
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revisiting the mechanism of the Fujiwara–Moritani reaction†
Christopher J.
Mulligan
a,
Jeremy S.
Parker
 b and
King Kuok (Mimi)
Hii
*a
b and
King Kuok (Mimi)
Hii
*a
aDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College London, 80, Wood Lane, London W12 0JQ, UK. E-mail: mimi.hii@imperial.ac.uk
bEarly Chemical Development, Pharmaceutical Sciences, R&D, AstraZeneca, Macclesfield, UK
First published on 13th May 2020
Abstract
The Fujiwara–Moritani reaction between p-methylacetanilide and n-butyl acrylate, catalysed by Pd(OAc)2 in the presence of toluenesulfonic acid and benzoquinone, was (re-)investigated using reaction calorimetry and complementary spectroscopic methods. The (most) active catalyst was identified and the catalytic turnover rate was found to be independent of all stoichiometric reagents. Catalyst regeneration and deactivation pathways are discussed.
Introduction
The Fujiwara–Moritani (F–M) reaction constitutes one of the earliest examples of a C–H activation reaction catalyzed by transition metals. The original report in 1967 described the reaction between a styrene–palladium chloride complex and benzene derivatives to form stilbenes.1 Catalytic reactions were later accomplished by using Pd(OAc)2 as a precursor for the olefination of simple (hetero)aromatic substrates (Scheme 1, eqn i and ii).2,3 In the early reports, oxygen gas was used in combination with copper or silver acetate to facilitate the dehydrogenative process, forming water as a by-product. In principle, the F–M reaction is more desirable than the Heck arylation reaction in terms of atom- and step-economies. In practice, however, the lack of a reactive centre poses problems such as poor regioselectivity and/or multiple substitutions of the olefin. Consequently, the F–M reaction was largely eschewed by the synthetic chemistry community, in favour of the Heck arylation reaction towards the end of the 20th century.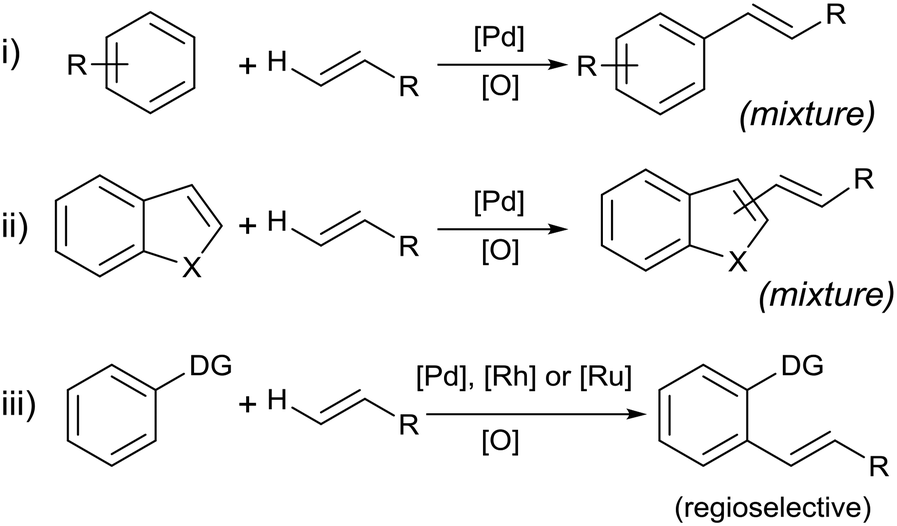 | ||
| Scheme 1 General examples of the Fujiwara–Moritani (F–M) reaction. | ||
In the last decade, a resurgence in research activity for C–H activation reactions has led to renewed interest in the F–M reaction.2,4–6 One of the most significant breakthroughs is to deploy Lewis basic entities (‘directing groups’) on the arene substrate to control the substitution at the ortho-position (Scheme 1, eqn iii).7–10 In recent years, directing scaffolds were also used very effectively in controlling selective olefination at distal positions.11,12 Moreover, the reaction is no longer exclusive to Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ru,13–18 Rh19–23 and Ir24 complexes have also been reported to be effective catalysts for certain systems.
:Ru,13–18 Rh19–23 and Ir24 complexes have also been reported to be effective catalysts for certain systems.
In 1979, Horino and Inoue reported that anilides could undergo regioselective ortho-palladation with Pd(OAc)2 to form palladacycles that can react with alkenes to form ‘Heck’ products.25 More than two decades later, the catalytic reaction was demonstrated successfully by an industrial research team,26 where the addition of benzoquinone (BQ) and toluenesulfonic acid (TsOH) effected the coupling between acetanilides (1) and acrylates (2) under mild reaction conditions (Scheme 2).
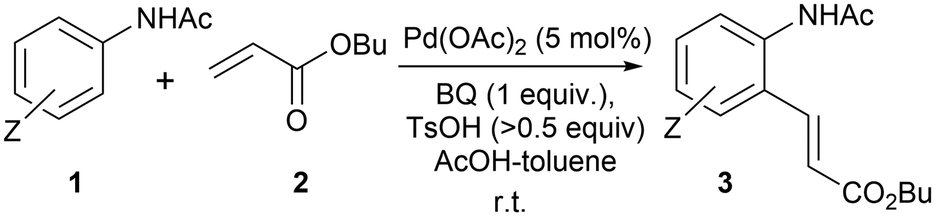 | ||
| Scheme 2 The first reported example of the F–M reaction of acetanilides with butyl acrylate, catalysed by Pd(OAc)2. | ||
The F–M reaction is generally believed to proceed via a redox cycle (Scheme 3): initiating with a reaction between the Pd(II) salt and the acetanilide to produce a palladacycle II.25 In the presence of an alkene, the complex undergoes migratory insertion and β-H elimination to afford the C–C coupled product and a hydridopalladium(II) intermediate (III). At this juncture, it is postulated complex III undergoes reductive elimination to produce acetic acid and a Pd(0) species, which is re-oxidised by BQ under acidic conditions,27 to regenerate the active Pd(II) catalyst.
