
Microfluidic immobilized enzyme reactors for continuous biocatalysis
Yujiao
Zhu
 abc,
Qingming
Chen
ab,
Liyang
Shao
d,
Yanwei
Jia
cef and
Xuming
Zhang
*ab
abc,
Qingming
Chen
ab,
Liyang
Shao
d,
Yanwei
Jia
cef and
Xuming
Zhang
*ab
aDepartment of Applied Physics, The Hong Kong Polytechnic University, Hong Kong, China. E-mail: apzhang@polyu.edu.hk
bThe Hong Kong Polytechnic University Shenzhen Research Institute, Shenzhen, China
cState Key Laboratory of Analog and Mixed Signal VLSI, Institute of Microelectronics, University of Macau, Macau, China
dDepartment of Electrical and Electronic Engineering, Southern University of Science and Technology, Shenzhen, China
eFaculty of Science and Technology, University of Macau, Macau, China
fFaculty of Health Sciences, University of Macau, Macau, China
First published on 16th September 2019
Abstract
Biocatalysis has attracted significant attention owing to its environmental-friendly nature, high efficiency, and remarkable selectivity for reactions. However, enzymes, which are powerful catalysts used in biocatalysis, suffer from low stability when used for long-term operations in solution and a gradual decrease in activity during storage. Microfluidic reactors are devices known for their smaller dimensions, large surface-to-volume ratios, and well-defined reaction times. Enzymes immobilized in such microfluidic reactors can exhibit distinct benefits, such as fast reaction rate, high storage stability, suppressed autolysis, and ease of use. The use of microfluidic immobilized enzyme reactors (μ-IMERs) offers several advantages over traditional technologies in performing biocatalytic reactions, such as low energy consumption, rapid heat exchange, fast mass transfer, high efficiency, and superior repeatability. In this review, the strategies of employing μ-IMERs for continuous biocatalysis have been investigated via a top-down approach. First, from the macroscopic perspective, the fabrication techniques of microfluidic reactors are presented encompassing materials, configurations, and technologies. Then, from the microscopic point of view, several strategies are discussed for the internal structural designs of microfluidic reactors. Moreover, when we move to the nanoscopic level, attention is paid to the choice of enzyme immobilization techniques for performance enhancement. Finally, the scalability of microfluidics that transfers biocatalysis from laboratory to industrial production was investigated. This review is intended to provide a guideline for using biocatalysis in microreactors and expediting the progress of this important research area.
 Yujiao Zhu | Yujiao Zhu received her B.Eng. in Materials Science and Engineering from the University of Science and Technology, Beijing, in 2014, and PhD in Applied Physics from the Hong Kong Polytechnic University in 2019. She is currently a Research Assistant in the Hong Kong Polytechnic University and University of Macau. Her research interests are bio-microfluidics, artificial photosynthesis, and green energy. |
 Qingming Chen | Qingming Chen received his BSc in optical information science and technology from the Huazhong University of Science and Technology, Hubei, China, in 2011, M.Eng. in optical engineering from Jinan University, Guangdong, China, in 2014, and PhD in applied physics from the Hong Kong Polytechnic University, Hong Kong, in 2018. Since 2018, he has been a Postdoctoral Research Fellow with Hong Kong Polytechnic University. His research interests mainly focus on optofluidics, lab-on-a-chip systems, and optical devices. |
 Li-Yang Shao | Li-Yang Shao received his PhD degree in Optical Engineering in 2008 from Zhejiang University. He was a research assistant/associate at the Hong Kong Polytechnic University (HKPU) from 2006 to 2009. Then, he worked as a Postdoctoral Fellow successively at Carleton University, HKPU, and University of Sydney. In 2013, he joined the Southwest Jiaotong University as a Professor. In 2017, he joined the Southern University of Science and Technology (SUST). In 2018, he became the Associate Dean of the School of Innovation and Entrepreneurship in SUST. His research interests include fiber grating, sensors, and smart sensing systems for the railway industry. |
 Yanwei Jia | Yanwei Jia received her PhD, MSc, and BSc degrees in Physics from the National University of Singapore (2006) and Hunan University in China (2002 and 1996), respectively. After her PhD, Jia worked as a Research Fellow in the National University of Singapore in 2006 before she moved to Brandeis University in the USA, working as a Postdoctoral Fellow, Research Associate, and Research Scientist (2006–2012). She is currently an Assistant Professor in the State Key Laboratory of Analog and Mixed Signal VLSI (AMSV), University of Macau, leading a group working on microfluidics for biological/chemical applications. |
 Xuming Zhang | Xuming Zhang is currently an Associate Professor with the Department of Applied Physics, the Hong Kong Polytechnic University. He received his B.Eng. degree in Precision Mechanical Engineering from the University of Science & Technology of China (USTC) in 1994, and his PhD from the School of Electrical & Electronic Engineering, Nanyang Technological University (NTU) in 2006. His research resulted in more than 100 journal papers with an h-index of 28 and citation count of 2900. His current research interests mainly include microfluidics, artificial photosynthesis, biomimetics, and green energy. |
1. Introduction
Biocatalysis is regarded as the most important green research area for sustainable manufacturing in the pharmaceutical and fine chemicals industries due to its low operating costs and high eco-efficiency.1 Enzymes are an important type of natural catalyst used for biocatalysis, which exhibit several excellent characteristics that are lacking in artificial catalysts, such as high efficiency, enhanced selectivity, environment-friendly, and ability to catalyze a reaction under milder conditions.2 The applications of enzymes for green and sustainable chemical synthesis in the industry have also infiltrated into our daily life. Despite this, certain aspects of enzymes still need to be improved upon prior to their applications in the mass production of industrialized products, such as reusability and activity recovery for economic effects, long-term operation and storage stability, inhibition of certain reaction products, and selectivity toward nonnative substrates.3 Moreover, the separation of enzymes from products after the completion of a reaction, although incurs time and effort, is always an indispensable part of work. The possible contamination of products should be avoided and the overall operational costs could be reduced.4 