
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcyl azide generation and amide bond formation in continuous-flow for the synthesis of peptides†
Alejandro
Mata
ab,
Ulrich
Weigl
c,
Oliver
Flögel
c,
Pius
Baur
c,
Christopher A.
Hone
 *ab and
C. Oliver
Kappe
*ab
*ab and
C. Oliver
Kappe
*ab
aCenter for Continuous Flow Synthesis and Processing (CCFLOW), Research Center Pharmaceutical Engineering GmbH (RCPE), Inffeldgasse 13, 8010 Graz, Austria. E-mail: christopher.hone@rcpe.at
bInstitute of Chemistry, University of Graz, NAWI Graz Heinrichstrasse 28, 8010 Graz, Austria. E-mail: oliver.kappe@uni-graz.at
cCilag AG, Hochstrasse 201, 8200 Schaffhausen, Switzerland
First published on 2nd March 2020
Abstract
The development of new protocols for peptide bond formation which avoid side reactions, including epimerization, are very important. The acyl azide method for the preparation of peptides is very good at maintaining chiral integrity, however; acyl azides are considered to be highly unstable and potentially explosive intermediates. Thus, the acyl azide method is underutilized by the synthetic community. Acyl azides were safely generated and reacted in situ within a continuous-flow system. The acyl azide was generated by using nitrous acid in water, and efficiently extracted into the organic phase containing the amine nucleophile for peptide coupling. The protocol has been used to prepare a number of dipeptides (5 examples) without epimerization (<1%). A tripeptide, D-Ala-D-Ala-L-Ala-OBn, was also synthesized.
Acyl azides are valuable synthetic intermediates for the preparation of amides, amines, isocyanates, ureas, ketenimines and carbodiimides.1 One particularly important application is their use for the synthesis of peptides and proteins.2 There are a number of synthetic strategies to form peptides from their corresponding amino acids.3 However, many of the traditional amide bond formation protocols do not reach the high requirements of enantioselectivity necessary for active pharmaceutical ingredient (API) synthesis. The acyl azide method is one of the best methods for racemization-free peptide segment condensation, which cannot be so readily achieved by many other amide coupling methods.2,4 A further benefit of the approach is that unprotected side chains can be used, such as for serine, threonine, and histidine derivatives.
The synthetic sequence commences with the preparation of an ester (1) from the corresponding amino acid. Ester 1 is then reacted with excess hydrazine hydrate (N2H2·H2O) to form hydrazide 2 (Scheme 1). The hydrazide 2 is converted to acyl azide 3 in the presence of nitrous acid (HNO2). A peptide coupling partner 4 is then introduced which reacts with the acyl azide to form an amide bond resulting in a dipeptide (5). Typically a low temperature (≤0 °C) is required to prevent decomposition of the azide which releases nitrogen gas. The coupling step typically takes several hours. The coupling step is typically performed under basic conditions which prevents the formation of hydrazoic acid (HN3). A limitation is that there are a number of side reactions that can occur, such as the Curtius rearrangement to form an isocyanate which can then undergo further reactions. Furthermore, there are safety concerns due to the stability of the azide intermediate since it is prone to undergo vigorous decomposition. The acyl azide method of coupling was developed over 100 years ago,5 but has been underutilized due to its associated challenges, particularly in terms of safety.6
 | ||
| Scheme 1 Acyl azide method for the preparation of dipeptides: 1) hydrazide formation; 2) acyl azide generation; and 3) peptide coupling. The Curtius rearrangement can result in the generation of an isocyanate as a side product. | ||
On first glance the hydrazide strategy looks unattractive for routine use, because of the additional steps when compared to other peptide coupling methods. However, there are three characteristic features that make it a popular option in specific cases. Firstly, it is the method that is most likely to guarantee the preservation of chiral integrity during peptide bond formation between segments.3a Secondly, specifically for fragment couplings the hydrazide method offers an advantage because in this case the acyl azides can be formed directly from the ester. The protocol obviates the need to remove an ester protecting group which is required for other approaches.7 Thirdly, minimum protection is necessary because the hydrazide strategy has been shown to work with unprotected serine, threonine and histidine derivatives.
