
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetrahydrofuran-based two-step solvent liquefaction process for production of lignocellulosic sugars†
Arpa
Ghosh
 a,
Martin R.
Haverly
b,
Jake K.
Lindstrom
a,
Patrick A.
Johnston
a and
Robert C.
Brown
*ac
a,
Martin R.
Haverly
b,
Jake K.
Lindstrom
a,
Patrick A.
Johnston
a and
Robert C.
Brown
*ac
aBioeconomy Institute, Iowa State University, 1140E Biorenewables Research Laboratory Building, Ames, Iowa 50011, USA. E-mail: rcbrown3@iastate.edu; Fax: +1 515 294 3091; Tel: +1 515 294 7934
bRenewable Energy Group, Ames, Iowa 50010, USA
cDepartment of Mechanical Engineering, Iowa State University, Ames, Iowa 50011, USA
First published on 22nd July 2020
Abstract
Large-scale production of biofuels and chemicals will require cost-effective, sustainable, and rapid deconstruction of woody biomass into its constituent sugars. Here, we introduce a novel two-step liquefaction process for producing fermentable sugars from red oak using a mixture of tetrahydrofuran (THF), water and dilute sulfuric acid. THF promotes acid-catalyzed solubilization of lignin and hemicellulose in biomass achieving 61% lignin extraction and 64% xylose recovery in a mild pretreatment step. The pretreatment opens the structure of biomass through delignification and produces a cellulose-rich biomass, which is readily solubilized at low temperature giving 65% total sugar yields in a subsequent liquefaction process employing the same solvent mixture. This process achieves competitive sugar yields at high volumetric productivity compared to conventional saccharification methods. THF, which can be derived from renewable resources, has several benefits as solvent including ease of recovery from the sugar solution and relatively low toxicity and cost.
1. Introduction
Lignocellulosic sugar has been proposed as feedstock for production of low carbon fuels from renewable feedstocks.1–3 Agricultural waste, forestry residue, and grasses are potential sources of lignocellulosic sugars.4 Cellulose, the most abundant component of biomass, can be depolymerized to its constituent monosaccharide, D-glucose, which could then be fermented to bioethanol or upgraded catalytically to other advanced biofuels such as 2,5-dimethylfuran.3,5 Similarly, hemicellulose can be converted to xylose, which is fermentable to ethanol by engineered microbes.6The inherent recalcitrance of lignocellulosic biomass represents a major barrier to commercial production of cellulosic sugars. Cellulose, hemicellulose and lignin intertwine to form a complex polymeric matrix. Cellulose is highly crystalline and has intensive intra- and intermolecular hydrogen bonding making it resistant to physical, chemical, or enzymatic degradation.7,8 Additionally, lignin sheaths the polysaccharides, protecting the carbohydrate from enzymatic and chemical decomposition.9,10 Clearly, the success of biomass conversion to produce fermentable sugars depends on the effectiveness of fractionating lignocellulose and depolymerizing the polysaccharides into processable sugars.
The conventional approach to producing cellulosic sugars employs physical and/or chemical pretreatment of biomass followed by enzymatic hydrolysis to yield monosaccharides.4 However, the estimated cost of enzyme production is as high as $1.47 per gallon of ethanol, based on typical glucose yields of around 70% and enzyme loadings of 20 mg g−1 of glucan.11 Glucose yields greater than 70% can take up to days and sometimes weeks to complete enzymatic hydrolysis.12–16 These slow rates translate into relatively high capital and operating costs. Potentially faster and more economical routes to saccharification of cellulosic biomass include pyrolysis, processing in molten salt hydrates, and solvent liquefaction, including acid hydrolysis, processing in ionic liquids and deep eutectic solvents.17–22 These processes have demonstrated cellulosic sugar yields ranging from 50–96% although each has unique challenges.23–26 Pyrolysis and processing in molten salt hydrates are accompanied by secondary reactions that degrade carbohydrates. In contrast, products from solvent liquefactions are diluted in the solvent, suppressing secondary reactions that degrade sugars. Solvent liquefaction has the ability to process wet biomass, which is challenging for most other thermochemical processes.27 However, many solvent liquefaction technologies pose practical challenges to lignocellulosic sugar production. For example, ionic liquids are extremely expensive and difficult to recover. Although deep eutectic solvents are relatively cheaper than ionic liquids, they may also involve complicated solvent recovery.28 Acid hydrolysis can lead to corrosion of equipment and can entail expensive wastewater treatment. Biomass processing in subcritical water can produce undesired humins by secondary repolymerization reactions of sugar derivatives. Use of supercritical water mitigates these secondary reactions, but adds the complexity and expense of high-pressure operation.29,30 Organic protic solvents such as primary alcohols lead to formation of functionalized sugars from biomass that needs additional upgrading strategies prior to production of biofuels.31
Polar aprotic solvents can overcome many of these challenges to producing sugars via solvent liquefaction. Many polar aprotic solvents are relatively low-cost, environmentally friendly, and can be derived from biomass. These solvents are capable of significantly reducing the apparent activation energy of cellulose depolymerization.32,33 With water as co-solvent, polar aprotic solvents can also reduce the apparent activation energy of hydrolysis of cellobiose (structural unit of cellulose) to glucose by 38% compared to water alone.34 In fact, these solvents enhance the reactivity of strong Brønsted acid catalysts (e.g. sulfuric acid), which are widely used in biomass conversion for sugar production.35 Sugar degradation and formation of humins, the product of secondary reactions of monosaccharides and/or their dehydration products,35–38 are less of a problem for polar aprotic solvents.
Luterbacher et al.39 demonstrated one of the first semi-continuous processes for deconstructing lignocellulosic biomass using a polar aprotic solvent. In their work, γ-valerolactone (GVL) with co-solvent water and acid catalyst was used as the solvent mixture to depolymerize and fractionate biomass at 157–217 °C to lead to soluble sugar monomers at 68% yield. Unfortunately, GVL is an expensive solvent, requiring almost quantitative recovery for economic viability of the process. The high boiling point of GVL (207–208 °C) necessitates complex recovery involving high pressure liquid CO2 extraction.40 Bai et al.41 has reported use of 1,4-dioxane, a relatively low boiling point polar aprotic solvent, in the solubilization of carbohydrates from switchgrass. Since this process employs high temperatures, operation at high pressures is required to prevent the solvent from boiling. It also entails a complicated solvent recovery process since the solvent forms an azeotrope with water.
We have recently shown that addition of dilute acid catalyst to aprotic solvents eliminates much of the performance difference among them, regardless of polarity and boiling points.42 This previous work points the way to developing a low temperature, and low pressure solvent liquefaction process.
In the present study, we report a new approach to solvent liquefaction that offers enhanced production of lignocellulosic sugars from red oak, which represents a hardwood type forestry residue, using tetrahydrofuran (THF), a low polarity aprotic solvent, with co-solvent water and dilute sulfuric acid as catalyst. The boiling point of THF is only 66 °C, offering the prospect of a one-step process that is efficient in deconstructing biomass and entails low-cost solvent recovery. THF, a relatively inexpensive solvent, is miscible with water at typical solvent liquefaction conditions. It can be produced from hemicellulose-sourced furfural through hydrogenation.43,44 THF is considerably less toxic than many widely used polar aprotic solvents such as dimethyl formamide, methylene chloride and dimethyl sulfoxide,45 with an Occupational Safety and Health Administration permissible exposure limit of 200 ppm.
In light of our recent work with acid-catalyzed cellulose solubilization in low polarity aprotic solvent and water, we hypothesize that a mixture of THF and water in the presence of dilute acid catalyst can facilitate biomass depolymerization at temperatures less than 300 °C.56 However, at these low temperatures, lignin is not readily dislodged from the polymeric matrix of biomass, an important requirement for effective carbohydrate solubilization. The present study further hypothesizes that biomass pretreatment also using a solution of THF, water, and acid can solubilize part of the lignin and hemicellulose to produce a cellulose-rich feedstock more susceptible to subsequent carbohydrate solubilization. Previous research shows that the high affinity of lignin and phenolic compounds to THF makes it an effective delignification agent.12,47–51 Nguyen et al.12 have showed that pretreatment of biomass via THF-based solvent processing improves enzymatic hydrolysis of lignocellulosic biomass. The goals of this study are two-fold: (1) evaluate a two-step THF solvent liquefaction process to enhance cellulosic sugar production at low temperature; and (2) demonstrate the effectiveness of a simple solvent recovery method.
