
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Arsenic immobilization as crystalline scorodite by gas-diffusion electrocrystallization†
Guillermo
Pozo
*ab,
Diane
van Houtven
a,
Jan
Fransaer
cb and
Xochitl
Dominguez-Benetton
 *ab
*ab
aSeparation and Conversion Technologies, VITO, Flemish Institute for Technological Research, Boeretang 200, 2400, Mol, Belgium. E-mail: xoch@vito.be
bSIM vzw, Technologiepark 935, BE-9052 Zwijnaarde, Belgium
cDepartment of Materials Engineering, Surface and Interface Engineered Materials, Katholieke Universiteit Leuven, Kasteelpark Arenberg 44 – box 2450, 3001 Leuven, Belgium. E-mail: gpozo17@gmail.com
First published on 15th May 2020
Abstract
The safe immobilization of arsenic present in liquids is a key environmental challenge due to the inherent toxicity of arsenic. This immobilization is mostly restricted by the application of chemicals and several stages of oxidation and precipitation. Although the formation of bioscorodite is a greener alternative, it is intensive in the use of energy for aeration, and it is costly due to nutrient addition. The electrochemically-driven crystallization of arsenic into scorodite is proposed here to overcome these limitations. We disclose gas-diffusion electrocrystallization (GDEx) for the immobilization of arsenic into highly crystalline scorodite (FeAsO4·2H2O) by the in situ production of oxidizing substances (i.e., H2O2) on gas-diffusion electrodes. GDEx yielded an exceptional arsenic immobilization efficiency of up to 70% without the use of any primary minerals or seed crystals. At 70 °C and using As3+ as the precursor, polydisperse micrometric scorodite particles were obtained (from fine particles of <1 μm to large particles of ∼5 μm). In contrast, fine micrometric particles of <1 μm were achieved using As5+ as the precursor. Using one-pot and one-step GDEx enabled the synthesis of scorodite that was 14 times less soluble than required for stable scorodite disposal. Current chemical oxidation–precipitation processes use two separate reactors, including the oxidation of As3+ to As5+, and then the precipitation of the As5+ with Fe3+ to generate scorodite at a temperature higher than 90 °C. In contrast, the new GDEx approach combines both reactors into one to produce crystalline scorodite at 50 °C, hence reducing energy requirements and chemical footprint.
Introduction
Non-ferrous base metals, such as copper and zinc, are frequently associated with chalcophile elements, such as arsenic and antimony. During smelting, arsenic-containing materials are enriched and accumulated in high volumes, which are barely marketable due to the inherent toxicity of arsenic.1 Among the four oxidation states of arsenic (+5, +3, 0, −3), the inorganic forms of arsenic, namely As3+ (arsenite; H3AsO3) and As5+ (arsenate; H3AsO4), are highly toxic and mobile in the environment, with their presence and mobility being controlled by redox potential, pH, biological activity, and adsorption/desorption reactions.2As environmental contamination is a serious global issue, many remediation technologies have been developed to remove arsenic from water, waste- and process-streams, and leachates. In either case—in nature or industrial remediation processes—the immobilization of arsenic into solid materials requires a two-step approach consisting of: (1) the oxidation of the soluble trivalent arsenic species into the pentavalent state, and (2) the precipitation into a stable solid product. The trivalent form is ten times more toxic than pentavalent compounds.3,4
The oxidation of soluble trivalent to pentavalent arsenic can occur under ambient conditions in the presence of oxygen; however, without a catalyst, the kinetics of this oxidation reaction is slow.5 Demopoulos et al.6 found that oxygen alone is not effective for oxidizing As3+ into As5+ in acidic solutions, even using elevated temperature and pressure and in the presence of a well-known Cu2+ catalyst. Aside from atmospheric oxygen, various chemicals have been used to directly oxidize arsenite in water, including hypochlorite, ozone, and permanganate.7 Following the oxidation of arsenite into arsenate, precipitation is often used to remove arsenic from waste streams.
The most commonly produced arsenic precipitates are arsenic sulfides, calcium arsenate, and ferric arsenate. However, their disposal and storage are not entirely safe, as they readily undergo physical and chemical changes with time, resulting in the resolubilization of arsenic in the environment.8 The precipitation of amorphous ferric arsenate requires a high ratio of iron consumption to arsenic, i.e., Fe/As > 4, and thus large amounts of waste material are produced.9 This gelatinous material of ferric arsenate contains no more than 6 wt% arsenic, with a maximum solid content of 20 wt%.4
In contrast, the immobilization of arsenic into crystalline scorodite (FeAsO4·2H2O) is the preferred route due to its low solubility and high arsenic content (30 wt%).10
Langmuir et al.7 compared the solubility in water of amorphous ferric arsenate (AFA) to that of crystalline scorodite. They showed that crystalline scorodite is about 100 times less soluble than the amorphous form. Thus, scorodite is usually produced hydrothermally, being, however, a costly solution. Furthermore, hydrothermal methods immobilize As5+ with Fe3+ as starting materials; this is inconvenient because most environmental and industrial streams contain As3+ and Fe2+, and thus an oxidation step is required a priori.11 In practice, precipitation is achieved either by autoclave hydrothermal precipitation of scorodite from acidic solutions (pH 1, 150 °C) containing Fe3+ and As5+12 or by ambient pressure precipitation from acidic solutions (pH ∼ 1, 90 °C) containing Fe3+ and As5+ or As3+. Scorodite has also been precipitated at 95 °C by the oxidation of ferrous ions and oxygen gas in the presence of As5+, with concentrations as high as 0.7 M in sulfuric acid solution.1 Biogenic scorodite has been made at 72 °C between 0.5 and 2.5 g As5+ L−1; in this case, thermophilic archaea were utilized to microbially-oxidize Fe2+ to Fe3+,13 though higher concentrations of arsenic were avoided due to arsenic inhibition of microorganisms. Moreover, the costs for aeration with the biomass and nutrient addition are elevated.
Electrochemical methods have also been explored for the removal of arsenic from aqueous streams, with electrocoagulation (EC) being the technique that offers a higher removal efficiency compared to the conventional approaches.14–18 In EC, a flocculating agent is generated by electrooxidation of a sacrificial anode, such as aluminum or iron, in a single electrochemical cell. Even though the EC process is very effective for the elimination of arsenic from aqueous media, the rate of passivation and frequency of the replacement of the electrodes is a significant drawback. Moreover, the by-products generated by EC consist of a mixture of crystalline and amorphous materials, including iron oxides, aluminum oxides, and mansfieldite (AlAsO4·2(H2O)). However, no one has investigated the immobilization of arsenic, even less so as crystalline scorodite, under cathodic conditions let alone using gas-diffusion electrodes. Therefore, our first aim was to demonstrate the gas-diffusion electrocrystallization (GDEx) process19,20 as an alternative to remove arsenic from solution, preferably forming scorodite without the use of any primary minerals or seed crystals.
Concisely, the GDEx process produces reactive intermediates that precipitate metal ions in solution, resulting from the oxygen reduction reaction (ORR) at a gas-diffusion cathode.19 Within this embodiment, GDEx has been described as a flexible and robust platform to synthesize numerous types of nanoparticles, including iron oxide nanoparticles19 and spin transition nanoparticles,20 as well as libraries of nanostructures.21 Yet, it can also be employed for the recovery of metals and metalloids, often as functional materials. The mechanism through which GDEx forms oxides and hydroxides is depicted in Fig. 1. It can vary as a function of the electrolyte and gas composition, as well as the reaction conditions.
