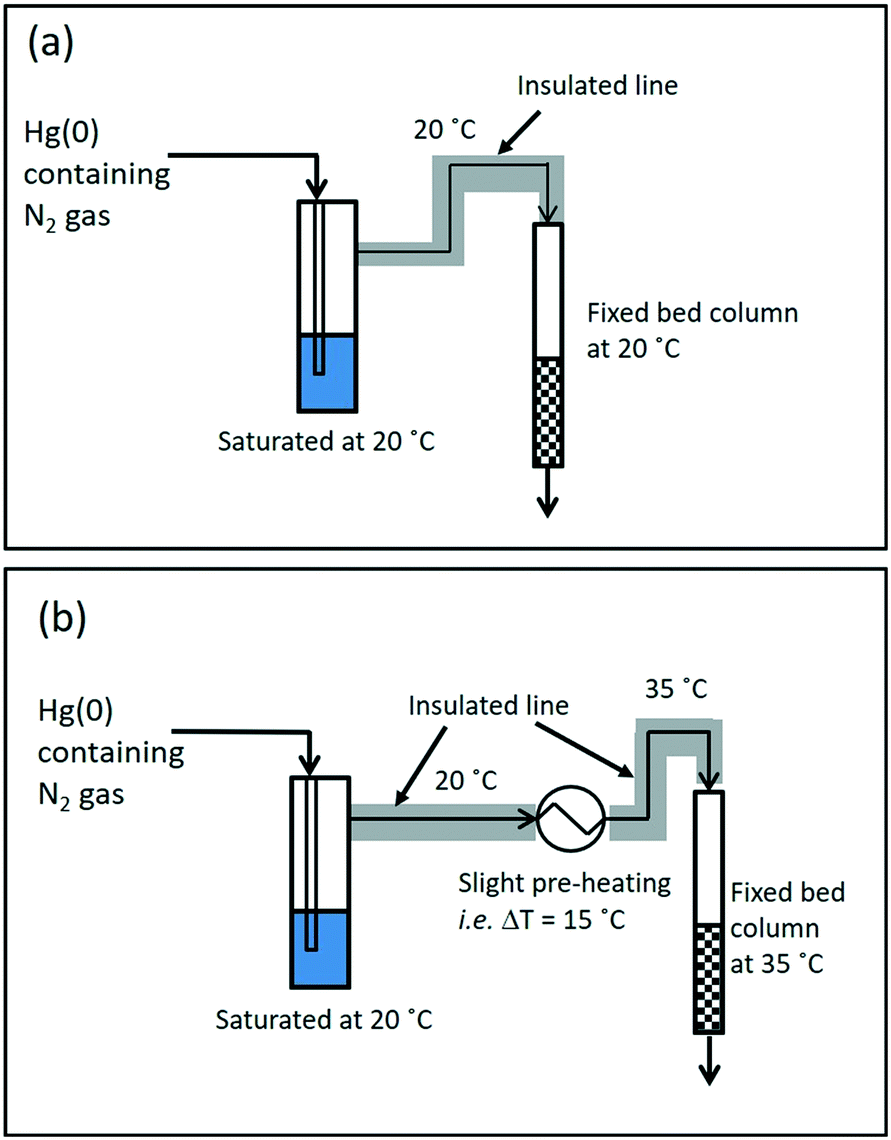 | ||
| Fig. 2 Wet-gas testing: (a) without pre-heating, and (b) with pre-heating (example ΔT of 15 °C). | ||
For the wet-gas testing setup, the generation and flow control of Hg(0)-containing nitrogen gas was the same as for the dry-gas testing setup, but the gas going into the fixed-bed column was pre-saturated to produce “wet-gas”, and the outlet gas from the fixed-bed column was also treated to remove moisture from the gas, since it might condense in the PSA Sir Galahad II Mercury analyser, and would affect the mercury analysis results. For the gas saturation, the incoming gas was passed through a H2O saturation unit at 20 °C (average ambient temperature in the laboratory). The saturated gas was then directed into the fixed bed column and the outlet gas from the fixed bed column was flowed through a series of moisture traps to maximise the moisture capture before entering the Sir Galahad II mercury analyser. As mentioned earlier, the wet-gas testing was carried out in two modes, with and without pre-heating. This can be further understood by referring to a preheated gas from say 20 °C to 35 °C, representing a 15 °C temperature increase, and the condition is represented as T3520.
The typical experimental conditions for dry and wet-gas testing are summarised in the caption for Fig. 1. These experimental conditions were selected to accelerate the breakthrough of the adsorbent samples tested and were used consistently across all the mercury extraction studies unless specified. The results for the mercury extraction studies are represented as a mercury breakthrough curve, where the outlet concentration of mercury are plotted against time. The breakthrough time for an extractant is defined as the time required from the start of the mercury extraction test until the time that the mercury outlet concentration reaches 5 ng L−1. All mercury breakthrough studies were carried out in duplicate for reproducibility.