2. Experimental section
2.1 Materials
As detailed in Table 1, the composition of the red oak used in this study includes 40.00 wt% glucan, 15.66 wt% xylan, 23.24 wt% lignin. The details of the analytical procedure for quantifying the biomass components are given in ESI† (section 1). The air-dried red oak sample was ground and sieved to particle size of 300–710 μm. D-Glucose (purity >99%) was obtained from Fisher Scientific and D-xylose (purity >99%) and D-sorbitol (purity >98%) were shipped from Acros Organics. Carbosynth was the source of levoglucosan (LG, purity >99.2%) and cellobiosan (purity >98.7%) for the study. Furfural (purity >99%) and 5-hydroxymethylfurfural (5-HMF, purity >99%) were purchased from Sigma Aldrich. Levoglucosan also served as the calibration standard for its isomer product, 1,6-anhydro-β-D-glucofuranose (AGF). HPLC grade and submicron filtered solvents tetrahydrofuran (THF), ethyl acetate, acetone and methanol were all purchased from Fisher Scientific. Additionally, 96.6 wt% sulfuric acid (H2SO4) was obtained from Fisher Scientific. Deionized (DI) water at 18.2 MΩ was supplied on-site in the laboratory.| Component | Composition (wt%) |
|---|---|
| Determined by Celignis Analytical. | |
| Glucan | 40.00 |
| Xylan | 15.66 |
| Mannan | 1.30 |
| Arabinan | 0.34 |
| Galactan | 0.92 |
| Klason lignin | 20.29 |
| Acid-soluble lignin | 2.95 |
| Extractives | 6.85 |
| Ash | 0.40 |
| Moisture | 7.30 |
| Total mass | 96.01 |
2.2 Two-stage solvent liquefaction process
The solvent liquefaction of red oak was conducted in a two-step process, a pretreatment step and a carbohydrate solubilization step, as illustrated in Fig. 1.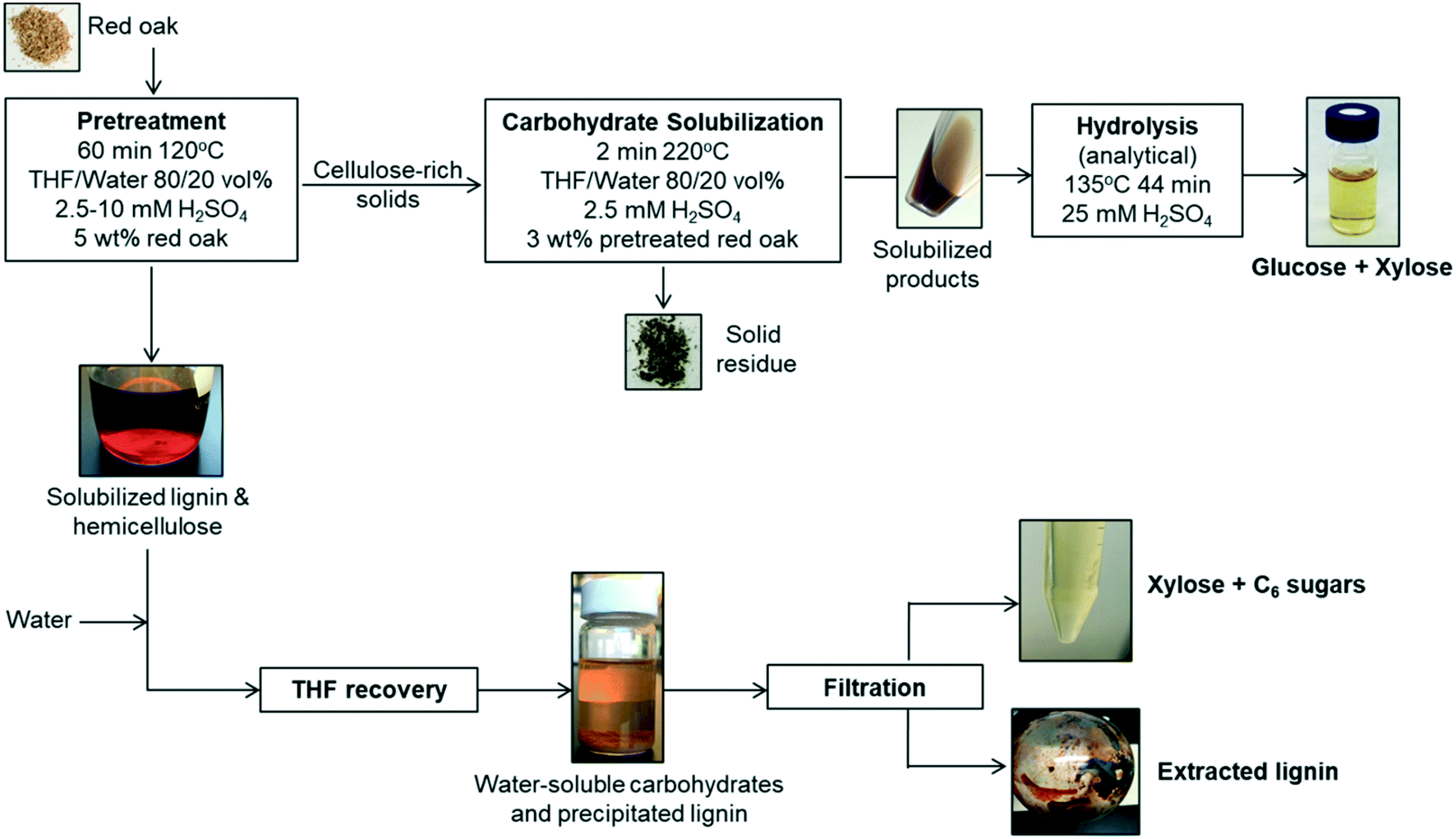 | ||
| Fig. 1 Process flow diagram of two-step solvent liquefaction of red oak for production of fermentable, solubilized carbohydrates using THF/water and sulfuric acid catalyst integrating lignin extraction and sugar recovery. | ||
Solids and liquid products from the pretreatment reaction were separated by vacuum filtration using a 0.45 μm filter paper. The filter paper and residuals were rinsed with acetone to wash out any residual solvent (THF, water). The filtrate was collected and used for recovery of a solubilized lignin and xylose-rich fraction. The solids collected on the filter paper were dried at 105 °C for approximately 4 h to drive off any residual acetone and subsequently weighed to determine the yield of pretreated red oak.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volumetric ratio, which caused the lignin to precipitate over 24 hours (see Fig. 1). This solid–liquid mixture was placed in a rotary evaporator (Heidolph, Germany) maintained at 25 °C and reduced pressure of 90 mbar to flash the THF to vapor. A chiller at −10 °C was used to condense the THF vapor in a collection flask. Upon removal of THF after 40 min of rotary evaporation operation, the remaining liquid separated into two distinct phases: a water-soluble (clear) solution and dark brown solid precipitate. The solid precipitate was dried at 50 °C for 2 days and then at 75 °C for 1 day. The dried solid was weighed for mass of extracted lignin. Lignin extraction was computed according to:
:1 volumetric ratio, which caused the lignin to precipitate over 24 hours (see Fig. 1). This solid–liquid mixture was placed in a rotary evaporator (Heidolph, Germany) maintained at 25 °C and reduced pressure of 90 mbar to flash the THF to vapor. A chiller at −10 °C was used to condense the THF vapor in a collection flask. Upon removal of THF after 40 min of rotary evaporation operation, the remaining liquid separated into two distinct phases: a water-soluble (clear) solution and dark brown solid precipitate. The solid precipitate was dried at 50 °C for 2 days and then at 75 °C for 1 day. The dried solid was weighed for mass of extracted lignin. Lignin extraction was computed according to: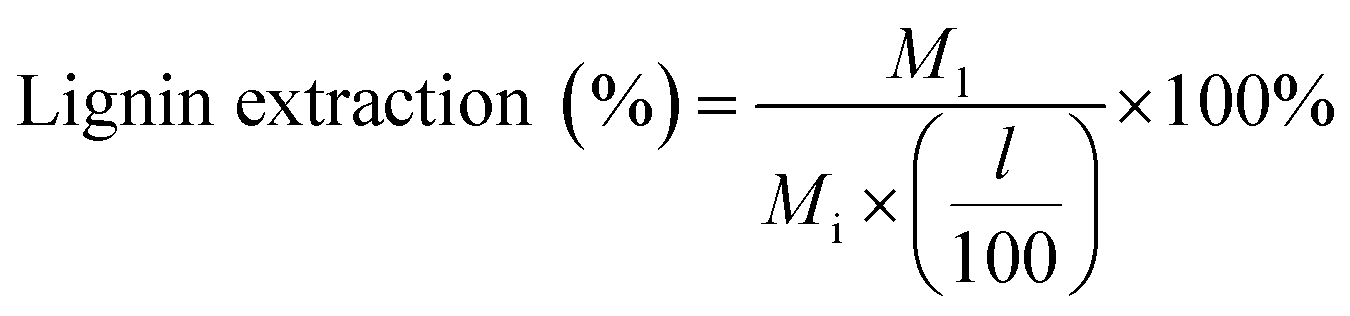 | (1) |
M l = mass of lignin extracted from pretreatment liquid product (mg)
l = lignin content of biomass feedstock on an extractives and ash free basis (wt%)
M i = initial mass of biomass feedstock (mg).
The water-soluble fraction of the liquid after lignin extraction was hydrolyzed using an analytical dilute acid hydrolysis method, as described in ESI† (section 2) to determine the total fermentable sugars present in the liquid. Whatman glass fiber syringe-filters of pore size 0.45 μm were used to sample the water-soluble fraction of the liquid for the purpose of quantifying carbohydrate products prior to the analytical hydrolysis method. The process of pretreatment was duplicated for one condition of pretreatment (60 min pretreatment with 10 mM sulfuric acid) and the lignin extraction and carbohydrate yields were reported based on the average results with standard error within 10% of mean.
Solids were filtered from the liquid and dried in an oven at 75 °C overnight. The separated liquid and dried solids were weighed using an analytical scale (Mettler Toledo, XPE205 DeltaRange® analytical balance, 220 g maximum capacity, readability 0.01 mg). Gas products were quantified by the difference in reactor mass before and after venting non-condensable gases from the cooled reactor:
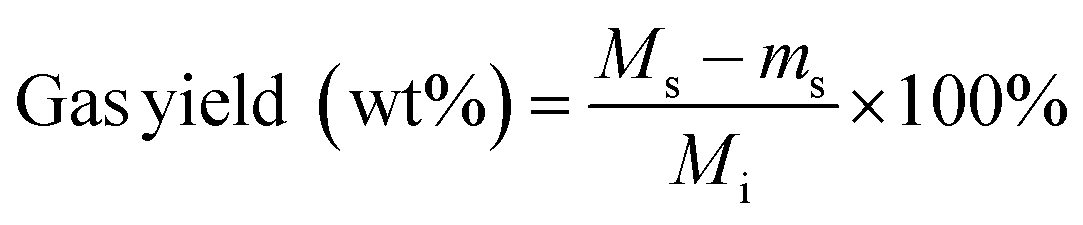 | (2) |
Solubilized product yields were calculated according to:
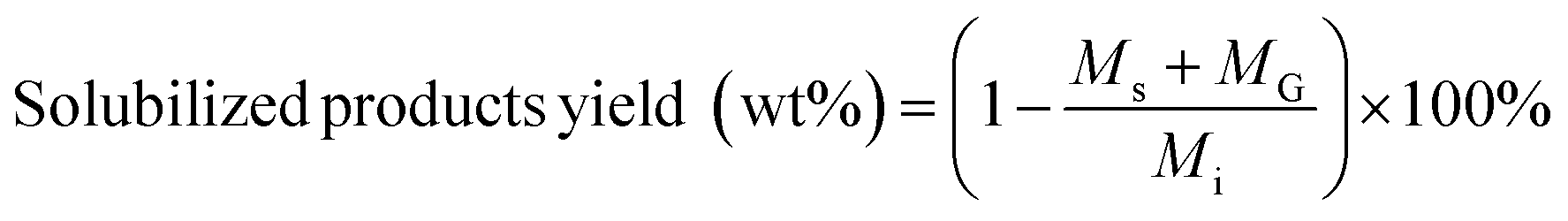 | (3) |
M s = change in mass of reactor content after processing feedstock and solvent (mg)
m s = change in mass of reactor content after processing only solvent (mg)
M s = mass of solid product of carbohydrate solubilization (mg)
M G = mass of gas product of carbohydrate solubilization (mg)
M i = initial mass of biomass feedstock (mg).
Each test was at least duplicated and standard errors of the presented yields in this work were within 10% of the mean yields.