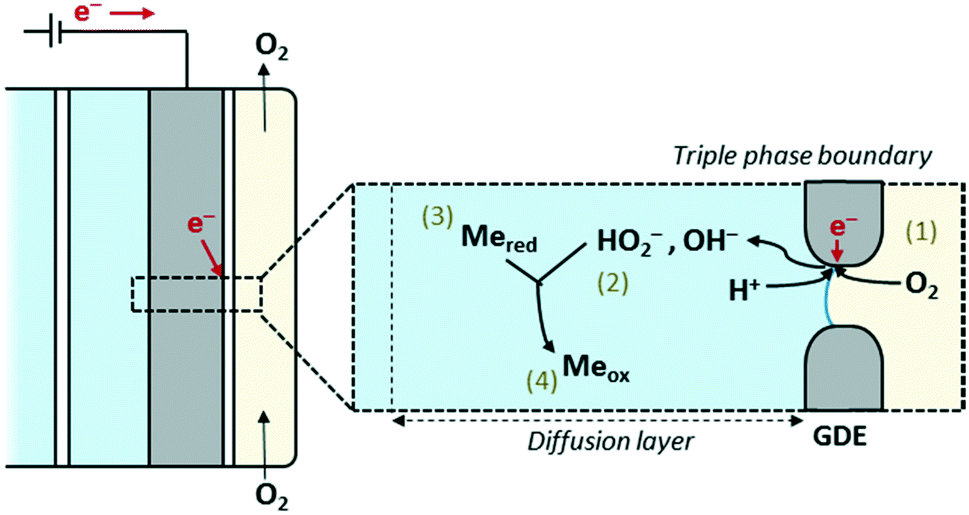 | ||
| Fig. 1 Schematic representation of the (half-cell) gas-diffusion electrocrystallization (GDEx) process. O2 is electrochemically reduced at the triple phase boundary of a gas-diffusion electrode (GDE), i.e., the cathode. The products of the oxygen reduction reaction (ORR) are oxidizing chemical species that turn metal ions into e.g., metal oxides or hydroxides. Alternative mechanisms are possible depending on the reaction conditions. | ||
Since As3+ is the predominant arsenic species in metallurgical operations, our second aim was to evaluate if scorodite could be formed via GDEx, by using only one step (simultaneous oxidation–precipitation) instead of two separate ones (i.e., oxidation followed by precipitation), thus enabling the simultaneous oxidation of As3+ and its immobilization as a crystalline scorodite product. In addition, the feasibility of doing this at temperatures lower than the state of the art was investigated. Different reactor configurations were also assessed in order to improve the process performance. Finally, to benchmark As-leachability from GDEx-scorodite, we prepared a reference material by chemical precipitation. To the best of our knowledge, there are no reports of scorodite synthesis using a one-pot one-step reactor without the use of seed crystals, setting a step towards greener arsenic-waste treatment.
Experimental
Gas-diffusion electrocrystallization reactor
The GDEx reactor employed for the synthesis of scorodite contained 3 chambers, as shown in Fig. 2. Through the first chamber, oxygen or air flow was maintained at a fixed rate. With a set overpressure (e.g., 26 mbar), they percolate through the hydrophobic layer of a VITO CORE® multi-layered PTFE-bonded carbon-based gas-diffusion cathode (GDE). The multi-layered electrodes consisted of a current collector, an active layer made of activated carbon and a hydrophobic, water-impervious, gas-diffusion layer. The current collector was made of a stainless-steel gauze, with a wire diameter of 100 μm and mesh 44 (316L, Solana, Belgium). The composition of the hydrophilic active layer was 80 wt% activated carbon with 20 wt% PTFE as the polymeric binder. Norit®SX1G (878 m2 g−1, Norit Americas Inc., USA) was employed as the active carbon source. The hydrophobic gas-diffusion backing was made of polytetrafluoroethylene (Teflon® PTFE-636-N, Dupont). The manufacturing method for the working electrode has been described elsewhere.22 The counter electrode (anode) consisted of a platinum/iridium (97 wt%/3 wt%) sheet, laser-welded to a titanium-plate current collector.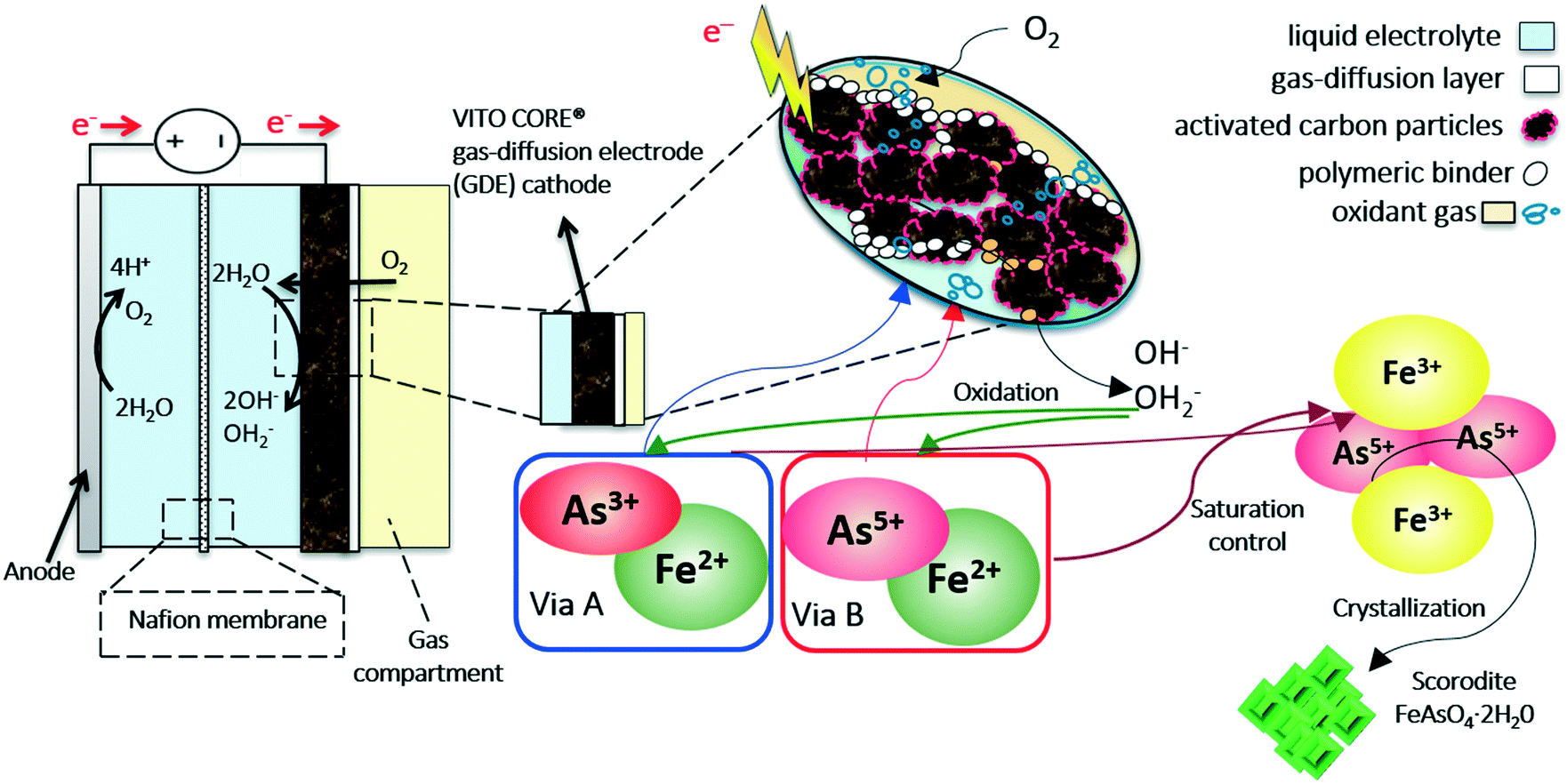 | ||
| Fig. 2 Schematic representation of the arsenic immobilization into crystalline scorodite by a one-pot, one step gas-diffusion electrocrystallization (GDEx) process. | ||
The anode and cathode compartments were separated by a proton exchange membrane (Nafion® N117, Ion power, Germany).
Both electrodes and the separator had a projected cross-section of 10 cm2. Acid pretreatment was carried out for Nafion® to enhance the ionic conductivity of the membrane by 3 wt% H2O2, 50 wt% HNO3 and 0.5 M H2SO4 as described elsewhere.23
A 3 M KCl saturated Ag/AgCl reference electrode (+200 mV vs. SHE) (REF321, Radiometer Analytical, Hach, USA) was inserted in proximity to the working electrode, via an external connector chamber, filled with 3 M KCl. All reported potentials referred to in this article are versus the standard hydrogen electrode (SHE).