Fortunately, enzyme immobilization is an impressive way to overcome these drawbacks. It considerably simplifies the separation and recovery of enzymes. The activity, stability, and selectivity of enzymes can also be improved after immobilization.5 Researchers have devoted considerable effort toward studying various enzyme immobilization techniques so far, including physical adsorption, affinity bonding, covalent binding, and encapsulation.6–9 Nevertheless, inappropriate immobilization can also cause conformational changes, blocking of active sites, and diffusion resistance to the enzyme, which, in turn, result in activity loss. Considering the structural diversity, complexity, and variability of enzymes, as well as their sensitivity toward environmental conditions, the selection of immobilization techniques should be very careful with specific analyses. Further, the exploration of simple, efficient, and widely adaptable enzyme immobilization methods should get increased attention.In laboratory studies involving enzyme immobilization, a higher yield can be obtained if the products are separated from the enzymes in time and the substrates are continuously supplemented.10 Therefore, the application of enzyme immobilization in continuous microfluidic reactors has attracted huge interest in industrial production, such as the syntheses of petrochemicals, active pharmaceutical ingredients, and value-added materials.11–13 Continuous microfluidic systems outperform batch systems accompanied by several advantages, such as smaller dimensions, low cost and energy consumption, high efficiency, rapid heat exchange, and fast mass transfer.14–16 In particular, a large surface-area-to-volume (SAV) ratio for microfluidic reactors is advantageous for enzyme loading. Different microchannel types (e.g., wall-coated type, packed-bed type, and monolithic type) also provide various possibilities for the integration of immobilization carriers into the micro space. When the enzyme is immobilized in microfluidic reactors, there is no need to separate the enzyme-loaded carriers from the reaction solution, which facilitates the recovery and reusability of the enzyme, therefore saving time and labor. Moreover, different catalytic reaction conditions (such as temperature, pH, residence time, and pressure) are easier to control in microfluidic reactors as compared to operation in batch systems.17 Higher temperature with low-boiling solvents, higher pressure, more uniform heat/pressure distribution, safer and easier reaction control, and less unwanted products can be achieved.18 Furthermore, reaction stoppages can be easily achieved by pumping the substrate out of the reactors without the need for the addition of an acid or base that may affect the detection accuracy or products. Further, microfluidic reactors can be directly incorporated into many instruments for real-time analysis and monitoring. Moreover, many natural biocatalytic reactions are cascaded multienzyme reactions.19 Immobilizing enzymes in microfluidic reactors facilitate control over their sequential order and relative positions, thereby reverting to natural cascaded reactions to the maximum extent. In addition, microfluidic reactors are easily scaled up or scaled out once careful design factors are taken into account.20 In general, these promising features of continuous microfluidic immobilized enzyme reactors (μ-IMERs) hold the key for their application in green, sustainable, economical, and large-scale industrial production.21–23
This review provides a comprehensive discussion on the factors that affect the performance of continuous biocatalysis in μ-IMERs using a top-down strategy. From the macroscopic point of view, the first thing to consider is the fabrication of microfluidic reactors in addition to materials' and configuration designs. Various materials have been developed in microfluidics, such as silicon, glass, polymers, and paper. The characteristics of fabrication materials are of vital importance to the performance of catalytic reactions. Special biocatalysis can also be achieved with a careful design of the device configuration. From the microscopic point of view, the capacity of the inner structures of the microreactors for enzyme loading also plays a significant role in the overall biocatalytic efficiency. In addition, the specific substrate diffusion path induced by different types of microreactors should also be taken into account. Moreover, from the nanoscopic point of view, different immobilization techniques, which dominate the performance of enzymes (e.g., activity, stability, and reusability) in the nanoenvironment of biocatalytic reactions, are also comprehensively studied. Finally, the scalability of microfluidics that can facilitate the transfer of biocatalysis in the laboratory to that in the industry for large-scale production is briefly reviewed. We hope that the discussion in this review can facilitate the understanding of the main characteristics of the rapidly developing μ-IMERs field that can be used to undertake continuous biocatalysis and can provide a clear guideline for future research.
2. Engineering of microfluidic reactors
2.1 Materials for microfluidic reactors
There are many materials that have been used for the fabrication of microfluidic reactors. The basic characteristics are stability and inertness.24 In the early days, silicon25 and glass were mostly used, which were directly inherited from the semiconductor industry and microelectromechanical systems (MEMS).26 They usually require surface salinization and the introduction of certain functional groups such as carboxyl groups or amino groups for further immobilization;27 however, high cost and complicated fabrication procedures usually limit their applications in microfluidics. Therefore, several polymers with easy fabrication and high compatibility for biocatalysis have become fairly popular for use in microfluidics.As a typical silicon-based organic polymer, polydimethylsiloxane (PDMS) has been very popular in biomicrofluidic applications. Its advantages are excellent biocompatibility, easy fabrication, low cost, and optical transparency, which are beneficial for the monitoring and optical detection of biocatalytic reactions.28 The flexibility feature also makes PDMS an excellent material to fabricate valves and pumps in microfluidic devices. Nevertheless, certain problems of PDMS are not negligible: swelling in certain organic solvents, changes in solution concentrations due to water evaporation, and hydrophobic surfaces that lead to nonspecific adsorption of biomolecules. Therefore, oxygen plasma or surface modification is usually required to make it hydrophilic and to introduce functional groups for enzyme immobilization.