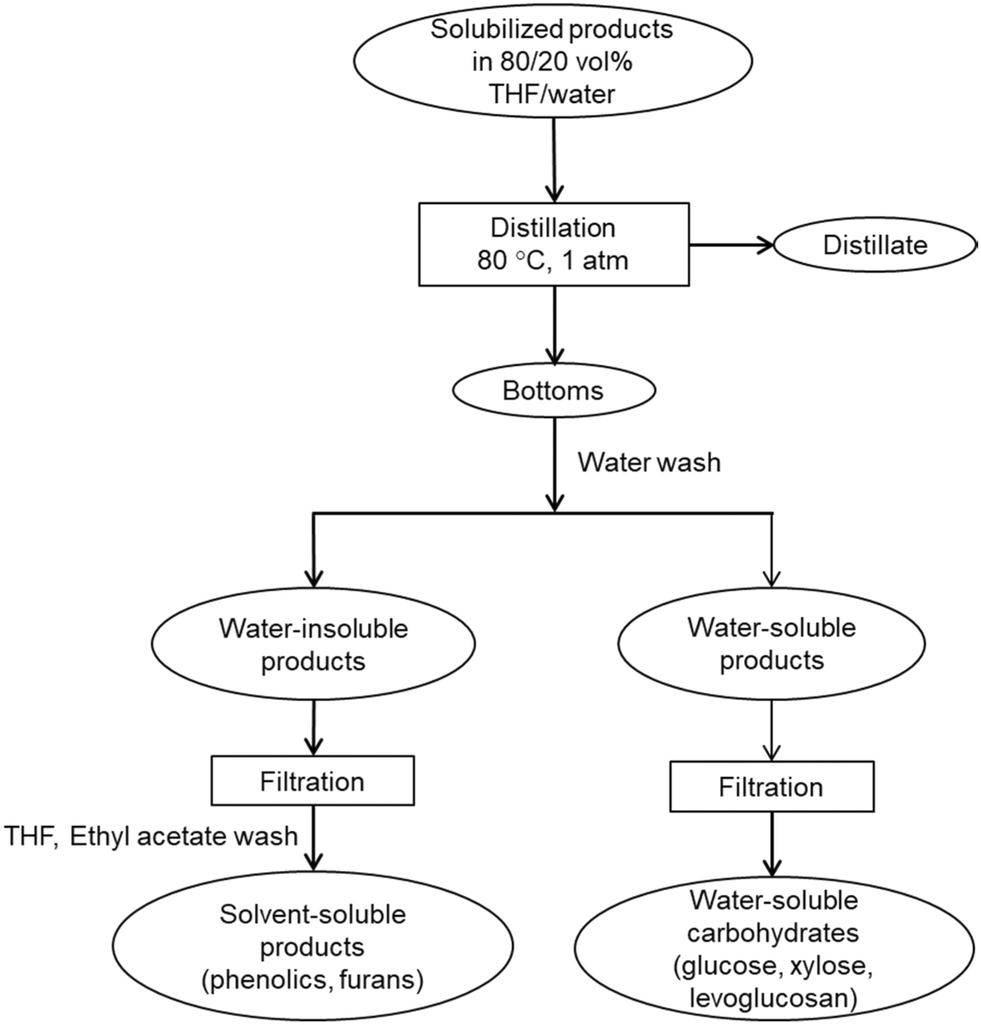 | ||
| Fig. 2 Schematic of distillation setup for recovery of THF and solubilized carbohydrates. | ||
The distillate was collected and analyzed for sugars and THF purity. Fresh DI water was added into the bottom fraction of distillation to dissolve the water-soluble products and precipitate water-insoluble products. The phenolic products were recovered by diluting in solvents, THF and ethyl acetate. The solvent-soluble products were filtered and analyzed for phenolic monomers and furan. Presence of phenolic oligomers was also investigated using molecular weight distribution of distillation bottoms. The water-soluble products were filtered and analyzed to determine recovery of solubilized carbohydrates and presence of trace amounts of furans and phenolic monomers.
2.3 Analytical methods
Carbohydrate products such as glucose, xylose, LG, sorbitol and cellobiosan from both pretreatment and carbohydrate solubilization steps were identified and quantified using high performance liquid chromatography (HPLC) system. For this study, HyperREZ XP Carbohydrate H+ 8 μm (300 × 7.7 mm) column was used at 8 bar pressure at 25 °C with ultrapure 18.2 MΩ deionized water as the mobile phase with 0.2 mL min−1 flow rate. Refractive index (RI) detector and appropriate calibration standards were used for identification and quantification of the sugars. Details of the above HPLC method, calibration and detector response characteristics are given in ESI† section 2 with Fig. S2 and Tables S1 and S2. All C5 and C6 carbohydrate products, detected in HPLC before and after analytical hydrolysis, including LG, cellobiosan and sorbitol were transformed into their equivalent glucose or xylose amounts before determining the theoretical yields of carbohydrates for a consistent basis of our analysis. Conversion factors of 1.111 for LG, 0.989 for sorbitol and 1.111 for cellobiosan were used for the above analysis. Solubilized carbohydrates yields were expressed as theoretical yields as follows: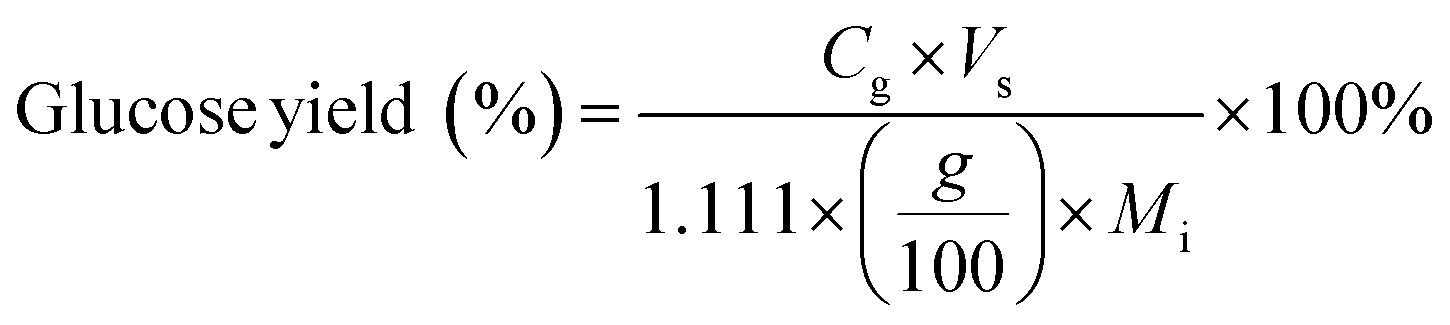 | (4) |
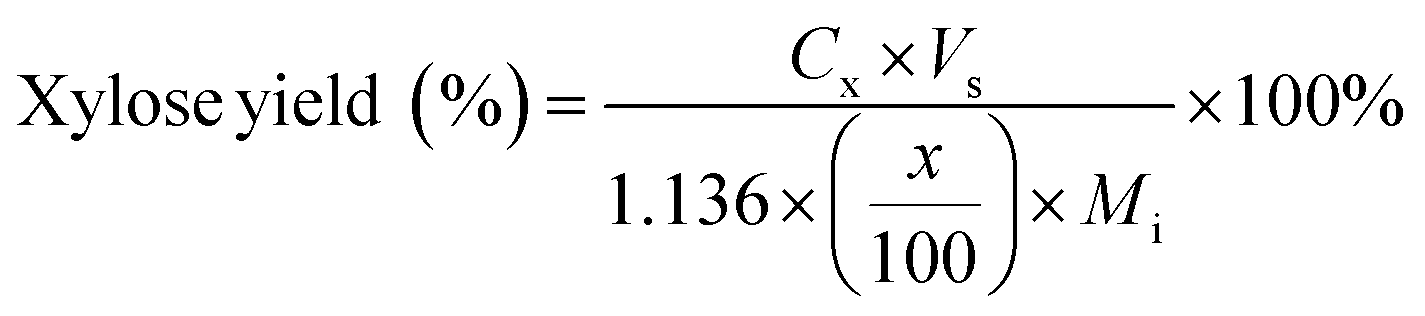 | (5) |
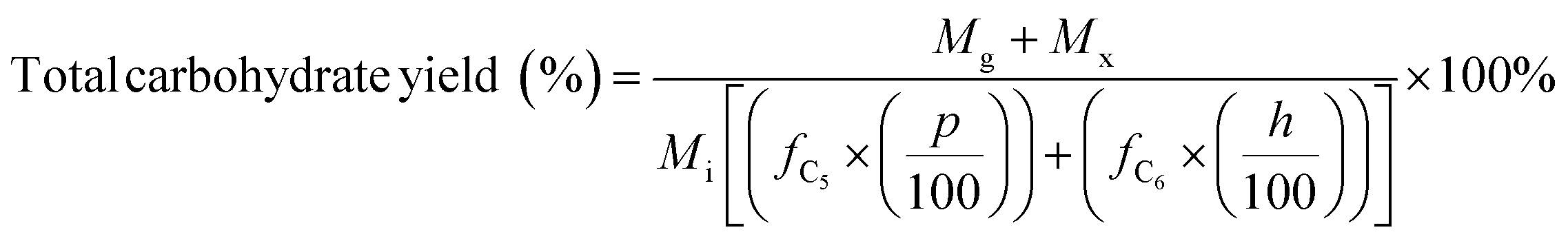 | (6) |
C g, Cx = concentration of glucose and xylose, respectively, in solution (mg mL−1)
V s = volume of solution (mL)
g, x = glucan and xylan content of biomass feedstock, respectively (wt%)
M i = initial mass of biomass feedstock (mg)
M g, Mx = mass of glucose and xylose product, respectively (mg)
f C5, fC6 = conversion factor for hydrolysis of pentosan (1.136) and hexosan (1.111), to C5 and C6 monosaccharides, respectively
p, h = pentosan and hexosan content of biomass feedstock, respectively (wt%).
A gas chromatograph with mass spectrometer and flame ionization detector (Agilent 7890B GC-MS/FID) was employed to analyze furfural and 5-HMF, carbohydrate monomer AGF, and phenolic monomers. Carbohydrate products and phenolic monomers were identified using the MS followed by quantification of only the carbohydrate products using the FID with appropriate standards. Details of this GC-MS/FID instrument is given elsewhere.32 Mass yields of furfural, 5-HMF and AGF were calculated according to:
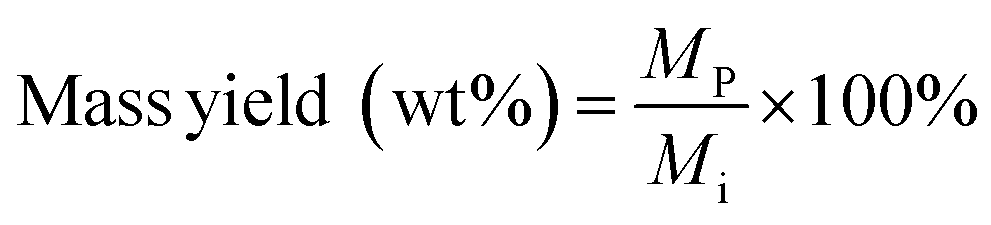 | (7) |
M p = mass of product (mg)
M i = initial mass of biomass feedstock (mg).