Operation of the gas-diffusion electrocrystallization (GDEx) reactor
The flow rate for the anolyte and catholyte solutions was 35 mL min−1. Their total volume was 250 mL, respectively, which was continuously stirred in a recirculation bottle at a rotation rate of 450 rpm using a polygonal PTFE-coated rotating magnetic stirring bar. Pure oxygen was fed through the cathodic gas compartment at a flow rate of 200 mL min−1. Although trials were conducted using air, the GDEx process was only effective when using pure oxygen. A mass gas flow meter and controller (GF40 Bronkhorst hi-tech B.V, Netherlands) were set in place. An overpressure of 26 mbar over a water column was applied at the gas exhaust. The anolyte and catholyte recirculation reservoirs were placed in an oil bath, which was supported on a heating plate with temperature control. The starting solutions did not contain any scorodite seeds.The anodic recirculation reservoir was filled with a 0.8 M H2SO4 solution, supplemented with 0.3 M Na2SO4. The cathodic recirculation reservoir was filled with a 0.8 M H2SO4 solution, supplemented with 0.3 M Na2SO4, 0.22 M As5+ in the form of As2O5 99.9% (Alfa Aesar, Thermo Fisher GmbH, Germany) or As3+ in the form of As2O3 99.9% (Alfa Aesar, Thermo Fisher GmbH, Germany), and 1.25 M Fe2+ in the form of FeSO4·7H2O (Sigma Aldrich, ≧99%). Low concentrations of arsenic and iron were considered to be out of scope from our proof of concept, due to the theoretical restrictions on scorodite stability at low concentrations (i.e., from dilute solutions, scorodite is only deemed to be stable, as predicted by E-pH diagrams, at high temperatures, as shown in the ESI†). Thus, the Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :As molar ratio was 5:1, which is in the appropriate range (>3:1) for the formation of scorodite through other methods.24,25
:As molar ratio was 5:1, which is in the appropriate range (>3:1) for the formation of scorodite through other methods.24,25
The initial pH was corrected to 0.0; therefore, the conductivity of the catholyte solution prior to the start of the electrochemical experiments was 122 mS cm−1 using As3+ and 153 mS cm−1 using As5+ as the arsenic precursor.
GDEx was operated chronoamperometrically (CA), at a constant polarization potential of −0.15 VSHE, using a multichannel potentiostat/galvanostat (VMP 3, Bio-Logic SAS, France) connected to an external high current booster unit (20 A) (VMP 3-80, Bio-Logic SAS, France). The booster was necessary due to the high concentration of electrolytes and metal ions in solution. Under these conditions, oxygen is electrochemically reduced to H2O2 in a 2 electron (2 e−) transfer process and H2O in a 4 electron (4 e−) transfer process at the GDE at non-catalyzed carbon electrodes.26
A photometric method was employed to determine the concentration of H2O2 in solution at pH 2.2 (Quantofix® Relax reflexion photometer, Macherey-Nagel, GmbH & Co. Germany). The peroxide determination was not possible under the GDEx operational conditions (pH = 0.0). In the pH range of 2–9, the accuracy of the determination is independent of the pH of the test solution.
Thus, a reference GDEx run was performed at pH 2. Scorodite cannot be formed at that pH. However, the H2O2 production rate was reckoned from the slope of this curve at relevant time intervals.
Three different GDEx modes of operation were evaluated using As3+ as the precursor at 70 °C (see Fig. 3 and Table ESI2†). The first configuration (a) corresponded to a two-chamber setup, wherein As3+ and Fe2+ were supplied to the cathode chamber (side of the GDE). Both chambers were here separated by a Nafion membrane. The second (b) corresponded to a one-chamber reactor without a Nafion membrane (aka, membrane-less-three electrode electrochemical cell), which combined the oxidation at the anode side with the OH2− production at the GDE.
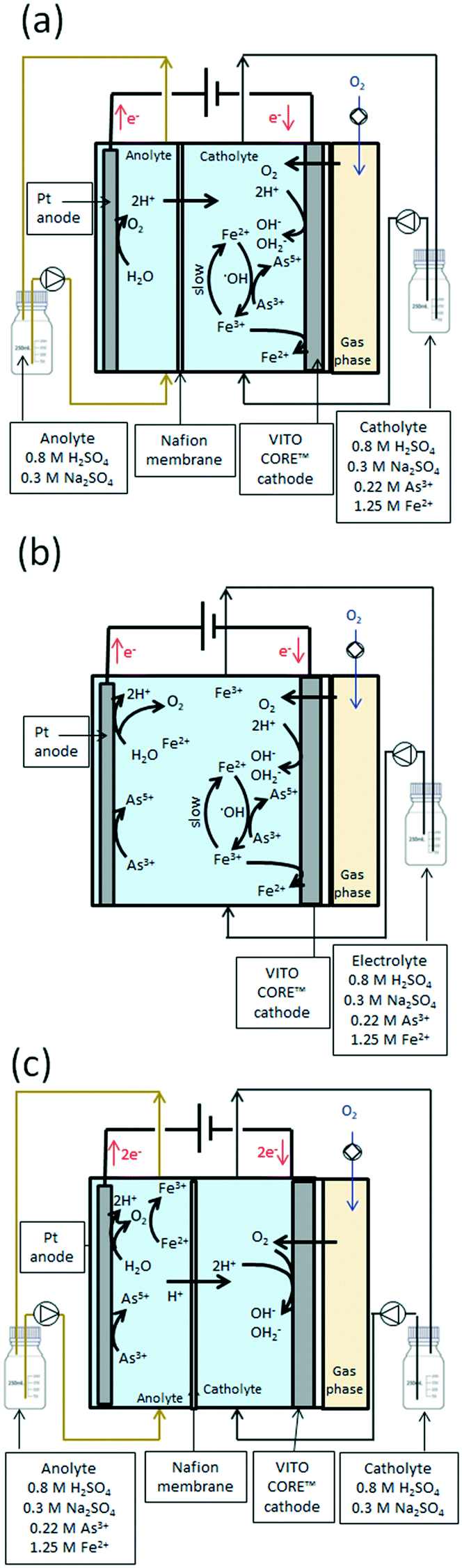 | ||
| Fig. 3 Different GDEx operation modes for the synthesis of scorodite: a) two-chamber setup, wherein As3+ and Fe2+ were supplied to the cathode chamber (side of the GDE), b) one-chamber setup without a Nafion membrane (aka, membrane-less-three electrode electrochemical cell), and c) two-chamber setup to elucidate the role of the GDE in the scorodite crystallization by feeding the precursor in the anode side without contact with the GDE. | ||
Finally, a two-chamber setup (c) was used to elucidate the role of the anode in scorodite crystallization using setup (b), by feeding the As3+ precursor in the anode side, without contact with the GDE.
A summary of the different operational conditions is provided in Table S2.† It is to be noted that in all instances, besides OH2−, OH− ions are generated at the cathode. However, due to the highly-acidic conditions, they are immediately neutralized. Thus, unlike other typical GDEx processing schemes in batch mode,19 pH evolution is not followed in this case. It is instead measured to be constant at 0.0.
