
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultitask prediction of site selectivity in aromatic C–H functionalization reactions†
Thomas J.
Struble‡
 ,
Connor W.
Coley‡
and
Klavs F.
Jensen
*
,
Connor W.
Coley‡
and
Klavs F.
Jensen
*
Department of Chemical Engineering, Massachusetts Institute of Technology, USA. E-mail: kfjensen@mit.edu
First published on 1st April 2020
Abstract
Aromatic C–H functionalization reactions are an important part of the synthetic chemistry toolbox. Accurate prediction of site selectivity can be crucial for prioritizing target compounds and synthetic routes in both drug discovery and process chemistry. However, selectivity may be highly dependent on subtle electronic and steric features of the substrate. We report a generalizable approach to prediction of site selectivity that is accomplished using a graph-convolutional neural network for the multitask prediction of 123 C–H functionalization tasks. In an 80/10/10 training/validation/testing pseudo-time split of about 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 aromatic C–H functionalization reactions from the Reaxys database, the model achieves a mean reciprocal rank of 92%. Once trained, inference requires approximately 200 ms per compound to provide quantitative likelihood scores for each task. This approach and model allow a chemist to quickly determine which C–H functionalization reactions – if any – might proceed with high selectivity.
000 aromatic C–H functionalization reactions from the Reaxys database, the model achieves a mean reciprocal rank of 92%. Once trained, inference requires approximately 200 ms per compound to provide quantitative likelihood scores for each task. This approach and model allow a chemist to quickly determine which C–H functionalization reactions – if any – might proceed with high selectivity.
1 Introduction
Aromatic and heterocyclic ring systems are ubiquitous in approved drugs and natural products. Decomposition of drugs into their ring components demonstrates the high representation of aromatic motifs.1 Because a substitution at any position of the aromatic ring can greatly alter the biological activity profile of a compound, it is imperative to investigate many substitution patterns during drug development to build a structure–activity relationship. The compounds that are often included in an analogue library are those that can be accessed from a common late-stage intermediate. A late stage functionalization is desired compared to an analogue of interest that would require carrying the substitution through the whole synthetic process. In an ideal scenario, we would be able to selectively functionalize an intermediate (or other member of the compound library) at a specific position of interest at any point during the synthetic route.Highly site selective reactions are more broadly useful for planning, prioritizing, and executing efficient routes in synthetic chemistry. Achieving high selectivity requires the use of conditions or substrates that can differentiate multiple similar reactive sites within the same molecule. Molecules or routes that include steps with unclear site selectivity may be discarded by chemists due to the lack of a robust method to access the target. This is especially true in synthesis campaigns where separation of isomers is difficult or prohibitively expensive. The concept of site selectivity has been highlighted recently with the development and application of synthetic methods for late stage C–H functionalization of drug targets2–6 which has spurred new methods research,7–11 HTS campaigns,12,13 and analytical techniques.14 Here, we focus on the subset of the prediction of functionalization reactions that target aromatic C–H motifs.
Historically, prediction of site selectivity for aromatic C–H bonds has focused on electrophilic aromatic substitution (EAS) reactions. EAS is acatalytic and proceeds through a relatively simple mechanism whereby selectivity is determined primarily by the stability of the intermediate cation, allowing for relatively straightforward predictivity.15 Early methods include using Hammett and Taft parameters to approximate the nucleophilicity of aromatic rings.16,17 Later, models using estimated 1H and 13C NMR shifts (using ChemDraw's linear additives rules), motivated by the same principle of estimating nucleophilicity, achieved 80% accuracy on a collection of 130 EAS reactions limited to electron rich aromatics and heterocycles.18 When supplemented with density functional theory (DFT) calculations,19 the accuracy of predictions was reported to be >95% on the same dataset and formed the basis for a follow-up semi-empirical quantum mechanical (SQM) method. The SQM model predicts selectivity based on estimated energies of carbocations generated from protonation at each site, meant to represent potential intermediates.20 Any carbocation that is within thresholds of 1 or 3 kcal mol−1 of the lowest energy structure is marked as a possible site for reactivity. The RegioSQM method reaches 90% accuracy within 1 kcal mol−1 and 96% accuracy within 3 kcal mol−1 on their test set of 525 reactions, although this definition of accuracy does not penalize the prediction of multiple reactive sites. More recently, a machine learning approach was applied to prediction of EAS site selectivity prediction using calculated descriptors, including RegioSQM carbocation calculations, and a random forest model to show 90% accuracy on an external validation set.21 These methods achieve good accuracy for EAS reactions but require several minutes per prediction, primarily because multi-conformer DFT or PM6 calculations are necessary as inputs.