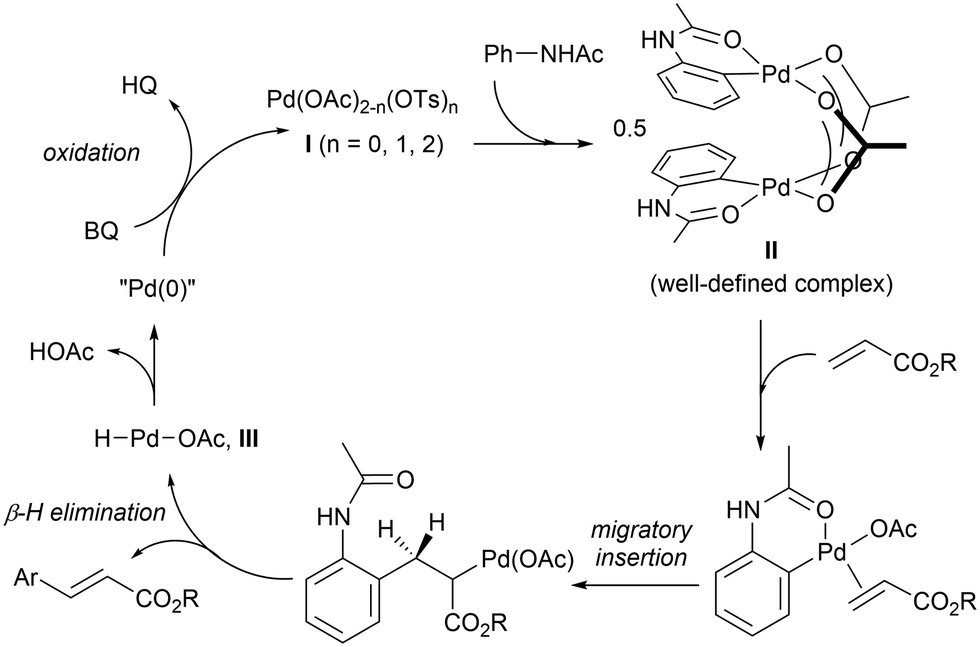 | ||
| Scheme 3 Redox catalytic cycle proposed for the F–M reaction. | ||
Results and discussion
In this work, we initiated a series of studies on the F–M reaction between 4-methylacetanilide (1, where Z = para Me) and butyl acrylate 2, under similar conditions28 to those reported by Boele et al.26 (Scheme 2), with the aim to delineate the roles of, and synergistic effects between, the TsOH and BQ in the catalytic cycle, as well as the deactivation process(es).Reaction order in Pd(OAc)2
In this study, reaction progresses were monitored using reaction calorimetry for reactions catalysed by ≥2.5 mol% of Pd(OAc)2. At lower catalyst loadings (or where reactions were slow), aliquots were extracted and analysed by HPLC.Pd(OAc)2 is known to exist in monomeric, dimeric, trimeric forms in solution, depending on the solvent, and amount of additives and water present.29 In acetic acid, the reaction solvent employed in F–M reaction, it is expected to be largely trimeric,30 which can dissociate into dimeric and monomeric species in a rapid equilibrating process (Scheme 4).31
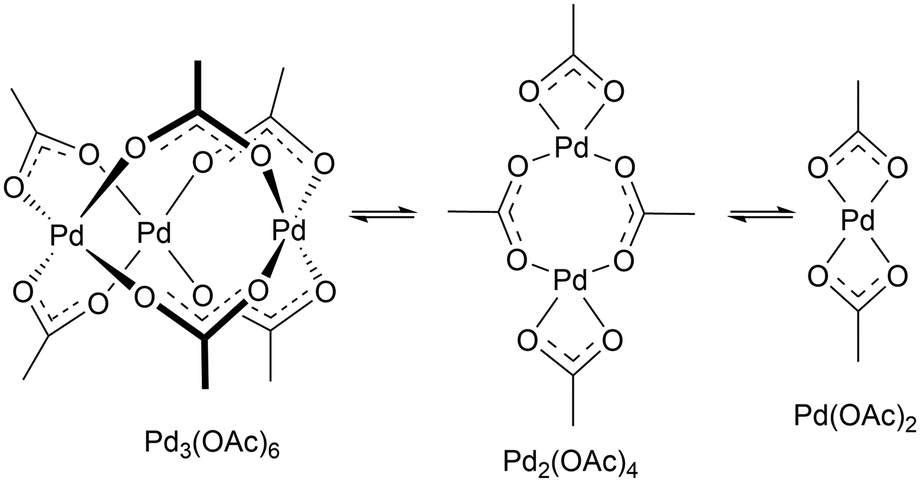 | ||
| Scheme 4 Equilibrating forms of Pd(OAc)2 in solution. | ||
To determine whether all three forms of Pd(OAc)2 could be catalytically active in the F–M reaction,32 the dependence of reaction rate on the [Pd] concentration was studied. By varying the catalyst loading between 2.5–10 mol% in the presence of 1 equivalent of TsOH, a linear plot of initial rates vs. [Pd(OAc)2] (Fig. 1A) and an overlay of the plots of νobs/[Pd(OAc)2] vs. [1] (Fig. 1B) were obtained, showing that the reaction is first order with respect to [Pd]. The zero-intercept observed in the Fig. 1A suggests there is only one catalytically active species that is kinetically significant.
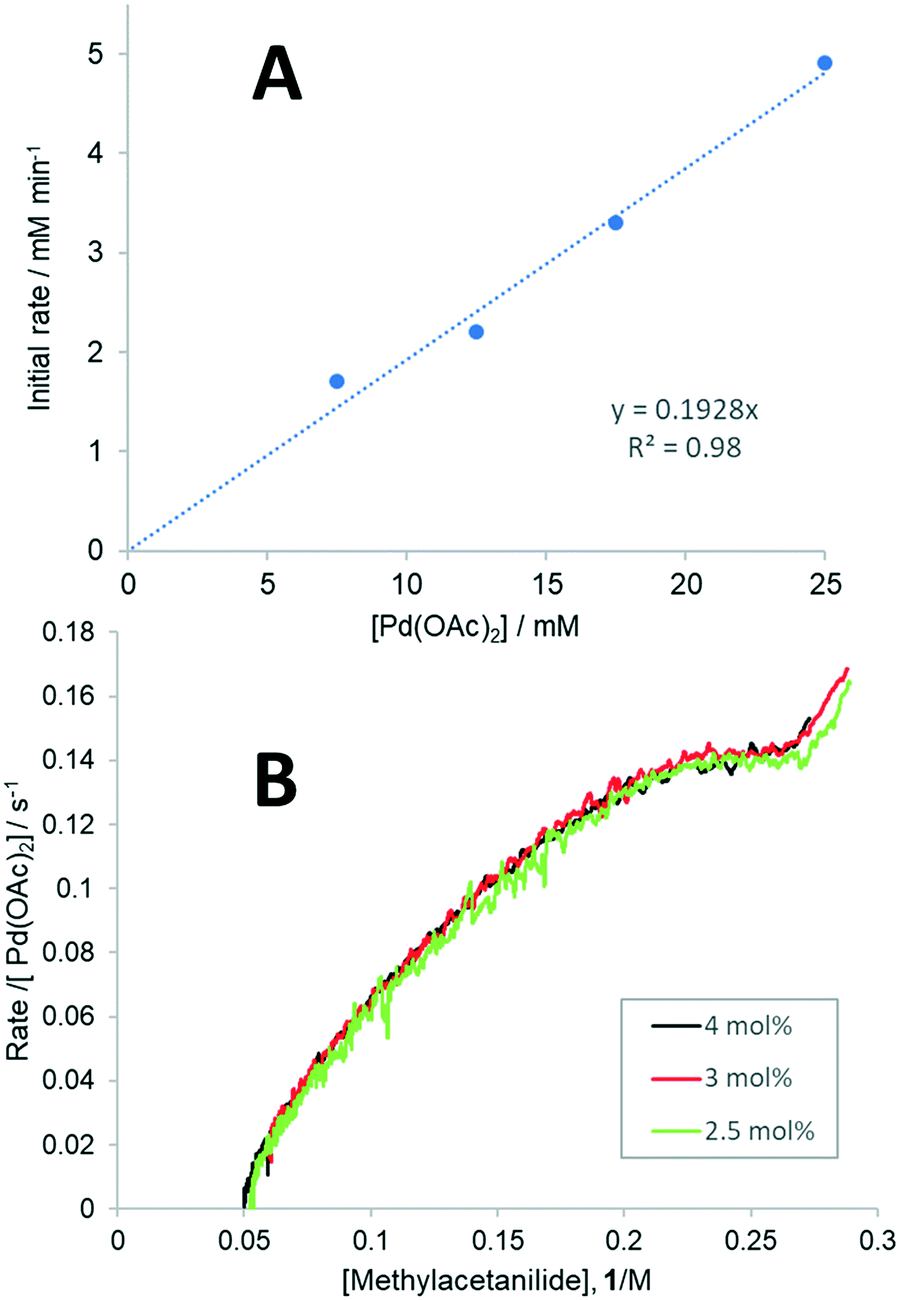 | ||
| Fig. 1 Establishing [Pd] order. A: Plot of initial rates (extracted from HPLC analysis) vs. Pd(OAc)2 concentrations between 2.5–10 mol%. B: Plots of rate/[Pd(OAc)2] vs. [1] (obtained by reaction calorimetry) at three different Pd(OAc)2 concentrations: 2.5–4 mol% (corresponding to 6.25–10 mM). Other reaction parameters: 1 (0.3 M), 2 (0.25 M), benzoquinone (0.25 M, 1 eq.) | ||
Effect of additive (p-toluenesulfonic acid)
Brønsted acid additives are commonly employed in many transition metal-catalysed C–H functionalisation reactions (for e.g. Cu,33 Ru,34 Co,35 Rh,36 Ir,37 as well as Pd (ref. 38 and 39)) for a variety of reasons, although their roles in the catalytic cycles are rarely studied. In the F–M reaction, the incorporation of 0.5–1 equivalent of p-toluenesulfonic acid (TsOH) has a dramatic effect on the catalytic turnover, allowing the reaction temperature to be lowered from 80 to 20 °C.26 Indeed, during this work, the calorimetric studies were initiated by the addition of TsOH to mixtures of 