Comparison of GDEx-made scorodite vs. chemically-made scorodite on As-leachability
Chemical precipitation experiments were carried out in reaction vessels of 100 mL with an operational volume of 70 mL. The precipitation reactor was filled with 0.8 M H2SO4 and 0.3 M Na2SO4 containing 0.22 M As5+ in the form of As2O5 99.9% (Alfa Aesar, Thermo Fisher GmbH, Germany). The reactor vessel was continuously stirred at a rotation rate of 450 rpm using a polygonal PTFE-coated rotating magnetic stirring bar. Pure oxygen was sparged into the solution of 0.22 M As5+ at a flow rate of 220 mL min−1 to achieve a redox potential of +610 mV. The solution was warmed up progressively. When the temperature reached 50 °C, As5+ fully dissolved (Fig. ESI1†), and then 1.25 M Fe2+ in the form of FeSO4·7H2O (Sigma Aldrich, ≥99%) was slowly added to the solution. The initial pH was corrected to 0.0, and the conductivity of the solution was 153 mS cm−1. The reactor was placed in an oil bath, which was supported on a heating plate. When the final temperature of 95 °C was reached and after completing the reaction time of 18 hours at a constant flow rate of O2, a green–gray precipitate was formed (see Fig. ESI1e†), which was dried at 60 °C during 12 h to be further analysed for arsenic leachability.Scorodite characterization
Irrespective of the synthesis method, the warm colloidal suspension was filtered using a 0.45 μm pore size Whatman PTFE membrane filter (GE Healthcare Life Sciences, USA) at a pressure of 4 bar to separate the solid. The solid precipitate was washed using demineralized water (10:1 w/w) to eliminate most of the remaining sulfuric acid of the precipitate,1 and then dried at 60 °C during 12 h. The dried precipitate was characterized by scanning electron microscopy (SEM) and X-ray powder diffraction (XRD). SEM analysis was performed using a Nova NanoSEM 450 instrument. The samples were mounted on a sticky carbon tab. A layer of about 2.5 nm of Pt/Pd (80–20 wt%) was sputtered on a stub (Cressington HR208), which was placed on the SEM. XRD analysis was obtained with a diffractometer (Empyrean, Malvern Panalytical, United Kingdom) using CuKα radiation (λ = 1.5405 Å) with a spinner at 40 mA–45 kV spending 4 s per step with a step size of 0.013° in the same scan range. Quantitative phase analysis (QPA) by the Rietveld refinement method with the HighScore Plus software (Empyrean, Malvern Panalytical, United Kingdom) was carried out for the quantitative analysis of the phase distribution using the measured diffraction profile and a calculated profile using the inorganic crystal structure database (ICSD). The crystallite size (D) was calculated using Scherrer's equation. This is expressed by eqn (1):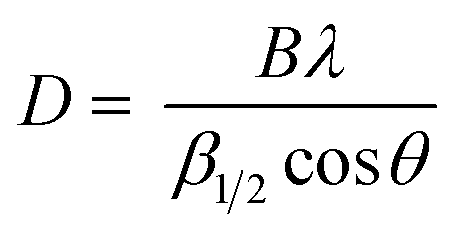 | (1) |
Mass balances and process efficiency
The yield of arsenic recovered into scorodite (Ysc/A, %) was calculated based on the initial amount of arsenic (As0, g) with respect to the quantitative mass of arsenic in scorodite produced at the end of the experiment (Assc, g), determined from the XRD and ICP analysis of powder samples, as expressed by eqn (2):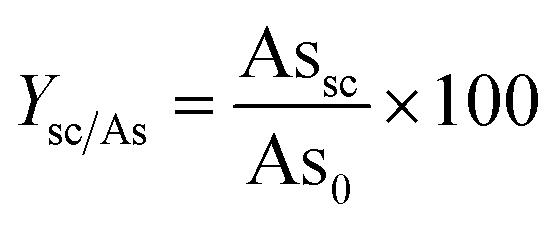 | (2) |
Leachate characterization
Leachate tests are the standard way to assess the stability of arsenic. The less arsenic is leached, the more stable is the scorodite. Stable scorodite should not leach As above 25 mg kg−1 of dry matter. The samples were analysed by extraction with Milli-Q water in a solid-to-liquid ratio of 1:10 (w/v) at 20 °C after 24 hours of a shaking speed of 200 rpm (Compact Shaker KS 15 A control, Edmund Bühler GmbH, Germany). The extraction solution was allowed to settle, and then the arsenic content was analyzed by ICP-OES. All extraction solutions were filtered with 0.45 μm filters (Millipore, USA) before ICP analysis.
Modeling electrochemical equilibria
In order to operate GDEx and chemical precipitation under the pH, ORP, temperature, and concentration of metal precursors conditions that could effectively yield scorodite, potential–pH (E–pH) diagrams were made, using HSC-Chemistry 9.0 software, considering the thermodynamic properties of the As–Fe–S system (Fig. 4) for the concentrations of precursors tested in this work, as well as for low concentrations (Fig. ESI2†). Based on the modeling results, the effect of the arsenic precursor (i.e., oxidation state, As3+vs. As5+) and temperature (at 50 °C or 70 °C) on producing crystalline scorodite was determined in batch mode with recirculation (see summary of the operational conditions described in Table ESI1†).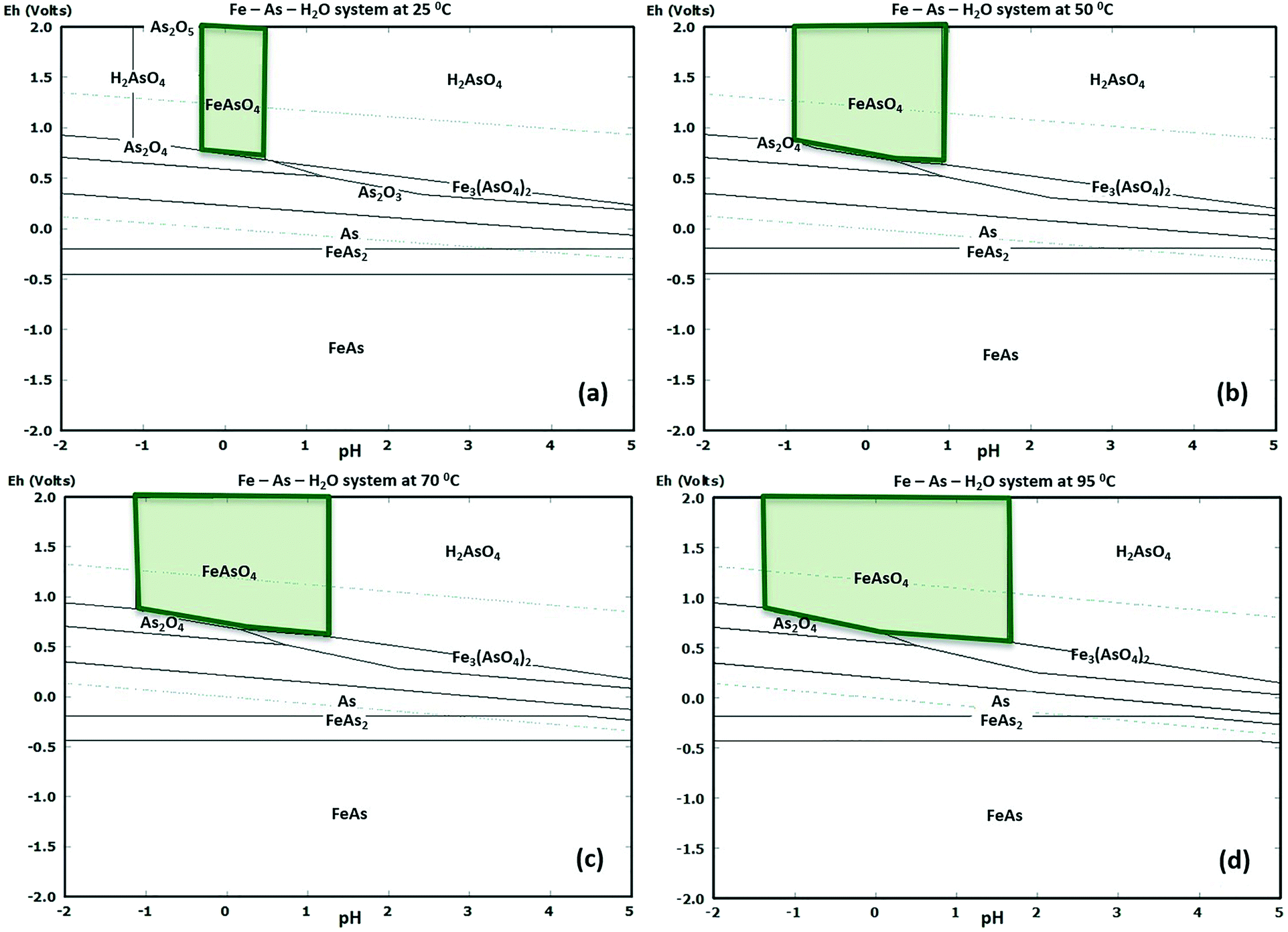 | ||
| Fig. 4 Thermodynamic equilibria for the As–Fe–H2O system at different temperatures: a) 25 °C, b) 50 °C, c) 70 °C, and d) 95 °C using high concentrations of As and Fe, i.e., 0.22 M As and 1.25 M Fe. These equilibria are shown for the case of low concentrations of As and Fe in Fig. ESI2.† | ||
Results and discussion
Electrochemical equilibria and stability of scorodite
The E–pH electrochemical equilibrium diagrams provide a useful guide for evaluating the limits of the thermodynamic predominance of solids and metal ions in aqueous solutions.27 The effect of temperature at a) 25 °C, b) 50 °C, c) 70 °C, and d) 95 °C, on these states, was calculated for the As–Fe–H2O system, as shown in Fig. 4. These diagrams were constructed at concentrations of ∑As = 0.22 M and ∑Fe = 1.25 M, and a pressure of 1 bar. The high concentrations of As and Fe were selected based on industrially-relevant waste streams. The upper and lower stability limits of water are also shown in the diagrams as blue dashed lines. Scorodite only appears under highly oxidizing conditions in aqueous media (e.g., >0.8 V). The pH stability region for scorodite shrinks as the temperature decreases from 95 °C to 25 °C.A decrease of ∑As to 0.04 M and ∑Fe to 0.3 M (e.g., relevant environmental conditions) also shrinks the stability region for scorodite when compared to the higher concentration system. Yet, at low concentrations, a small stability region is only found at temperatures above 70 °C; below this temperature, scorodite is not thermodynamically favourable (see Fig. ESI2†). Lu and Zhu28 have shown that decreasing the ∑As from 10−3 M to 10−8 M further decreases the stability range of scorodite, where the ∑Fe was set to 10−6 M.