An alternative one-pot method that starts from the acid is to use diphenylphosphonic azide (DPPA) as reagent, but this approach is more expensive.8 Even more significant is that DPPA contains an azide moiety which will release hydrazoic acid upon reaction with water, thus when DPPA is stored at room temperature for a long time it can slowly and partially hydrolyze with moisture in air to produce diphenyl phosphate and hydrazoic acid (HN3).9 HN3 is a volatile and explosive liquid at room temperature.
Continuous-flow reactors have been demonstrated to address many of these challenges.10 In particular, continuous-flow reactors have emerged as an enabling technology for accessing “forgotten” and forbidden chemistry.11 Microreactor systems display improved mass and heat transfer characteristics, and also enable precise control over the reaction parameters.12 Unstable and hazardous intermediates can be generated in situ, with only a small inventory formed at any one time, which can then be used immediately in a subsequent reaction. The capability to perform reaction telescoping and multistep transformations within a single process has been demonstrated.13 Moreover, the addition of in-line work-up strategies for the quenching of reagents and products can improve the inherent safety. A review of continuous-flow methods for the synthesis of peptides has recently been published.14
The main alternative to the acyl azide method for maintaining chiral integrity is the triphosgene approach of coupling.15 Recent studies have reported the high enantioselectivity achieved when combining this method with continuous-flow technologies.16–18 Fuse and co-workers reported a microflow protocol for the synthesis of dipeptides through the coupling of carboxylic acids with amines by using inexpensive and less-toxic triphosgene as a coupling reagent.16 The protocol avoided epimerization as a side reaction, with ≤3% in most cases. The same group subsequently reported the total synthesis of feglymycin, an anti-HIV and antimicrobial 13-mer peptide, by using triphosgene as coupling reagent within a microreactor system.18 Very recently, Fuse and co-workers also reported a mixed carbonic anhydride-based amidation with unprotected amino acids within microflow technology with very little racemization observed.19
Along similar lines, recent efforts have focused on the reaction of acyl azides to form isocyanates, which can then be treated with nucleophiles to form products such as amines, carbamates and amides.20–23 Ley and co-workers utilized an azide-containing monolith in a flow system for performing the Curtius rearrangement with acid chloride precursors.20 Wille and co-workers reported the in situ preparation of a diacyl azide as an intermediate for the Curtius rearrangement within a microreactor.21 Watts and co-worker reported the continuous-flow synthesis of acyl azides under monophasic and biphasic regimes.22 There are also examples of azide formation using polymer-bound and monolith azide sources.24
We envisaged that a continuous-flow protocol could overcome many of the limitations associated with the acyl azide method for peptide bond formation. We herein report the development of an efficient and safe continuous-flow procedure for the in situ preparation of acyl azides from the corresponding hydrazide and the subsequent peptide coupling.
Our studies commenced with the batch preparation of the hydrazide derivatives 2a–2d from commercially-available t-butoxy carbonyl (Boc) group protected amino acid methyl esters 1a–1d. In general, Boc-protected esters are stable towards hydrazinolysis.25 However, the hydrazide strategy is incompatible with benzyl ester protected amino acids because the benzylesters are cleaved during the hydrazide formation. Our protocol was based on modifying a literature protocol by De Souza and co-workers.26 The treatment of the methyl ester Boc-protected amino acids 1a–1d with 3 equivalents of hydrazine monohydrate at ambient temperature provided full conversion to the corresponding acyl hydrazide derivatives 2a–2d after 24 hour reaction time (see ESI†). Quenching reagents such as bleach, hydrogen peroxide and phthalates were trialed for the safe removal of the excess hydrazine, but unfortunately they also reacted with the hydrazide derivatives. Thus, the remaining hydrazine, along with other volatiles, was carefully removed under reduced pressure (see Caution Note 1, ESI†) to afford clean hydrazide products 2a–2d in quantitative yields. This procedure was used for the synthesis of the D- and L-enantiomers.