Gel permeation chromatography (GPC) was employed to determine molecular weight distribution of phenolic compounds in solubilized products of pretreatment and carbohydrate solubilization, extracted lignin, and the bottoms and water-soluble products from distillation. GPC was conducted using Dionex Ultimate 3000 series HPLC system equipped with a Shodex refractive index (RI) detector and diode array detector (DAD) where THF was used as the eluent with a flow rate of 1 mL min−1. Since phenolic monomers could co-elute with levoglucosan during GPC, they were analyzed using GC-MS. The GPC spectrum was only used for analyzing phenolic dimers and phenolic oligomers. Details of the GPC methodology and product analysis is found in section 3 of the ESI.†
Field-emission scanning electron microscopy (FEI Quanta FEG 250 SEM) was used to generate images of untreated and pretreated red oak. Due to a highly heterogeneous structure of lignocellulosic biomass, areas of common features were compared between biomass particles. Air-dried samples were mounted with carbon tape. The samples were sputter coated with Ir. A beam acceleration voltage of 10 kV was used for imaging the surface structures of the samples.
3. Results and discussion
3.1 Enhanced carbohydrate production from two-step liquefaction process
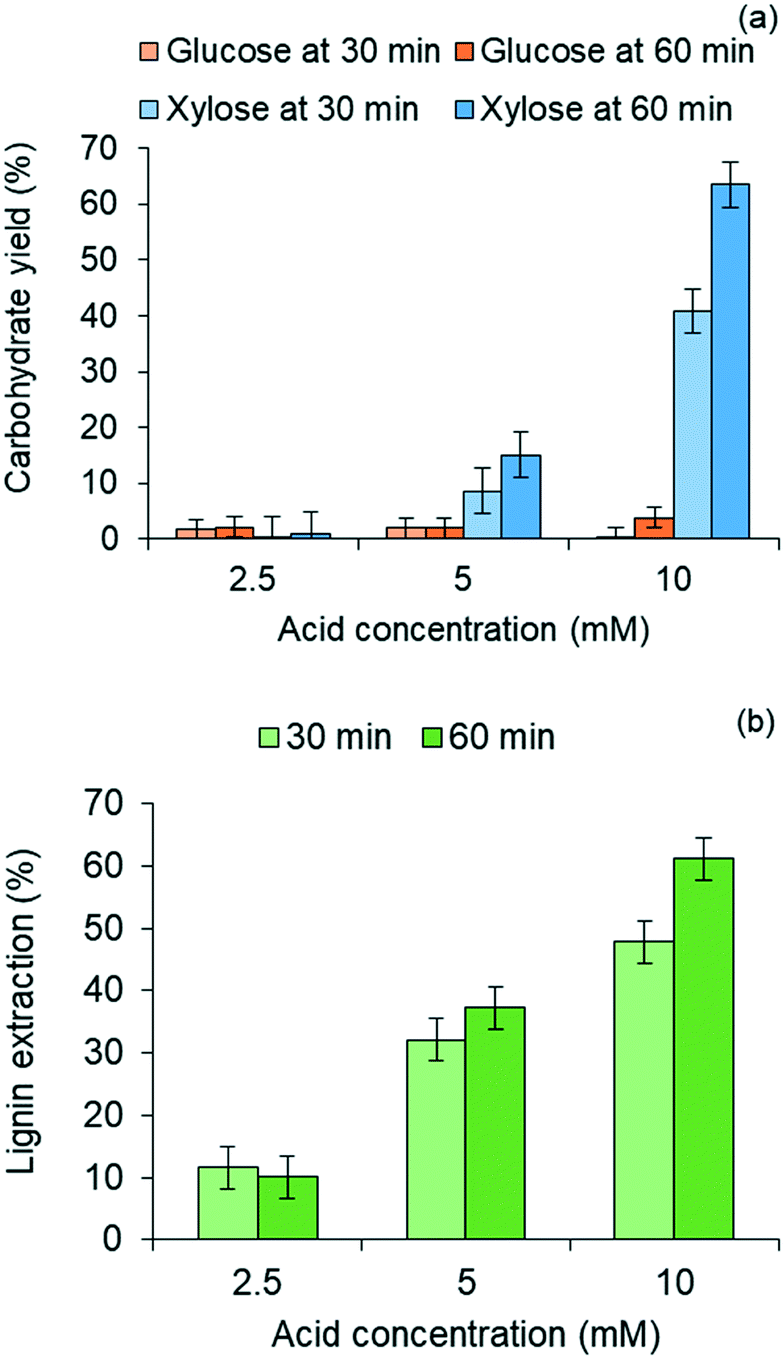 | ||
| Fig. 3 Results from pretreatment of red oak in 80/20 vol% THF/water mixture at 120 °C as a function of reaction time and sulfuric acid concentration (a) C5 and C6 carbohydrate yields; (b) lignin extraction. | ||
Lignin residues extracted from the water-insoluble fraction of the solubilized product of red oak pretreatment is illustrated in Fig. 3(b) as a function of acid catalyst concentration and reaction times of pretreatment. Like xylose recovery, higher acid concentration and longer pretreatment time gave rise to higher lignin extraction. The effect of acid concentration on xylose yields was greater than the effect of pretreatment time. Maximum lignin extraction of 61.1% corresponded to the maximum xylose yield. The pretreated red oak solids yields decreased with increasing pretreatment temperature and time due to increased solubilization of hemicellulose and lignin, and were collected at 56 wt% yields at 10 mM acid concentration and 60 min of pretreatment (ESI,† section 4, Fig. S3).
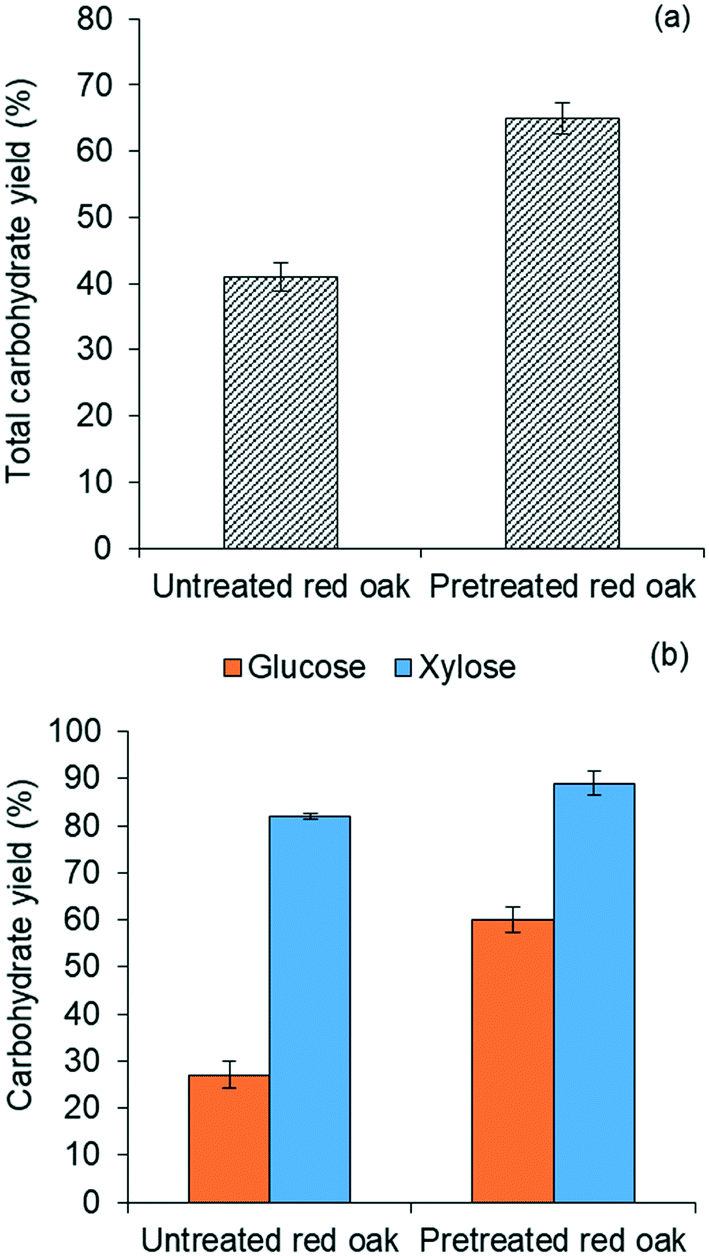 | ||
| Fig. 4 (a) Total solubilized carbohydrates yield and (b) glucose and xylose yields from combined pretreatment and carbohydrate solubilization using 80/20 vol% THF/water and 2.5 mM H2SO4 at 220 °C for 2 min reaction time. | ||
Fig. 4(b) further illustrates that yields of both glucose and xylose from carbohydrate solubilization of pretreated red oak were higher than for untreated red oak. However, glucose yields demonstrated a dramatic improvement, increasing from 27% to 60% as a result of biomass pretreatment while xylose yield as a result of pretreatment only increased from 82% to 89%. Thus, the enhancement of total carbohydrate yield from carbohydrate solubilization of pretreated biomass could be chiefly attributed to increased glucose production.
3.2 Product distribution of combined pretreatment and carbohydrate solubilization
| Productsa | Yieldb,c (wt%) |
|---|---|
| a Reaction was performed at 220 °C for 2 min using 80/20 vol% THF/water and 2.5 mM H2SO4. b Yields of products from combined pretreatment and carbohydrate solubilization. c Based on initial red oak. | |
| C6 sugars | 29.03 |
| C5 sugars | 14.78 |
| 5-HMF | 5.10 |
| Furfural | 4.30 |
| Lignin extracted | 14.20 |
| Solid residue | 14.30 |
| Other | 14.55 |
| Total products | 96.26 |
The molecular weight of lignin extracted by the pretreatment, as shown in Table S3,† was 1593 Da, a value that falls between those for milled and organosolv red oak lignin.54,55 The average molecular weight of THF-extracted lignin was also in the range reported for lignin extracted using GVL.54 This suggests that lignin extracted by THF/water pretreatment was less modified than occurs during organosolv processing. Furthermore, analytical pyrolysis of the extracted lignin from pretreatment revealed a large proportion of phenolic compounds, furfural, and a small amount of levoglucosan (see ESI,† section 7, Fig. S6(d)). The furfural in extracted lignin was likely derived from the water-insoluble xylo-oligomers, co-precipitated during lignin extraction after pretreatment. Analytical pyrolysis of the solid residue from carbohydrate solubilization showed a relatively reduced proportion of phenolic compounds (see ESI,† section 7, Fig. S6(c)). Most of the products from the analytical pyrolysis of the solid residue are typical of pyrolysis of carbohydrate, arising from unconverted C5 and C6 polysaccharides in the biomass. The above results show an incomplete but successful fractionation of lignin and carbohydrates of biomass in the two-step THF liquefaction process.