Results and discussion
Surface area and porosity analyses were conducted for the blank solid supports and the synthesized reactive material-supported adsorbent samples, where the results obtained are tabulated in Table 1.| Samples | Surface area BETa/m2 g−1 | Pore volume BJHb (average)/cm3 g−1 | Pore size BJHb (average)/nm |
|---|---|---|---|
| a BET = Brunauer–Emmett–Teller method.45 b Barrett–Joyner–Halenda method.46 | |||
| Blank support | |||
| Silica | 210 | 0.88 | 15.2 |
| Activated carbon, AC | 1220 | 0.78 | 1.8 |
| γ-Alumina | 235 | 0.68 | 9.6 |
| Supported sample | |||
| CuCl2 on silica | 200 | 0.80 | 14.6 |
| NaCl–CuCl2 on silica | 220 | 0.84 | 13.8 |
| NaCl–CuCl2 on silica (χCuCl2 = 0.67) | 220 | 0.83 | 14.0 |
| NaCl on silica (2 wt% Na) | 205 | 0.82 | 11.5 |
| [C4mim]Cl–CuCl2 on silica | 190 | 0.74 | 13.6 |
| CuCl2 on AC | 1150 | 0.39 | 2.5 |
| NaCl–CuCl2 on AC | 1100 | 0.37 | 2.5 |
| NaCl–CuCl2 on γ-alumina | 215 | 0.60 | 9.4 |
| [C4mim]Cl–CuCl2 on γ-alumina | 180 | 0.50 | 8.6 |
Activated carbon (AC) is the most widely used support due to its large micropore and mesopore volumes, and the resulting high surface area that can range from 300 to ∼4000 m2 g−1.43 Silica is known to be amorphous and an ordered mesoporous silicate that can be synthesized by the hydrothermal formation of silica gels in the presence of surfactant templates.43,44 Alumina, on the other hand, is crystalline and its source materials, i.e. gibbsite and diaspora, are transformed into different forms, where γ-alumina is most commonly used for adsorption and catalysis.43 Selected chemicals were impregnated on the solid supports and are presented in Table 1. Impregnation resulted in the reduction of both surface area and porosity, except for showing an increase in pore size on activated carbon, and was observed possibly due to the blockage of the micropores by the reactive materials. Microanalysis results for the synthesized adsorbent samples are listed in the ESI.†
All the synthesized supported adsorbent samples were tested for mercury extraction under dry-gas conditions. Prior to that, blank supports were tested for the potential removal of Hg(0), and the results showed that all the solid supports, i.e. activated carbon, silica, and γ-alumina, had no inherent ability to remove Hg(0) showing immediate mercury breakthrough. The results for mercury extraction under dry-gas conditions for the synthesized supported adsorbent samples are summarised in Fig. 3.
From the results obtained, the best performance was observed for the CuCl2-impregnated activated carbon support, and NaCl–CuCl2-impregnated activated carbon showing comparable performance. This might be due to the nature of activated carbon that possesses a higher surface area resulting from its large micropore and mesopore volumes.35,43 It was reported that CuCl2-impregnated activated carbon showed good ability for the oxidation and adsorption of mercury.28 It was also found that an increase in CuCl2 loading in the CuCl2-AC adsorbent resulted in an increased Hg(0) oxidation but did not enhance the Hg adsorption. This is because an increase in the CuCl2 loading decreased the pore availability of carbon sites, resulting in the poor adsorption of any oxidized mercury species.35
Samples on γ-alumina gave the lowest performance for mercury removal. γ-Alumina was used due to its high surface area but it was identified to have multiple copper species when impregnated with CuCl2.31 At low CuCl2 loading (<10 wt%), Cu species on γ-alumina were found to be copper aluminate and paratacamite, Cu2(OH)3Cl, where paratacamite is formed by the hydrolysis of CuCl2·2H2O catalysed by basic sites of the alumina surface.28,31,47,48 Cu species other than CuCl2 on γ-alumina, i.e. copper aluminate, and paratacamite, are reported to be inactive for Hg(0) oxidation.31,48 On the contrary, α-alumina was reported to be inert towards reactions with CuCl2, and CuCl2 was retained in crystalline form on α-alumina as opposed to amorphous form on γ-alumina.28 Copper aluminate and paratacamite species were not detected on CuCl2/α-alumina due to the lack of active hydroxyl groups on the α-alumina surface.28,31
CuCl2 impregnated on silica showed almost half the mercury removal performance when compared to that impregnated on the activated carbon. This might be due to the high surface area with porosity ranging from micropores to mesopores and macropores on activated carbon as opposed to the lower surface area of mesoporous silica. When doping with NaCl, it was found to have a longer mercury breakthrough time in comparison to CuCl2 on silica. It was reported that the supported (on silica) and unsupported forms of NaCl-CuCl2 mixtures showed no new copper(II) complex formation, and it was composed entirely of a mixture (refer to the powder X-ray diffraction data in the ESI†) of NaCl and CuCl2.49 The enhancement of the mercury extraction performance was previously discussed to be possibly due to dispersion effects, supported by the scanning electron microscopy studies.50 For bulk [C4mim]Cl–CuCl2 (χCuCl2 = 0.5), it formed the chlorocuprate(II) ion, which is likely to be [Cu2Cl6]2–,38,41 and this chlorocuprate(II) is expected to function differently than the CuCl2. The species of [C4mim]Cl–CuCl2 (χCuCl2 = 0.5) on the silica support is unknown, but the electronic absorption spectra, i.e. the d–d ligand field transition, suggested a different Cu(II) ion coordination in comparison to the CuCl2 on silica.41 For NaCl on silica, it was found to have an immediate breakthrough, and this is in line with the thermodynamics of chemical reactions, where the reactivity of NaCl and Hg(0) is thermodynamically infeasible via the Ellingham diagram plot.51–54
Based on these results, selected samples on activated carbon and silica were chosen for testing under the wet-gas conditions, that are the preferred conditions for mercury removal in this work. The results for mercury extraction under wet-gas conditions in comparison to the respective adsorbent samples under dry-gas conditions are summarised in Fig. 4.
