
In-vacuum electropolymerization of vapor-deposited source molecules into polymer films in ionic liquid†
Keisuke
Okawara
a,
Tomonobu
Nishimura
a,
Shingo
Maruyama
 a,
Masaki
Kubo
a and
Yuji
Matsumoto
*b
a,
Masaki
Kubo
a and
Yuji
Matsumoto
*b
aDepartment of Applied Chemistry, School of Engineering, Tohoku University, 6-6-07, Aramaki Aza Aoba Aobaku, Sendai, Miyagi 980-8579, Japan
bDepartment of Chemical Engineering, School of Engineering, Tohoku University, 6-6-07, Aramaki Aza Aoba Aobaku, Sendai, Miyagi 980-8579, Japan. E-mail: y-matsumoto@tohoku.ac.jp
First published on 1st November 2019
Abstract
In this communication, a new in-vacuum electropolymerization process combined with vapor-phase deposition using ionic liquid (IL) is proposed, in which source molecules are sequentially introduced into an IL layer coated on a patterned electrode substrate under the control of applied electrode potential. Based on this process concept, the direct polymerization of terthiophene into polythiophene films in IL was successfully demonstrated. The process can be numerically simulated to reproduce well the dynamic time-response of the oxidative current to the vapor deposition sequence and to prove its capability to precisely control electropolymerization by the saturated oxidative current density proportional to the vapor deposition rate.
Developing new synthesis methods for polymer materials (e.g., introducing some new technology into the existing polymer processes) is still important for not only realizing the cost-effective mass-production of known polymer materials, but also enabling to fabricate novel structures and functional polymers. Among many synthesis processes for polymer films, we, in this study, have particularly focused on the vapor deposition polymerization, which is a method combined with the vacuum deposition technology, notable for its solvent-free process of polymerization,1,2 as well as in its direct preparation of orientation-controlled polymer thin films.3,4 In most vapor deposition polymerization processes, however, further post-annealing treatments are required to polymerize the monomer deposits. For example, Yamazaki and Usui et al. reported the vapor deposition polymerization of polyimide thin films.5 The monomer molecules of 1,12-diaminododecane (DADD) and pyromellitic dianhydride (PMDA) were co-deposited to form a thin film of polyamic acid, but post-annealing in a nitrogen atmosphere at 100 to 250 °C is needed for the polyamic acid to be further imidized. Another example is the case of polyurea thin films.6–8 The monomers will not be polymerized during the room temperature deposition, and a post-annealing treatment at a high temperature of 200 °C in vacuum is needed for their polymerization, similar to the case of polyimide thin films.
To solve this problem, we have proposed a new vapor deposition polymerization with ionic liquid (IL).9 ILs are so stable in liquids, even under high-vacuum conditions up to about 120 °C, that we can use them as organic solvents not only for organic crystallizations,10–12 but also for polymerization reactions in vacuum deposition. In fact, we have demonstrated that ILs promote the substantial polymerization of urea films, even at room temperature (RT,) for vapor deposition polymerization in a vacuum. Furthermore, similar porous structures to those of bulk polyurea synthesized at the IL/water interface13 were found.9,14
Polythiophene is one of the most well-known conducting polymers that has been synthesized by the electropolymerization of thiophene monomer molecules or oligomers such as bithiophene (2T) and terthiophene (3T) in IL,15–17 as well as the conventional organic electrolytes. However, there have been no reports on the direct synthesis of polythiophene films in a vacuum (dry-process). In our previous work, we vacuum-deposited 3T molecules via IL but unfortunately, no polymerization occurred directly, even in the IL, unlike the case of polyurea (Fig. S1†).18 From the viewpoint of the crystal growth of 3T, which has two possible polymorphs of high- and low-temperature phases,19 the results themselves are somewhat interesting: large 3T platelet single crystals (several tens μm in size) of the low-temperature phase were obtained in a bulky IL droplet, while the high-temperature phase, which is normally difficult to obtain in its pure phase, was greatly stabilized in IL only a few nm-thick. In contrast, a key to realizing the electropolymerization in vapor deposition is the electrolyte nature of ILs, which are composed only of anion and cation pairs, enabling the use of ILs as electrolytes for electrochemical reactions combined with vacuum deposition. In fact, we have verified that this idea does work: metallic Cu particles can be directly electrodeposited in IL coated on a three-electrode patterned substrate by depositing CuCl, an inorganic salt, as a Cu source in a vacuum.20
In this communication, based on the above preliminary result of the electrochemical redox-driven vapor deposition with IL, we report on our attempt at the direct electropolymerization of polythiophene (P3T) films in IL by depositing 3T molecules into the IL in a vacuum, and its process of analysis by numerical simulations. As a result, the vapor-deposited 3T was found to be directly electropolymerized in the IL under steady-state conditions with a clear response of the oxidative current density originating from its electropolymerization to the variation of the 3T deposition rate.