Prior to the development of a continuous-flow protocol, we were interested in conducting an extensive solvent screen under batch conditions (Table 1). Initial experiments used N-Boc-L-Ala-NHNH2 (2a) as a model substrate and L-Ala-OBn (4a) as a coupling partner to enable the identification of conditions that resulted in good conversion and selectivity. We were also interested in selecting conditions that resulted in minimal solid formation to ensure the chemistry was amenable for transfer to continuous-flow. Dichloromethane and chloroform were not investigated during the solvent screening due to their corresponding reactions with azide anions to form the highly dangerous di- and triazidomethane respectively.27 The use of water as a solvent resulted in a 90% NMR yield for 5a with <1% of the epimer 6a based on chiral HPLC analysis (entry 1). Racemization is not observed because an oxazolone is not generated as an intermediate from the acyl azide, the main cause of epimerization. A limitation was that a considerable amount of solid was formed when employing standard aqueous conditions found in the literature. The use of tetrahydrofuran (THF), methanol (MeOH), and dimethylacetamide (DMA) as solvents resulted in full conversion but very poor selectivity. Overall, the experiments conducted with organic solvents under a monophasic regime showed either poor selectivity and/or solubility issues.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Time [min] | 2a Conv.b [%] | Yield 5a (epimer 6a)c [%] |
| a Reaction conditions: a flask submerged within an ice-batch (0 °C) was charged with N-Boc-L-Ala-NHNH2 (2a) (0.492 mmol) and organic solvent (1.5 mL). 3.5 M aqueous solution of NaNO2 (0.5 mL) was added. For entries 6–10, H2O (1 mL) was added. Subsequently, a 1 M HCl aqueous solution (1 mL) was added. The mixture was stirred for 7 min at 0 °C. A solution of L-Ala-OBn (4a) (0.516 mmol) in H2O (0.5 mL) and neat Et3N (103 μL) were added. The reaction mixture was stirred for 2 hours. Phases were separated, organic layer dried and evaporated. b Conversion was measured by integration of desired product with remaining L-Ala-OBn (4a). c Yield was determined by NMR against dioxane as an internal standard and epimerization by chiral HPLC. d Significant solid formation was observed. N. D. = not detected. | ||||
| 1d | H2O | 1.5 | >99 | 90 (<1%) |
| 2 | MeOH | 1.5 | >99 | 25 |
| 3 | THF | 1.5 | >99 | N.D. |
| 4 | DMA | 1.5 | >99 | N.D. |
| 5 | Dioxane | 15 | 70 | 50 |
| 6 | MeTHF/H2O | 1.5 | >99 | N.D. |
| 7d | Et2O/H2O | 7 | >99 | 40 |
| 8 | EtOAc/H2O | 7 | 60 | 42 |
| 9 | MTBE/H2O | 7 | 75 | 41 |
| 10 | PhMe/H2O | 7 | >99 | 70 (<1%) |
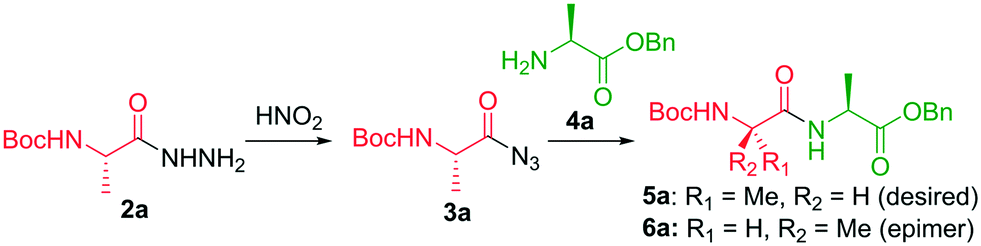
More promising results, in terms of solubility of all components, were achieved by using a biphasic regime (Table 1, entries 6–10). We also proposed that the utilization of a biphasic regime would enable a safe quench of any azide anions remaining in the aqueous phase, whilst avoiding any interference with the desired product within the organic phase. Full conversion and only moderate yields were achieved when diethyl ether (Et2O), ethyl acetate (EtOAc) and methyl tert-butyl ether (MTBE) were used as the organic phase (entries 7–9). A toluene (PhMe)/water system provided the dipeptide in 70% NMR yield and <1% epimerization was observed (entry 10). Although a slight amount of precipitation was observed, we thought that this would be manageable on transfer to continuous-flow conditions. A study of the reaction time when using PhMe/water (Table S2†) as a biphasic mixture indicated that the acyl azide was formed within 90 seconds.