1, 2 and Pd(OAc)2 which were thermally-equilibrated at 30 °C. It was suggested that the addition of the strong acid protonates an acetate ligand to generate catalytically active Pd(OAc)+; a hypothesis subsequently supported by a computational study by MacGregor and co-workers, who proposed an energetically-feasible acetate-assisted, concerted, metalation–deprotonation (CMD) mechanism for the C–H metalation step.40 More recently, it has also been shown that the addition of carboxylic acids to Pd(OAc)2 promotes the C–H activation step kinetically and thermodynamically.41Interestingly, the F–M reaction can also proceed in the absence of carboxylate ions. In 2010, Nishikata and Lipshutz reported that oxidative ortho-olefination of electron-rich acetanilides can be effected at room temperature using cationic [Pd(MeCN)4][BF4]2 as a catalyst precursor.42,43 In the same year, Brown and co-workers also reported that an isolated cationic cyclometalated complex containing a η1-bound toluenesulfonate anion is catalytically active in the F–M reaction between acetanilides and methyl acrylate.44 This led the authors to propose that [PdOTs]+ is able to activate the C–H bond of an acetanilide in the absence of acetate, via a ‘general base catalysis’ (GBC).45
TsOH (pKa −7) is a much stronger acid than AcOH (pKa 4.7). Hence, we envisaged that TsOH can protonate Pd-bound acetate ligands to generate a mixture of 1:146 and 1:247 complexes in a facile equilibrium (Scheme 5). To delineate the role of the anionic ligands in the F–M reaction, kinetic experiments were performed with different amounts of [TsOH]. Between 15–37.4 mol% (Fig. 2 and S4, ESI†), the reaction displayed first order dependency on [TsOH]. Once again, the observation of a near-zero intercept in Fig. 2 suggests that while there may be more than one catalytically active species (Scheme 5), there is only one that may be kinetically relevant.
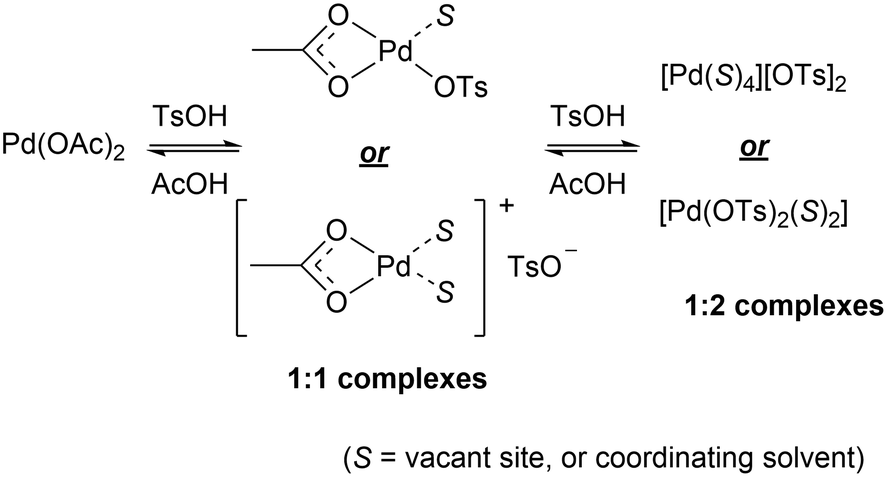 | ||
| Scheme 5 Ligand exchange between acetate and toluenesulfonate ions. | ||
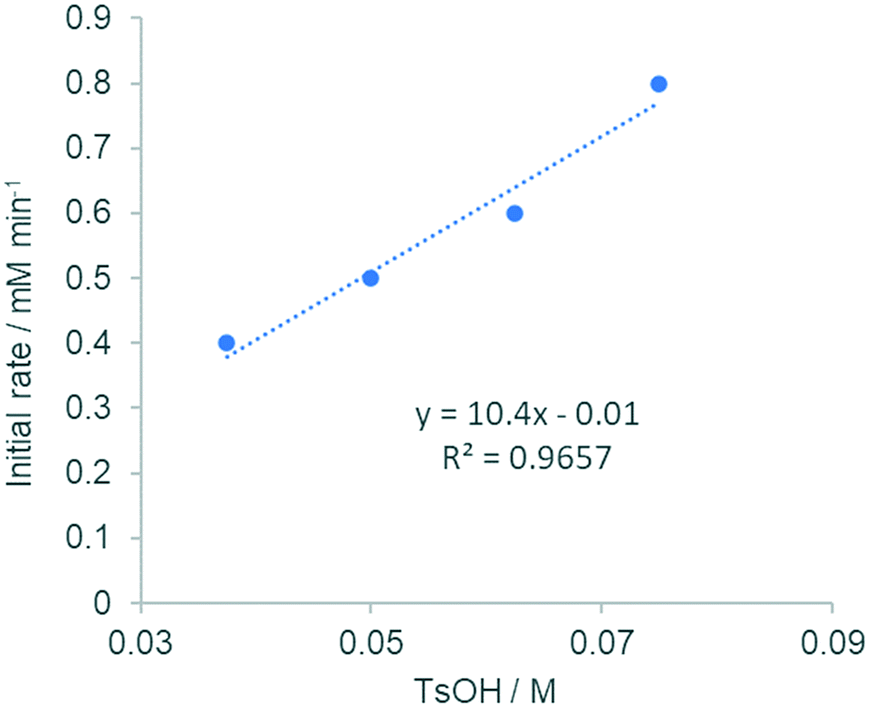 | ||
| Fig. 2 Plot of initial rates vs. [TsOH] (between 15–30 mol%, extracted from HPLC analysis). For an RPKA analysis (between 2.5–37.5 mol%), see Fig. S2 (ESI†). | ||
Above 50 mol% (reaction conditions), however, the reaction rate became independent of [TsOH] (saturation kinetics), implying a rapid equilibrium is reached between these homoleptic and heteroleptic species, compared to the turnover-limiting step.26
Substituting TsOH with different para-substituted aryl sulfonic acids p-Z–C6H4SO3H (where Z = CF3, Me, OMe, 1 equivalent) has no discernible effects on the reaction rate (Fig. S1, ESI†). The addition of p-trifluoromethyl benzenesulfonic acid (Z = CF3) to Pd(OAc)2 in acetone-d6 induced a δ19F shift (Fig. S6, ESI†), which can be used to construct a Job plot (continuous variation method): the resultant plot of Δδ vs. the mole fraction of the reactants displayed a maximum at 0.5 (Fig. 3), suggesting that the formation of a 1:1 complex is the most thermodynamically favourable; the curvature of the plot suggests a Keq value > 100.48