Computational prediction of reaction outcomes has a plethora of approaches through many years. Early methods used hand coded heuristics to determine reaction outcomes and include the programs CAMEO,22,23 EROS,24 IGOR,25 SOPHIA,26 and Robia.27 Later, machine learning methods have addressed the issue of reaction prediction by using synthetically-generated mechanistic data,28–30 by scoring predictions based on heuristically extracted templates from synthetic31 or experimental data,32,33 or by making direct predictions of product species also using experimental data.34–36 These approaches can generalize across a large range of different reaction types and reaction outcomes, but more subtle aspects of site, stereo, and regio-selectivity are lost.
A chemist's intuition for site selectivity is based on structure and rarely reliant on precise calculations. There have been many approaches to directly learn molecular function from structure for various prediction tasks, without relying on fixed descriptors, fingerprints, or other feature engineering. One such example is the Weisfeiler-Lehman network, a type of graph convolutional neural network model, used by Coley et al.36 and Jin et al.35 This model, based on the Weisfeiler-Lehman graph kernel, operates directly on molecular graphs containing atom- and bond-level features to learn a meaningful numerical representation of structure.
We sought to address prediction of site selectivity using two basic hypotheses. 1) Selectivity can be learned from two-dimensional structure without calculated atom features or an explicit 3D conformer, thus reducing the computational overhead. 2) Any reaction class can be learned simultaneously using multitask learning, expanding the scope of predictions beyond EAS reactions.
1.1 Methods
The EAS dataset was extracted by identifying all reactions where the sole structural difference between reactants and products is the replacement of an aromatic C–H with a bromo, chloro, nitro, or sulfonyl group. Matching reactions were further filtered to only include ones likely to proceed through EAS by only allowing certain reagents. Brominations were restricted to reactions using Br2 or N-bromosuccinimide; chlorinations using N-chlorosuccinimide, sulfuryl chloride, phosphorus oxychloride, Cl2, or thionyl chloride; nitrations using nitric acid; sulfonylations using chlorosulfonic acid or sulfuric acid.
The more diverse dataset was extracted by identifying all bimolecular reactions where the difference between one reactant and the product is the replacement of an aromatic C–H with a single-bonded heavy atom (preserving the aromaticity of the ring). Matching reactions were categorized into distinct “tasks” based on the identity of the other reactant species. Note that tasks are not defined based on the fragment contributed by the other reactant species, but by its full identity; this is to separate different means of introducing the same functional group, as there may be different selection criteria. Only tasks with at least 100 precedent examples were kept.
Both datasets were further filtered to exclude any reactions with <50% yield, due to our inability to know with certainty that the reported product was the major product (i.e., the site of C–H activation was the most favorable site). The data was further filtered to include only reactions that have more than one aromatic C–H site in the reactant which left 58k examples. It is important to note that the training data is not uniform in its distribution across tasks. For example, of the 127 tasks there are 16k bromination examples with the next highest task having 5.7k examples and many only have 100 examples; a detailed description of the different tasks can be found in the ESI† (Table S1). Reactant symmetry was taken into account when preparing ground truth labels of the most favorable site. We use an 80:10:10 split for training, validation, and testing within each task using a pseudo-time split37 validation based on the date each reaction appeared in Reaxys; this is intended to simulate a prospective prediction of site selectivity based on our current body of knowledge. Performance on the validation set was used for early analysis of hyperparameter settings as well as early stopping during training.
P and molar refractivity,39 total polar surface area, accessible surface area, electrotopological state,40 and Gasteiger partial charges.41 These descriptors are all atom features which would give a richer description to the initial atom feature vectors. Furthermore, they can be calculated quickly using the open source RDKit package and do not significantly slow down inference. For each bond, its bond order and ring status are included in a feature vector fuv.