Fujita et al.1,29 produced crystalline scorodite at 70 °C and small particles of scorodite at 50 °C using O2 as the oxidizing agent at high concentrations of As5+ (∑As = 0.67 M). Based on the studies of Fujita et al.1,29 and our thermodynamic modeling results, we inferred that the synthesis of scorodite by GDEx would be feasible at temperatures ≥50 °C. To validate this, we conducted experiments at 50 °C and 70 °C.
Mechanism of scorodite formation by GDEx
After polarizing the GDE at a constant potential of −0.15 VSHE, the current response was recorded for the four experimental conditions shown in Table ESI1.† The current response was similar in magnitude for the cases with As3+ (circa −10 mA cm−2), as well as for As5+ as a precursor (circa −40 mA cm−2), at the steady state, correspondingly. The variation in current densities is attributed to the different conductivities of the initial arsenic solutions at pH 0.0. Charge consumption was relative to the form of As supplied, i.e., lower charge consumption for As5+ than for As3+. Indeed, when As3+ is provided, it first needs to be oxidized to As5+, before precipitating as scorodite. Fig. ESI3† shows the representative current and charge profiles obtained for the different As precursors. During the CA experiments at −0.15 VSHE, the production of H2O2 by electrochemical O2 reduction (eqn (3)), in acid media, takes place with the electrodes and electrolyte used in this work as described by Šljukić et al.30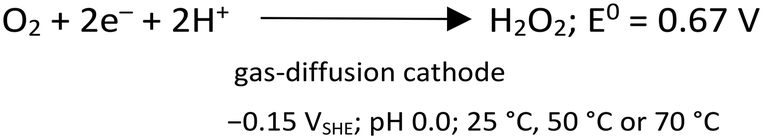 | (3) |
 | (4) |
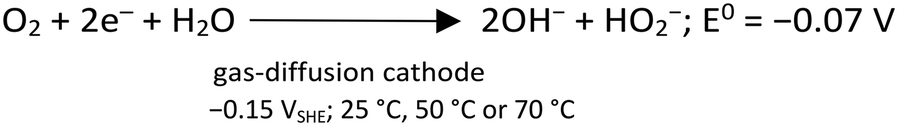 | (5) |
However, we do not see such abrupt pH change in this case, as the base immediately reacts with the abundantly available protons in the very acidic solution. It can be thus inferred that at the steady-state, a pH gradient develops throughout the hydrodynamic diffusion layer. Either within the electrode pores or at the diffusion layer, this may set enough precedent for local saturation conditions at the electrochemical interface.
The cumulative hydrogen peroxide production at −0.15 VSHE and at different temperatures (25 °C, 50 °C and 70 °C) are plotted in Fig. ESI4.† On the basis of the charge consumed, the production of H2O2 reached 3.4 ± 0.03 mg L−1 at 70 °C and 8.1 ± 0.07 mg L−1 at 50 °C, respectively, facilitating the oxidation of Fe2+ (eqn (6)) and As3+ (eqn (7)) and reactive precipitation at the electrochemical interface. After three hours of chronoamperometry test for the oxygen reduction reaction at 70 °C, a peroxide production of 0.14 ± 0.04 mmol cm−2 per day was measured, with no significant differences in production rates between duplicates. A 1.5-fold increase in cathodic H2O2 production rate to 0.22 ± 0.02 mmol cm−2 per day was obtained at 50 °C in duplicate experiments. A higher H2O2 concentration was found at 25 °C, which reached 17 ± 0.9 mg L−1, which corresponds to a production rate of 0.5 ± 0.1 mmol cm−2 per day.
After polarizing the GDE at −0.15 VSHE using the GDEx mode of operation, as shown in Fig. 3a, the oxidation of Fe2+ takes place through the well-known electro-Fenton reaction in acidic medium31,33 (eqn (6))
| Fe2+ + H2O2 + H+ → Fe3+ + H2O + ˙OH | (6) |
As3+ oxidation by H2O2 has been studied at neutral and acidic pH, showing slow reaction rates, because only H2AsO3− and HAsO32− react with H2O2, but not H3AsO3.34 However, the authors explained the As3+ oxidation via ˙OH radicals (eqn (7)) produced in eqn (6).
| As3+ + ˙OH → As4+ | (7) |
| As4+ + O2 → As5+ + O2˙− | (8) |
The involvement of ˙OH radicals was consistent with quenching experiments in which a 2-propanol scavenger quenched the As3+ oxidation at acidic pH.35 In the electro-Fenton reaction system, Fe2+ can be regenerated via homogeneous and heterogeneous Fenton reactions. The homogeneous reaction rate is four orders of magnitude lower than the Fe2+ oxidation reaction (eqn (9)), and produces Fe2+ and hydroperoxyl radicals (HO2˙).33
| Fe3+ + H2O2 + H+ → Fe2+ + HO2˙ + H+ | (9) |
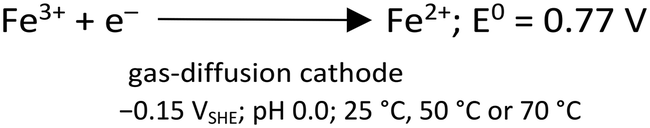
In the heterogeneous Fenton reaction, Fe3+ can be directly reduced to Fe2+ at the GDE (eqn (10)), decreasing the Fe3+ available for scorodite precipitation.
 | (10) |
As3+ can be also oxidised directly to As5+ at the anode (eqn (11)), after polarizing the GDE at −0.15 VSHE using the membrane-less-electrochemical reactor shown in Fig. 3b and the GDEx reactor shown in Fig. 3c. Under these conditions, the anode typically evolves a potential of 2 VSHE, which is consistent with the potentials found in the E–pH diagrams in Fig. 4. Scorodite only appears under highly oxidizing conditions in aqueous media (e.g., >0.8 V).
 | (11) |
Faceted scorodite particles then grow from the precursors in solution (Fe2+ and H3AsO3) after polarizing the GDE at −0.15 VSHE, in agreement with eqn (12) described by Fujita et al.29
| Fe3+ + H3AsO4 + 2H2O → FeAsO4·2H2O↓ + 3H+ | (12) |
The charge density (Qt, C cm−2) consumed by the oxidation of Fe2+ and As3+ is described in Table ESI1.†Qt is based on the amount of charge required to oxidize a high concentration of Fe2+ (∑Fe = 1.25 M) and As3+ (∑As = 0.22 M).