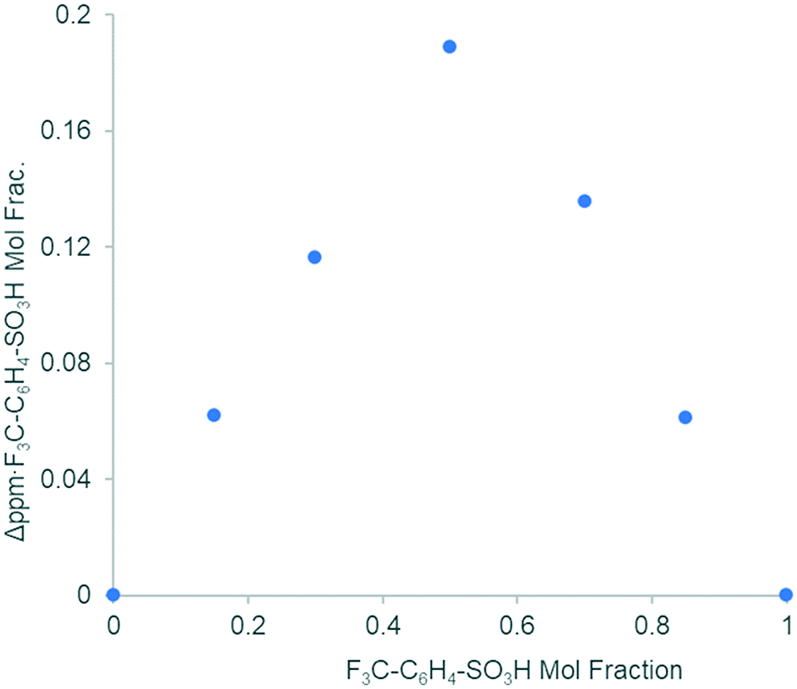 | ||
| Fig. 3 Response in δ19F induced by the addition of p-trifluoromethanesulfonic acid to Pd(OAc)2 in acetone (Job plot). | ||
As may be expected, an attempt to reproduce the titration experiment in acetic acid did not produce any noticeable change in δ19F, as the equilibrium will favour the diacetate complex. Hence, in order for either Pd(OAc)+ or Pd(OTs)+ to operate as active catalysts under the reaction conditions (where acetic acid is employed as a reaction solvent), they will need to be substantially more reactive than Pd(OAc)2 (Curtin–Hammett principle). This is further supported by comparing kinetic rates, where the reaction catalysed by Pd(OTs)+ under acetate-free conditions (reported to be 0.115 mM min−1 in acetone-d6)44 is approximately 20 times slower than the reaction performed with Pd(OAc)2/TsOH in acetic acid (2 mM min−1, present work). Combined with the result of the Job plot, and the absence of any para-substituent effect on the aryl sulfonate anion, we conclude that Pd(OAc)+ is likely to be a more active catalyst than Pd(OTs)+ under the reaction conditions.
Catalyst deactivation
A series of ‘same excess’ experiments49 were performed to probe the catalyst stability towards different reaction components (Table 1).| Components | [1]/M | [2]/M | [3]/M | [HQ]/M |
|---|---|---|---|---|
| a Pd(OAc)2 (12.5 mM, 5 mol%), TsOH (0.125 M, 0.5 eq.), BQ (0.25 M, 1 eq.), 30 °C, AcOH. | ||||
| Expt 1: initial conditions | 0.3 | 0.25 | 0 | 0 |
| Expt 2: ‘same excess’ | 0.175 | 0.125 | 0 | 0 |
| Expt 3: ‘same excess’ + HQ | 0.175 | 0.125 | 0 | 0.125 |
| Expt 4: ‘same excess’ + [3] | 0.175 | 0.125 | 0.125 | 0 |
| Expt 5: ‘same excess’ + HQ + [3] | 0.175 | 0.125 | 0.125 | 0.125 |
Experiments 1 and 2 were performed with different initial concentrations of acrylate 2 (0.25 and 0.175 M, respectively), maintaining the same excess of the acetanilide 1 (0.05 M). The resultant plots (Fig. 4A) showed that experiment 2 proceeds with a faster reaction rate than experiment 1, indicative of the catalyst deactivation. This can be attributed entirely to the presence of products (experiment 5, Fig. 4B). Subsequent addition of each of the two reaction products, hydroquinone (HQ) and 3 (experiments 3 and 4, respectively), showed that the HQ has a stronger inhibitory effect than the substituted alkene 3 (Fig. S5, ESI†).
 | ||
| Fig. 4 Results of ‘same excess’ experiments (Table 1). | ||
The good overlap between the curves in Fig. 4B also shows that other catalyst deactivation pathways, such as the formation of Pd black, is not significant under these reaction conditions.