The atom-centered feature vectors are iteratively updated L times by sum-pooling a learned embedding of neighboring atoms at each iteration. The final representation of local atomic structure is calculated using another learned network. An attention mechanism42 is included to capture the influence of atoms further than L bonds away, including atoms on disconnected molecules; it is used to calculate a weighted sum of all reactant atoms is to give a global representation of each atom. Together, the local and global atom features are used to predict atom reactivity scores for each task, scaled to between zero and one by a sigmoid activation function. The sigmoid activation function is used so that multiple sites can be predicted as in the case with symmetry or non-selective reactions and does not force the model to predict a site if it is not likely as would be the case with a softmax activation. The WLN and the multitask predictor are optimized together by minimizing the cross entropy loss of
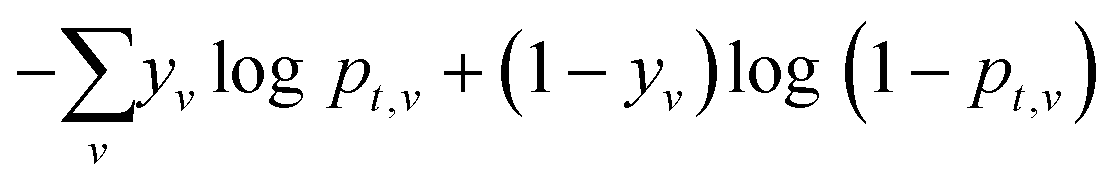
During inference, a prediction for every atom per task is made and the overall architecture is shown in Fig. 1.
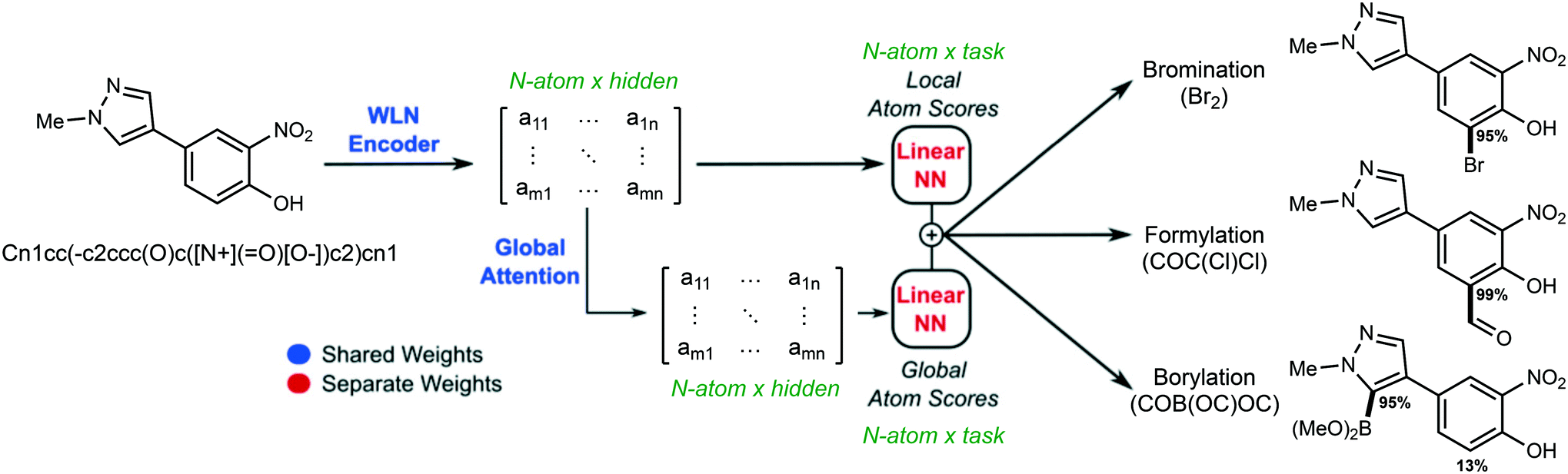 | ||
| Fig. 1 Overall model workflow. A Weisfeiler-Lehman network (WLN) learns to encode a molecular structure into atom-level feature vectors, which are used as the basis for a multitask prediction of site-selectivity for many reaction types simultaneously. | ||
As a baseline model, we include a multilayer feed forward neural network that operates only on atom feature vectors fv to make reactivity predictions in isolation. This model does not contain any pooling of information from neighboring atoms.