3.3 Impact of delignification in carbohydrate solubilization of biomass
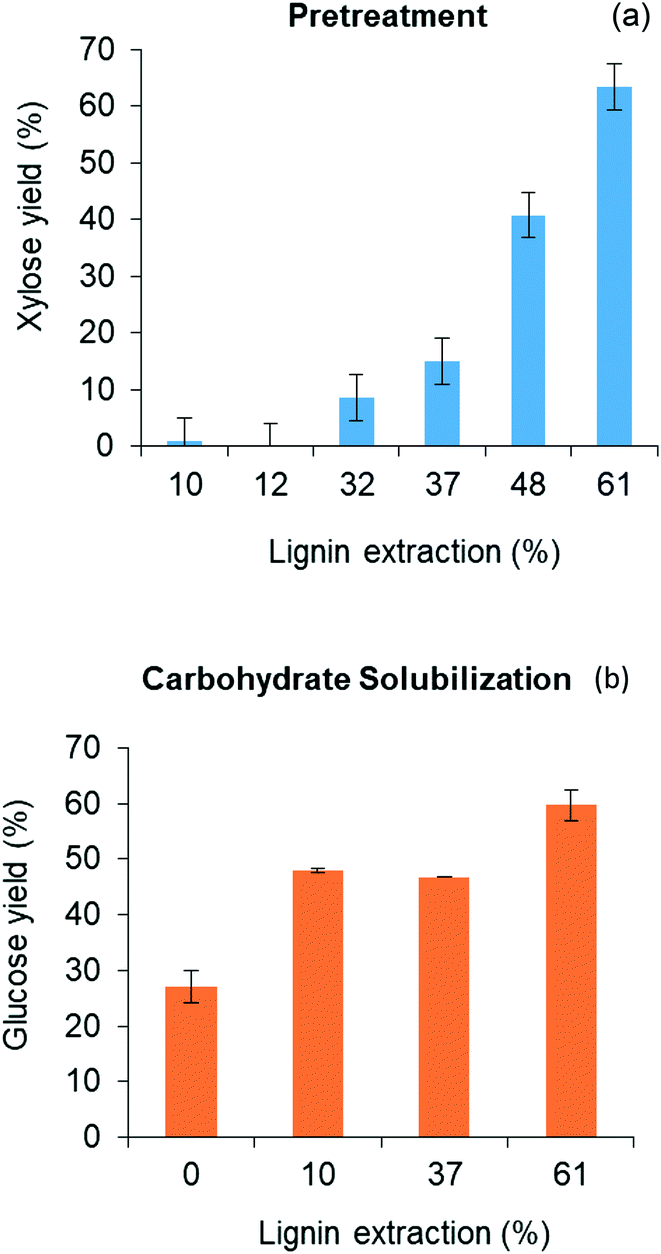 | ||
| Fig. 5 Sugar yields from two-step solvent liquefaction process: (a) xylose from pretreatment step; (b) glucose carbohydrate solubilization step. | ||
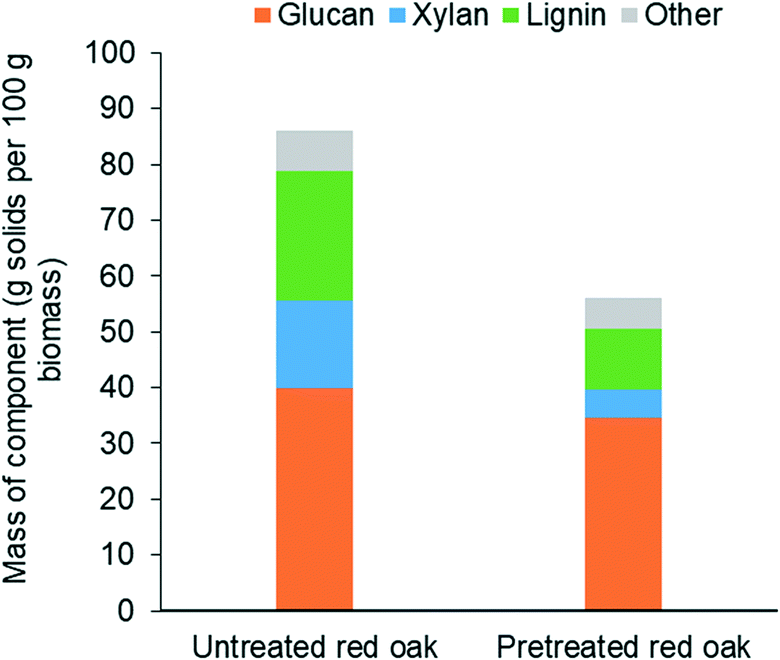 | ||
| Fig. 6 Composition of untreated and pretreated red oak solids on dry basis. | ||
Fig. 6 shows that THF/water/acid pretreatment substantially altered the composition of the biomass. While glucan content reduced by only 14% due to pretreatment, loss of xylan and lignin during pretreatment were as high as by 67% and 53%, respectively. This clearly indicates that THF/water/acid pretreatment could effectively solubilize hemicellulose and lignin fractions of biomass, rendering the pretreated biomass highly enriched with glucan, representing over 60 wt% of the delignified biomass. This cellulose-rich substrate is expected to be readily deconstructed during carbohydrate solubilization in low polarity solvents.46
 | ||
| Fig. 7 SEM images of red oak from different stages of process (a) untreated red oak, (b) pretreated red oak (example 1) and (c) pretreated red oak area (example 2). | ||
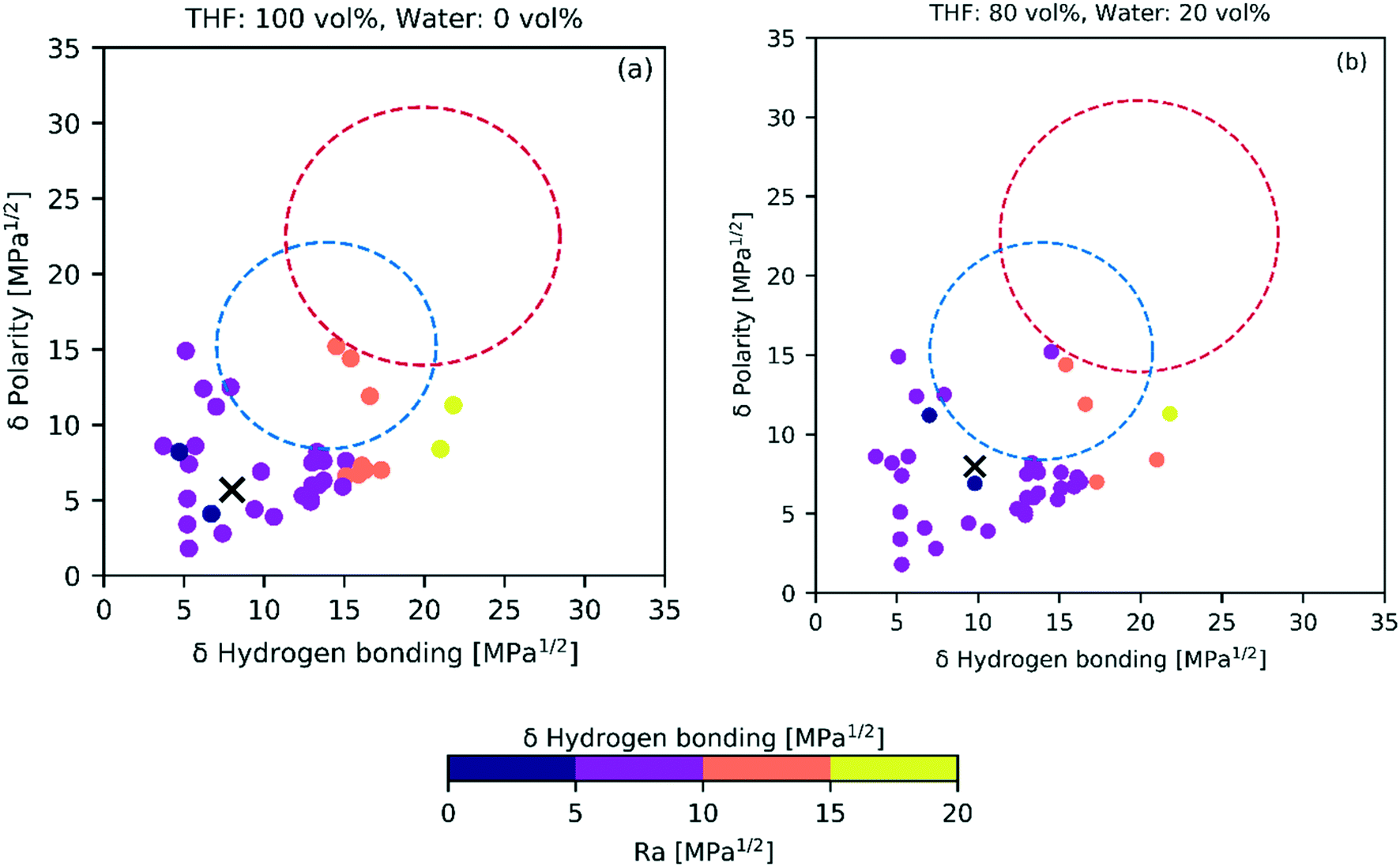 | ||
| Fig. 8 The HSP distance (Ra) between solvent (cross mark) and biomass products (colored circles) indicates potential solubility in the solvent, where the shorter distances (darker colors) likely have better solvation. This solvation potential changes as tetrahydrofuran (a) is blended in an 80:20 vol% ratio with water (b). Blue and red dashed circles represent lignin and cellulose, respectively. | ||
3.4 Solvent separation and sugar recovery
A simple distillation technique readily separated THF and facilitated recovery of carbohydrate products from the liquid product from carbohydrate solubilization of pretreated red oak. Fig. 9(a) and (b) illustrate that glucose, xylose and levoglucosan in the distillation feed partitioned to the bottom fraction of the distillation products. On the other hand, the distillate fraction contained no solubilized carbohydrates.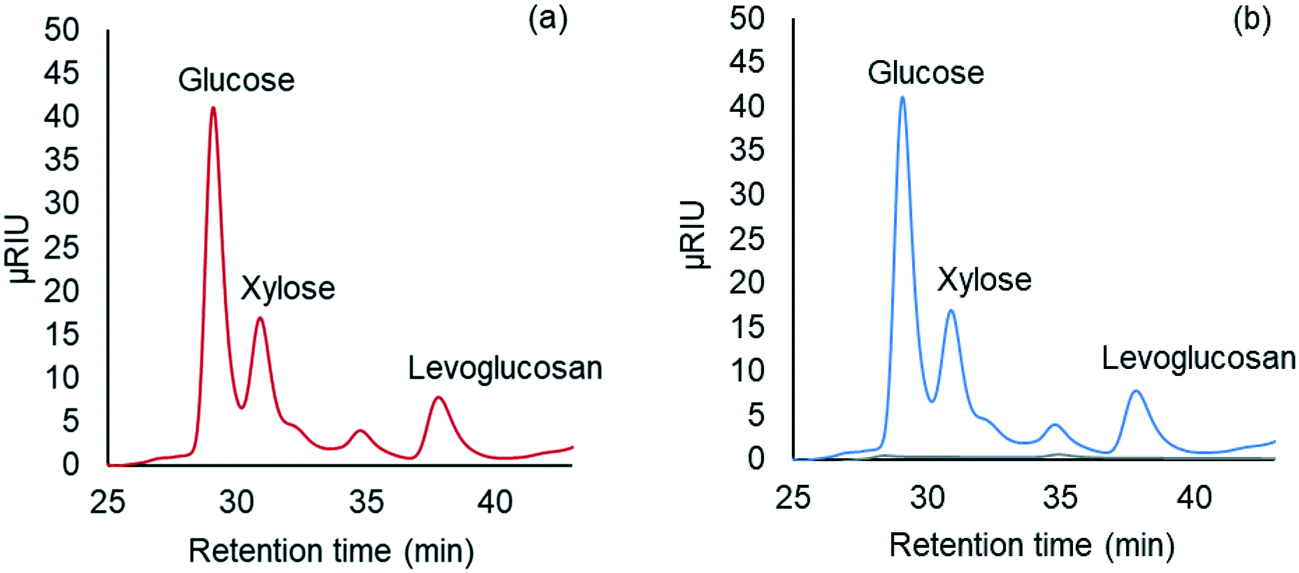 | ||
| Fig. 9 HPLC chromatogram of (a) solubilized products fed to distillation (red line), and of (b) products recovered after distillation. Blue and grey lines represent bottom products of distillation containing water-soluble products and distillate containing THF, respectively. | ||
As shown in Table 3 recovery of glucose, xylose and levoglucosan in the bottom fraction individually was 106%, 101%, and 83%, respectively while the total recovery of these above sugars was 99% in the same fraction. Loss of xylose and levoglucosan could be attributed to minor hydrolysis or degradation occurring during distillation at bench-scale setup. This effect could easily be avoided in a flashing unit at large-scale operation for THF separation from the solubilized products of biomass.