Fig. 1 shows a schematic representation of a patterned three-electrode substrate with a working electrode (WE), a counter electrode (CE) and a reference electrode (RE), respectively, and the deposition of 3T in a vacuum chamber by a continuous wave (CW) infrared (IR) laser deposition method18,21,22 together with the polymerization scheme of P3T from 3T, and their chemical structures of IL and 3T used in this study.
 | ||
| Fig. 1 Electrochemical set-up of CW-IR laser deposition, along with the electropolymerization scheme of 3T to P3T. | ||
The average thickness of the IL dIL was about 10 μm in the experiments. The solubility of 3T in 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)amide ([emim][TFSA]) at RT was estimated to be 74 mM in bulk (Fig. S2†).
Fig. 2(a) shows a set of cyclic voltammetry (CV) curves (plotted in red) scanned in the potential range between −0.5 and +0.3 V vs. Au at a scan rate of 0.05 V s−1 during deposition of 3T at RT, where the deposition rate was 5 nm min−1. The inset is 10 cycles of CV curves (plotted in black) for a pure IL taken before the deposition of 3T. As compared to the CV curves before the deposition of 3T, the redox currents, as indicated by the arrows in Fig. 2(a), significantly increased with the cycle time. This is more clearly seen in Fig. 2(b), where the increase in the current density at an electrode potential of +0.3 V vs. Au from the initial value before starting the deposition is plotted as a function of the cycle number of CV, which has a substantial response to on–off switching of the CW-IR laser. These results suggest that the observed redox current change before and after depositing 3T originated from its electropolymerization.
 | ||
| Fig. 2 (a) Set of CV curves (plotted in red) scanned in a potential range between −0.5 and +0.3 V vs. Au at a scan rate of 0.05 V s−1 during the deposition of 3T at RT, where the deposition rate was 5 nm min−1. The inset shows 10 cycles of CV curves (plotted in black) for a pure IL taken before the deposition of 3T. (b) The current density increased at an electrode potential of +0.3 V vs. Au from the initial value before starting the deposition plotted as a function of the cycle number of CV. | ||
Next, by attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectroscopy, we examined the chemical structure of the deposit found in the sample of Fig. 2, obtained after the CV measurement during the deposition of 3T; the deposition amount of 3T was, in total, 50 nm thick. It should be pointed out that no nucleation of 3T crystals occurred when the 50 nm-thick 3T was just deposited into the bulky IL without application of electrode potential. This is because of the relatively high solubility of 3T in [emim][TFSA] as already discussed. Therefore, the deposit found in the sample of Fig. 2 is the result of some electrochemical reactions.
Fig. 3(a) shows a typical ATR-FTIR spectrum of the deposit after removing the remaining IL by vacuum annealing (red). It also shows the spectra of a 3T film (2.1 μm thick) deposited without IL on an Au substrate (green), the IL that we used (blue), and a bare Au substrate (yellow), just for comparison. In the ATR-FTIR spectrum of the 3T film deposited without IL, the CH out-of-plane bending vibrations, the wave-numbers of which are characteristic of the positions of substitution, appeared in the 900–600 cm−1 region.23 The strongest band at around 797 cm−1 resulted from the 2,5-disubstituted thiophene ring (2,5-T), while the second strongest band at around 687 cm−1 was from the 2-monosubstituted thiophene ring (2-T). On the other hand, in the ATR-FTIR spectrum of the deposit obtained after the CV cycles during the deposition of 3T, the corresponding 2,5-T band appeared at 791 cm−1, lower than that of the 3T film by 6 cm−1. In general, the 2,5-T bands of polythiophene polymers are known to shift to lower wavenumbers by 12–7 cm−1 as compared to those of the monomer and small oligomers. Furthermore, the band at around 629 cm−1 was clearly observed, which was assigned to the C–S stretching vibration characteristic of polythiophene.24 Therefore, the deposit after the CV cycles during the deposition of 3T was identified to be polythiophene (P3T), synthesized by the electropolymerization of 3T in IL. In fact, the area intensity of the 2-T band at 692 cm−1 found in the deposit is much smaller than that of the 3T film; this is because the 2-monosubstituted thiophene rings exist only at the ends of a polymer chain in polythiophene. On the other hand, a characteristic band of 2,4-disubstituted thiophene (2,4-T) about 740 cm−1 (ref. 23) is not so obvious in the deposit, indicating that the electrochemical polymerization occurs mainly at the α,α′-positions. The degree of polymerization Ndp is thus obtained from eqn (1):