Other polymer materials, such as polymethyl methacrylate (PMMA),29–31 polystyrene (PS),32 polycarbonate (PC),33 poly(ethylene terephthalate) (PET),34 and polytetrafluoroethylene (PTFE),35,36 have also been widely used for microfluidics fabrication. Although they possess excellent chemical, electrical, mechanical, optical, and thermal properties,37–40 they usually require additional surface modifications due to the lack of functional groups on the surfaces. Sometimes, stainless steel and ceramics are used for reactor fabrication if the reaction is operated under higher temperatures and higher pressures.41 However, their high fabrication cost largely restricts their broader applications. Ogończyk et al. firstly used PC microchannels for enzyme immobilization in 2012.33 The PC microfluidic chip could immobilize different kinds of enzymes, such as alkaline phosphatase (ALP) and urease, by the physical–chemical method. Further, enzymatic microfluidic chips also exhibit attractive operation reproducibility, storage stability, and higher conversion rates.
Paper is another promising material for the fabrication of microfluidic reactors. Paper-based microfluidics generally have porous and open channels, which provide larger surface areas for enzyme immobilization as compared to conventional microfluidics, which only have hollow channels. However, paper-based microfluidics are mainly used for biochemical analysis, medical diagnostics, and forensic diagnostics,42,43 which are not in the scope of this review.
2.2 Configuration design of microfluidic reactors
The configuration of microfluidic reactors varies under different situations. Four representative configuration designs are shown in Fig. 1. A single-channel microfluidic chip is the simplest. It has only one straight channel for the immobilization of enzymes and substrate transport (Fig. 1a). Open-tubular capillaries can also be classified under this category. Nevertheless, the volume of such a chip is generally limited for enzyme loading. A serpentine channel (or curved channel) is accordingly designed by folding up a single channel into a serpentine (curved) shape to increase the effective volume for immobilization (Fig. 1b). A multichannel microfluidic chip is a more advanced design that multiplies the effective immobilization volume.44 As shown in Fig. 1c, the microchannels are divided into an array on the input side and a similar unit on the output side. For further volume increase, a planar microfluidic chip is presented by simply enlarging the channel into a planar chamber in the lateral direction (Fig. 1d). | ||
| Fig. 1 Representative configuration designs of microfluidic chips: (a) single-channel chip, (b) serpentine-channel chip, (c) multi-channel chip and (d) planar chamber chip. | ||
The configuration of microfluidic reactors should be well designed when applied to continuous biocatalysis. In the bulk system, the substrate solutions react with enzymes by mixing and diffusion. The reaction performance is unlikely influenced by the container shape. However, in a microfluidic chip, the substrate solutions are driven by external forces to react with enzymes immobilized on the chip. If the immobilization amount of the enzyme and residence time of biocatalysis are fixed, the configuration design can have a substantial impact on the accessibility of the substrate to the immobilized enzyme, which then affects the overall biocatalytic reaction performance.45 Hoffmann et al. designed four HRP-immobilized microreactors with different configurations: full surface, half surface, fine checkerboard, and coarse checkerboard.45Fig. 2a shows product absorbance at the reactor outlet for each pattern. The fine and coarse checkerboard structures exhibit an increased efficiency, with 81% and 56% higher absorbance per active area than the fully modified surface. Different configurations can yield different velocities across the reactors, thereby influencing mass transport and fluid mixing. Consequently, the accessibility of the substrate for the enzyme near the surface is affected, leading to a difference in the reaction performance.
 | ||
| Fig. 2 (a) Comparison of experimentally determined ABTS˙+ absorbance at 414 nm for per mm2 of the modified area at the reactor outlet under the steady-state condition (blue) and ABTS˙+ outlet concentration obtained from CFD simulations in mM mm−2 (red) for the different surface patterns: reference (horseradish peroxidase (HRP) adsorption on a nonmodified surface; empty squares), half-modified surface, coarse checkerboard, fine checkerboard, and fully modified surface; (b) illustrations of the product concentrations on the top surface of the microreactor (under steady-state simulations) dimensions using the fully modified surface (b1), half-modified surface (b2), fine checkerboard structure (b3), and coarse checkerboard structure (b4), where the red surfaces illustrate high and blue surfaces denote low product concentrations. These two figures were reproduced from ref. 45 with the permission from Elsevier Ltd. (c)–(g) Reaction performances of the five prepared reactors using 0.1% β-lactoglobulin solution as the substrate. (h) Pressure drops of the five prepared reactors. These 6 figures were reproduced from ref. 46 with permission from Elsevier Ltd. | ||
Further, Nakagawa et al. proved that the channel shape has a substantial impact on the backpressure, which further affects enzyme activity.46 Five microreactors with different numbers and lengths of elbows and straight sections were prepared. Protease was immobilized in a freeze-dried polyvinyl alcohol (PVA) micromonolith prepared in the microreactors. The proteolytic reaction yields of the five reactors obtained under the same residence time were significantly different due to the changes in the elbow and straight sections, as shown in Fig. 2c–g. The microreactor with the least number of elbow sections had the highest reaction yield, but the smallest pressure drop (Fig. 2h). The pressure drop is related to the resultant fluid resistance, which is determined by microchannel patterning. Therefore, the accessibility of the substrate to the immobilized enzyme via diffusion is largely affected, resulting in differences in the reaction yields. Then, the microreactor with the smallest pressure drop would have the highest enzyme activity.