| Distillation fraction | Contents | THF recovery | Sugar recovery |
|---|---|---|---|
| Distillate | THF (almost pure, with low level of contaminants) | 106.3% | 0.0% |
| Bottom | Water and recovered products of carbohydrate solubilization | 0.3% | Glucose: 106 ± 4% |
| Xylose: 101 ± 10% | |||
| Levoglucosan: 83 ± 5% | |||
| Total sugars: 99 ± 7% |
Table 3 also shows that THF could be recovered at high efficiency by one-step simple distillation. Overall recovery of the solvent in distillate fraction was 106.3% while a trace amount of 0.3% remained in the bottom fraction after distillation. The recovered THF was almost pure and should be easy to recycle to the pretreatment and carbohydrate solubilization units for large-scale applications. The detailed mass balance of THF distillation and images of distillation fractions are presented in ESI† (section 8, Table S4 and Fig. S7). The bottom fraction of distillation appeared dark brown in color, which could be due to presence of phenolic species. In this work, GPC of bottom product indicates that phenolic oligomers were likely present in this fraction (Fig. S5†). However, after washing the bottom products with water, no phenolic oligomers were found in the water-soluble products (filtered) based on GPC analysis (ESI,† section 6, Fig. S5(e)). Additionally, GC-MS of filtered water-soluble products showed no evidence of phenolic monomers and furanic compounds in it. For a more detailed analysis of the phenolics, an analytical Folin–Ciocalteu method can also be employed to further confirm the presence of the phenolic species and determine their mass fraction in the distillation bottom fraction.57
3.5 Prospects for sugar production via THF-based solvent liquefaction of lignocellulose
Two-step THF-based solvent liquefaction of biomass offers several advantages for cellulosic sugar production. THF is a relatively inexpensive, low-toxicity, and biomass-derived polar aprotic solvent that is miscible with water at typical solvent liquefaction conditions and can be readily recovered at low cost owing to its low boiling point.58–60 We also investigated thermal stability of THF at the reaction conditions employed. GC-FID peak area analysis indicates that only 0.1% of THF decomposed during each cycle of carbohydrate solubilization at 220 °C producing mainly 1,4-butanediol. Thus, the acid catalyzed THF/water system represents a robust, recyclable and sustainable solvent medium for producing lignocellulosic sugars.From a biorefinery perspective, the THF liquefaction process could enable successful fractionation and utilization of biomass components by producing high-purity streams of monomeric C5 and C6 sugars via rapid and selective solubilization and depolymerization of hemicellulose and cellulose, respectively, and a high-quality technical lignin as the byproduct that can be further upgraded to fuels or chemicals.61 One of the unique advantages of THF liquefaction process is its prospects for high volumetric sugar productivity, defined as the mass production rate of sugar per unit of reactor volume. Volumetric sugar productivity in these laboratory-scale experiments reached 2334 g L−1 h−1, orders of magnitude greater than is possible with conventional enzymatic hydrolysis and other solvent liquefaction processes. Enzymatic hydrolysis, despite >70% sugar yields, exhibits volumetric sugar productivity of only 0.7 g L−1 h−1 for sugar production owing to a considerably slow rate of bioconversion.62 Even the non-enzymatic GVL liquefaction process only achieves 23 g L−1 h−1 in the production of cellulosic sugars.39 Whether the impressive volumetric sugar yields achieved with this two-step process in the laboratory can be replicated at commercial-scale would appear to be heat transfer limited for this endothermic process rather than chemical reaction rate limited.
Our work further demonstrated total solubilized carbohydrates and glucose could be produced at concentrations of 125 g L−1 and 91 g L−1, respectively. The glucose concentration are well within the titer requirement for fermentation to ethanol.
A concept for cellulosic ethanol production using the proposed two-step process for solvent liquefaction of lignocellulose into sugars is illustrated in Fig. 10. Wet biomass is directly loaded in a high-solids reactor (e.g. 20 wt% solids loading) that is heated to pretreatment temperature by steam injection in a manner similar to a steam explosion reactor.63 This ensures rapid heating and uniform temperature. Additionally, this type of reactor is able to use larger particles of biomass and hence obviates the need for energy-intensive comminution processes.64 Moreover, pressure release at the end of the process assures rapid cooling of products. The pretreated biomass is separated and transferred to the carbohydrate solubilization unit.
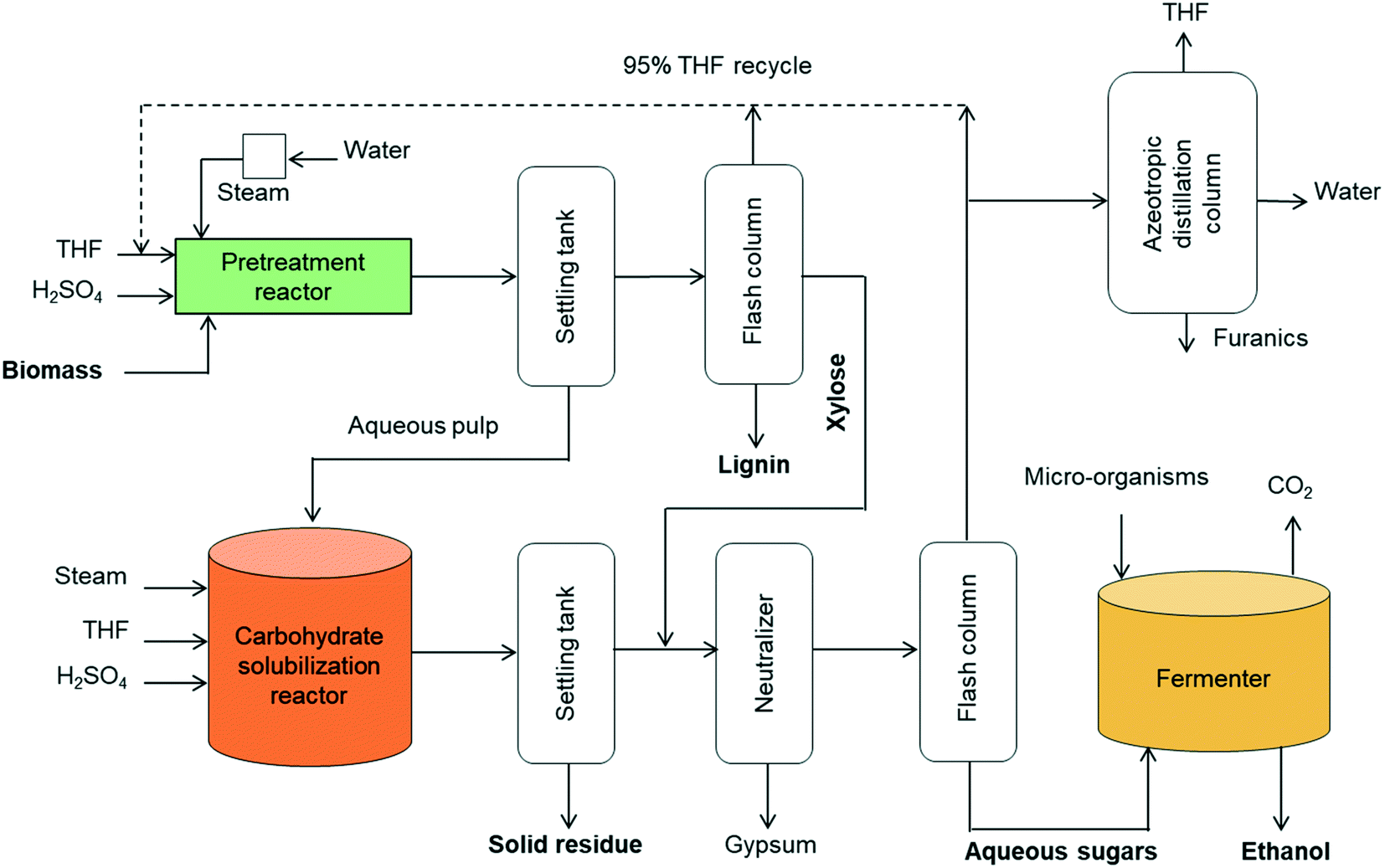 | ||
| Fig. 10 Block diagram of cellulosic ethanol plant based on two-stage THF-based solvent liquefaction of lignocellulose. | ||
The liquid from the settling tank (top fraction) is flashed to recover THF, which also precipitates lignin. The recovered lignin is clean and only moderately modified, making it attractive for manufacture of chemicals and fuels or burned as solid fuel for stationary power generation. The water-soluble stream from the distillation column is rich in xylose, which can be blended with the products from the carbohydrate solubilization reactor for subsequent fermentation as a mixture of C6 and C5 sugars.