2 Results and discussion
2.1 Single-task performance
Previous studies employing the Weisfeiler-Lehman graph convolutional neural network32,35,36,43 formed the basis for using structure, in the form of a molecular graph, as the input to site predictions. The Weisfeiler-Lehman neural network (WLN) is perfectly suited to this task; a local representation for each atom is calculated and a global context based using an attention mechanism can be employed. As proof of concept that the WLN encoder would allow for learning site selectivity, a single task network was constructed to predict a score for each atom in a molecule for four EAS reactions: bromination, chlorination, nitration, and sulfonylation.We first evaluate the performance of our model when training on each selectivity task individually. Table 1 shows the accuracy of the 4 different EAS reactions on the validation and test sets. Overall, average to good performance is seen for each reaction. The highest accuracy was seen with bromination and nitration reactions which correlates to both of these reactions having more training data than chlorination or sulfonylation. The benefit of the 2D graph representation is that it circumvents the need for conformer generation and energy minimization which is both time intensive and does not scale with increasing molecular size. The results in 1 demonstrate that using the SMILES as an input to construct the molecular graph allows the WLN network to learn from the molecular structure without the need for computationally expensive atom features (e.g., DFT estimates of partial charge require a workflow of conformer generation and energy minimization which takes minutes to hours depending on the size of the molecule).
2.2 Multi-task performance
2.3 Cross-task performance
Since the four previous EAS reactions operate under a similar mechanism, a model with better performance should transfer to the other similar tasks. Sulfonylation was removed from this study since the performance was only 50% for its own test set. A separate network was trained for each task (reaction type) and then tested on the validation set for all the other tasks. Results in Table 2 demonstrate that the accuracy varies when a model is transferred to another task's validation set but that these tasks are clearly not independent. Overall, bromination has the most data and seems to generalize the best across tasks but nitration with the second most amount of data does not seem to follow this trend. Additionally, aromatic scaffolds in each task might not be very diverse since they are a subset of C–H functionalization reactions and each task would not generalize well to others.| Trained on | ||||
|---|---|---|---|---|
| Br (N = 13028) |
Cl (N = 2264) | NO2 (N = 4660) | ||
| Tested on | Br (N = 1629) | 89.5 | 75.8 | 70.3 |
| Cl (N = 283) | 79.9 | 75.6 | 66.1 | |
| NO2 (N = 583) | 81.3 | 64.3 | 88.7 | |
To further demonstrate that single task network training/testing was not a viable option for a larger corpus of reactions, all 127 tasks were trained/tested on one another. A heat-map shown in Fig. 2 demonstrates that direct transfer across tasks is not achievable. A small cross-section is blown up to show that there are reactions that might not operate under the same mechanism in the dataset. This is due to two main factors, 1) not all the tasks are similar in mechanism and 2) some tasks only have around 100 examples which might not be sufficient for the structure encoding to be learned and generalized.