One-pot, one-step GDEx enabled the synthesis of scorodite from As3+ solutions
Under the conditions tested of temperature and arsenic precursors, different green precipitates were obtained after the GDEx process, without adding seeds for crystallization (Fig. 5b). The precipitates had the characteristic pale-green color of scorodite produced hydrothermally at 95 °C, 125 °C, and 150 °C.36 Although the only exception was a dark green precipitate obtained using As5+ at 70 °C, all XRD patterns can be assigned to crystalline scorodite (Fig. 5a). Rietveld refinements from the diffractograms in Table ESI3† yielded the same lattice constants of those powdered samples synthesized from As5+ and Fe2+ by the chemical precipitation method.37 In previous studies, similar pale-green precipitates were obtained under microbial oxidation of As3+ and Fe2+, also identified as crystalline scorodite as per the corresponding XRD patterns.38 GDEx-scorodite precipitated with As3+ and Fe2+ at 70 °C yielded the largest crystallite sizes (108 nm) vs. those precipitated at a lower temperature, i.e., 50 °C (71 nm) and using As5+ as the arsenic precursor (66 nm) at 70 °C.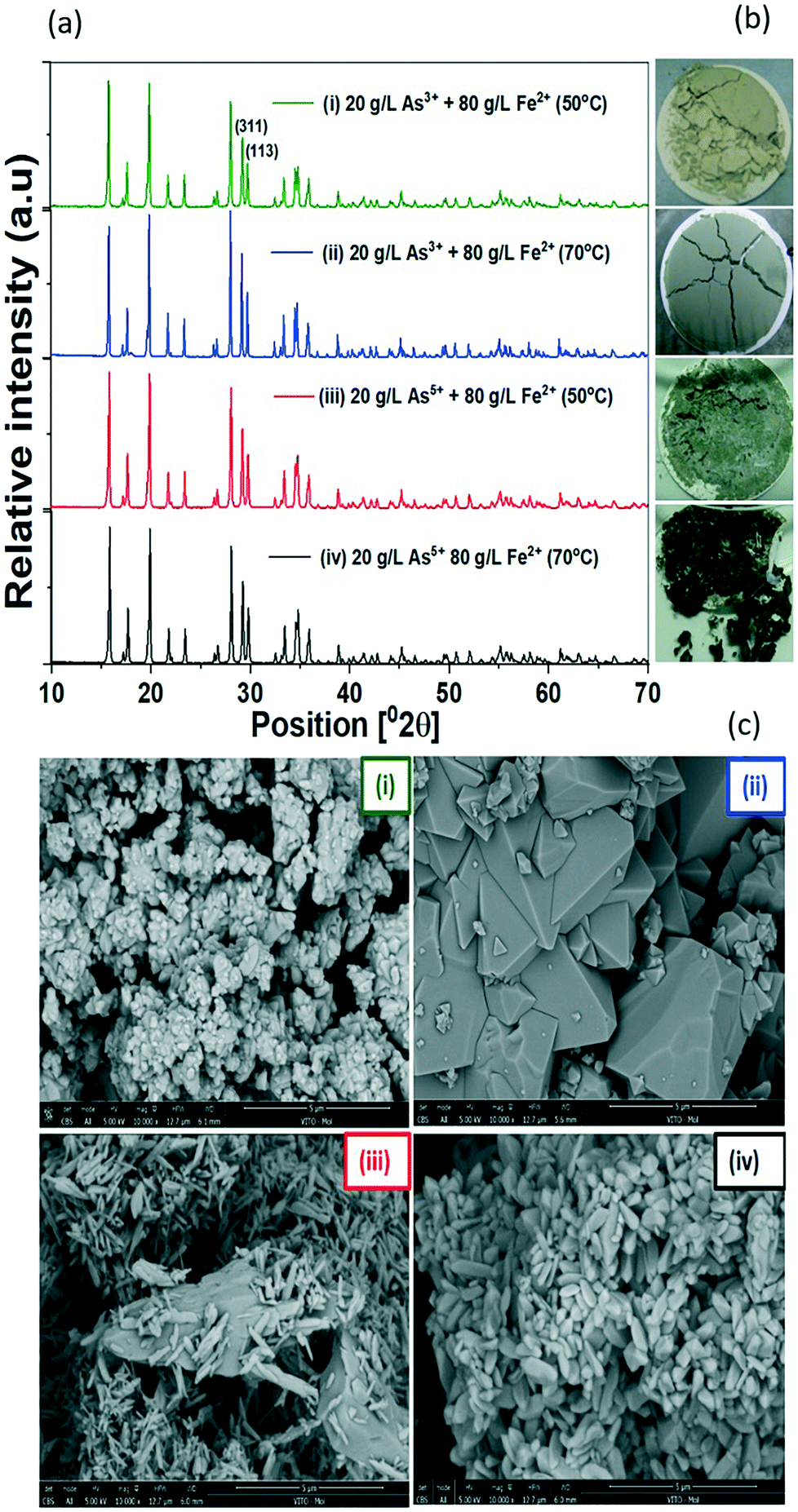 | ||
| Fig. 5 (a) XRD patterns Cu-Kα (λ = 1.5405 Å) of scorodite produced by GDEx under different processing conditions. (b) Macroscopic view of the precipitate cakes collected after filtration from the GDEx process using As3+ as the precursor at (i) 50 °C and (ii) 70 °C and As5+ as the precursor at (iii) 50 °C and (iv) 70 °C. (c) SEM images of scorodite samples synthesized from different precursors and at different temperatures of synthesis. The scale bars are 5 μm. | ||
The SEM images also revealed morphological variations in scorodite under the different tested conditions (Fig. 5c) of GDEx processing. Scorodite formed using As3+ at 50 °C (i) results in less homogeneous particles <1 μm, when compared to the case at 70 °C (ii), wherein significantly bigger particles were found (5 μm). Homogeneous particles of 1 μm were found using As5+ as the precursor at 50 °C and 70 °C. The product formed using As3+ at a temperature of 70 °C was significantly bigger than in the other instances tested, which is desirable for lower leachability, because leaching is a heterogeneous process and thus the rate of leaching is directly proportional to the area of the product that is being leached out.39
The larger particle size of scorodite produced at 70 °C is partly attributed to the higher synthesis temperature, which is one of the conditions to induce crystallinity in a precipitate. Yet, a higher temperature alone is not enough to induce crystallinity in scorodite; it requires the simultaneous control and maintenance of a low supersaturation environment.40 Scorodite precipitation by GDEx from As3+ as the precursor hinges on the oxidation of Fe2+ and As3+ to Fe3+ and As5+ while imposing a cathodic polarization condition of −0.15 VSHE, as the latter condition triggers the production of oxidant species. However, it competes with direct reduction reactions at the cathode, especially due to the high concentrations of the metal ions. Thus, the yield of arsenic recovered into scorodite depends on the speed with which oxidants were produced vs. the speed at which reduction takes place directly at the cathode (both determined by the total current exchanged). The control of H2O2 concentration and pH as critical factors has been reported to ensure crystalline scorodite.6 If the pH is raised to quickly or H2O2 is added too fast beyond the critical supersaturation limit, instantaneous heterogeneous nucleation and precipitation of amorphous compounds occur.41 Another explanation of the crystal size obtained under the different conditions was the operation time, which affects the crystal growth period. The GDEx-operation time using As3+ was 300 hours vs. 50 hours using As5+, due to the higher charge required to oxidise As3+.