The continuous-flow setup consisted of 5 feeds (Scheme 2) containing an aqueous solution of the hydrazide substrate 2a–2d and HCl (1.2 equivalents) (feed 1), an aqueous solution of NaNO2 (feed 2), neat toluene (feed 3), an aqueous solution of the amine coupling partner 4a–4b (feed 4), and a solution of Et3N in toluene (feed 5). The Boc-protected hydrazide derivatives 2a–2d were stable at room temperature for 24 h within the feed solution. Feeds 1, 2 and 3 were mixed by using a PEEK cross-assembly (0.5 mm i.d.), prior to entering the first reactor (4 mL internal volume, 0.5 mm i.d.). Feed 4 and 5 were mixed using a PEEK T-mixer (0.5 mm i.d.) resulting in a biphasic segmented flow regime. The small internal channel dimensions of continuous-flow reactors maximize the interfacial area between the aqueous and organic phases.12 The biphasic mixture was introduced to the effluent from reactor 1 by using a PEEK T-mixer (1 mm i.d.), prior to entering reactor 2 (38 mL internal volume, 1 mm i.d.). The whole setup, excluding pumps, was submerged within an ice bath to maintain a temperature of 0 °C. A back pressure regulator (BPR) was not installed prior to the outlet due to concern over gradual solid formation within the BPR which would cause a pressure rise over operation time.
 | ||
| Scheme 2 Continuous-flow setup for the acyl azide formation and subsequent amide coupling for N-Boc-L-Ala-NHNH2 as substrate and amide coupling partner. | ||
An initial set of experiments were performed using N-Boc-L-Ala-NHNH2 (2a) as model substrate within the continuous-flow setup. The reaction is acidic within the first reactor. Once the acyl azide is formed then the pH is carefully adjusted so it is mildly basic (pH ∼ 8) for the peptide coupling within the second reactor. Mildly basic conditions prevents the formation of hydrazoic acid. The azide is extracted to an organic phase for reaction with an amine nucleophile for amide bond formation. The collection vessel was contained within an ice bath. We could achieve similar results by using the same reagent equivalents as explored in the batch optimization. Furthermore, the residence time was reduced to 16 min for the amide coupling step. Gravity-separation within a separating funnel was used to separate the aqueous and organic phases. At the outlet, the aqueous phase was treated with hydrochloric acid solution to reach a pH of 1, which in combination with the remaining NaNO2 in solution, quenched any azide anions present (see Caution Note 2, ESI†). Within the quench there could be a small amount of hydrazoic acid that forms at low concentrations.28 However, it will not accumulate since it is immediately destroyed by HNO2. A study of the aqueous layer by NMR did show peaks associated with the hydrazide starting material and small amounts of desired product, which partly explains the loss in yield. However, attempts to conduct further extractions to obtain more desired product resulted in a drop in purity. The organic layer was then dried and evaporated to afford the crude product. The separation of the organic phase and aqueous phase could also be achieved in a continuous fashion by using an in-line liquid–liquid separator (PTFE membrane with 0.45 μm pore size, Zaiput). The system was demonstrated to be stable for 1 h with product collected for 30 min. We were concerned that in certain cases the formation of a fine solid suspension could cause problems with an in-line liquid–liquid separation, thus gravity separation is used herein.