Completing the cycle: regeneration of the active catalyst
BQ is believed to have dual roles in this process,51 acting as an oxidant and a ligand to stabilize the low valent-Pd, preventing it from degradation to Pd black. It was also proposed that following β-hydride elimination, the resultant AcO–Pd–H intermediate III (Scheme 3) is reduced to a BQ-stabilised Pd(0) species, which is regenerated to Pd(II) by BQ in the presence of acetic acid – a computational study of this process by López and Maseras showed that the transformation of (ηE-BQ)Pd(AcOH)2 to Pd(OAc)2 can occur via consecutive hydrogen transfer from AcOH to BQ (Scheme 6) – the energies were computed to be 92.1 kJ mol−1, and 65.7 kJ mol−1 for the reverse (dehydrogenative) process.50 The low energy associated with the dehydrogenative step will account for the observed HQ inhibition of the catalyst.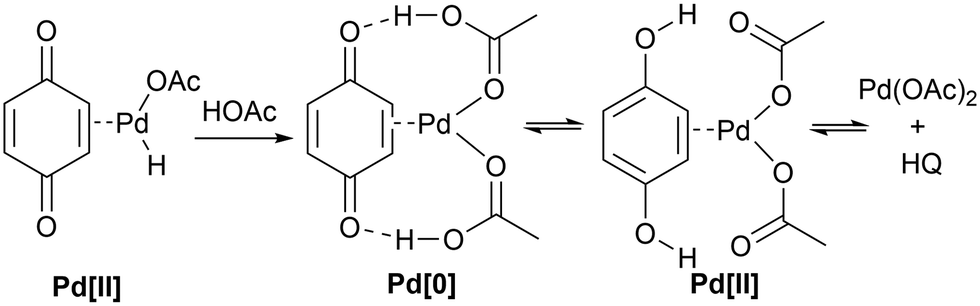 | ||
| Scheme 6 Regeneration of Pd(OAc)2via a redox pathway involving H-transfer, proposed by López and Maseras.50 | ||
Rate dependence on the concentration of reactants
In the previous study by the de Vries group,26 the electrophilic attack of the cationic Pd(OAc)+ on the arene ring was proposed to be the turnover-limiting step, based on the following observations: i) the F–M reaction proceeds faster with electron-rich acetanilides (Hammett correlation); ii) observation of a significant kinetic isotope effect (kD/kH = 3; established by comparing kobs obtained from using acetanilide-h5 and d5); and iii) the reaction between the isolated palladacycle complex II with the olefin is an order of magnitude faster than the equivalent reaction using the catalyst generated in situ. In the study by Brown et al., conducted in the absence of acetate-ions,44 the reaction also exhibited first order dependency on acetanilide, zero order in BQ and a modest effect of the alkenes, which also supports a turnover-limiting metalation step.Thus, we were rather surprised to find that the rate of the reaction is, in fact, independent of the initial acetanilide concentration (Fig. 5). Subsequently, the reaction rate was also shown to be independent of the initial concentrations of the alkene and BQ (Fig. S2 and S3, ESI†).
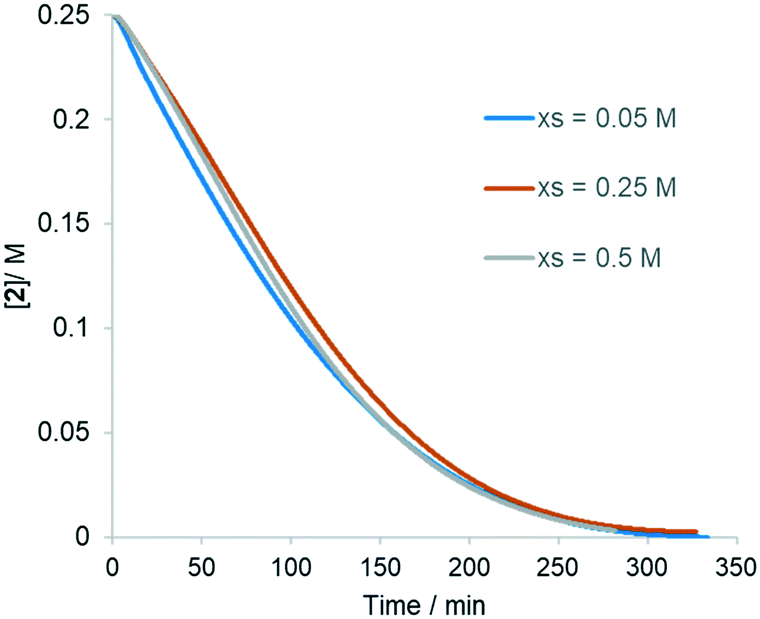 | ||
| Fig. 5 Rate curve showing that the reaction rate is independent of [1]. Reaction conditions: butyl acrylate 2 (0.25 M), BQ (0.25 M), TsOH (0.125 M), Pd(OAc)2 (12.5 mM, 5 mol%). Legend: 0.30 M (0.05 M excess), 0.50 M (0.25 M excess) and 0.75 M (0.5 M excess) of [1]. | ||
The apparent zero-order in acetanilide may be explained by the existence of an acetanilide-bound complex (IV) immediately prior to the rate-limiting cyclometalation step (Scheme 7). The proposal will also explain why the heteroleptic [Pd(OAc)][OTs] complex is more active than the corresponding ditosylate or diacetate salts, as it contains a labile OTs that can be displaced easily by the acetanilide substrate to form complex IV.
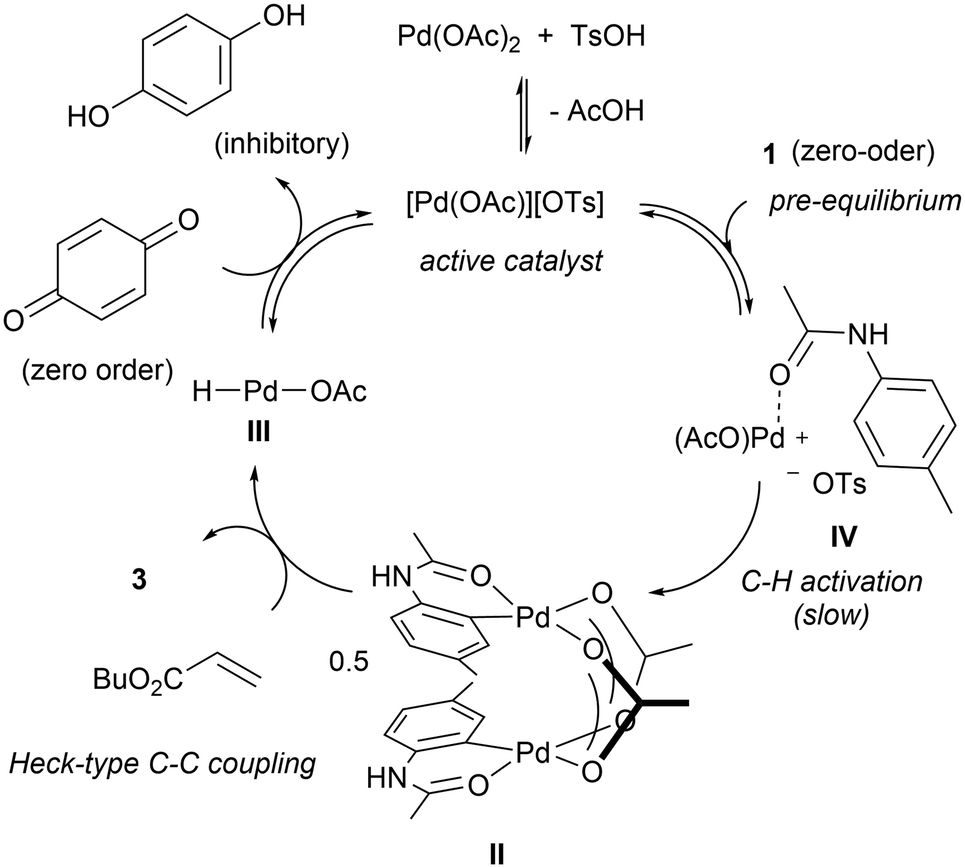 | ||
| Scheme 7 Revised catalytic cycle for the F–M reaction. | ||
Kinetic modelling
The initiating steps of the updated catalytic cycle shown in Scheme 7 is reminiscent of the Michaelis–Menten mechanism, where the reaction is dominated by a reversible binding of the substrate (acetanilide) to the active metal centre (K1) to form complex IV, prior to slow, irreversible C–H bond cleavage to form the cyclometalated complex II (k2), which undergoes spontaneous C–C coupling (zero order in alkene) to generate the product 3 and the hydridopalladium intermediate (k3). BQ then participates in the regeneration of the active catalyst, which can be inhibited by the HQ by-product (K4).A microkinetic model of these elementary steps can be constructed from the reaction intermediates and reactants, incorporating six discreet rate constants (Scheme 8). Using an ordinary differential equation (ODE) solver, the data gathered from 16 independent kinetic experiments were fitted to the model, from which the rate constants can be extracted (Fig. S7–S9, ESI†).