Electrosynthesis of peroxide on GDE drives the oxidation of As3+ and scorodite crystallization
Scorodite crystallization was studied in three different operation modes by recirculating the arsenic and iron solution with and without contact with the GDE. The results of such configurations are shown in Table 1 and Fig. ESI5.† The effect of the temperature and arsenic precursor was evaluated in a two-chamber setup separated by a Nafion membrane, wherein As3+ or As5+ and Fe2+ were supplied to the cathode chamber (side of the GDE). A clear trend of increasing yield of arsenic recovered into scorodite was observed when the temperature was increased from 50 °C to 70 °C as shown in Table 1. The use of As5+ increased the yield of arsenic recovered into scorodite to 25%, which was five times higher than when starting with As3+ at 70 °C. This is attributed to a lower As3+ oxidation rate, i.e., 0.01 mmol cm−2 per day at 50 °C vs. 0.02 mmol cm−2 per day at 70 °C. These rates were calculated based on the amount of arsenic found in the precipitated scorodite (see mass balance in Table 1). The efficiency to oxidize Fe2+ and As3+ corresponds to 23% H2O2 produced at 50 °C, which reveals that some Fe3+ ions are probably reduced back to Fe2+ on the GDE surface. A 1.5-fold increase in the efficiency to produce hydrogen peroxide to 34% was attained at 70 °C, where 16% accounted for oxidizing As3+ and 18% to oxidize Fe2+ (based on the H2O2 production rate calculated from Fig. ESI4† and the amount of arsenic and iron in scorodite). Although the yield of arsenic recovery into scorodite was improved at 70 °C, arsenic recovery into scorodite was low (5%) using a two-chamber setup. Increasing the amount of reacted arsenic can be solved by increasing the retention time in the electrochemical cell so that less unreacted precursors remain. Thus, a configuration without the membrane was evaluated to improve the arsenic recovery rate into scorodite. The yield of arsenic recovered into scorodite increased to 70% using As3+ at 70 °C in a membrane-less-three electrode electrochemical cell (Fig. 3b).| Two-chamber setup | One-chamber setup | ||||
|---|---|---|---|---|---|
| Precursor | As3+, Fe2+ | As3+, Fe2+ | As5+, Fe2+ | As5+, Fe2+ | As3+, Fe2+ |
| Temperature (°C) | 50 | 70 | 50 | 70 | 70 |
| Arsenic influent (g) | 4.1 | 4.1 | 4.1 | 4 | 4 |
| Arsenic transport to the anode (g) | 0.7 | 0.8 | 0.3 | 0.5 | — |
| Arsenic unreacted in the catholyte (g) | 3.2 | 3.1 | 3.6 | 2.5 | 1.2 |
| Arsenic in scorodite (g) | 0.1 | 0.2 | 0.2 | 1 | 2.8 |
| Yield of arsenic recovery into scorodite (%) | 1.6 | 5 | 5.2 | 25 | 70 |
| Crystallite size (nm) | 71 ± 2 | 108 ± 0 | 70 ± 6 | 66 ± 4 | 79 ± 3 |
| As-Leachability (mg kg−1) | 1320 | 9 | 4321 | 5606 | 191 |
| Fe-Leachability (mg kg−1) | 4220 | 173 | 30814 |
34096 |
308 |
| Power consumption (kW h m−3 water treated) | 14320 |
6448 | 5136 | 992 | 400 |
| Power consumption (kW h kg−1 scorodite recovered) | 895 | 403 | 321 | 62 | 25 |
Given that As3+ oxidation can also take place on the anode side (eqn (9)), the oxidation rate reached 0.3 mmol As3+ per cm2 per day based on the arsenic content in scorodite, which was 16 times higher than the two-chamber reactor separated by a Nafion membrane. Thanks to the extra oxidation on the anode, the mass of scorodite precipitated increased from 1.6 mg cm−2 d−1 to 26 mg cm−2 d−1 using an electrolyte solution with 0.22 M As3+ and 1.25 M Fe2+. The SEM images and XRD patterns in Fig. ESI5† showed a well-crystalline scorodite product with an orthorhombic morphology and revealed no significant size variation in two different modes of GDEx operation with and without membrane.
Ultimately, a two-chamber setup was used to elucidate the role of the GDE in the scorodite crystallization by feeding the precursor in the anode side without contact with the GDE (Fig. 3c). No scorodite formation was found using this feeding strategy (see Fig. ESI5†), which strongly supports the hypothesis that the electrosynthesis of H2O2 on GDE drives the oxidation of As3+ and scorodite crystallization in a low supersaturation environment. A slow oxidation rate is critical for achieving large-sized scorodite crystals, which is driven by the reductive polarization of the GDE. The feeding strategy in the anode side without contact with the GDE gave faster oxidation of 3.5 mmol Fe2+ per cm2 per day without being reduced back in the cathode compartment, which was >8 times higher than in the one-chamber reactor. A fast oxidation rate increased the degree of supersaturation, thereby enhancing nucleation rather than crystal growth.2,11,41
Effect of the GDEx operation on arsenic leachability and crystal growth
The scorodite crystal growth was strongly dependent on the mode of GDEx operation, as shown in Fig. 4c. The product with the largest particle size exhibited the smallest arsenic leachability, as shown in Table 1. A larger particle size is preferred because a low surface-to-volume ratio makes it difficult to dissolve scorodite in an acidic solution.42 Larger particle sizes, including non-homogeneous particles (from fine particles <1 μm to large particles >5 μm), were generated by a slow H2O2 production rate at 70 °C, together with an enhanced dissolution caused by the more extended electrode polarization periods using As3+ as the precursor (304 hours) vs. 50 hours using As5+ as the precursor. Sun et al.39 demonstrated that scorodite undergoes crystal shape changes during aging, from a laminar to polyhedral to octahedral structure. This was here observed, and it seems to have contributed significantly to decrease As-leachability. The arsenic content after the leaching test of GDEx-made scorodite at 70 °C with As3+ as a precursor was 9 mg kg−1 dry matter vs. 120 mg kg−1 of dry matter formed by chemical precipitation. The lowest temperature at which scorodite could be achieved through the chemical precipitation method was 95 °C, with this being the reason for our benchmarking conditions for scorodite stability. The norm for dangerous waste category determines the limit at 25 mg kg−1 of dry matter;43 therefore, the scorodite produced here by GDEx would be considered safe for disposal.The most relevant GDEx operational costs are: electricity and electrode replacement (∼80% of OPEX costs). Table 1 also shows power consumption under the three different operation strategies. All operation modes could amount for less than 100€ per kg of scorodite recovered, considering the Belgian prices for industrial electricity use and electrode replacement every 2 years.
Conclusions
This work demonstrates the potential of gas-diffusion electrocrystallization (GDEx) as a new route to remove arsenic from solution and its immobilization as a crystalline scorodite using a one-pot and one-step reactor at a relatively low temperature (50 °C) and under ambient pressure conditions. GDEx successfully enables the simultaneous oxidation of Fe2+ and As3+ by the production of H2O2 issued from the oxygen reduction reaction (ORR) at a gas-diffusion cathode, forming scorodite without the use of any primary minerals or seed crystals. Additionally, increasing the temperature improves the arsenic recovery rate (24% in the solid) at 70 °C. We expected to synthesize scorodite without the GDE using the oxidation from the anodic reaction at 70 °C. However, the As3+ oxidation rate was faster, which enhanced nucleation rather than crystal growth; therefore, scorodite was not formed without the GDE at 70 °C. A slow oxidation rate was critical for achieving large-sized scorodite crystals, which was driven by the reductive polarization of the GDE. Using a one-pot, one-step, two compartment GDEx-setup enabled the synthesis of scorodite that was 21 times more stable than the synthesis in the one-chamber reactor. However, the scorodite precipitation rate was 14 times slower under these conditions. At this early stage of discovery, there is still plenty of room for improvement in future studies. Future research should focus on seeding scorodite conducted using GDEx to increase the scorodite precipitation rate in a low supersaturation environment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. Pozo acknowledges the funding from the European Union's Horizon 2020 research and Innovation Programme MSCA-IF-2017, under grant agreement no. 796320 (MAGDEx: Unmet MAGnetic properties in micro and nano-particles by synthesis through gas diffusion electrocrystallization (GDEx)). X. Dominguez-Benetton thank VITO's strategic research funds and management for the possibility of conducting this pioneering research. GP, JF, and XDB thank the support from the Flemish SIM MaRes programme, under grant agreement no. 150626 (Get-A-Met project). XDB and JF thank the funding from the European Union's Horizon 2020 research and Innovation Programme under grant agreement no. 654100 (CHPM2030 project). We would also like to acknowledge Myriam Mertens for fruitful discussions and her support on XRD analysis as well as Kristof Tirez and Wilfried Brusten for assistance with analytical measurements. We would like to especially thank Karolien Vanbroekhoven and Sofie van Ermen for the fruitful discussions that helped us to improve this work.References
- T. Fujita, R. Taguchi, M. Abumiya, M. Matsumoto, E. Shibata and T. Nakamura, Hydrometallurgy, 2008, 90, 92–102 CrossRef CAS.