For the wet-gas without the pre-heating test, the initial immediate breakthrough was observed for the samples supported on silica. This is due to the nature of the silica support, which is hydrophilic, and it is used as a desiccant due to its large capacity for water (∼40 wt%),43 and the adsorption properties are due to the surface hydroxyl groups known as silanol groups.55 This high affinity for water molecules is anticipated to lead to capillary condensation or pore blocking on silica, thereby resulting in immediate mercury breakthrough. The effect of water on silica is severe but the effect on activated carbon is milder and it is incorrect to say that carbon is hydrophobic since the sorption of water vapour on activated carbon follows an S-shaped curve on the adsorption isotherm.43 The adsorption was reported to be slow at low vapour pressure because of weak van der Waals interactions but once a few water molecules are adsorbed, adsorbate–adsorbate interactions commence, leading to cluster formation that will eventually result in pore filling or capillary condensation in the micropores.43 This can be seen from the mercury breakthrough test result of the CuCl2 supported on activated carbon, where the breakthrough time is 5 h.
The idea of preheating the gas was mainly to avoid the capillary condensation effect or shifting away from the dew point, and to retain the superior performance for mercury removal as observed in the dry-gas system, achieving the ‘dry’ nature of the gas via pre-heating. However, the effect of pre-heating as anticipated to retain performance as in the dry-gas system was not observed in all silica-supported adsorbent samples. The CuCl2-silica and NaCl–CuCl2-silica adsorbent samples showed enhanced performance when compared to the dry-gas results. The mercury breakthrough time could be prolonged more than two-fold by a slight pre-heating of ΔT = 15 °C. For [C4mim]Cl–CuCl2-silica and NaCl–CuCl2-AC, the results obtained were comparable to the results from dry-gas conditions. The CuCl2-silica adsorbent sample was chosen for further optimisation of ΔT, and the results are presented in Fig. 5.
From Fig. 5, for the wet-gas tests, it was observed that at ΔT = 10 °C (T3020), the best performance was observed, with almost 20 h mercury breakthrough time. In contrast, for ΔT = 5 °C (T2520), the performance of the adsorbent sample plummeted to a breakthrough time of 0.1 h, which is almost comparable to the isothermal adsorbent sample at 20 °C (without pre-heating) that had a breakthrough time of 0 h. For the dry-gas testing, changing the temperature from 20 to 35 °C made no significant difference to the performance of the adsorbent (CuCl2 on silica). The calculated amounts of Hg loading on the adsorbent after mercury breakthrough were 1.32, 1.27 and 1.2 wt% at temperatures of 20 °C, 30 °C and 35 °C, respectively. Inlet Hg used for the experiment was 2000 ng L−1 and the targeted outlet was <5 ng L−1 (∼99.75% removal) and the actual outlet observed was always around 0.5 ng L−1. It could be that the performance slightly deteriorated with increased temperature, but the difference was too small to reach a definitive conclusion. However, if the effect was real, it would agree with the literature data on the sulfur-impregnated activated carbon performance, which was not affected by temperature.56
Chemistry investigation impacting performance
From the mercury removal performance results, it can be inferred that the effect of pre-heating the gas in the wet-gas stream had a significant influence as it increased the mercury breakthrough time by almost 4-fold in comparison to the dry-gas conditions at the optimum ΔT of 10 °C. As a first step to carefully investigate the reason for this superior performance, the characteristics of CuCl2-silica as a starting material needed to be understood. For this, the adsorbent sample was prepared by impregnating CuCl2 on a silica support using a calculated amount of water as the solvent and the adsorbent sample was dried in the oven under ambient conditions for 15 hours at 90 °C. The dried adsorbent sample was used for mercury removal testing, where it was stored in a closed vial but intermittently exposed to air during handling. The first step was to understand whether the starting material was either anhydrous CuCl2 on silica, or hydrated CuCl2 on silica. It was reported that the electronic absorption spectra of crystalline CuCl2 at 25 °C showed a band at 9000 cm−1 and a shoulder at 6300 cm−1.57 For CuCl2·2H2O and aqueous solutions of CuCl2, the d–d ligand field transition band was reported to be at around 12500 cm−1, while the solution of CuCl2 in 3M HCl showed a band at around 11111 cm−1.47 It was also reported that [Cu(H2O)6]2+ has an absorption band above 12000 cm−1.58 A range of CuCl2-silica adsorbent samples was prepared to mimic the scenario of different possibilities of handling the adsorbent sample as the starting material, as summarised in Table 2. The UV-visible spectra for all adsorbent samples are shown in Fig. 6.