2.3 Fabrication technologies of microfluidic reactors
The fabrication process of microfluidic reactors includes microchannel fabrication and microfluidic chip bonding. The techniques for both these steps should be carefully selected by considering the reactor material, reactor configuration, and fabrication cost and time. For microchannel fabrication, most techniques are adopted and improved from the MEMS, such as photolithography, etching, soft lithography, thermoforming, and so on. In contrast, the techniques for the bonding process are generally different from those used in MEMS. They may be divided into indirect and direct bonding. | ||
| Fig. 3 Process flow of photolithography and soft lithography with PDMS. | ||
As an extension of photolithography, soft lithography is a collection of techniques to fabricate microstructures in a wide range of soft elastomer materials, such as polymers, gels, and organic monolayers for microfluidic applications.47 Basically, it uses a patterned elastomeric polymer layer as a mask, mold, or stamp to emboss, mold, or print the pattern to another soft substrate.48 The patterned elastomeric polymer is usually a layer of PDMS that is fabricated from a solid master produced by photolithography (as shown in steps 5b and 6b, Fig. 3). The basis of soft lithography includes microcontact printing, replica molding, microtransfer molding, micromolding in capillaries, and a large number of patterning techniques.49
Thermoforming techniques are usually employed to pattern semifinished thermoplastic foils by stretching or stamping with pressure and heat.50,51 Injection molding and hot embossing can also be classified in this category. They have the advantages of cost-effective high-volume fabrication and high-frequency manufacturing.44 However, thermoforming techniques are less precise for controlling the aspect ratio than lithography techniques.52
3. Internal structural designs of microfluidic channels
The amount and activity of immobilized enzymes in a microfluidic chip can considerably impact the biocatalytic reaction rate. Further, the internal structure of microfluidic channels can strongly affect these two factors that can be achieved by adjusting the SAV ratio of the microchannel and the diffusion pressure of the solution passing through it. Generally, the internal structures of μ-IMERs are classified into three types: wall-coated type, monolithic type, and packed-bed type (Fig. 4). A comparison of the properties of these three types of microchannels is provided in Table 1. | ||
| Fig. 4 Typical designs for the internal structure of microfluidic channels: (a) wall-coated type channel, (b) packed-bed type channel and (c) monolithic type channel. | ||
| Microchannel type | Wall-coated | Packed-bed | Monolithic |
|---|---|---|---|
| SAV ratio | Small | Large | Large |
| Pressure drops | Low | High | Low |
| Diffusion length | Large | Small | Small |
| Heat transfer | Large | Small | Moderate |
| Mechanical stability | High | Low | Moderate |
3.1 Wall-coated-type channel
For wall-coated-type μ-IMERs (Fig. 4a), the enzyme is directly immobilized onto the inner wall of the microchannel.61–63 Notably, the available surface areas of the microfluidic walls are fairly limited, resulting in low enzyme-loading capacity. In addition, the substrate diffusion path is relatively large in this case, leading to a low biocatalytic conversion rate. Researchers have been dedicated toward increasing the SAV ratio of microchannels, thereby enhancing the enzyme-loading capacity. One efficient method to achieve this is to modify the inner wall with certain biocompatible nanostructured materials such as dopamine,64 gold nanoparticles,65 graphene,66,67 graphene oxide,68,69 nanosprings,70 or MXenes.71Recently, Valikhani et al. designed a borosilicate microchannel with silica nanosprings attached to the surface for the immobilization of sucrose phosphorylase (Fig. 5a).70 It has been demonstrated that the enzyme-loading amount can be increased by an order of magnitude or more as compared to that of enzyme loaded on uncoated microchannel walls. Moreover, nanospring microreactors showed enhanced conversion efficiency involving the synthesis of α-glucose-1-phosphate and improved reusability and stability when compared with a plain microreactor. Another feasible solution to increase the active enzyme-loading amount is to multiply the immobilization layers, which is also referred to as the layer-by-layer (LBL) assembly approach.34,35,72–74 A representative work was conducted by Bi et al. who alternatively absorbed polyethyleneimine (PEI) and Candida antarctica lipase B (CAL-b) on the microreactor surface (Fig. 5b).35 Lipase loading was enlarged as the number of layers increased. It reached saturation at the eighth layer. The microreactor was also demonstrated to have high conversion efficiency and excellent stability for producing wax ester.
 | ||
| Fig. 5 Examples of wall-coated microchannels for enzyme immobilization. (a) Schematic of sucrose phosphorylase immobilized on nanospring microreactors with enhanced enzyme activity. Reprinted with permission from ref. 70. Copyright 2017 American Chemical Society. (b) Process of immobilization of CAL-b based on the self-oxidation of dopamine and LBL method. Long chains with positive charges represent PEI, and circles represent lipase. Reproduced from ref. 35 with permission from The Royal Society of Chemistry. | ||
3.2 Packed-bed-type channels
Even though many methods have been developed to increase the SAV ratio of wall-coated-type μ-IMERs, the room for enhancement is very small. In order to maximize the space utilization of size-limited channels and therefore maximizing the enzyme-loading amount, enzymes are designed to be immobilized on polymeric or inorganic particles. The enzyme-immobilized particles are then packed into the microchannel of μ-IMERs, which is regarded as a packed-bed-type channel (Fig. 4b). The higher SAV ratios of a packed-bed channel also ensure relatively shorter diffusion distances between substrates and enzymes when compared with those in wall-coated channels.75,76 Many polymeric particles have already been commercially available for packing.76–79 Certain inorganic materials such as glass,80,81 silica,82 and Fe2O3 microparticles83–85 have also been explored. These packed-bed-type μ-IMERs can be easily fabricated and exhibit an extremely high enzyme-loading capacity.Kundu et al. designed a microreactor packed with commercially available mesoporous PMMA beads, where CAL-b was physically immobilized to study the polymerization of polycaprolactone from ε-caprolactone in the continuous mode (Fig. 6a–c).77 It was demonstrated that faster polymerization and higher molecular mass could be obtained in the microreactors as compared to that in the batch reactors. Another example is the packing of GOx-modified MNPs in microfluidic channels for the electrochemical detection of glucose (Fig. 6d).85 The performance of MD could be optimized by changing the packing length of MNPs, which was difficult to achieve in other types of devices. The device also showed good reproducibility, favorable stability, and promising potential in glucose detection without the need for pretreatment of serum samples. However, due to the densely packed particles, the fluid passing through the channel is difficult to control and heat transfer inside the channel is very limited.86 Moreover, there may be substantial pressure drops when the substrate solution flows along the channel. These may negatively impact the enzyme activity.