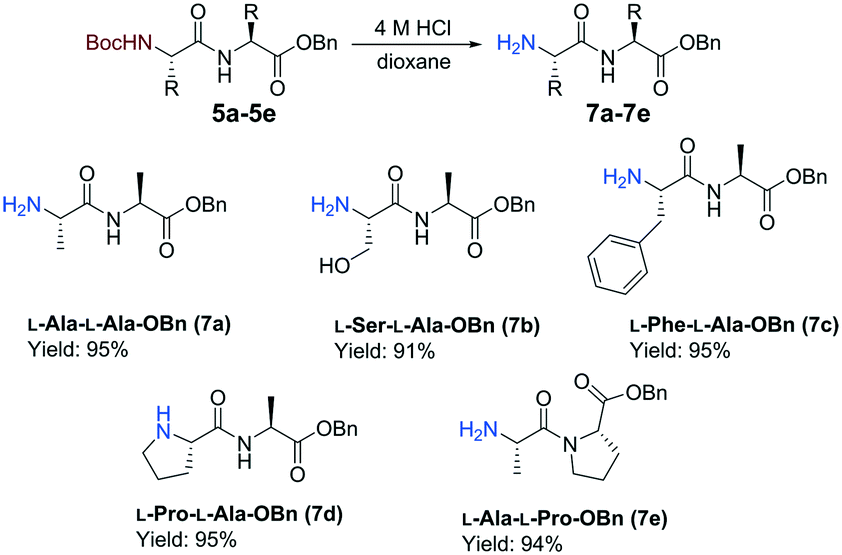
The continuous-flow reactions were performed on the L-enantiomer of each hydrazide substrate 2a–2d (Scheme 3). Batch experiments were conducted for comparison between the flow and batch results. To enable epimerization studies, batch reactions were conducted with the D-enantiomer as starting material. A residence time of 32 min was necessary for the amide coupling step when using the hydrazide substrates 5b–5d. Furthermore, pleasing results were also obtained when changing the amine coupling partner to L-Pro-OBn (4b) to give peptide 5e in a comparable yield. All reactions displayed very low epimerization (<1%). A Boc-cleavage reaction was performed in batch on all the dipeptides synthesized using the continuous-flow protocol. We selected not to modify an acid free Boc deprotection flow protocol at high temperature (300 °C) and high pressure (100 bar), which was reported by Djuric and co-workers, due to concern over decomposition.29 Each N-Boc-protected dipeptide 5a–5e was treated with a solution of 4 M HCl in dioxane based on a literature batch protocol (Scheme 4).30 The Boc-group was successfully cleaved at room temperature within 1 h reaction time. The Boc-cleavage acted as a method to further remove impurities formed during the acyl azide formation and coupling stages. Good combined yields between 60 to 70% over 4 steps were obtained for the formation of benzyl ester dipeptides 7a–7e from the corresponding N-Boc-protected methyl ester amino acids 1a–1d. The feasibility of the Boc-deprotection was also assessed in flow for the preparation of 7a. The flow setup consisted of two syringe pumps, feed 1 was 0.13 M 5a in PhMe and feed 2 was 4 M HCl in dioxane. The two feeds were mixed within a T-piece prior to a coil reactor (4 mL, 0.5 mm i.d.). The deprotection proceeded with quantitative conversion within 10 min at ambient temperature.
 | ||
| Scheme 3 Scope of dipeptides formation employing the acyl azide continuous-flow method. Yields were determined by NMR against an internal standard, see ESI† for more details. Epimerization was determined by chiral HPLC. | ||
 | ||
| Scheme 4 Boc-cleavage to give free benzyl ester dipeptides. | ||
The robustness and stability of the acyl azide generation and peptide coupling continuous-flow protocol was demonstrated by performing a long-run experiment for 2 hours of operation time. A 5 g sample of N-Boc-D-Ala-NHNH2 (9) was processed throughout the run. The system was stable for the duration of the run with no pressure rise observed. The coupling was achieved in 70% yield, which is comparable to the results from the smaller-scale experiments, thus demonstrating the scalability of the flow protocol.