| Samples | Sample treatment |
|---|---|
| A | Aqueous CuCl2 solution was impregnated on silica, followed by drying in air at 90 °C for 15 h. The dried sample was crushed and sieved and the UV-vis spectrum immediately recorded (4.2 wt% CuCl2) |
| B | Aqueous CuCl2 solution was impregnated on silica, followed by drying in air at 90 °C for 15 h. The dried sample was crushed and sieved and exposed to the air for 2 days. After 2 days, the UV-visible spectrum (4.2 wt% CuCl2) of the sample was recorded |
| C | Aqueous CuCl2 solution was impregnated on silica, followed by drying in air at 90 °C for 15 h. The dried sample was crushed and sieved and kept sealed (no exposure) for 2 days. After 2 days, the UV-visible spectrum (4.2 wt% CuCl2) of the sample was recorded |
| D | Aqueous CuCl2 solution was impregnated on silica, followed by drying in air at 90 °C for 15 h. The dried sample was crushed and sieved, followed by another drying in air at 90 °C for 15 hours. After drying, the UV-visible spectrum (4.2 wt% CuCl2) of the sample was recorded |
Fig. 6 shows that all adsorbent samples exhibited bands quite similar to CuCl2·2H2O and the aqueous solution of the CuCl2 band at ca. 12300–12600 cm−1; however, the band at ca. 10600 cm−1 was inconclusively compared with literature values, where it was reported as a poorly resolved shoulder.47 Therefore, the CuCl2-silica adsorbent sample as the starting material presumably showed the presence of largely hydrated copper(II) species, of CuCl2.47,58–60 Silica is a known water adsorbent via its surface silanol functionality,43,55 which correlated with the TGA analysis and showed an approximately 3 wt% H2O loss at around 150 °C (see TGA results in ESI†). Based on the amount of water and CuCl2 loaded onto silica, the mol ratio of H2O:Cu in the supported sample was estimated to be around 5:1, and this showed that this amount of water was sufficient to cause full hydration of the copper(II) metal centre. Furthermore, the scanning electron microscopy (SEM) micrograph and the corresponding EDX spectra for CuCl2/SiO2 (see Fig. 2 in ESI†) showed good dispersion of CuCl2 on the surface of the support. The above investigation enabled us to understand the nature of CuCl2 on silica, which was mainly in the form of hydrated copper(II) species. As discussed earlier, the mercury removal enhancement effect due to gas pre-heating in the wet-gas rig was observed for CuCl2 and NaCl–CuCl2 on silica; however, a similar enhancement was not observed on the [C4mim]Cl–CuCl2 on silica and on NaCl–CuCl2 on activated carbon. The copper centre on CuCl2 with silica was more reactive to incoming mercury(0), which caused the enhancement of the removal performance (see Fig. 3). Fig. 7 shows the change in the colour of the adsorbent in the fixed-bed column during the mercury removal process when pre-heating was applied under wet-gas conditions (i.e. in the presence of moisture and in a range of temperatures enhanced by ΔT).
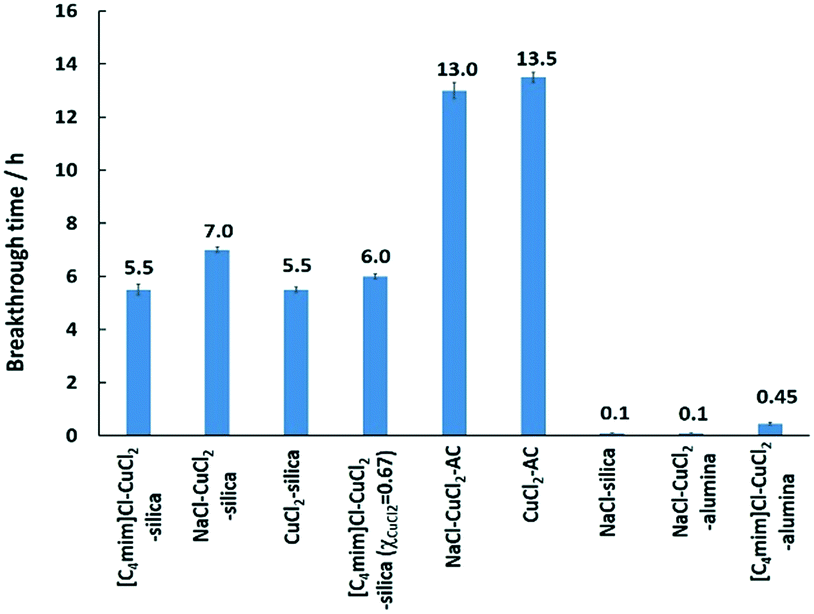 | ||
| Fig. 3 Summary of the mercury extraction breakthrough time for all the adsorbent samples tested under dry-gas conditions at 20 °C. All adsorbent samples had 2 wt% Cu loading with χCuCl2 = 0.5, unless otherwise specified. Typical experimental conditions for mercury extraction studies in the fixed bed column were as follows: (i) adsorbent weight, 30 mg; (ii) adsorbent size, 300–500 μm; (iii) column internal diameter, 2 mm; (iv) gas flow rate, 600 cm3 min−1; gas linear velocity, 31.8 m s−1, (v) contact time, ca. 0.005 s; (vi) inlet Hg(0) concentration, 2000 ng L−1; (vii) carrier gas, N2 (O2 free, 99.998% purity). | ||
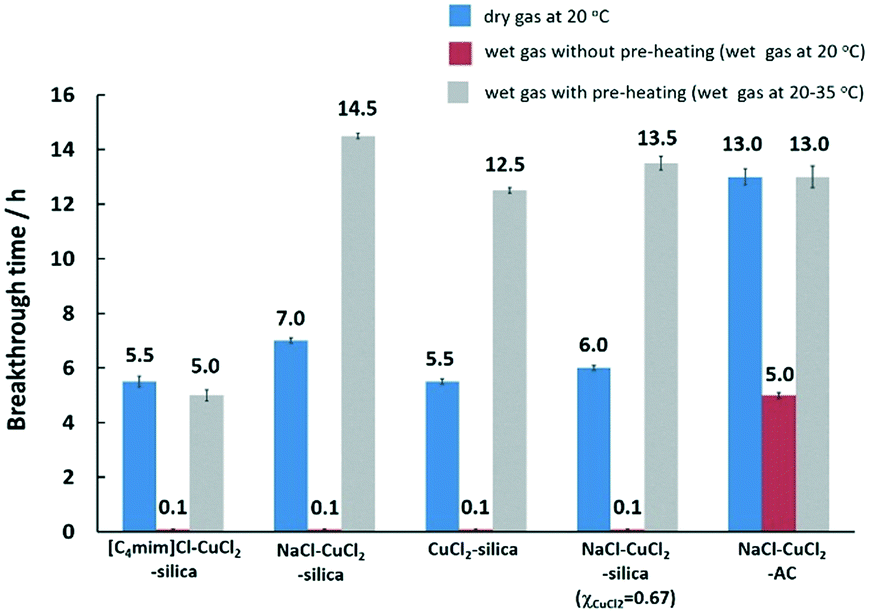 | ||