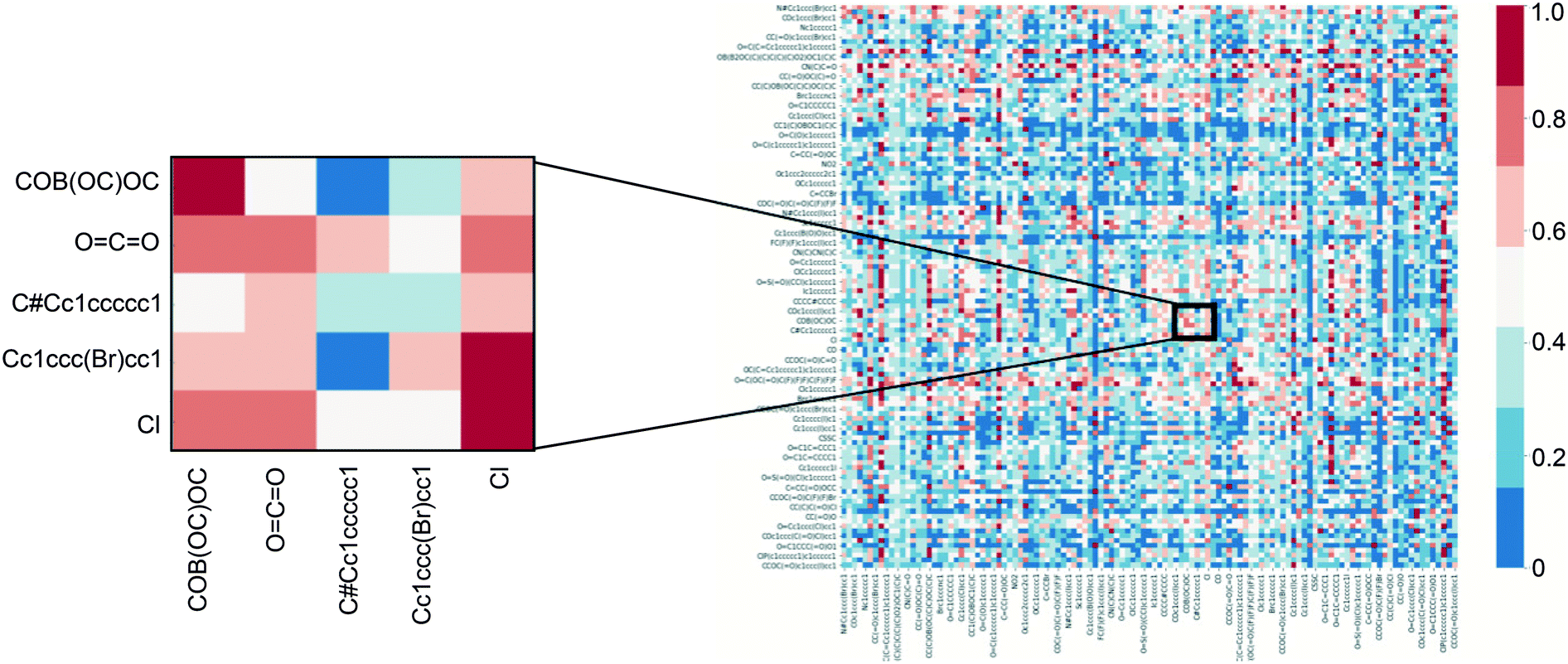 | ||
| Fig. 2 Top-1 accuracy when training on a single C–H functionalization task and testing on another. Columns correspond to the training task; rows correspond to the testing task. | ||
To address the issue of low data availability for some tasks, a multitask network was constructed so that the encoder weights are shared. A multitask network imitates the chemists intuition that within the broad scope of all C–H functionalization reactions, there are shared mechanisms driving selectivity (e.g., nucleophilicity, electrophilicity, steric hindrance, catalyst-directing groups). After calculating learned atom representation, the representations are fed through a fully-connected linear layer, summed, and separated into the individual tasks. A baseline model was also constructed that performs the atom embedding without the graph convolution step. Without iterative updating of neighboring atoms, the baseline model cannot directly learn structure and results are poor (Table 3). To demonstrate a baseline model benefits from atom descriptors, simple features calculated by RDKit§ were included in the input representation and indeed a significant boost in accuracy is observed (Table 3). The hypothesis that the atom embedding calculated by the WLN encoder are a good representation of the local area around the atom is supported by the similar performance regardless of whether additional atom features are included in the input of the WLN encoder (i.e., pre-calculating these atom properties provides little benefit in performance).
| Model | Without atom features | With atom features | ||
|---|---|---|---|---|
| Top-1 acc. (%) | MRR (%) | Top-1 acc. (%) | MRR (%) | |
| Baseline (no WLN) | 21.3 | 45.9 | 47.6 | 64.9 |
| Single task | 81.8 | 88.6 | 81.0 | 88.3 |
| Multitask | 83.1 | 89.5 | 84.0 | 90.1 |
Table 3 shows that the overall accuracy for single task prediction is close to or the same as the multitask predictor. Generally, within each task, the operating mechanism that defines selectivity is inherent to the task and there is not a wide diversity of conditions that would lead to differing selectivity. For example, in the task of coupling bromobenzene (task labeled Brc1ccccc1), some form of palladium is listed in the reagents for over 70% of the reactions. Similarly, reaction conditions (reagents, catalysts, solvents, etc.) are relatively consistent within a task, so adding the reagents to the input does not greatly improve the model performance for most tasks (see ESI† for complete details). Including reagents can help improve accuracy and is desirable for cases such a virtual screens or if the chemist already knows the conditions, but for a chemist's idea generation it is beneficial to also have a model that would not require the additional input of reagents.
Examples of the poor predictions are presented in the ESI.† The only clear trend that arises is due to the time-split validation we use for evaluation. There are only nine tasks that have accuracy below 50%; of these, many errors can be attributed to new chemistry. That is, reports of new methods often incorporating catalysts/ligand combinations to achieve unprecedented selectivity. The model is designed to understand chemical structure and should generalize to new aromatic cores, but cannot generalize to new chemistry that significantly alters reactivity and selectivity.