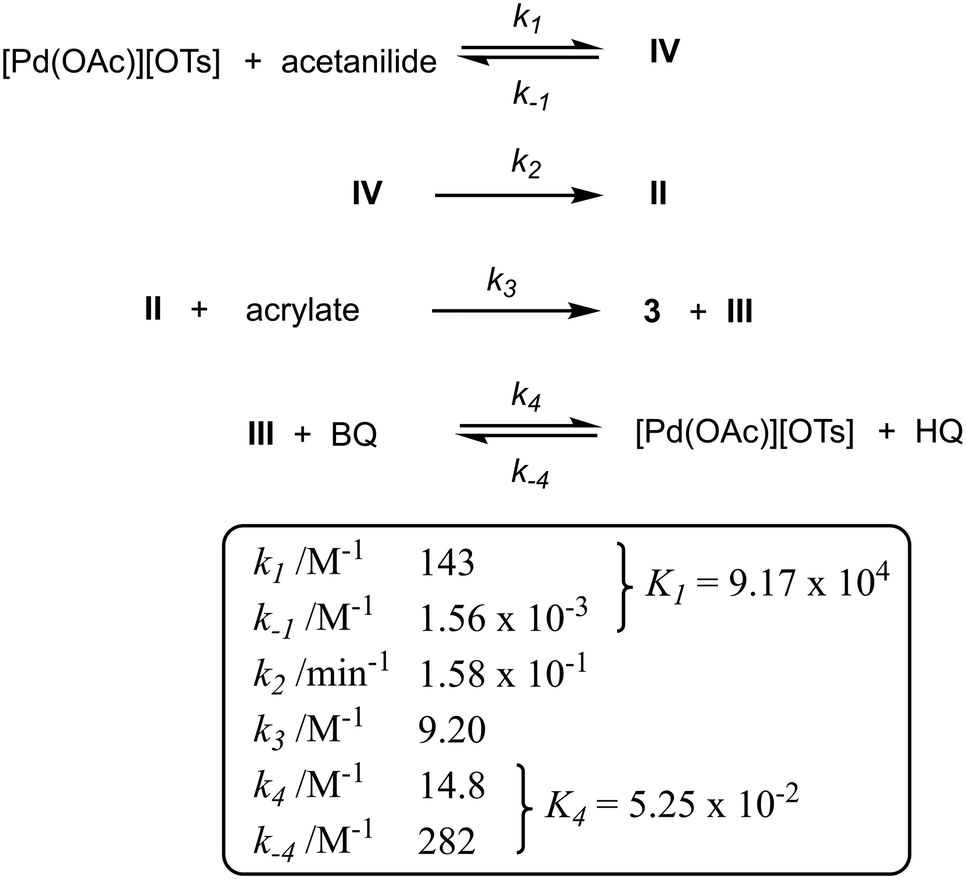 | ||
| Scheme 8 Kinetic modelling and kinetic constants (with reference to Scheme 7). | ||
A good fit between the simulated and experimental data suggests that catalyst decomposition (e.g. formation of inactive Pd black) is not a significant process under these reaction condition, as was previously verified by the ‘same excess’ experiments (Fig. 4 and S5†). While the rate constants confirmed k2 as the turnover-limiting step (as expected), it is interesting to note the magnitudes of the two equilibrium processes: K4 (0.05) ≪ K1 (9.17 × 104), suggesting that the stronger binding of the acetanilide (compared to HQ) to the [Pd(OAc)][OTs] provides the driving force for the turnover. Towards the end of the reaction, as the concentration of acetanilide is depleted, the binding of the HQ by-product become competitive, leading to catalyst inhibition.
Role of the terminal oxidant (BQ vs. O2)
Benzoquinone is widely implicated in the catalyst regeneration step. It has an important role in stabilising the monometallic Pd species, thus preventing the aggregation of Pd that leads to the formation of catalytically inactive Pd black. In the studies described above, a stoichiometric amount of BQ was employed. Under these conditions, the formation of Pd black was not a competitive process.However, the use of BQ as a stoichiometric reagent is unattractive in terms of the atom-economy. BQ and HQ are known irritants and possible carcinogens, so it will be desirable to reduce or replace them with safer oxidants. Previously, Pd-catalyzed F–M reactions have been reported to proceed under high O2 pressures (>8 bar) and elevated temperatures with an excess of the arene reactant.52–54 Encouraged by these accounts, we attempted to replace BQ with O2. However, our efforts were thwarted by highly capricious results: in the absence of BQ, a large variation of reaction yields between 11 to >95% were obtained from twenty-two individual, but identical reaction mixtures conducted under aerobic conditions. Speculating that diffusion of gaseous O2 into the liquid-phase may be the cause of the irreproducibility, reactions were repeated up to five times, in parallel, on a larger scale with mechanical (overhead) stirrers (STEM Integrity10 Reaction Station), varying the stirring rate between 350–1200 rpm. In all cases, no correlation between reaction yield and stirring speed/reaction vessel can be established. In this part of the study, the formation of Pd black was observed in the early stages of the reaction, irrespective of the reaction outcome. This led us to postulate that, in the absence of BQ, Pd aggregation occurs readily to form a ‘cocktail’ of interchanging Pd species, including complexes, colloids/clusters and nanoparticles55 – a highly stochastic system that is difficult to control and replicate using stirred glass reactors and ambient temperature and pressure.56 Indeed, reproducibility of the reaction could only be achieved by the addition of at least 50 mol% of BQ or 20 mol% of Cu(OAc)2 (as a redox mediator).
Conclusions
The work provides new insights to the kinetic profile of the F–M reaction (Schemes 7vs.3). In the presence of TsOH, a heteroleptic [Pd(OAc)][OTs] complex is generated, which is catalytically more active than either Pd(OAc)2 or Pd(OTs)2. The F–M reaction was found to operate under saturation (Michaelis–Menten) kinetics, where zero order rate dependencies were observed for all the stoichiometric reagents (acetanilide, acrylate and BQ). This is attributed to the reversible binding of the acetanilide substrate to the catalytically active Pd species, prior to a slow intramolecular C–H activation step. Subsequent kinetic modelling revealed that competitive binding between the acetanilide and HQ to the active catalyst is also important in achieving catalytic turnover.Irreversible catalyst deactivation occurs through the Pd aggregation, which can be prevented by the addition of BQ or Cu salts. In the present work, the coupling between 1 and 2 were attempted under aerobic conditions (in the absence of BQ or redox mediators). Under ambient conditions, quantitative conversions can be obtained in some instances, but the reaction outcome was found to be hugely irreproducible, possibly due to the highly stochastic nature of the Pd–Pd aggregation process and the difficulty in controlling the delivery of O2 to the solution under ambient conditions. Understanding the causes and control of these processes will be important in the future development of atom-economical and sustainable oxidative coupling reactions under ambient, aerobic conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC and AstraZeneca for studentship support. Part of this work (calorimetry and parallel reaction screening) was conducted during CJM's industrial placement at AstraZeneca's R&D site at Macclesfield, UK. We are grateful to Dr Jordi Bures (University of Manchester) for helpful comments on this manuscript.Notes and references
- I. Moritani and Y. Fujiwara, Aromatic substitution of styrene-palladium chloride complex, Tetrahedron Lett., 1967, 8, 1119–1122 CrossRef.