Carbohydrate solubilization of the pretreated biomass can also be performed in a steam-injected explosion reactor with fast heating and cooling capabilities. Solubilized products from this reactor are delivered to a neutralization unit where lime neutralizes the sulfuric acid. The liquid is flashed to produce a sugar-rich aqueous fraction and THF-rich fraction. The THF fraction includes a small amount of water, but further drying is unnecessary before recycling to the pretreatment and carbohydrate solubilization reactors because these employ THF/water mixtures containing 20 vol% water. However, further separation of THF from water and furanics (furfural, 5-HMF) could be achieved via azeotropic distillation.
The water-soluble fraction from the flash column containing glucose, levoglucosan and xylose are highly concentrated in the aqueous phase after removal of THF. This sugar stream is delivered to the fermenter for production of ethanol. Conventional yeast (e.g. Saccharomyces cerevisiae) can be employed for conversion of glucose whereas engineered microorganisms would typically be employed to ferment xylose and levoglucosan.6,65 In some cases, separate fermenters for C5 and C6 carbohydrates may be desired. Alternatively, anhydrosugars in the solubilized carbohydrates could be hydrolyzed to fully hydrated sugars (glucose and xylose) prior to sending them to the fermenter.
Conclusions
We have developed a novel pathway for saccharification of hardwood biomass using THF/water and dilute acid to overcome current challenges in biological and thermochemical conversion of lignocellulose to sugars. The pretreatment step in this two-step process removed up to 61% of the lignin and 64% of the hemicellulose, producing a cellulose-rich solid that was rapidly converted in a second solvent process at relatively moderate temperatures to fermentable carbohydrates at yields up to 65%. This two-step solvent liquefaction process achieved average glucose and xylose yields of 60% and 89% xylose, respectively. These yields are comparable to biological and other kinds of thermochemical saccharification processes while achieving volumetric sugar productivity orders of magnitude higher than possible through enzymatic hydrolysis or the GVL process. Furthermore, THF was not significantly degraded in the pretreatment or carbohydrate solubilization step and was readily recovered by one-step distillation. The solubilized carbohydrates were extracted at 99% recovery in water with no phenolic and furanic contaminants present after distilling THF.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support of the Iowa Energy Center. We would like to thank Nicholas Chierici for his help in conducting pretreatment and carbohydrate solubilization experiments as well as analyzing products. The authors also greatly appreciate the contribution of Kelley Okoren, Jordan Funkhouser, Tannon Daugaard, Preston Gamble, Lysle Whitmer, Taylor Schulz, Patrick Johnston, Marjorie Rover, Patrick Hall, Ryan Smith, Daniel Hayes (Celignis Analytical) and Warren Straszheim (Materials Analysis and Research Laboratory at Iowa State University) in setting up the experimental apparatus, analyzing products, generating SEM images, and providing help with preparing biomass samples. We sincerely thank Dr. Jeremy Luterbacher (École polytechnique fédérale de Lausanne), Dr. Peter Ciesielski (National Renewable Energy Laboratory), Dr. Gregg T. Beckham (National Renewable Energy Laboratory), and Dr. Brent H. Shanks (Iowa State University) for insightful remarks and constructive suggestions on this work.References
- R. D. Perlack, L. L. Wright, A. F. Turhollow and R. L. Graham, Biomass as Feedstock for A Bioenergy and Bioproducts Industry, 2005, DOI:10.1007/s13398-014-0173-7.2.
- P. January, R. L. Graham, R. Nelson, J. Sheehan, R. D. Perlack and L. L. Wright, Current and Potential U.S. Corn Stover Supplies, Agron. J., 2007, 99(1), 1–11, DOI:10.2134/agronj2005.0222.
- P. McKendry, Energy Production from Biomass (Part 1): Overview of Biomass, Bioresour. Technol., 2002, 83(1), 37–46 CrossRef CAS PubMed.
- R. C. Brown and T. Brown, Biorenewable Resources: Engineering New Products from Agriculture, John Wiley & Sons Inc., 2nd edn, 2014, DOI:10.1002/9781118524985.
- M. Chidambaram and A. T. Bell, A Two-Step Approach for the Catalytic Conversion of Glucose to 2,5-Dimethylfuran in Ionic Liquids, Green Chem., 2010, 12(7), 1253–1262, 10.1039/c004343e.
- M. Kuyper, A. A. Winkler, J. P. Van Dijken and J. T. Pronk, Minimal Metabolic Engineering of Saccharomyces Cerevisiae for Efficient Anaerobic Xylose Fermentation: A Proof of Principle, FEMS Yeast Res., 2004, 4(6), 655–664, DOI:10.1016/j.femsyr.2004.01.003.
- M. Jarvis, Chemistry: Cellulose Stacks Up, Nature, 2003, 426(6967), 611–612, DOI:10.1038/426611a.
- Y. Nishiyama, J. Sugiyama, H. Chanzy and P. Langan, Crystal Structure and Hydrogen Bonding System in Cellulose Iβ from Synchrotron X-Ray and Neutron Fiber Diffraction, J. Am. Chem. Soc., 2003, 125(47), 14300–14306, DOI:10.1021/ja037055w.
- W. O. S. Doherty, P. Mousavioun and C. M. Fellows, Value-Adding to Cellulosic Ethanol: Lignin Polymers, Ind. Crops Prod., 2011, 33(2), 259–276, DOI:10.1016/j.indcrop.2010.10.022.
- M. E. Himmel, S.-Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Biomass Recalcitrance: Engineering Plants and Enzymes for Biofuels Production, Science, 2007, 315(5813), 804–807, DOI:10.1126/science.1137016.
- D. Klein-Marcuschamer, P. Oleskowicz-Popiel, B. A. Simmons and H. W. Blanch, The Challenge of Enzyme Cost in the Production of Lignocellulosic Biofuels, Biotechnol. Bioeng., 2012, 109(4), 1083–1087, DOI:10.1002/bit.24370.
- T. Y. Nguyen, C. M. Cai, R. Kumar and C. E. Wyman, Co-Solvent Pretreatment Reduces Costly Enzyme Requirements for High Sugar and Ethanol Yields from Lignocellulosic Biomass, ChemSusChem, 2015, 8(10), 1716–1725, DOI:10.1002/cssc.201403045.
- W. R. Grous, A. O. Converse and H. E. Grethlein, Effect of Steam Explosion Pretreatment on Pore Size and Enzymatic Hydrolysis of Poplar, Enzyme Microb. Technol., 1986, 8(5), 274–280, DOI:10.1016/0141-0229(86)90021-9.
- T. A. Lloyd and C. E. Wyman, Combined Sugar Yields for Dilute Sulfuric Acid Pretreatment of Corn Stover Followed by Enzymatic Hydrolysis of the Remaining Solids, Bioresour. Technol., 2005, 96(18 SPEC. ISS), 1967–1977, DOI:10.1016/j.biortech.2005.01.011.
- K. Öhgren, R. Bura, J. Saddler and G. Zacchi, Effect of Hemicellulose and Lignin Removal on Enzymatic Hydrolysis of Steam Pretreated Corn Stover, Bioresour. Technol., 2007, 98(13), 2503–2510, DOI:10.1016/j.biortech.2006.09.003.
- Y. Zhao, Y. Wang, J. Y. Zhu, A. Ragauskas and Y. Deng, Enhanced Enzymatic Hydrolysis of Spruce by Alkaline Pretreatment at Low Temperature, Biotechnol. Bioeng., 2008, 99(6), 1320–1328, DOI:10.1002/bit.21712.
- P. Lenihan, A. Orozco, E. O'Neill, M. N. M. Ahmad, D. W. Rooney and G. M. Walker, Dilute Acid Hydrolysis of Lignocellulosic Biomass, Chem. Eng. J., 2010, 156(2), 395–403, DOI:10.1016/j.cej.2009.10.061.
- S. Sadula, A. Athaley, W. Zheng, M. Ierapetritou and B. Saha, Process Intensification for Cellulosic Biorefineries, ChemSusChem, 2017, 10(12), 2566–2572, DOI:10.1002/cssc.201700183.
- Y. P. Wijaya, R. D. D. Putra, V. T. Widyaya, J.-M. Ha, D. J. Suh and C. S. Kim, Comparative Study on Two-Step Concentrated Acid Hydrolysis for the Extraction of Sugars from Lignocellulosic Biomass, Bioresour. Technol., 2014, 164, 221–231, DOI:10.1016/j.biortech.2014.04.084.
- J. B. Binder and R. T. Raines, Fermentable Sugars by Chemical Hydrolysis of Biomass, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(10), 4516–4521, DOI:10.1073/pnas.0912073107.
- A. Procentese, E. Johnson, V. Orr, A. Garruto Campanile, J. A. Wood, A. Marzocchella and L. Rehmann, Deep Eutectic Solvent Pretreatment and Subsequent Saccharification of Corncob, Bioresour. Technol., 2015, 192, 31–36, DOI:10.1016/j.biortech.2015.05.053.
- N. Kuzhiyil, D. Dalluge, X. Bai, K. H. Kim and R. C. Brown, Pyrolytic Sugars from Cellulosic Biomass, ChemSusChem, 2012, 5(11), 2228–2236, DOI:10.1002/cssc.201200341.
- X. Bai, P. Johnston and R. C. Brown, An Experimental Study of the Competing Processes of Evaporation and Polymerization of Levoglucosan in Cellulose Pyrolysis, J. Anal. Appl. Pyrolysis, 2013, 99, 130–136, DOI:10.1016/j.jaap.2012.10.012.
- F. Ronsse, X. Bai, W. Prins and R. C. Brown, Secondary Reactions of Levoglucosan and Char in the Fast Pyrolysis of Cellulose, Environ. Prog. Sustainable Energy, 2012, 31(2), 256–260, DOI:10.1002/ep.11633.
- S. M. Sen, J. B. Binder, R. T. Raines and C. T. Maravelias, Conversion of Biomass to Sugars via Ionic Liquid Hydrolysis: Process Synthesis and Economic Evaluation, Biofuels, Bioprod. Biorefin., 2012, 6(4), 444–452, DOI:10.1002/bbb.1336.
- M. J. Taherzadeh and K. Karimi, Acid-Based Hydrolysis Processes for Ethanol from Lignocellulosic Materials: A Review, BioResources, 2007, 2(3), 472–499 CAS.
- F. Behrendt, Y. Neubauer, M. Oevermann, B. Wilmes and N. Zobel, Direct Liquefaction of Biomass, Chem. Eng. Technol., 2008, 31(5), 667–677, DOI:10.1002/ceat.200800077.
- A. Procentese, F. Raganati, G. Olivieri, M. E. Russo, L. Rehmann and A. Marzocchella, Deep Eutectic Solvents Pretreatment of Agro-industrial Food Waste, Biotechnol. Biofuels, 2018, 11(37), 37, DOI:10.1186/s13068-018-1034-y.
- F. Salak Asghari and H. Yoshida, Acid-Catalyzed Production of 5-Hydroxymethyl Furfural from D-Fructose in Subcritical Water, Ind. Eng. Chem. Res., 2006, 45(7), 2163–2173, DOI:10.1021/ie051088y.