2.4 Comparison to other methodology
Previous methodology, namely RegioSQM,20 achieves high site prediction accuracy based on enumeration and calculation of protonated carbocation intermediates in the EAS pathway.¶ comparison of the WLN methodology to RegioSQM was limited to bromination reactions for a fair comparison to the intended application of RegioSQM. 500 reactions were selected at random from our test set for comparison. 6 of the 500 had either had a structure that failed to converge or had a proton transfer during optimization, and were thrown out leaving 494 total reactions for comparison. The RegioSQM methodology outputs the lowest energy carbocation and any other of enumerated carbocations that are within a set threshold of 1 kcal mol−1 as the top predictions. This means that there can be multiple predictions by RegioSQM. In contrast, analysis of the WLN uses only the top 1 pick to calculate accuracy. To overcome this disparity in evaluation between the two methodologies in how accuracy is defined, analysis was performed by ranking the predictions. With RegioSQM, the predictions were ranked by lowest to highest energy conformer and in the WLN ranking was based on the final atom scores for bromination (a comparison based on how the authors of RegioSQM performed analysis is detailed in the ESI†). The accuracy of the top 1, 2, and 3 choices and the mean reciprocal rank are calculated and reported.In Table 4 are the results of this comparison which shows that the WLN predictions for bromination are more accurate than those of RegioSQM methodology. Finally and most important is that 300 predictions by RegioSQM takes over 10 days to complete while the neural network model makes the same predictions in 6.3 seconds both using 12 CPU cores. Even when training is factored in for the WLN, the total time is less than 4 hours for all 130 tasks and 58k total examples (train/valid/test) on a single GTX 1080 GPU. Although not surprising that a neural net will make predictions faster than semi-empirical methods, our approach now makes application to thousands of molecules tractable.
| Top 1 (%) | Top 2 (%) | Top 3 (%) | MRR (%) | Time (12 CPU's) | |
|---|---|---|---|---|---|
| RegioSQM19 | 79.7 | 89.2 | 93.3 | 87.1 | >10 days |
| WLN | 85.0 | 92.3 | 95.7 | 90.5 | 6.3 s |
Some examples of predictions for both methodologies are shown in Fig. 3. Only the top 1 prediction is drawn for the WLN and only sites that are within 1 kcal mol−1 of the lowest energy conformer in the RegioSQM method are drawn. The first two examples show that the WLN is effective at finding the correct site and often that correlates with the prediction from RegioSQM. An advantage of the WLN is demonstrated by the sites chosen for compound 1 where the WLN chooses one site and RegioSQM gives 4 predictions one of which is correct. Predictions for compounds 3 and 4 show that when the sites with the lowest calculated energies are not the true reactive site, the WLN can recognize the structure and furnish the correct position. An interesting pattern is seen when both methodologies predict the incorrect site where the WLN agrees with the RegioSQM prediction demonstrating that the structure is indeed learned using the multitask network. The final examples is one that was shown in the original publication of RegioSQM methodology where presumably sterics on the phenolic oxygen disfavor the lowest energy site from reacting and the WLN correctly identifies the reactive site.