| Fig. 4 Comparison of the tests performed in dry-gas and wet-gas with pre-heating. All adsorbent samples had 2 wt% Cu loading with χCuCl2 = 0.5, unless otherwise specified. Typical experimental conditions for mercury extraction studies in the fixed-bed column are as follows: (i) adsorbent weight, 30 mg; (ii) adsorbent size, 300–500 μm; (iii) column internal diameter, 2 mm; (iv) gas flow rate, 600 cm3 min−1; gas linear velocity, 31.8 m s−1; (v) contact time, ca. 0.005 s; (vi) inlet Hg(0) concentration, 2000 ng L−1; (vii) carrier gas, N2 (O2 free, 99.998% purity). | ||
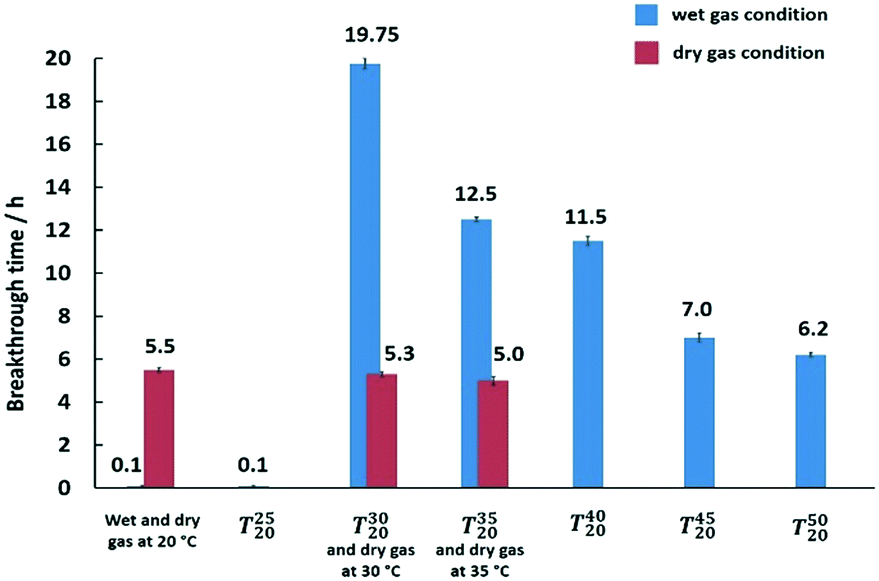 | ||
| Fig. 5 Optimisation of ΔT for wet-gas conditions plotted together with dry-gas conditions tested at different temperatures for the CuCL2-silica adsorbent sample. Typical experimental conditions for mercury extraction studies in the fixed bed column are as follows: (i) adsorbent weight, 30 mg; (ii) adsorbent size, 300–500 μm; (iii) column internal diameter, 2 mm; (iv) gas flow rate, 600 cm3 min−1; gas linear velocity, 31.8 m s−1; (v) contact time, ca. 0.005 s; (vi) inlet Hg(0) concentration, 2000 ng L−1; (vii) carrier gas, N2 (O2 free, 99.998% purity). | ||
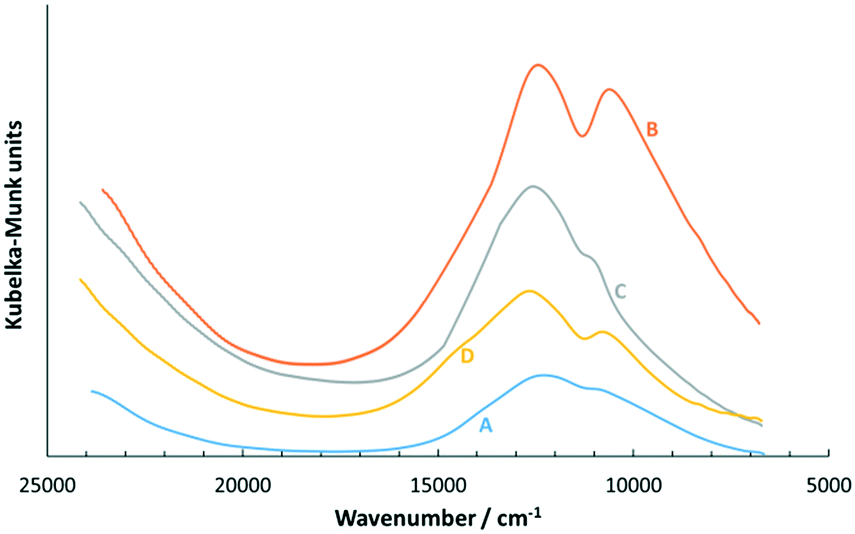 | ||
| Fig. 6 Electronic absorption spectra utilising the visible-near IR region for adsorbent samples A–D. | ||
 | ||
| Fig. 7 Colour changes in the CuCl2 impregnated on silica tested in the wet-gas conditions under different pre-heating temperatures, ΔT, where the pictures were taken after 15 min of the experiment. | ||
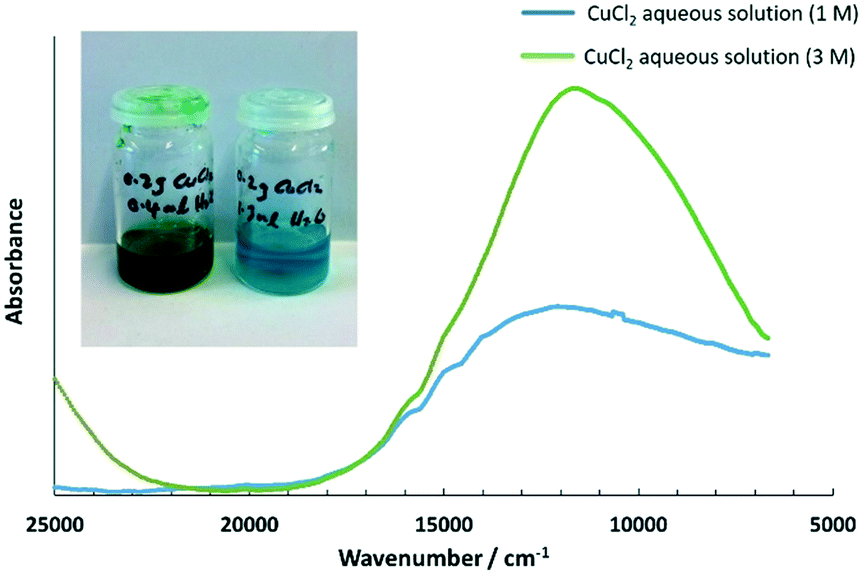 | ||
| Fig. 8 UV-vis-near IR spectra for aqueous solutions of CuCl2 at 1 and 3 mol L−1. | ||
These observations indicate a correlation between the adsorbent sample performance and colour, i.e., the enhancement of mercury removal performance and colour changes, and this effect was more chemical than physical. This may be attributed to the different Cu(II) coordination spheres under the pre-heating environment. It is speculated that without preheating (wet-gas at 20 °C), [Cu(H2O)6]2+ was the main species formed and as the incoming wet-gas feed was gradually preheated, this complex started to lose water ligands, which were gradually substituted by the weaker chloride ligand.
In a related report, the UV-visible spectra for aqueous Cu2+ solutions (concentration 0.1 mol L−1) in a solvent mixture of cholinium chloride and water,61 were presented. A similar trend was observed, where the colour of the aqueous Cu2+ solutions (concentration 0.1 mol L−1) changed gradually from pale blue, to green and greenish-yellow to yellow, with increasing concentrations of cholinium chloride, and the absorption maxima shifted to shorter wavelengths with increasing water content. To verify this in the solution state, 1 mol L−1 of CuCl2 aqueous solution and 3 mol L−1 of CuCl2 aqueous solution were prepared and analysed using UV-visible spectroscopy (see Fig. 8), for comparison with the absorption bands of CuCl2 on silica under pre-heating at different ΔT. Since the above CuCl2 solutions were highly concentrated, a thin film of the solution was used for UV-visible spectroscopy studies by sandwiching between two quartz plates. This study enabled us to understand the absorption maxima qualitatively and the absorption bands, are shown in Table 3. It can be seen in Table 3 that the absorption maxima shifted towards lower energy with decreasing water content (higher Cu2+ concentration), and this was attributed to chloride being a weaker ligand.
| Absorption maxima/cm−1 | |
|---|---|
| CuCl2 aqueous solution (1 M) | 12100 |
| CuCl2 aqueous solution (3 M) | 11530 |