 | ||
| Fig. 6 Examples of packed-bed microchannels for enzyme immobilization. (a) Reaction scheme for ring-opening polymerization of ε-caprolactone to polycaprolactone. (b) Schematic of the microreactor setup. (c) Photograph of a typical microreactor used in this study. CAL-b-immobilized solid beads (macroporous PMMA) were filled in the channel. These three figures were reprinted with permission from ref. 77. Copyright 2011 American Chemical Society. (d) Schematic representation of the construction and analytical procedure of glucose oxidase (GOx)-magnetic nanoparticles (MNPs) microfluidic device (MD). BR: buffer reservoir; SR: sample reservoir; DR: detection reservoir; WE: working electrode; RE: reference electrode; AE: auxiliary electrode. Reproduced from ref. 85 with permission from Elsevier Ltd. | ||
3.3 Monolithic-type channel
To overcome the drawbacks of a packed-bed-type channel, such as high pressure drops, limited heat transfer, and possible leakage at higher flow rates, a monolithic-type channel (Fig. 4c) has been developed. In such a case, the channel is filled by monolithic material with interconnected meso- or microporous structures. Such a structure exhibits higher void fractions, facilitating fluid flow. Consequently, relatively higher flow rates, lower pressure drops, and higher productivities can be achieved as compared to those obtainable from packed-bed-type channels in monoliths.1,87–89 Higher backpressure can facilitate the rapid diffusion of substrate to the immobilized enzyme. As a result, the enzyme activity is increased, which is reflected from an increase in the turnover number (kcat) and decrease in the Michaelis constant (KM).90 It also possesses the advantages of high mechanical durability and reduced diffusion path length over a wall-coated-type channel. Qiao et al. immobilized L-asparaginase (L-ASNase) in both monolithic microreactor and coated reactor (i.e., the wall-coated type) by the same immobilization method.91Fig. 7a–d show the SEM images of monolithic and coated microreactors. The monolithic microreactor was demonstrated to have lower KM than that of the coated microreactor, showing that better affinity between the substrate and enzyme can be obtained in monolithic structures as a result of the lower diffusion path length in monoliths (Fig. 7e). However, a higher maximal velocity was observed in the coated microreactor due to its relatively lower flow pressure. | ||
| Fig. 7 SEM images of monolithic and coated microreactors in capillary: (a and b) monolithic, (c and d) coated. (e) Lineweaver–Burk plots of L-ASNase immobilized on monolithic (A, ■) and coated (B, ●) enzymatic microreactors. Reproduced from ref. 91 with permission from The Royal Society of Chemistry. | ||
The monolithic materials can be organic,92–96 inorganic,97–100 or hybrid.66,101–103 This selection should be carefully made for different enzyme immobilizations and different reaction environments. Generally, organic monoliths are copolymerized from many monomers; sometimes, one of the monomers is an enzyme. They usually have good biocompatibility and pH resistance, but may be damaged by certain organic solvents. An example is the immobilization of amylase in PVA foam by mixing an amylase solution with the PVA solution before they were put in a cylindrical sample case for freeze-drying.93 The amylase-immobilized microreactor was demonstrated to successfully conduct continuous starch hydrolysis reactions over 8 days. For inorganic monoliths, silica-based monoliths are the most widely used due to their higher binding capacity, promising biocompatibility, chemical and thermal stabilities, and easy functionalization.104 However, when compared with organic polymer monoliths, the preparation of inorganic monoliths is relatively difficult.105 Therefore, monoliths used for enzyme immobilization are mostly hybrids of organic and inorganic materials. For example, Ma et al. developed an organic–inorganic-hybrid silica monolith with immobilized trypsin and demonstrated its excellent enzyme activity and long-term stability in proteome analyses.101 However, there is still the possibility that the pores are blocked, leading to nonuniform permeability along the channel. Further, the fabrication of monolithic materials is usually time-consuming and poorly reproducible. Each portion of the internal structure has its own advantages and disadvantages. All the aspects should be taken into account as much as possible when designing the structure. In particular, attention should be paid to economy, sustainability, and green chemistry for industrial applications.