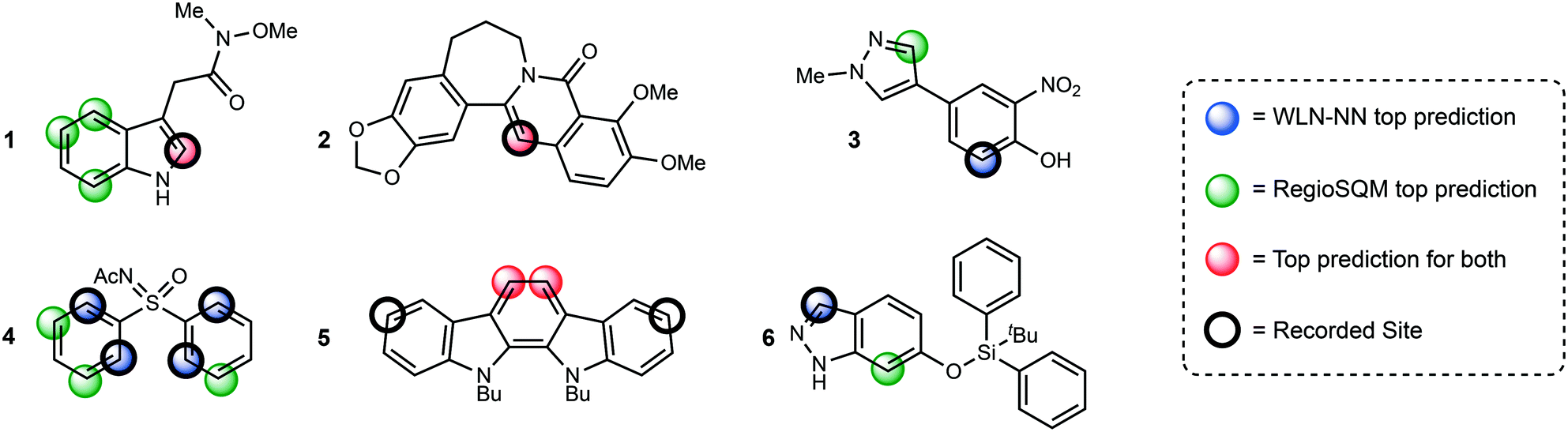 | ||
| Fig. 3 Comparative examples between the WLN and RegioSQM predictions. All RegioSQM predictions within 1 kcal mol−1 are highlighted. | ||
3 Conclusion
In conclusion, a multitask network was developed to predict site selectivity of aromatic C–H functionalization reactions using a Weisfeiler-Lehman encoder for learning predictions based on structure. Top 1 accuracy of the model on the test set is 84% with a mean reciprocal rank of 90%. The WLN achieves similar or better accuracy when compared to semi-empirical methods and drastically reduces the time for predictions from minutes to less than a second per molecule. In addition, the tool can be used to prioritize compounds or intermediates in a synthetic route that could be accessed selectively, leading to a more diversified collection of compounds. Finally, a large corpus of general reactions was previously used with the WLN architecture for reaction prediction and this study extends the WLN's capability to learn very specific site selectivity and reactivity.Conflicts of interest
Authors declare no competing interests.Acknowledgements
We thank the DARPA Make-It program under contract ARO W911NF-16-2-0023 and the Machine Learning for Pharmaceutical Discovery and Synthesis consortium and for funding this research. We also thank Elsevier for the invaluable curation of the Reaxys data set.References
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- W. Joanna and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef PubMed.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2015, 45, 546–576 RSC.
- J. Boström, D. G. Brown, R. J. Young and G. M. Keserü, Nat. Rev. Drug Discovery, 2018, 17, 709 CrossRef PubMed.
- H. Yao, Y. Liu, S. Tyagarajan, E. Streckfuss, M. Reibarkh, K. Chen, I. Zamora, F. Fontaine, L. Goracci and R. Helmy, et al. , Eur. J. Org. Chem., 2017, 7122–7126 CrossRef CAS.
- L. J. Durak, J. T. Payne and J. C. Lewis, ACS Catal., 2016, 6, 1451–1454 CrossRef CAS PubMed.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- H. M. L. Davies and D. Morton, ACS Cent. Sci., 2017, 3, 936–943 CrossRef CAS PubMed.
- S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed.
- F. D. Toste, M. S. Sigman and S. J. Miller, Acc. Chem. Res., 2017, 50, 609–615 CrossRef CAS PubMed.
- K. Feng, R. E. Quevedo, J. T. Kohrt, M. S. Oderinde, U. Reilly and M. C. White, Nature, 2020, 1–11 Search PubMed.
- S. B. Boga, M. Christensen, N. Perrotto, S. W. Krska, S. Dreher, M. T. Tudge, E. R. Ashley, M. Poirier, M. Reibarkh and Y. Liu, et al. , React. Chem. Eng., 2017, 2, 446–450 RSC.
- S. W. Krska, D. A. DiRocco, S. D. Dreher and M. Shevlin, Acc. Chem. Res., 2017, 50, 2976–2985 CrossRef CAS PubMed.
- J. Richardson, G. Sharman, F. Martínez-Olid, S. Cañellas and J. E. Gomez, React. Chem. Eng., 2020, 5, 779–792 RSC.
- G. A. Olah, Acc. Chem. Res., 1971, 4, 240–248 CrossRef CAS.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59(1), 96–103 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- M. Kruszyk, M. Jessing, J. L. Kristensen and M. Jørgensen, J. Org. Chem., 2016, 81, 5128–5134 CrossRef CAS PubMed.
- M. Liljenberg, T. Brinck, B. Herschend, T. Rein, G. Rockwell and M. Svensson, J. Org. Chem., 2010, 75, 4696–4705 CrossRef CAS PubMed.