Finally, we applied the protocol for the synthesis of a tripeptide, D-Ala-D-Ala-L-Ala-OBn (12) (Scheme 5). The continuous-flow protocol successfully combined D-Ala-L-Ala-OBn (6a) as the dipeptide partner with Boc-D-Ala-NHNH2 (9) to form the N-Boc-protected tripeptide 11. The deprotection protocol was then applied to afford the unprotected tripeptide D-Ala-D-Ala-L-Ala-OBn 12, corresponding to a combined yield of 49% over the 7 steps.
 | ||
| Scheme 5 Full synthesis of a tripeptide, D-Ala-D-Ala-L-Ala-OBn, employing the acyl azide method. | ||
The described flow protocol establishes a proof of concept for acyl azide formation and peptide coupling within a flow reactor. In the cases where a particular compound is incompatible with hydrazide synthesis then the hydrazide formation conditions have to be changed or alternative strategies should be applied. One can envisage the possibility to link three steps (acyl azide formation, coupling step and boc deprotection) within an integrated flow process. The acyl hydrazide formation could not be conducted inline due to the incompatibility of the benzyl group to the excess hydrazine. A potential limitation of a flow protocol is that it is reliant on the solubility of all the reaction components within the biphasic mixture to achieve a smooth and high yielding continuous flow process. With other amino acids or longer chain peptides it may be necessary to adjust concentrations or to modify the solvent system The next step would be to perform the tripeptide synthesis without human intervention in an automated iterative fashion in a similar manner as described elsewhere for other peptide synthesis approaches.31
The existing protocol has been developed within small tubular reactors. These reactors show excellent performance at a small scale but cannot be used to synthesize large quantities of material. Numbering-up (parallelization of reactor units) is one strategy for scale-up. It is an attractive option for liquid–liquid reactions as there is a limit to the dimension of a tube before mass transfer limitations and droplet coalescence are observed.32 Another strategy to address the challenge of scaling-up is to apply “smart dimensioning” by selecting the correct structure geometry for the necessary liquid–liquid mixing function and also by using the appropriate reactor modules at the right locations.33
In conclusion, we have developed a continuous-flow protocol for the safe application of the acyl azide method for peptide bond formation. The acyl azide is formed in small quantities at any one time, thus ensuring the process is inherently safe. Subsequently, in a second reactor coil the acyl azide reacted in situ with the amine coupling partner. The protocol utilizes a biphasic flow regime: the amide is extracted into the organic phase whereas the azide anions (N3−) can be safely quenched within the aqueous phase. The accumulation is avoided by destroying any remaining azide immediately after the coupling. The flow protocol was applied to the synthesis of 5 dipeptides and a tripeptide in moderate to good yields. Epimerization was observed to be <1% in all cases. The protocol needs to be further validated on more challenging amino acids in their unprotected form. Overall, the flow protocol enables the safe utilization of the acyl azide method for peptide bond formation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CCFLOW project (Austrian Research Promotion Agency FFG No. 862766) is funded through the Austrian COMET Program by the Austrian Federal Ministry of Transport, Innovation and Technology (BMVIT), the Austrian Federal Ministry of Digital and Economic Affairs (BMDW) and by the State of Styria (Styrian Funding Agency SFG).Notes and references
- (a) M. Balci, Synthesis, 2018, 50, 1373–1401 CrossRef CAS; (b) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
- For a comprehensive overview of the acyl azide method for peptide synthesis, see: (a) J. Meienhofer, in The Peptides: Analysis, Synthesis, Biology, Academic Press, London, UK, 1979, ch. 4, vol. 1, pp. 197–239 Search PubMed; (b) Y. S. Klausner and M. Bodanszky, Synthesis, 1974, 549–559 CrossRef CAS.