| Ndp = 2Ro/R + 2 | (1) |
 | ||
| Fig. 3 (a) Typical ATR-FTIR spectrum of the deposit after removing the remaining IL by vacuum annealing (red), together with the spectra of a 3T film (2.1 μm-thick) deposited without IL on a Au substrate (green), the same IL as used (blue), and a bare Au substrate (yellow), just for comparison. (b) Schematic of a P3T film grown on an electrode surface with the assumption of its polymer chain direction normal to the surface, along with a typical AFM image of the film after the removal of the IL. | ||
The polythiophene deposits obtained on WE have different colours of red or blue-black, depending on the CV conditions. It is known that polythiophene can be electrochemically reduced or oxidized by changing the applied electrode potential, accompanied by a colour change from red to blue.26 The cathode and anode polythiophene films were doped/de-doped with cations and anions, respectively, which were originally contained in the electrolyte solution.27 In fact, similar electrochemical redox reactions were confirmed in CV measurements between −2 and +1 V vs. Au, performed for our P3T deposit in air with IL: the colour changed from red to blue-black at about +0.7 V vs. Au in a positive potential sweep and back again to red at about −0.4 V vs. Au in its return negative potential sweep, respectively (Fig. S3†). The observed electrochemically controlled colour change was also evidence for the deposit being polythiophene.
In order to understand this electropolymerization in the vapor deposition process, the time-response of the oxidative (anodic) current originating from electropolymerization to the vapor deposition of 3T was investigated at a constant electrode potential. Fig. 4(a) is a typical result of chronoamperometry (CA) at 0 V vs. Au for a deposition rate of 15 nm min−1. Upon turning the IR laser on, i.e., on starting to deposit 3T molecules, the oxidative current was found to sharply increase, approaching a constant value, and then gradually decreased after turning it off, i.e., stopping the deposition. The saturated oxidative current density depends on the deposition rate of 3T, and if limited to the data set of the first measurements, a better linear relationship between them was found as shown in Fig. 4(b). Here, it is noted that the ratio of the amount of electric charge for electro-oxidation to the deposition amount of monomers is close to 1. This estimation suggests that most of the deposited monomers are electro-oxidized and incorporated into the polymer formation, even taking into account some of the electric charge possibly coming from the electro-oxidation of IL. The linear relationship between the saturated oxidative current density and the deposition rate of 3T also suggests that a steady-state condition is reached when the oxidative current density becomes constant in the vapor deposition–electropolymerization process, as illustrated in Fig. 4(c).28 In the steady-state conditions, the supply rate of 3T molecules at the IL-vacuum interface, which corresponds to its deposition rate from the gas phase υDEP, the diffusion rate of 3T molecules in IL υDiff and the electropolymerization rate at the electrode-IL interface υEP, all become balanced to the same value with the establishment of a constant concentration gradient in IL. In order to confirm this steady-state model, we attempted to numerically simulate the time-development and -decay of the concentration profile c(x, t) of 3T molecules in IL. For simplification, we employed a one-dimensional diffusion model, assuming first-order reaction kinetics for the electropolymerization. This is because it would be reasonable that the rate-limiting step throughout the polymerization reaction is the electro-oxidation of monomers since the monomers are supplied from the gas phase at deposition rates less than 50 nm min−1 in thickness and their concentration in IL should, therefore, be very low. Accordingly, the differential equations with the boundary conditions at x = 0 and x = dIL are described as follows:
 | (2) |
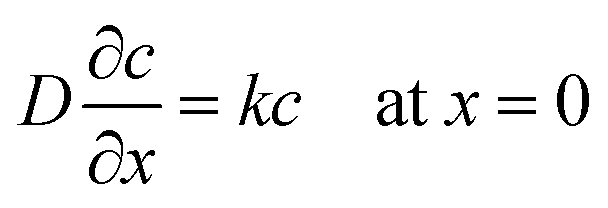 | (3) |
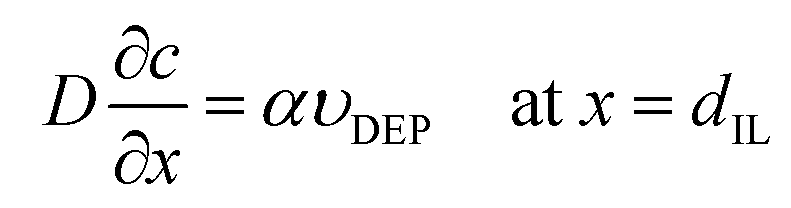 | (4) |
 | ||
| Fig. 4 (a) Time-response of the oxidative (anodic) current originating from electropolymerization to the vapor deposition of 3T at a constant electrode potential of 0 V vs. Au for a deposition rate of 15 nm min−1. (b) The saturated oxidative current density plotted against the deposition rate. (c) Schematic of a model proposed for the vapor deposition–electropolymerization process under steady-state conditions. | ||
 | ||
| Fig. 5 Numerical simulation results based on a one-dimensional diffusion model. The time-development (a) and -decay (b) of the concentration profile c(x, t) of 3T molecules in the IL. (c) A constant concentration gradient is established in the IL in steady-state conditions for different deposition rates. (d) Concentration at x = 0 in steady-state conditions, representing the saturated oxidative current density, linearly increases with the deposition rate. | ||
In the simple numerical simulation, the time-development and -decay of the concentration at x = 0 are also able to well reproduce the time-response of the measured oxidative current density as displayed in Fig. 4(a) and furthermore, it was found that the time required for reaching a steady-state condition seemed almost constant, irrespective of the deposition rate (Fig. S4†). In fact, according to the numerical simulation, the time constant τ, as expressed by eqn (5), does not depend on the deposition rate:
 | (5) |
From this equation, we can roughly estimate the diffusion coefficient D of 3T molecules in the IL; for example when dIL ∼ 10 μm and τ ∼ 400 s in the first measurement are put into the equation, the value of D is calculated to be ∼0.2 μm2 s−1, which is smaller by about one order of magnitude than those reported for some organic molecules in ILs,29,30 but acceptable for this rough estimation. In addition, the diffusion coefficient in ILs is known to generally increase by the inclusion of water impurity.31 Unlike the previously reported cases where most measurements were carried out under atmospheric conditions, our measurement was performed in a high vacuum with negligible water effects, probably giving a smaller diffusion coefficient.
Conclusions
In this communication, a novel vapor deposition electropolymerization with IL was demonstrated in the fabrication of polythiophene films starting from 3T as a monomer source. IL coated on a patterned electrode substrate was found to work as the electrolyte and play similar roles even in vacuum electropolymerization under the control of an applied electrode potential as in the bulk IL. The degree of polymerization was about 10 thiophene rings, i.e., three or four 3T molecules were successively polymerized at the α and α′-positions. The time-response of the oxidative current to the vapor deposition of 3T was observed at a constant electrode potential, which was reproduced by well numerical simulation and it was then confirmed that the vapor deposition electropolymerization proceeded under steady-state conditions with the saturated oxidative current density well proportional to the 3T vapor deposition rate. The present results indicate new possibilities for the nanoscale control of electropolymerization for new polymer films, independent of the electrode potential, in a similar way to the molecular beam deposition process for inorganic thin films.Conflicts of interest
There are no conflicts to declare.Notes and references
- J. R. Salem, F. O. Sequeda, J. Duran, W. Y. Lee and R. M. Yang, J. Vac. Sci. Technol., A, 1986, 4, 369–374 CrossRef CAS.
- Y. Takahashi, M. Iijima, K. Inagawa and A. Itoh, J. Vac. Sci. Technol., A, 1987, 5, 2253–2256 CrossRef CAS.
- A. Kubono, H. Higuchi, S. Umemoto and N. Okui, Thin Solid Films, 1993, 229, 133–136 CrossRef CAS.
- A. Kubono, N. Okui, K. Tanaka, S. Umemoto and T. Sakai, Thin Solid Films, 1991, 199, 385–393 CrossRef CAS.
- T. Yamazaki, C. Mahapun, S. Usui, K. Tanaka and H. Usui, J. Phys.: Conf. Ser., 2017, 924, 012017 CrossRef.
- T. Yanase, T. Hasegawa, T. Nagahama and T. Shimada, Jpn. J. Appl. Phys., 2012, 51, 041603 CrossRef.
- Y. Takahashi, S. Ukishima, M. Iijima and E. Fukuda, J. Appl. Phys., 1991, 70, 6983–6987 CrossRef CAS.