- T. Kitamura and Y. Fujiwara, Dehydrogenative Heck-type reactions: the Fujiwara-Moritani reaction, RSC Green Chem. Ser., 2015, 26, 33–54 CAS.
- Y. Fujiwara, I. Moritani, S. Danno, R. Asano and S. Teranishi, Aromatic substitution of olefins. VI. Arylation of olefins with palladium(II) acetate, J. Am. Chem. Soc., 1969, 91, 7166–7169 CrossRef CAS.
- C. Liu, J. W. Yuan, M. Gao, S. Tang, W. Li, R. Y. Shi and A. W. Lei, Oxidative coupling between two hydrocarbons: an update of recent C-H functionalizations, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS.
- E. M. Ferreira, H. Zhang and B. M. Stoltz, in The Mizoroki-Heck Reaction, ed. M. Oestreich, John Wiley & Sons, 2009, ch. 9, pp. 345–382 Search PubMed.
- L. H. Zhou and W. J. Lu, Towards ideal synthesis: alkenylation of aryl C-H bonds by a Fujiwara-Moritani reaction, Chem. – Eur. J., 2014, 20, 634–642 CrossRef CAS PubMed.
- S. I. Gorelsky, Origins of regioselectivity of the palladium-catalyzed (aromatic)C-H bond metalation-deprotonation, Coord. Chem. Rev., 2013, 257, 153–164 CrossRef CAS.
- H. Choi, M. Min, Q. Peng, D. Kang, R. S. Paton and S. Hong, Unraveling innate substrate control in site-selective palladium-catalyzed C-H heterocycle functionalization, Chem. Sci., 2016, 7, 3900–3909 RSC.
- W. Ma, P. Gandeepan, J. Li and L. Ackermann, Recent advances in positional-selective alkenylations: removable guidance for twofold C–H activation, Org. Chem. Front., 2017, 4, 1435–1467 RSC.
- H. Chen, P. Wedi, T. Meyer, G. Tavakoli and M. van Gemmeren, Dual ligand-enabled nondirected C-H olefination of arenes, Angew. Chem., Int. Ed., 2018, 57, 2497–2501 CrossRef CAS PubMed.
- S. Bag and D. Maiti, Palladium-catalyzed olefination of aryl C-H bonds by using directing scaffolds, Synthesis, 2016, 48, 804–815 CrossRef CAS.
- R. Sharma and U. Sharma, Remote C-H bond activation/transformations: A continuous growing synthetic tool; Part II, Catal. Rev.: Sci. Eng., 2018, 60, 497–565 CrossRef CAS.
- R. Manikandan, P. Madasamy and M. Jeganmohan, Ruthenium-catalyzed ortho alkenylation of aromatics with alkenes at room temperature with hydrogen evolution, ACS Catal., 2016, 6, 230–234 CrossRef CAS.
- R. Das and M. Kapur, Fujiwara-Moritani reaction of Weinreb amides using a ruthenium-catalyzed C-H functionalization reaction, Chem. – Asian J., 2015, 10, 1505–1512 CrossRef CAS PubMed.
- R. Das and M. Kapur, Product control using substrate design: ruthenium-catalysed oxidative C-H olefinations of cyclic weinreb amides, Chem. – Eur. J., 2016, 22, 16984–16988 Search PubMed.
- K. Graczyk, W. Ma and L. Ackermann, Oxidative alkenylation of aromatic esters by ruthenium-catalyzed twofold C-H bond cleavages, Org. Lett., 2012, 14, 4110–4113 CrossRef CAS PubMed.
- Y. Y. Ping, Z. B. Chen, Q. P. Ding, Q. Zheng, Y. Q. Lin and Y. Y. Peng, Ru-catalyzed ortho-oxidative alkenylation of 2-arylbenzo d thiazoles in aqueous solution of anionic surfactant sodium dodecylbenzenesulfonate (SDBS), Tetrahedron, 2017, 73, 594–603 CrossRef CAS.
- Y. C. Yuan, C. Bruneau, T. Roisnel and R. Gramage-Doria, Site-selective Ru-catalyzed C-H bond alkenylation with biologically relevant isoindolinones: a case of catalyst performance controlled by subtle stereo-electronic effects of the weak directing group, Catal. Sci. Technol., 2019, 9, 4711–4717 RSC.
- X. Xue, J. Xu, L. Zhang, C. Xu, Y. Pan, L. Xu, H. Li and W. Zhang, Rhodium(III)-catalyzed direct c-h olefination of arenes with aliphatic olefins, Adv. Synth. Catal., 2016, 358, 573–583 CrossRef CAS.
- X.-G. Liu, H. Gao, S.-S. Zhang, Q. Li and H. Wang, N–O bond as external oxidant in group 9 Cp*M(III)-catalyzed oxidative C–H coupling reactions, ACS Catal., 2017, 7, 5078–5086 CrossRef CAS.
- C. Xia, A. J. P. White and K. K. Hii, Synthesis of isoindolinones by Pd-catalyzed coupling between N-methoxybenzamide and styrene derivatives, J. Org. Chem., 2016, 81(17), 7931–7938 CrossRef CAS PubMed.
- W. D. Lin, W. Q. Li, D. D. Lu, F. Su, T. B. Wen and H. J. Zhang, Dual effects of cyclopentadienyl ligands on Rh(III)-catalyzed dehydrogenative arylation of electron-rich alkenes, ACS Catal., 2018, 8, 8070–8076 CrossRef CAS.
- Q. S. Han, X. M. Guo, Z. Y. Tang, L. Su, Z. Z. Yao, X. F. Zhang, S. Lin, S. C. Xiang and Q. F. Huang, Rhodium-catalyzed regioselective ortho C-H olefination of 2-arylindoles via nh-indole-directed C-H bond cleavage, Adv. Synth. Catal., 2018, 360, 972–984 CrossRef CAS.
- P. Cooper, G. E. M. Crisenza, L. J. Feron and J. F. Bower, Iridium-catalyzed alpha-selective arylation of styrenes by dual C-H functionalization, Angew. Chem., Int. Ed., 2018, 57, 14198–14202 CrossRef CAS PubMed.
- H. Horino and N. Inoue, Ortho-vinylation of anilines via cyclopalladation - new route to nitrogen-heterocycles, Tetrahedron Lett., 1979, 2403–2406 CrossRef CAS.
- M. D. K. Boele, G. P. F. van Strijdonck, A. H. M. de Vries, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, Selective Pd-catalyzed oxidative coupling of anilides with olefins through C-H bond activation at room temperature, J. Am. Chem. Soc., 2002, 124, 1586–1587 CrossRef CAS.
- B. P. Babu, X. Meng and J.-E. Bäckvall, Aerobic oxidative coupling of arenes and olefins through a biomimetic approach, Chem. – Eur. J., 2013, 19, 4140–4145 CrossRef CAS.
- The reaction was conducted isothermally at 30 °C to counteract fluctuations in ambient temperature. A slight excess of the acetanilide was also employed to suppress the formation of the di-olefinated product. .