Copper(II) complexes are extremely labile and the ligand exchange rates are of the order 10−6 to 10−9 M−1 s−1 and are therefore challenging to determine unless in situ UV-visible measurements are carried out during the mercury adsorption process.62 On preheating at ΔT = 10 °C, it was anticipated that the hexaaquacopper(II) centre was converted to the tetraaquadichlorocopper(II) centre, which may be more reactive to incoming elemental mercury with better kinetics when compared to the fully chlorinated copper centre, thereby enhancing the mercury removal performance to more prolonged durations. Additionally, the fate of mercury captured on the adsorbent was expected to be in complex form rather than as mercury(II) chloride that can leach under high pressure or accelerated conditions.33,63,64 The possible mercury reaction mechanism is not conclusive enough to be reported without further in situ analytical work and needs to be probed for both copper and mercury centres. We anticipate that mercury fixation under these wet-gas conditions was via the formation of more stable mercury complexes on the silica support rather than in a molecular state of HgCl2, which was reported to be formed under dry-gas conditions.65 Further, the comproportionation reaction between Hg(II) in the complex with the incoming Hg(0), leading to the formation of stable compounds as calomel, is also feasible.41
Conclusions
By introducing a simple process design, an enhanced effect on elemental mercury removal from a complex natural gas stream dominated by moisture in the feed (wet-gas) was achieved by utilising the redox activity of copper(II) to mercury(0) in copper(II) chloride-impregnated adsorbents. It was shown that slightly preheating the simulated gas entering into the fixed bed mercury adsorbent column at ΔT = 10 °C resulted in performance enhancement of almost 4-fold when using CuCl2-silica as an adsorbent in comparison to dry-gas conditions. The enhancement of performance may be both chemical and physical, i.e. due to the dynamic copper coordination sphere under the pre-heating environment (with moisture in the gas feed) being more reactive towards elemental mercury and simultaneously avoiding capillary condensation caused by water vapour at slightly elevated temperatures. Fresh CuCl2 on silica before loading in the fixed bed reactor was found to be in the form of largely hydrated copper(II) species. [Cu(H2O)6]2+ was anticipated to be formed at the initial stage of the experiment without pre-heating, and as the incoming wet-gas feed was gradually preheated to different temperatures, fully hydrated [Cu(H2O)6]2+ started to lose water ligands which were gradually substituted by a weaker ligand, Cl−, at various ΔT. ΔT = 10 °C was optimised to provide the best performance in mercury extraction when compared to dry-gas conditions. The captured mercury on the adsorbent was expected to be in a more stable complex form than in molecular HgCl2 and led to better fixation. As authors with experience in developing sustainable chemical technologies in full industrial scale, we believe that this work will demonstrate the use of known redox chemistry from commercially viable CuCl2 for the selective removal of mercury contaminants from a complex hydrocarbon stream by integrating with appropriate engineering solutions. Eventually, the waste management of spent adsorbents may be by traditional incineration or by underground storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Prof Kenneth Seddon (deceased), Ex-Director, Queen's University Ionic Liquid Laboratories, Queen's University Belfast for useful academic discussions and PETRONAS, Malaysia for funding this work.References
- S. M. Wilhelm and N. Bloom, Fuel Process. Technol., 2000, 63, 1–27 CrossRef CAS.