- G. P. Demopoulos, Hydrometallurgy, 2009, 96, 199–214 CrossRef CAS.
- A. M. Nazari, R. Radzinski and A. Ghahreman, Hydrometallurgy, 2017, 174, 258–281 CrossRef CAS.
- Y. Jang, Y. Somanna and H. Kim, Int. J. Appl. Environ. Sci., 2016, 11, 559–581 Search PubMed.
- M. Bissen and F. H. Frimmel, Acta Hydrochim. Hydrobiol., 2003, 31, 97–107 CrossRef CAS.
- G. P. Demopoulos, TMS (The Miner. Met. Mater. Soc.) Annu. Meet., 2005, pp. 25–50.
- D. Langmuir, J. Mahoney and J. Rowson, Geochim. Cosmochim. Acta, 2006, 70, 2942–2956 CrossRef CAS.
- X. B. Min, Y. P. Liao, L. Y. Chai, Z. H. Yang, S. Xiong, L. Liu and Q. Z. Li, Trans. Nonferrous Met. Soc. China, 2015, 25, 1298–1306 CrossRef CAS.
- D. Paktunc, J. Dutrizac and V. Gertsman, Geochim. Cosmochim. Acta, 2008, 72, 2649–2672 CrossRef CAS.
- P. González-Contreras, J. Weijma and C. J. N. Buisman, Water Res., 2012, 46, 5883–5892 CrossRef PubMed.
- G. Demopoulos, D. Droppert and G. Van Weert, Hydrometallurgy, 1995, 38, 245–261 CrossRef CAS.
- M. A. Gomez, L. Becze, J. N. Cutler and G. P. Demopoulos, Hydrometallurgy, 2011, 107, 74–90 CrossRef CAS.
- P. Gonzalez-Contreras, J. Weijma and C. J. N. Buisman, Appl. Microbiol. Biotechnol., 2012, 93, 1295–1303 CrossRef CAS PubMed.
- J. A. G. Gomes, P. Daida, M. Kesmez, M. Weir, H. Moreno, J. R. Parga, G. Irwin, H. McWhinney, T. Grady, E. Peterson and D. L. Cocke, J. Hazard. Mater., 2007, 139, 220–231 CrossRef CAS PubMed.
- N. Balasubramanian, T. Kojima, C. A. Basha and C. Srinivasakannan, J. Hazard. Mater., 2009, 167, 966–969 CrossRef CAS PubMed.
- T. Banerji and S. Chaudhari, J. Environ. Chem. Eng., 2016, 4, 3990–4000 CrossRef CAS.
- P. V. Nidheesh and T. S. A. Singh, Chemosphere, 2017, 181, 418–432 CrossRef CAS PubMed.
- X. Zhao, B. Zhang, H. Liu and J. Qu, J. Hazard. Mater., 2010, 184, 472–476 CrossRef CAS PubMed.
- R. A. Prato, V. Van Vught, S. Eggermont, G. Pozo, P. Marin, J. Fransaer and X. Dominguez-Benetton, Sci. Rep., 2019, 9, 15370 CrossRef PubMed.
- G. Pozo, P. De Presa, R. Prato, I. Morales, P. Marin, J. Fransaer and X. Dominguez-benetton, Nanoscale, 2020, 12, 5412–5421 RSC.
- R. A. Prato, V. Van Vught, K. Chayambuka, G. Pozo, S. Eggermont, J. Fransaer and X. Dominguez-Benetton, J. Mater. Chem. A, 2020 10.1039/d0ta00633e.
- Y. Alvarez-Gallego, X. Dominguez-Benetton, D. Pant, L. Diels, K. Vanbroekhoven, I. Genné and P. Vermeiren, Electrochim. Acta, 2012, 82, 415–426 CrossRef CAS.
- R. Kuwertz, C. Kirstein, T. Turek and U. Kunz, J. Membr. Sci., 2016, 500, 225–235 CrossRef CAS.
- P. González Contreras, J. Weijma and C. J. N. Buisman, Adv. Mater. Res., 2009, 71–73, 629–632 Search PubMed.
- S. Sorlini and F. Gialdini, Water Res., 2010, 44, 5653–5659 CrossRef CAS PubMed.
- H.-H. Yang and R. L. McCreery, J. Electrochem. Soc., 2000, 147, 3420 CrossRef CAS.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, National A., Texas, 1974 Search PubMed.
- P. Lu and C. Zhu, Environ. Earth Sci., 2011, 62, 1673–1683 CrossRef CAS.
- T. Fujita, R. Taguchi, M. Abumiya, M. Matsumoto, E. Shibata and T. Nakamura, Hydrometallurgy, 2008, 90, 85–91 CrossRef CAS.
- B. Šljukić, C. E. Banks and R. G. Compton, J. Iran. Chem. Soc., 2005, 2, 1–25 CrossRef.
- E. Brillas, I. Sirés and M. A. Oturan, Chem. Rev., 2009, 109, 6570–6631 CrossRef CAS PubMed.
- X. Dominguez Benetton, Y. Alvarez, C. Porto-Carrero, K. Gijbels and S. Rajamani, US2018/0023201A1, 2018, 40 Search PubMed.
- S. Qiu, D. He, J. Ma, T. Liu and T. D. Waite, Electrochim. Acta, 2015, 176, 51–58 CrossRef CAS.
- M. Pettine, L. Campanella and F. Millero, Geochim. Cosmochim. Acta, 1999, 63, 2727–2735 CrossRef CAS.
- S. J. Hug and O. X. Leupin, Environ. Sci. Technol., 2003, 37, 2734–2742 CrossRef CAS PubMed.
- T. Fujita, S. Fujieda, K. Shinoda and S. Suzuki, Hydrometallurgy, 2012, 111–112, 87–102 CrossRef CAS.
- K. Shinoda, T. Tanno, T. Fujita and S. Suzuki, Mater. Trans., 2009, 50, 1196–1201 CrossRef CAS.
- N. Okibe, M. Koga, S. Morishita, M. Tanaka, S. Heguri, S. Asano, K. Sasaki and T. Hirajima, Hydrometallurgy, 2014, 143, 34–41 CrossRef CAS.
- Y. Sun, Q. Yao, X. Zhang, H. Yang, N. Li, Z. Zhang and Z. Hao, RSC Adv., 2018, 8, 19560–19569 RSC.
- A. Mersmann and M. Löffelmann, Chem. Eng. Technol., 2000, 23, 11–15 CrossRef CAS.
- D. Filippou and G. P. Demopoulos, JOM, 2007, 49, 52–55 CrossRef.
- M. C. Harvey, M. E. Schreiber, J. D. Rimstidt and M. M. Griffith, Environ. Sci. Technol., 2006, 40, 6709–6714 CrossRef CAS PubMed.
- 1999 Landfill Directive Council for the EU, Off. J. Eur. Communities, 1999, 19.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00054j |
| This journal is © The Royal Society of Chemistry 2020 |