- W. A. Carole and T. J. Colacot, Understanding palladium acetate from a user perspective, Chem. – Eur. J., 2016, 22, 7686–7695 CrossRef CAS.
- R. N. Pandey and P. M. Henry, Interaction of palladium(II) acetate with sodium and lithium acetate in acetic-acid, Can. J. Chem., 1974, 52, 1241–1247 CrossRef CAS.
- L. A. Adrio, B. N. Nguyen, G. Guilera, A. G. Livingston and K. K. Hii, Deconvolution of the mechanism of homogeneous gold-catalyzed reactions, Catal. Sci. Technol., 2012, 2, 316–323 RSC.
- J. Vana, J. Hanusek and M. Sedlak, Bi and trinuclear complexes in palladium carboxylate-assisted C-H activation reactions, Dalton Trans., 2018, 47, 1378–1382 RSC.
- K.-S. Masters, T. R. M. Rauws, A. K. Yadav, W. A. Herrebout, B. Van der Veken and B. U. W. Maes, On the importance of an acid additive in the synthesis of pyrido[1,2-a]benzimidazoles by direct copper-catalyzed amination, Chem. – Eur. J., 2011, 17, 6315–6320 CrossRef CAS PubMed.
- J. B. C. Mack, J. D. Gipson, J. Du Bois and M. S. Sigman, Ruthenium-catalyzed C–H hydroxylation in aqueous acid enables selective functionalization of amine derivatives, J. Am. Chem. Soc., 2017, 139, 9503–9506 CrossRef CAS PubMed.
- X. X. Shi, W. Y. Xu, R. C. Wang, X. F. Zeng, H. Y. Qiu and M. Wang, Ketone-directed cobalt(III)-catalyzed regioselective C2 amidation of indoles, J. Org. Chem., 2020, 85, 3911–3920 CrossRef CAS PubMed.
- Y. Sun and N. Cramer, Enantioselective synthesis of chiral-at-sulfur 1,2-benzothiazines by Cp*RhIII-catalyzed C−H functionalization of sulfoximines, Angew. Chem., Int. Ed., 2018, 57, 15539–15543 CrossRef CAS PubMed.
- D. Xing, X. T. Qi, D. Marchant, P. Liu and G. B. Dong, Branched-selective direct-alkylation of cyclic ketones with simple alkenes, Angew. Chem., Int. Ed., 2019, 58, 4366–4370 CrossRef CAS PubMed.
- X.-H. Liu, H. Park, J.-H. Hu, Y. Hu, Q.-L. Zhang, B.-L. Wang, B. Sun, K.-S. Yeung, F.-L. Zhang and J.-Q. Yu, Diverse ortho-C(sp2)–H Functionalization of Benzaldehydes Using Transient Directing Groups, J. Am. Chem. Soc., 2017, 139, 888–896 CrossRef CAS PubMed.
- Y. Xiong, T. Besset, D. Cahard and X. Pannecoucke, Palladium(II)-catalyzed directed trifluoromethylthiolation of unactivated C(sp3)-H bonds, J. Org. Chem., 2015, 80, 4204–4212 CrossRef PubMed.
- D. L. Davies, S. M. A. Donald and S. A. Macgregor, Computational study of the mechanism of cyclometalation by palladium acetate, J. Am. Chem. Soc., 2005, 127, 13754–13755 CrossRef CAS PubMed.
- J. Vana, J. Bartacek, J. Hanusek, J. Roithova and M. Sedlak, Bi and trinuclear complexes in palladium carboxylate-assisted C-H activation reactions, J. Org. Chem., 2019, 84, 12746–12754 CrossRef CAS PubMed.
- Note that in this case, both BQ (1 equiv.) and AgNO3 (2 equiv.) were employed as oxidants. .
- T. Nishikata and B. H. Lipshutz, Cationic Pd(II)-catalyzed Fujiwara-Moritani reactions at room temperature in water, Org. Lett., 2010, 12, 1972–1975 CrossRef CAS PubMed.
- W. Rauf, A. L. Thompson and J. M. Brown, Anilide activation of adjacent C-H bonds in the palladium-catalyzed Fujiwara-Moritani reaction, Dalton Trans., 2010, 39, 10414–10421 RSC.
- W. Rauf and J. M. Brown, Palladium-catalyzed directed C-H activation by anilides and ureas; water participation in a general base mechanism, Org. Biomol. Chem., 2016, 14, 5251–5257 RSC.
- Attempts to isolate this mixed-ligand Pd species resulted in its decomposition to Pd black. .
- C. E. Houlden, C. D. Bailey, J. G. Ford, M. R. Gagné, G. C. Lloyd-Jones and K. I. Booker-Milburn, Distinct reactivity of Pd(OTs)2: the intermolecular Pd(II)-catalyzed 1,2-carboamination of dienes, J. Am. Chem. Soc., 2008, 130, 10066–10067 CrossRef CAS PubMed.
- J. S. Renny, L. L. Tomasevich, E. H. Tallmadge and D. B. Collum, Method of continuous variations: applications of Job plots to the study of molecular associations in organometallic chemistry, Angew. Chem., Int. Ed., 2013, 52, 11998–12013 CrossRef CAS PubMed.
- D. G. Blackmond, Kinetic profiling of catalytic organic reactions as a mechanistic tool, J. Am. Chem. Soc., 2015, 137, 10852–10866 CrossRef CAS PubMed.
- J. J. Plata, M. García-Mota, A. A. C. Braga, N. López and F. Maseras, Vinyl acetate synthesis on homogeneous and heterogeneous Pd-based catalysts: a theoretical analysis on the reaction mechanisms, J. Phys. Chem. A, 2009, 113, 11758–11762 CrossRef CAS PubMed.
- A. Vasseur, J. Muzart and J. Le Bras, Ubiquitous benzoquinones, multitalented compounds for palladium-catalyzed oxidative reactions, Eur. J. Org. Chem., 2015, 2015, 4053–4069 CrossRef CAS.
- R. S. Shue, Catalytic coupling of aromatics and olefins by homogeneous palladium(II) compounds under oxygen, J. Chem. Soc. D, 1971, 1510–1511 RSC.
- J. Hajek, M. Dams, C. Detrembleur, R. Jerome, P. A. Jacobs and D. E. De Vos, Heterogeneous alkenylation of aromatics under oxygen, Catal. Commun., 2007, 8, 1047–1051 CrossRef CAS.
- M. Dams, D. E. De Vos, S. Celen and P. A. Jacobs, Toward waste-free production of Heck products with a catalytic palladium system under oxygen, Angew. Chem., Int. Ed., 2003, 42, 3512–3515 CrossRef CAS PubMed.
- D. B. Eremin and V. P. Ananikov, Understanding active species in catalytic transformations: From molecular catalysis to nanoparticles, leaching, "Cocktails" of catalysts and dynamic systems, Coord. Chem. Rev., 2017, 346, 2–19 CrossRef CAS.
- High temperatures and pressures for aerobic processes were avoided for safety reasons, see: A. Gavriilidis, A. Constantinou, K. Hellgardt, K. K. Hii, G. Hutchings, G. Brett, S. Kuhn and S. P. Marsden, React. Chem. Eng., 2016, 1, 595–612 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional data. See DOI: 10.1039/d0re00133c |
| This journal is © The Royal Society of Chemistry 2020 |