- For comprehensive reviews of peptide coupling reagents, see: (a) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed; (b) C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS.
- For examples using the acyl azide method, see: (a) K. Hofmann, A. Jöhl, A. E. Furlenmeier and H. Kappeler, J. Am. Chem. Soc., 1957, 79, 1636–1641 CrossRef CAS; (b) I. A. L. Ali, ARKIVOC, 2008, 78–89 Search PubMed; (c) S. M. El Rayes, I. A. I. Ali and W. Fathalla, ARKIVOC, 2008, 86–95 Search PubMed; (d) W. Fathalla and I. A. I. Ali, Heteroat. Chem., 2007, 18, 637–643 CrossRef CAS.
- T. Curtius, Ber. Dtsch. Chem. Ges., 1890, 23, 3023–3033 CrossRef.
- J.-P. Hagenbuch, Chimia, 2003, 57, 773–776 CrossRef CAS.
- M. M. Joullié and K. M. Lassen, ARKIVOC, 2010, 189–250 Search PubMed.
- (a) T. Shioiri, K. Ninomiya and S. Y. Yamada, J. Am. Chem. Soc., 1972, 94, 6203–6205 CrossRef CAS PubMed; for a flow protocol using DPPA as reagent, see: (b) M. Baumann, I. R. Baxendale, S. V. Ley, N. Nikbin, C. D. Smith and J. P. Tierney, Org. Biomol. Chem., 2008, 6, 1577–1586 RSC.
- T. Shioiri, TCIMAIL, 2007, 134, 2–19 CAS.
- For reviews in flow chemistry see: (a) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed; (b) M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC; (c) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed; (d) R. Gérardy, N. Emmanuel, T. Toupy, V. Kassin, N. N. Tshibalonza, M. Schmitz and J. M. Monbaliu, Eur. J. Org. Chem., 2018, 2301–2351 CrossRef.
- B. Gutmann and C. O. Kappe, J. Flow Chem., 2017, 7, 65–71 CrossRef CAS.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed.
- (a) J. Britton and C. L. Raston, Chem. Soc. Rev., 2017, 46, 1250–1271 RSC; (b) H. R. Sahoo, J. G. Kralj and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 5704–5708 CrossRef CAS PubMed.
- For a review of continuous-flow peptide coupling strategies, see: (a) S. Fuse, Y. Otake and H. Nakamura, Chem. – Asian J., 2018, 13, 3818–3832 CrossRef CAS PubMed; for a recent important contribution, see: (b) C. P. Breen and T. F. Jamison, Chem. – Eur. J., 2019, 25, 14527–14531 CrossRef CAS PubMed.
- H. Eckert and B. Forster, Angew. Chem., Int. Ed. Engl., 1987, 26, 894–895 CrossRef.
- S. Fuse, N. Tanabe and T. Takahashi, Chem. Commun., 2011, 47, 12661–12663 RSC.
- S. Fuse, Y. Mifune and T. Takahashi, Angew. Chem., Int. Ed., 2014, 53, 851–855 CrossRef CAS PubMed.
- S. Fuse, Y. Mifune, H. Nakamura and H. Tanaka, Nat. Commun., 2016, 7, 13491–13497 CrossRef CAS PubMed.
- S. Fuse, K. Masuda, Y. Otake and H. Nakamura, Chem. – Eur. J., 2019, 25, 15091–15097 CrossRef CAS PubMed.
- M. Baumann, I. R. Baxendale, S. V. Ley, N. Nikbin and C. D. Smith, Org. Biomol. Chem., 2008, 6, 1587–1593 RSC.