4. Enzyme immobilization techniques
Since Nillson and Griffin in 1916 firstly reported that invertase retained its activity after physical adsorption onto charcoal,106 various enzyme immobilization techniques have been developed and studied. Most of these techniques can be directly used for enzyme immobilizations in microfluidic chips for biocatalysis. A combination of enzyme immobilization and microfluidic chips provides the advantages of higher stability and reusability, high enzyme-to-substrate ratio, and rapid catalytic reactions.107 Basically, the techniques of enzyme immobilization in microfluidic chips can be classified into two types: surface binding and encapsulation, as shown in Fig. 8. The inner surfaces of microfluidic reactors can offer support for enzyme immobilization when the surface binding technique is used. Moreover, microchannels with special microstructures can entrap relatively larger structures; then, the encapsulation of enzymes can be employed. For the surface binding method, it is usually subdivided into noncovalent binding and covalent binding. Noncovalent binding includes nonspecific physical adsorption, ionic bonding, His-tag/metal binding, and affinity binding.3,108 However, covalent binding involves the immobilization of enzymes on the surface via covalent forces between certain functional groups, such as amino, carboxyl, hydroxyl, or sulfhydryl groups.109 With regard to encapsulation immobilization, enzymes are confined into smaller spaces built by polymeric networks, membranes, or nanochannels. Different immobilization methods have different advantages as well as disadvantages (see Table 2). Therefore, careful consideration should be made before any immobilization strategy is adopted. Some representative examples of μ-IMERs for biocatalysis are summarized in Table 3. | ||
| Fig. 8 Different enzyme immobilization techniques. | ||
| Characteristics | Physical adsorption | Ionic binding | Affinity binding | Covalent binding | Cross-linking | Entrapment and encapsulation |
|---|---|---|---|---|---|---|
| Preparation | Simple | Simple | Moderate | Difficult | Moderate | Difficult |
| Cost | Low | Low | Moderate | High | Moderate | Moderate |
| Applicability | Wide | Wide | Wide | Selective | Selective | Wide |
| Binding force | Weak | Moderate | Moderate | Strong | Strong | Strong |
| Stability | Low | Moderate | Moderate | High | High | High |
| Enzyme leakage | Yes | Possible | Possible | No | No | Possible |
| Enzyme activity | Moderate | High | High | Low | Low | Moderate |
| Protection from microbial | No | No | No | No | Possible | Yes |
| Diffusional limitation | Low | Low | Low | Low | Moderate | High |
| Immobilization techniques | Enzyme | Platform | Biocatalysis performance | Ref. |
|---|---|---|---|---|
| Physcial immobilization | CAL-b | Macroporous PMMA microbeads packed aluminum microreactor | Faster polymerization and higher molecular mass | 77 |
| Ionic binding | Angiotensin-converting enzyme | Fused silica capillary column | High activity, stability, reusability, renewability and reduced costs | 110 |
| Sucrose phosphorylase | Nanosprings-coated Borosilicate glass | 10-fold activity enhancement, 11-fold operational stability increase, 85% conversion rate retaining after 840 reactor cycles | 70 | |
| Layer-by-layer ionic binding | Trypsin | Wall-coated PET microfluidic chip | Short digestion time and small volume of protein samples, a potential solution for low-level protein analysis | 34 |
| CAL-b | PTFE open-tubular microreactor | Production efficiency reached to 95% within 35 min, 83% initial activity retained after 144 hours usage | 35 | |
| His-tag/Ni-NTA binding | PikC hydroxylase | Agarose beads packed PDMS microfluidic reactor | High enzyme loading and conversion rate | 111 |
| Transketolase | Wall-coated PMMA microfluidic chip | The 1-step-immobilization method without the pre-amination of PMMA surface showed higher specific activity | 112 | |
| Streptavidin/biotin | ALP, GOx and HRP | Phospholipid bilayer-coated PDMS microchannels and borosilicate microcapillary tubes | The feasibility of using the microchannels to obtain kinetic data and the potential application for multistep chemical synthesis were demonstrated | 113 |
| HRP and β-galactosidase | PDMS microchip reactor packed with commercial microbeads | Similar kinetic analysis results were obtained in the microfluidic-based assays as that obtained in solution, reduced cost, reagent economy and increased throughput were observed | 79 | |
| ALP, Gox and HRP | Protein coated PDMS/glass microchannel | Photoimmobilization of multiple, well-defined enzymes were developed for both single-enzyme and multi-enzyme systems | 114 | |
| DNA directed immobilization | CAL-b and HRP | Fused silica capillaries with polymer coated | High reusability and renewability, the reaction time available for glucose oxidase could be independently and modularly varied by the distance between two enzymes | 115 |
| Covalent binding | Trypsin | Porous polymer monolithic microfluidic capillaries and chips | Very short digestion time compared to the traditional approach and great potential for broader application in various protein mapping | 96 |
| GOx | Magnetic nanoparticles packed microreactor | Low detection limit of glucose, high reproducibility and storage stability, availability of direct detection of serum samples | 85 | |
| β-Gal and GOx | Au coated PDMS microfluidic chip | 5 times of the reaction yield could be obtained if the gap distance decreased from 100 to 50 μm | 116 | |
| Encapsulation | Trypsin | PMMA microchip filled with sol–gel | Analytic time was shortened and operation stability was increased, digestion of protein with multiple cleavage sits and separation of digest fragments are applicable | 117 |
| Trypsin | Titania and alumina sol–gel based PDMS microfluidic reactors | Short digestion time and increased operation stability | 118 | |
| Lipase | Mesoporous silica coated PDMS/glass microreactor | Higher activity compared to that in batch system | 119 | |
| Cross-linking | Aminoacylase | Wall-coated PTFE microtubes | Higher stability against heat and organic solvents, applicable to various enzymes with low isoelectric points | 36 |
| Cross-linking/encapsulation | ALP and urease | PDMS microfluidic device with PEG-based hydrogel structures | Enzyme-catalyzed reactions were able to reach 90% conversion within 10 min | 120 |
4.1 Surface binding
However, nonspecific forces are generally very weak and highly dependent on environmental and surface conditions. As a result, enzymes can easily fall off from the surface, particularly in fluidic systems or high ionic and pH solutions. This would cause the contamination of reaction systems and reduction of enzyme activity. In addition, since the enzymes adsorbed on the support are randomly oriented, the activity of enzymes can also be affected by the hindering of active sites to the support due to the random orientation after adsorption. Moreover, certain other problems, such as diffusion resistance, denaturation of enzyme, and overloading, could also cause enzyme activity loss when physical adsorption is adopted.121 Therefore, a combination of other immobilization methods with the adsorption method is usually applied to overcome these shortcomings and to enhance the enzyme activity, stability, and overall efficiency.