- X.-S. Xian-Shan Wang, M. Iijima, Y. Takahashi and E. Fukuda, Jpn. J. Appl. Phys., 1993, 32, 2768–2773 CrossRef.
- Y. Ohsawa, R. Takahashi, S. Maruyama and Y. Matsumoto, ACS Macro Lett., 2016, 5, 1009–1013 CrossRef CAS.
- Y. Takeyama, S. Maruyama and Y. Matsumoto, Cryst. Growth Des., 2011, 11, 2273–2278 CrossRef CAS.
- Y. Takeyama, S. Maruyama, H. Taniguchi, M. Itoh, K. Ueno and Y. Matsumoto, CrystEngComm, 2012, 14, 4939–4945 RSC.
- K. Kuroishi, S. Maruyama, N. Ohashi, M. Watanabe, K. Naito and Y. Matsumoto, J. Cryst. Growth, 2016, 453, 34–39 CrossRef CAS.
- L. Zhu, C.-Y. Huang, Y. H. Patel, J. Wu and S. V. Malhotra, Macromol. Rapid Commun., 2006, 27, 1306–1311 CrossRef CAS.
- R. Takahashi, S. Maruyama, Y. Ohsawa and Y. Matsumoto, Chem. Lett., 2018, 47, 1460–1463 CrossRef CAS.
- K. Nishihata, K. Tsunashima, Y. Ono and M. Matsuyama, ECS Trans., 2017, 75, 99–103, DOI:10.1149/07552.0099ecst.
- J. M. Pringle, M. Forsyth, D. R. MacFarlane, K. Wagner, S. B. Hall and D. L. Officer, Polymer, 2005, 46, 2047–2058 CrossRef CAS.
- K. Sekiguchi, M. Atobe and T. Fuchigami, J. Electroanal. Chem., 2003, 557, 1–7 CrossRef CAS.
- K. Okawara, S. Maruyama and Y. Matsumoto, Jpn. J. Appl. Phys., 2019, 58, 085503 CrossRef.
- S. Guillaume, P. Nicolas, R. Claire, A. Roland, K. Markus, T. Samuel, L. Vincent, C. Jerome, G. Yves and G. Gabin, Cryst. Growth Des., 2011, 11, 3663–3672 CrossRef.
- Y. Sato, S. Maruyama and Y. Matsumoto, J. Vac. Sci. Technol., A, 2018, 36, 031516 CrossRef.
- Y. Takeyama, S. Maruyama and Y. Matsumoto, Sci. Technol. Adv. Mater., 2011, 12, 054210 CrossRef.
- S. Yaginuma, K. Itaka, M. Haemori, M. Katayama, K. Ueno, T. Ohnishi, M. Lippmaa, Y. Matsumoto and H. Koinuma, Appl. Phys. Express, 2008, 1, 015005 CrossRef.
- M. Akimoto, Y. Furukawa, H. Takeuchi and I. Harada, Synth. Met., 1986, 15, 353–360 CrossRef CAS.
- D. Zhang, J. Qin and G. Xue, Synth. Met., 1999, 100, 285–289 CrossRef CAS.
- J. L. Sauvajol, D. Chenouni, J. P. Lere-Porte, C. Chorro, B. Moukala and J. Petrissans, Synth. Met., 1990, 38, 1–12 CrossRef CAS.
- K. Kaneto, K. Yoshino and Y. Inuishi, Solid State Commun., 1983, 46, 389–391 CrossRef CAS.
- K. Kaneto, K. Yoshino and Y. Inuishi, Jpn. J. Appl. Phys., 1983, 22, L567–L568 CrossRef.
- Y. Matsumoto and S. Maruyama, Ionic Liquid Devices, Smart Materials Series, No. 28, ed. A. Eftekhari, RSC, London, UK, 2018, ch. 6, p. 136 Search PubMed.
- D. K. Sasmal, A. K. Mandal, T. Mondal, K. J. H. Bhattachary, S. N. Baker and G. A. Baker, J. Phys. Chem. B, 2011, 115, 7781–7787 CrossRef CAS.
- Y. Nishiyama, M. Fukuda, M. Terajima and Y. Kimura, J. Chem. Phys., 2008, 128, 164514 CrossRef CAS PubMed.
- J. H. Werner, S. N. Baker and G. A. Baker, Analyst, 2003, 128, 786–789 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9re00343f |
| This journal is © The Royal Society of Chemistry 2020 |