- M. H. Keating, Mercury study report to congress. Volume II: Inventory of anthropogenic mercury emissions in the United States, Report EPA-452/R-97-004, Environmental Protection Agency, Research Triangle Park, NC (United States). Office of Air Quality Planning and Standards and Office of Research and Development, 1997 Search PubMed.
- S. M. Wilhelm, Process Saf. Prog., 1999, 18, 178–188 CrossRef CAS.
- T. Tsubaki and K. Irukayama, Minamata Disease: Methylmercury Poisoning in Minamata and Niigata, Kodansha Ltd., Tokyo, 1977 Search PubMed.
- N. J. Langford and R. E. Ferner, J. Hum. Hypertens., 1999, 13, 651–656 CAS.
- W. W. Bodle, A. Attari and R. Serauskas, presented in part at the 6th International Conference on Liquefied Natural Gas, Kyoto, 1980.
- S. M. Wilhelm, A. McArthur and R. D. Kane, presented in part at the 73rd Annual Convention - Gas Processors Association, New Orleans, April, 1994.
- Mercury and Air Toxics Standards (MATS), https://www.epa.gov/mats, (accessed May 2020).
- Controlling Air Pollution from Boilers and Process Heaters, https://19january2017snapshot.epa.gov/boilers_.html, (accessed May 2020).
- S. Mokhatab, W. A. Poe and G. Zatzman, Handbook of Natural Gas Transmission and Processing, Gulf Professional Publishing, Oxford, 2012 Search PubMed.
- N. Eckersley, D. Radtke, L. Rogers and S. Brennan, Mercury removal process comparison, presented at the 63rd LRGCC Conf. Proc., Norman, 2013 Search PubMed.
- E. J. Granite, H. W. Pennline and R. A. Hargis, Ind. Eng. Chem. Res., 2000, 39, 1020–1029 CrossRef CAS.
- P. J. H. Carnell and V. A. Row, presented in part at the 15th International Conference & Exhibition on Liquefied Natural Gas, Barcelona, April, 2007.
- M. J. C. Cousins, World Pat., WO2008020250A1, Johnson-Matthey PLC, 2008 Search PubMed.
- M. J. Cousins, C. J. Young and R. Logan, World Pat., WO2009101429A1, Johnson-Matthey PLC, 2009 Search PubMed.
- M. J. Cousins, D. Davis, P. Rafferty, S. Ridley and A. G. Tapster, World Pat., WO2013136046A1, Johnson-Matthey PLC, 2013 Search PubMed.
- A. Fish and M. D. G. Lunn, World Pat., WO2014016561A1, Johnson-Matthey PLC, 2014 Search PubMed.
- D. Simonetti, K. V. Ivanov and T. Traynor, World Pat., WO2013119357A1, UOP LLC, 2013 Search PubMed.
- J. C. Candelon, A. Pucci and C. Jubin, US Pat., US9011676B2, Axens, 2015 Search PubMed.
- D. Bazer-Bachi, D. Chiche, J. Lopez, T. Serres, T. Frising, O. Ducreux and P. Euzen, US Pat., US20180353929A1, Axens, IFP Energies Nouvelles, 2018 Search PubMed.
- Y. El-Shoubary, R. Maes and S. C. Seth, US Pat., US6589318B2, Merck & Co. Inc., 2003 Search PubMed.
- Y. El-Shoubary, R. Maes and S. C. Seth, US Pat., US6638347B2, Merck & Co. Inc., 2003 Search PubMed.
- Y. El-Shoubary and R. Maes, US Pat., US6841513B2, Merck & Co. Inc., 2005 Search PubMed.
- C. Chao and S. J. Pontonio, US Pat., US2006/0205592A1, Praxair, Inc., 2006 Search PubMed.
- C. Chao and S. J. Pontonio, World Pat., WO2006/099291A2, Praxair, Inc., 2006 Search PubMed.
- S.-S. Lee, J.-Y. Lee and T. C. Keener, Environ. Sci. Technol., 2009, 43, 2957–2962 CrossRef CAS PubMed.
- X. Li, Z. Liu and J.-Y. Lee, J. Hazard. Mater., 2013, 252, 419–427 CrossRef PubMed.
- X. Li, Z. Liu, J. Kim and J.-Y. Lee, Appl. Catal., B, 2013, 132, 401–407 CrossRef.
- S.-S. Lee, J.-Y. Lee and T. C. Keener, J. Chin. Inst. Chem. Eng., 2008, 39, 137–142 CrossRef CAS.
- X. Li, J.-Y. Lee and S. Heald, Fuel, 2012, 93, 618–624 CrossRef CAS.
- Z. Liu, X. Li, J.-Y. Lee and T. B. Bolin, Chem. Eng. J., 2015, 275, 1–7 CrossRef CAS.
- J. Yang, Y. Zhao, J. Zhang and C. Zheng, Fuel, 2016, 164, 419–428 CrossRef CAS.