- S. Saka and T. Ueno, Chemical Conversion of Various Celluloses to Glucose and Its Derivatives in Supercritical Water, Cellulose, 1999, 6(3), 177–191, DOI:10.1023/A:1009232508644.
- Y. Ishikawa and S. Saka, Chemical Conversion of Cellulose as Treated in Supercritical Methanol, Cellulose, 2001, 8(3), 189–195, DOI:10.1023/A:1013170020469.
- A. Ghosh, R. C. Brown and X. Bai, Production of Solubilized Carbohydrate from Cellulose Using Non-Catalytic, Supercritical Depolymerization in Polar Aprotic Solvents, Green Chem., 2016, 18(4), 1023–1031, 10.1039/C5GC02071A.
- H. Kawamoto, W. Hatanaka and S. Saka, Thermochemical Conversion of Cellulose in Polar Solvent (Sulfolane) into Levoglucosan and Other Low Molecular-Weight Substances, J. Anal. Appl. Pyrolysis, 2003, 70(2), 303–313, DOI:10.1016/S0165-2370(02)00160-2.
- M. A. Mellmer, D. Martin Alonso, J. S. Luterbacher, J. M. R. Gallo and J. A. Dumesic, Effects of γ-Valerolactone in Hydrolysis of Lignocellulosic Biomass to Monosaccharides, Green Chem., 2014, 16(11), 4659–4662, 10.1039/C4GC01768D.
- M. A. Mellmer, C. Sener, J. M. R. Gallo, J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Solvent Effects in Acid-Catalyzed Biomass Conversion Reactions, Angew. Chem., Int. Ed., 2014, 53(44), 11872–11875, DOI:10.1002/anie.201408359.
- C. Sievers, I. Musin, T. Marzialetti, M. B. Valenzuela Olarte, P. K. Agrawal and C. W. Jones, Acid-Catalyzed Conversion of Sugars and Furfurals in an Ionic-Liquid Phase, ChemSusChem, 2009, 2(7), 665–671, DOI:10.1002/cssc.200900092.
- D. M. Alonso, J. M. R. Gallo, M. A. Mellmer, S. G. Wettstein and J. A. Dumesic, Direct Conversion of Cellulose to Levulinic Acid and Gamma-Valerolactone Using Solid Acid Catalysts, Catal. Sci. Technol., 2013, 3(4), 927–931, 10.1039/C2CY20689G.
- X. Fu, J. Dai, X. Guo, J. Tang, L. Zhu and C. Hu, Suppression of Oligomer Formation in Glucose Dehydration by CO2 and Tetrahydrofuran, Green Chem., 2017, 19(14), 3334–3343, 10.1039/C7GC01115F.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Nonenzymatic Sugar Production from Biomass Using Biomass-Derived –Valerolactone, Science, 2014, 343(6168), 277–280, DOI:10.1126/science.1246748.
- J. Han, J. S. Luterbacher, D. M. Alonso, J. A. Dumesic and C. T. Maravelias, A Lignocellulosic Ethanol Strategy via Nonenzymatic Sugar Production: Process Synthesis and Analysis, Bioresour. Technol., 2015, 182, 258–266, DOI:10.1016/j.biortech.2015.01.135.
- X. Bai, R. C. Brown, J. Fu, B. H. Shanks and M. Kieffer, The Influence of Alkali and Alkaline Earth Metals and the Role of Acid Pretreatments in Production of Sugars from Switchgrass Based on Solvent Liquefaction, Energy Fuels, 2014, 28(2), 1111–1120, DOI:10.1021/ef4022015.
- A. Ghosh, X. Bai and R. C. Brown, Solubilized Carbohydrate Production by Acid-Catalyzed Depolymerization of Cellulose in Polar Aprotic Solvents, ChemistrySelect, 2018, 3(17), 4777–4785, DOI:10.1002/slct.201800764.
- R. Xing, W. Qi and G. W. Huber, Production of Furfural and Carboxylic Acids from Waste Aqueous Hemicellulose Solutions from the Pulp and Paper and Cellulosic Ethanol Industries, Energy Environ. Sci., 2011, 4(6), 2193, 10.1039/c1ee01022k.
- K. Yan, G. Wu, T. Lafleur and C. Jarvis, Production, Properties and Catalytic Hydrogenation of Furfural to Fuel Additives and Value-Added Chemicals, Renewable Sustainable Energy Rev., 2014, 38, 663–676, DOI:10.1016/j.rser.2014.07.003.
- R. P. Pohanish, Sittig's Handbook of Toxic and Hazardous Chemicals and Carcinogens, William Andrew, 7th edn, 2012, DOI:10.1016/C2009-0-64361-0.
- A. Ghosh and R. C. Brown, Factors Influencing Cellulosic Sugar Production during Acid-Catalyzed Solvent Liquefaction in 1,4-Dioxane, ACS Sustainable Chem. Eng., 2019, 7(21), 18076–18084, DOI:10.1021/acssuschemeng.9b05108.
- A. T. W. M. Hendriks and G. Zeeman, Pretreatments to Enhance the Digestibility of Lignocellulosic Biomass, Bioresour. Technol., 2009, 100(1), 10–18, DOI:10.1016/j.biortech.2008.05.027.
- C. Li, G. Cheng, V. Balan, M. S. Kent, M. Ong, S. P. S. Chundawat, L. daCosta Sousa, Y. B. Melnichenko, B. E. Dale and B. A. Simmons, et al. Influence of Physico-Chemical Changes on Enzymatic Digestibility of Ionic Liquid and AFEX Pretreated Corn Stover, Bioresour. Technol., 2011, 102(13), 6928–6936, DOI:10.1016/j.biortech.2011.04.005.
- C. E. Wyman, B. E. Dale, R. T. Elander, M. Holtzapple, M. R. Ladisch and Y. Y. Lee, Coordinated Development of Leading Biomass Pretreatment Technologies, Bioresour. Technol., 2005, 96(18 SPEC. ISS), 1959–1966, DOI:10.1016/j.biortech.2005.01.010.
- B. Yang and C. E. Wyman, Effect of Xylan and Lignin Removal by Batch and Flowthrough Pretreatment on the Enzymatic Digestibility of Corn Stover Cellulose, Biotechnol. Bioeng., 2004, 86(1), 88–95, DOI:10.1002/bit.20043.
- S.-J. Kim, S.-H. Jung and J.-S. Kim, Fast Pyrolysis of Palm Kernel Shells: Influence of Operation Parameters on the Bio-Oil Yield and the Yield of Phenol and Phenolic Compounds, Bioresour. Technol., 2010, 101(23), 9294–9300, DOI:10.1016/j.biortech.2010.06.110.
- M. R. Haverly, A. Ghosh and R. C. Brown, The Effect of Moisture on Hydrocarbon-Based Solvent Liquefaction of Pine, Cellulose and Lignin, J. Anal. Appl. Pyrolysis, 2020, 146, 104758, DOI:10.1016/j.jaap.2019.104758.
- J. Zhang, J. B. Li, S. B. Wu and Y. Liu, Advances in the Catalytic Production and Utilization of Sorbitol, Ind. Eng. Chem. Res., 2013, 52(34), 11799–11815, DOI:10.1021/ie4011854.
- J. Zhang, K. H. Kim, Y. S. Choi, A. H. Motagamwala, J. A. Dumesic, R. C. Brown and B. H. Shanks, Comparison of Fast Pyrolysis Behavior of Cornstover Lignins Isolated by Different Methods, ACS Sustainable Chem. Eng., 2017, 5(7), 5657–5661, DOI:10.1021/acssuschemeng.7b01393.
- G. A. Kraus, R. C. Brown and A. D. Sadow, Center for Sustainable Aromatic Molecules, National Science Foundation SCI & TECH CTRS (INTEG PTRS), 2014, p NSF Proposal Number 1522278.
- A. Ghosh and R. C. Brown, Factors Influencing Cellulosic Sugar Production during Acid-Catalyzed Solvent Liquefaction in 1,4-Dioxane, ACS Sustainable Chem. Eng., 2019, 7(21), 18076–18084, DOI:10.1021/acssuschemeng.9b05108.
- M. R. Rover and R. C. Brown, Quantification of Total Phenols in Bio-Oil Using the Folin–Ciocalteu Method, J. Anal. Appl. Pyrolysis, 2013, 104, 366–371, DOI:10.1016/j.jaap.2013.06.011.
- R. Xing, W. Qi and G. W. Huber, Production of Furfural and Carboxylic Acids from Waste Aqueous Hemicellulose Solutions from the Pulp and Paper and Cellulosic Ethanol Industries, Energy Environ. Sci., 2011, 4(6), 2193, 10.1039/c1ee01022k.
- K. Yan, G. Wu, T. Lafleur and C. Jarvis, Production, Properties and Catalytic Hydrogenation of Furfural to Fuel Additives and Value-Added Chemicals, Renewable Sustainable Energy Rev., 2014, 38, 663–676, DOI:10.1016/j.rser.2014.07.003.
- R. P. Pohanish, Sittig's Handbook of Toxic and Hazardous Chemicals and Carcinogens, Elsevier, 2012, DOI:10.1016/C2009-0-64361-0.
- J. G. Linger, D. R. Vardon, M. T. Guarnieri, E. M. Karp, G. B. Hunsinger, M. A. Franden, C. W. Johnson, G. Chupka, T. J. Strathmann and P. T. Pienkos, et al. Lignin Valorization through Integrated Biological Funneling and Chemical Catalysis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(33), 12013–12018, DOI:10.1073/pnas.1410657111.
- D. B. Hodge, M. N. Karim, D. J. Schell and J. D. McMillan, Soluble and Insoluble Solids Contributions to High-Solids Enzymatic Hydrolysis of Lignocellulose, Bioresour. Technol., 2008, 99(18), 8940–8948, DOI:10.1016/j.biortech.2008.05.015.
- Y. Lu, Y. Wang, G. Xu, J. Chu, Y. Zhuang and S. Zhang, Influence of High Solid Concentration on Enzymatic Hydrolysis and Fermentation of Steam-Exploded Corn Stover Biomass, Appl. Biochem. Biotechnol., 2010, 160(2), 360–369, DOI:10.1007/s12010-008-8306-0.
- J. Zhang, W. Hou and J. Bao, Reactors for High Solid Loading Pretreatment of Lignocellulosic Biomass, in Bioreactor Engineering Research and Industrial Applications II, Springer-Verlag, 2015, vol. 123, pp. 75–90, DOI:10.1007/10_2015_307.
- L. R. Jarboe, Z. Wen, D. Choi and R. C. Brown, Hybrid Thermochemical Processing: Fermentation of Pyrolysis-Derived Bio-Oil, Appl. Microbiol. Biotechnol., 2011, 91(6), 1519–1523, DOI:10.1007/s00253-011-3495-9.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00192a |
| This journal is © The Royal Society of Chemistry 2020 |