- J. C. Kromann, J. H. Jensen, M. Kruszyk, M. Jessing and M. Jørgensen, Chem. Sci., 2017, 9, 660–665 RSC.
- A. Tomberg, M. J. Johansson and P. Norrby, J. Org. Chem., 2019, 84, 4695–4703 CrossRef CAS PubMed.
- T. D. Salatin and W. L. Jorgensen, J. Org. Chem., 1980, 45, 2043–2051 CrossRef CAS.
- M. G. Bures, B. L. Roos-Kozel and W. L. Jorgensen, J. Org. Chem., 1985, 50, 4490–4498 CrossRef CAS.
- J. Gasteiger, M. G. Hutchings, B. Christoph, L. Gann, C. Hiller, P. Löw, M. Marsili, H. Saller and K. Yuki, Organic Synthesis, Reactions and Mechanisms, Berlin, Heidelberg, 1987, pp. 19–73 Search PubMed.
- I. Ugi, J. Bauer, K. Bley, A. Dengler, A. Dietz, E. Fontain, B. Gruber, R. Herges, M. Knauer, K. Reitsam and N. Stein, Angew. Chem., Int. Ed. Engl., 1993, 32, 201–227 CrossRef.
- H. Satoh and K. Funatsu, J. Chem. Inf. Comput. Sci., 1995, 35, 34–44 CrossRef CAS.
- I. M. Socorro, K. Taylor and J. M. Goodman, Org. Lett., 2005, 7, 3541–3544 CrossRef CAS PubMed.
- M. A. Kayala, C.-A. Azencott, J. H. Chen and P. Baldi, J. Chem. Inf. Model., 2011, 51, 2209–2222 CrossRef CAS PubMed.
- M. A. Kayala and P. Baldi, J. Chem. Inf. Model., 2012, 52, 2526–2540 CrossRef CAS PubMed.
- K. Molga, E. P. Gajewska, S. Szymkuć and B. A. Grzybowski, React. Chem. Eng., 2019, 4, 1506–1521 RSC.
- J. N. Wei, D. Duvenaud and A. Aspuru-Guzik, ACS Cent. Sci., 2016, 2, 725–732 CrossRef CAS PubMed.
- C. W. Coley, R. Barzilay, T. S. Jaakkola, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2017, 3, 434–443 CrossRef CAS PubMed.
- M. H. S. Segler and M. P. Waller, Chem. – Eur. J., 2017, 23, 5966–5971 CrossRef CAS PubMed.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. Bekas and A. A. Lee, arXiv e-prints, 2018, arXiv:1811.02633.
- W. Jin, C. W. Coley, R. Barzilay and T. S. Jaakkola, NIPS, 2017 Search PubMed.
- C. W. Coley, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jensen, Chem. Sci., 2019, 10(2), 370–377 RSC.
- R. P. Sheridan, J. Chem. Inf. Model., 2013, 53, 783–790 CrossRef CAS PubMed.
- G. Landrum, RDKit: Open-source cheminformatics, http://www.rdkit.org Search PubMed.
- S. A. Wildman and G. M. Crippen, J. Chem. Inf. Comput. Sci., 1999, 39, 868–873 CrossRef CAS.
- L. H. Hall and L. B. Kier, J. Chem. Inf. Comput. Sci., 1995, 35, 1039–1045 CrossRef CAS.
- J. Gasteiger and M. Marsili, Tetrahedron, 1980, 36, 3219–3228 CrossRef CAS.
- D. Bahdanau, K. Cho and Y. Bengio, CoRR, 2014, arXiv:1409.0473.
- T. Lei, W. Jin, R. Barzilay and T. S. Jaakkola, ICML, 2017 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00071j |
| ‡ Equal contribution. |
| § Added atom features to the initial Weisfeiler-Lehman implementation include atomic number, atom contributions to Crippen logP, Crippen molar refractivity, total polar surface area, accessible surface area, Gasteiger charge, and atom electronegativity. |
| ¶ Other methodology by Kruszyk et al. using neural networks was not implemented for comparison due to the restriction of using inaccessible software such as Gaussian for DFT inputs. RegioSQM uses free software that is easily obtained and deployed and furthermore is an input in Kruszyk et al. neural network implementation. |
| This journal is © The Royal Society of Chemistry 2020 |