- W. Du, L. Yin, Y. Zhuo, Q. Xu, L. Zhang and C. Chen, Ind. Eng. Chem. Res., 2014, 53, 582–591 CrossRef CAS.
- V. Sriram, C. Li, Z. Liu, M. Jafari and J.-Y. Lee, Chem. Eng. J., 2018, 343, 244–257 CrossRef CAS.
- S.-S. Lee, J.-Y. Lee, S.-J. Khang and T. C. Keener, Ind. Eng. Chem. Res., 2009, 48, 9049–9053 CrossRef CAS.
- X. Zhou, W. Xu, H. Wang, L. Tong, H. Qi and T. Zhu, Chem. Eng. J., 2014, 254, 82–87 CrossRef CAS.
- Z. Zhou, X. Liu, Y. Hu, Z. Liao, S. Cheng and M. Xu, Fuel, 2018, 216, 356–363 CrossRef CAS.
- M. Abai, M. P. Atkins, A. Hassan, J. D. Holbrey, Y. Kuah, P. Nockemann, A. A. Oliferenko, N. V. Plechkova, S. Rafeen, A. A. Rahman, R. Ramli, S. M. Shariff, K. R. Seddon, G. Srinivasan and Y. Zou, Dalton Trans., 2015, 44, 8617–8624 RSC.
- M. Abai, M. P. Atkins, A. Hassan, J. D. Holbrey, Y. Kuah, P. Nockemann, A. A. Olifrenko, N. V. Plechkova, S. Rafeen, A. A. Rahman, K. R. Seddon, S. M. Shariff, G. Srinivasan and Y. Zou, presented in part at the International Gas Union Research Conference, Seoul, October, 2011.
- M. Abai, M. P. Atkins, K. Y. Cheun, J. Holbrey, P. Nockemann, K. Seddon, G. Srinivasan and Y. Zou, World Pat., WO2012046057A3, The Queen's University of Belfast, 2012 Search PubMed.
- R. Boada, G. Cibin, F. Coleman, S. Diaz-Moreno, D. Gianolio, C. Hardacre, S. Hayama, J. D. Holbrey, R. Ramli, K. R. Seddon, G. Srinivasan and M. Swadźba-Kwaśny, Dalton Trans., 2016, 45, 18946–18953 RSC.
- ISO/IEC 14532: 2014, Natural gas -- Vocabulary, https://www.iso.org/obp/ui/#iso:std:iso:14532:ed-2:v1:en, (accessed May 2020).
- R. T. Yang, Adsorbents: Fundamentals and Applications, John Wiley & Sons, Inc., Hoboken, 2003 Search PubMed.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard and S. B. McCullen, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- C. Zipelli, J. C. J. Bart, G. Petrini, S. Galvagno and C. Cimino, Z. Anorg. Allg. Chem., 1983, 502, 199–208 CrossRef CAS.
- W. Yang, Y. Adewuyi, A. Hussain and Y. Liu, Environ. Chem. Lett., 2019, 17, 19–47 CrossRef CAS.
- M. P. Vorobei and O. V. Skiba, Zh. Neorg. Khim., 1970, 15, 1414–1417 CAS.
- K. Yongcheun, PhD Thesis, The Queen's University Belfast, 2012, Unpublished work Search PubMed.
- H. J. T. Ellingham, J. Soc. Chem. Ind., London, 1944, 63, 125–133 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press LLC, Boca Raton, 2002 Search PubMed.
- NIST-JANAF Thermochemical Tables, http://kinetics.nist.gov/janaf/, (accessed May 2020).
- CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC Press, Boca Raton, 2014 Search PubMed.
- A. A. Christy, Int. J. Eng. Technol., 2012, 4, 484 CrossRef CAS.
- M. D. Bingham, SPE Prod. Eng., 1990, 5, 120–124 CrossRef CAS.
- S. V. Volkov, N. I. Buryak and O. B. Babushkina, Russian Journal of Chemistry, 1981, 26, 1093–1095 Search PubMed.
- D. Nicholls, Complexes and First-row Transition Elements, Macmillan Education Ltd, Hampshire, 1974 Search PubMed.
- G. Leofanti, M. Padovan, M. Garilli, D. Carmello, A. Zecchina, G. Spoto, S. Bordiga, G. T. Palomino and C. Lamberti, J. Catal., 2000, 189, 91–104 CrossRef CAS.
- G. Leofanti, M. Padovan, M. Garilli, D. Carmello, G. L. Marra, A. Zecchina, G. Spoto, S. Bordiga and C. Lamberti, J. Catal., 2000, 189, 105–116 CrossRef CAS.
- P. De Vreese, N. R. Brooks, K. Van Hecke, L. Van Meervelt, E. Matthijs, K. Binnemans and R. Van Deun, Inorg. Chem., 2012, 51, 4972–4981 CrossRef CAS PubMed.
- R. R. Wright, N. R. Walker, S. Firth and A. J. Stace, J. Phys. Chem. A, 2001, 105, 54–64 CrossRef CAS.
- L. Bernard, K. O. Awitor, J. P. Badaud, O. Bonnin, B. Coupat, J. P. Fournier and P. Verdier, J. Phys. III, 1997, 7, 311–319 CrossRef CAS.
- K. O. Awitor, L. Bernard, B. Coupat, J. P. Fournier and P. Verdier, New J. Chem., 2000, 24, 399–401 RSC.
- L. F. Kozin and S. C. Hansen, Mercury Handbook: Chemistry, Applications and Environmental Impact, Royal Society of Chemistry, Cambridge, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00240b |
| This journal is © The Royal Society of Chemistry 2020 |