- H. Sprecher, M. N. P. Payán, M. Weber, G. Yilmaz and G. Wille, J. Flow Chem., 2012, 1, 20–23 CrossRef.
- (a) C. R. Sagandira and P. Watts, Eur. J. Org. Chem., 2017, 6554–6565 CrossRef CAS; (b) C. R. Sagandira and P. Watts, J. Flow Chem., 2018, 8, 69–79 CrossRef CAS.
- For additional microreactor studies investigating acyl azide formation in flow, see: (a) C. R. Sagandira and P. Watts, Beilstein J. Org. Chem., 2019, 15, 2566–2589 Search PubMed; (b) P. Filipponi, C. Ostacolo, E. Novellino, R. Pellicciari and A. Gioiello, Org. Process Res. Dev., 2014, 18, 1345–1353 CrossRef CAS; (c) M. M. E. Delville, P. J. Nieuwland, P. Janssen, K. Kock, J. C. M. Van Hest and F. P. J. T. Rutjes, Chem. Eng. J., 2011, 167, 556–559 CrossRef CAS.
- For examples of azide formation using polymer-bound and monolith azide sources, see: (a) L. Kupracz, J. Hartwig, J. Wegner, S. Ceylan and A. Kirschning, Beilstein J. Org. Chem., 2011, 7, 1441–1448 CrossRef CAS PubMed; (b) C. J. Smith, C. D. Smith, N. Nikbin, S. V. Ley and I. R. Baxendale, Org. Biomol. Chem., 2011, 9, 1927–1937 RSC.
- L. Moroder, in Houben-Weyl Methods in Organic Chemistry: Synthesis of Peptides and Peptidomimetics, Thieme, Stuttgart, Germany, 2002, ch. 3, vol. 24a, pp. 425–442 Search PubMed.
- C. F. Da Costa, A. C. Pinheiro, M. V. De Almeida, M. C. S. Lourenço and M. V. N. De Souza, Chem. Biol. Drug Des., 2012, 79, 216–222 CrossRef CAS PubMed.
- A. Hassner, M. Stern, H. E. Gottlieb and F. Frolow, J. Organomet. Chem., 1990, 55, 2304–2306 CAS.
- It is known that aqueous solutions of up to ca. 10% HN3 are safe to handle, see: S. Bräse, M. Mende, H. H. Jobelius and H.-D. Scharf, Hydrazoic Acid and Azides in Ullmann's Encyclopedia of Industrial Chemistry, Encyclopedia of Inorganic Chemistry, ed. R. B. King, Wiley-VCH, Weinheim, 2nd edn, 2015 Search PubMed.
- A. R. Bogdan, M. Charaschanya, A. W. Dombrowki, Y. Wang and S. W. Djuric, Org. Lett., 2016, 18, 1732–1735 CrossRef CAS PubMed.
- G. Han, M. Tamaki and V. J. Hruby, J. Pept. Res., 2001, 58, 338–341 CrossRef CAS PubMed.
- For automated iterative amidation reactions in flow, see: (a) L. K. Rahbek Knudsen, M. Ladlow, Z. Bandpey and S. V. Ley, J. Flow Chem., 2014, 4, 18–21 CrossRef; (b) D. Lücke, T. Dalton, S. V. Ley and Z. E. Wilson, Chem. – Eur. J., 2016, 22, 4206–4217 CrossRef PubMed.
- A. Ufer, M. Mendorf, A. Ghaini and D. W. Agar, Chem. Eng. Technol., 2011, 34, 353–360 CrossRef CAS.
- (a) P. Plouffe, D. M. Roberge and A. Macchi, Chem. Eng. J., 2016, 300, 9–19 CrossRef CAS; (b) E. Mielke, D. M. Roberge and A. Macchi, J. Flow Chem., 2016, 6, 279–287 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00034e |
| This journal is © The Royal Society of Chemistry 2020 |