Generally, ionic binding is stronger than nonspecific physical adsorption, which can subsequently guarantee higher enzyme stability and reusability. However, ionic binding is highly dependent on environmental pH and ionic strength. This may affect the enzyme-loading amount and pH stability of the enzymes. Therefore, the selection of suitable chemicals with an appropriate isoelectric point is the focus of this method. In biochemistry, typical positively charged functional groups are protonated amines (NH3+) and quaternary ammonium cations (NR4+). Negatively charged functional groups are usually carboxylic acid (–COO−) and sulfonic acid (–RSO3−).27 PEI is a popular polycation, which has multiple cation groups with strong anion exchange capacity for enzyme immobilization by ionic binding.35,122–125 Some other polycations have also been used for ionic binding, such as chitosan,34,72 hexadimethrine bromide (HDB),110,126 and poly(diallyldimethylammonium chloride) (PDDA).127,128 Sometimes, polyanions such as alginate,122,123 hyaluronic acid (HA),34,72 poly(Lys),36 and functionalized graphene oxide69 have also been employed to form multilayers to stabilize immobilization and to increase the enzyme-loading amount.
Certain schematic representations of enzyme immobilization by ionic binding via different polycations and polyanions are shown in Fig. 9. The reversibility of enzyme immobilization by ionic binding is also an important advantage over other immobilization methods. The support surface can be washed without damage by simply changing the ionic strength of the environment. Then, new enzymes can be immobilized onto the same support. In this way, the microfluidic chip can be reused, thereby saving labor and cost.
 | ||
| Fig. 9 Schematic representations of enzyme immobilization by ionic binding via different polycations and polyanions. (a) Adenosine deaminase (ADA) immobilization by PEI and alginates. Reproduced from ref. 122 with permission of Elsevier Ltd. (b) Trypsin immobilization by HA and chitosan. Reprinted with permission from ref. 34. Copyright 2006 American Chemical Society. (c) Trypsin immobilization by PDDA and negatively charged graphene oxide. Reproduced from ref. 69 with permission from The Royal Society of Chemistry. | ||
A) His-tag/metal binding. Metal binding needs enzymes and supports to be bonded together by coordination with metals in between them. Generally, highly active, stable, and specific immobilized enzymes can be obtained by using this method.129–131 The enzyme-loading amount by this method is also usually higher than that required in other methods.111,130 In such cases, polyhistidine linkers can be genetically tagged to the enzyme and then connected to nitrilotriacetic acid (NTA) attached on the support for enzyme immobilization. Recently, Kulsharova et al. immobilized transketolase (TK) in PMMA microfluidic devices by two methods using His-tag/nickel-NTA interactions: the 1-step-immobilization method (see Fig. 10a) and 3-step-immobilization method.112 The device fabricated by the 1-step-immobilization method exhibited higher specific activity and reusability than that fabricated using the 3-step method. The 1-step method also required fewer chemicals and lesser time duration. Moreover, it was also demonstrated that His-tag/Ni binding had higher reversibility, facilitating the reuse of the microreactor.132
 | ||
| Fig. 10 Schematic representations of enzyme immobilization by His-tag/metal binding. (a) Diagram of enzyme immobilized by His-tag/Ni-NTA binding; reproduced from ref. 112 with permission from Elsevier Ltd. (b) Chemical structures of the building blocks and schematic of the stepwise assembly process. Ethylene-glycol-based mono-adamantyl linker (b1) for minimizing nonspecific protein adsorption, biotinylated bisadamantyl linker (b2) for the first assembly step, and streptavidin (b3) as the second assembly step. Biotinylated ALP (bt-ALP, 4) is immobilized onto these streptavidin–biotin surfaces. Reproduced with permission from ref. 136. Copyright 2004 American Chemical Society. (c) Schematic diagram of the photoimmobilization process. (Top) Enzyme patches are formed on the top and bottom of a microchannel using the following procedure: (c1) Passivation of the surface with a fibrinogen monolayer is followed by (c2) biotin-4-fluorescein surface attachment. This is accomplished by photobleaching with a 488 nm laser line. (c3) Next, the binding of streptavidin-linked enzymes that can be exploited to immobilize catalysts and (c4) to monitor the reaction processes on-chip. Reproduced from ref. 114 with permission from John Wiley & Sons, Inc. | ||
However, this method also suffers from several intrinsic drawbacks. Sometimes, this method cannot be easily reproduced due to the formation of nonuniform adsorption sites and metal ion leakage.133 Therefore, it is usually combined with covalent bonding or crosslinking to get a more stable formation of adsorption sites and chelation.
B) Avidin/biotin binding. Avidin/biotin binding is one of the most popular affinity binding techniques with high affinity and specificity. The interaction is regarded as the strongest noncovalent interaction134 having the advantages of rapid fabrication and insensitivity to pH, temperature, proteolysis, and denaturing agents.83,113,135 Moreover, avidin or biotin can be easily modified by other chemicals, enabling more effective enzyme immobilization or certain other interesting functions.114,136
González-Campo et al. developed a supramolecular platform with a combination of orthogonal supramolecular interactions of the host (β-cyclodextrin)–guest (adamantane) and biotin–streptavidin interactions for the site-selective immobilization of enzymes in a microchannel (Fig. 10b).136 A microfluidic chip with a supramolecular platform was demonstrated to exhibit considerable reproducibility and reusability in enzyme reactions when calf intestine alkaline phosphatase (ALP) was used as the model enzyme. The site-selective immobilized ALP could also maintain comparable activity in other environments (free in solution or immobilization by other methods). Holden et al. also presented a study for the photoimmobilization of multiple enzymes in PDMS/glass microfluidic channels by site-specific immobilization (Fig. 10c).114 A biotin-linked dye solution was used to immobilize streptavidin-linked enzymes on select photopatterning positions. The patterning of enzymes in a sequence inside microfluidic channels could be achieved by photobleaching instead of using valves.
C) DNA-directed immobilization. DNA-directed immobilization (DDI) is based on the Watson–Crick pairing mechanism between the single-strand DNA (ssDNA) attached on enzymes and complementary DNA (cDNA) attached to the supports. The attachment of ssDNA to enzymes can be usually accomplished by covalent binding or avidin/biotin binding.115,137,138 Generally, binding by DDI is more stable and robust than other methods, thereby yielding high immobilization efficiency and site-specificity. The DDI method is superior to others methods, mainly because of its ability of precisely controlling the relative positions of different enzymes,19 which is critical to cascaded enzyme reactions.
 | ||
| Fig. 11 Covalent binding for enzyme immobilization. (a) Commonly used covalent bonds: mechanisms of Schiff chemistry and carbodiimide chemistry; reproduced from ref. 10 with permission from Elsevier Ltd. (b) Preparation of laccase-immobilized membrane on the inner wall of a PTFE microtube. (c) Parabolic velocity profile characteristic of the laminar flow inside a microtube and (d) confocal acquisition of the sectional view of a laccase-immobilized microreactor (dry state). These three figures were reproduced from ref. 139 with permission from Elsevier Ltd. | ||
Typically, only one layer of covalently immobilized enzyme can be formed on the surface of the support. Then, the loading amount can become very limited. Therefore, a crosslinker may be used to form enzyme polymerization to facilitate this increase. Lloret et al. prepared a laccase-immobilized microreactor by the formation of an enzyme–polymer membrane on the inner wall of the microtubes (Fig. 11b–d).139 This membrane was formed by the crosslinking polymerization reaction between laccase and crosslinkers (paraformaldehyde and glutaraldehyde). The microreactor with crosslinked laccase not only exhibited important biotransformation efficiency when compared with conventional bioreactors, but also exhibited excellent pH, temperature, inactivating agent, storage, and long-term stabilities.
4.2 Encapsulation
The encapsulation of an enzyme is defined as the enzyme entrapped inside a small space that allows the substrates and products to pass through but retains the enzyme. It mainly includes matrix entrapment and membrane encapsulation, as shown in Fig. 8. For matrix entrapment, the enzymes are entrapped into a matrix, which is usually formed by polymers (like alginate122,123 and PEG hydrogels120,140,141) or other inorganic materials (like titania118 and silica sol–gels97,117,142). When a semipermeable membrane, like a hollow fiber143 or microencapsulate,144 is used to encapsulate the enzymes, it is classified as membrane encapsulation. When compared with physical adsorption, the entrapment method is more stable and can immobilize a larger amount of enzyme. In addition, this entrapment does not need the chemical modification of the enzyme, which not only saves time but also avoids any conformational changes in the enzyme. However, the slower diffusion of the substrate to the enzyme in this case may severely restrict biocatalytic production. Further, there are possibilities of enzyme leakage and enzyme contamination by the matrix. Moreover, the microenvironments of the matrix are difficult to control, which may lead to a reduction in the enzyme activity and stability. However, there is a great opportunity to reduce the impact of these problems by carefully choosing the polymer materials with proper modification and by adjusting the pore size or capsule size. Moreover, capsules can imitate the multicompartment structures of cellular architectures to encapsulate enzymes in a controllable number, type, and spatial arrangement, thereby maintaining or even enhancing the overall catalytic activity.145,146Mizukami developed a microreactor containing lipases encapsulated in folded-sheet mesoporous (FSM) silicas with two different pore diameters (Fig. 12a and b).119 FSM with larger diameters (∼7 nm) (FSM7) indicated a higher enzyme-loading amount than FSM with a smaller diameter (∼4 nm) (FSM4). The two types of lipase FSM silicas were loaded in the microreactors for the hydrolysis of a triglyceride; both of them exhibited higher enzyme activity than that by the batch reaction. The results also revealed that the enzyme activity in lipase-FSM7 was slightly higher than that in lipase-FSM4. This may be attributed to the larger pore size of FSM7, which facilitates the access of the substrate to the encapsulated enzyme. Blanchette et al. entrapped the active particulate methane monooxygenase (pMMO) and associated lipids in a PEG-based hydrogel (Fig. 12c).140 The hybrid materials were then suspended between the gas and liquid reservoirs in a flow-through reactor (Fig. 12d). With this configuration, a methane/air gas mixture and NADH could be constantly introduced into the reactor while continuously removing and collecting methanol in a buffer. The native conformation and physiological activity of pMMO are completely retained by this encapsulation method. In addition, this strategy enables the reuse and continuous use of pMMO. Moreover, this method allows the facile fabrication of immobilization structures in 3D structures from the micro to millimeter scales, which guarantees the higher loading of pMMO when compared with that obtainable through surface immobilization.
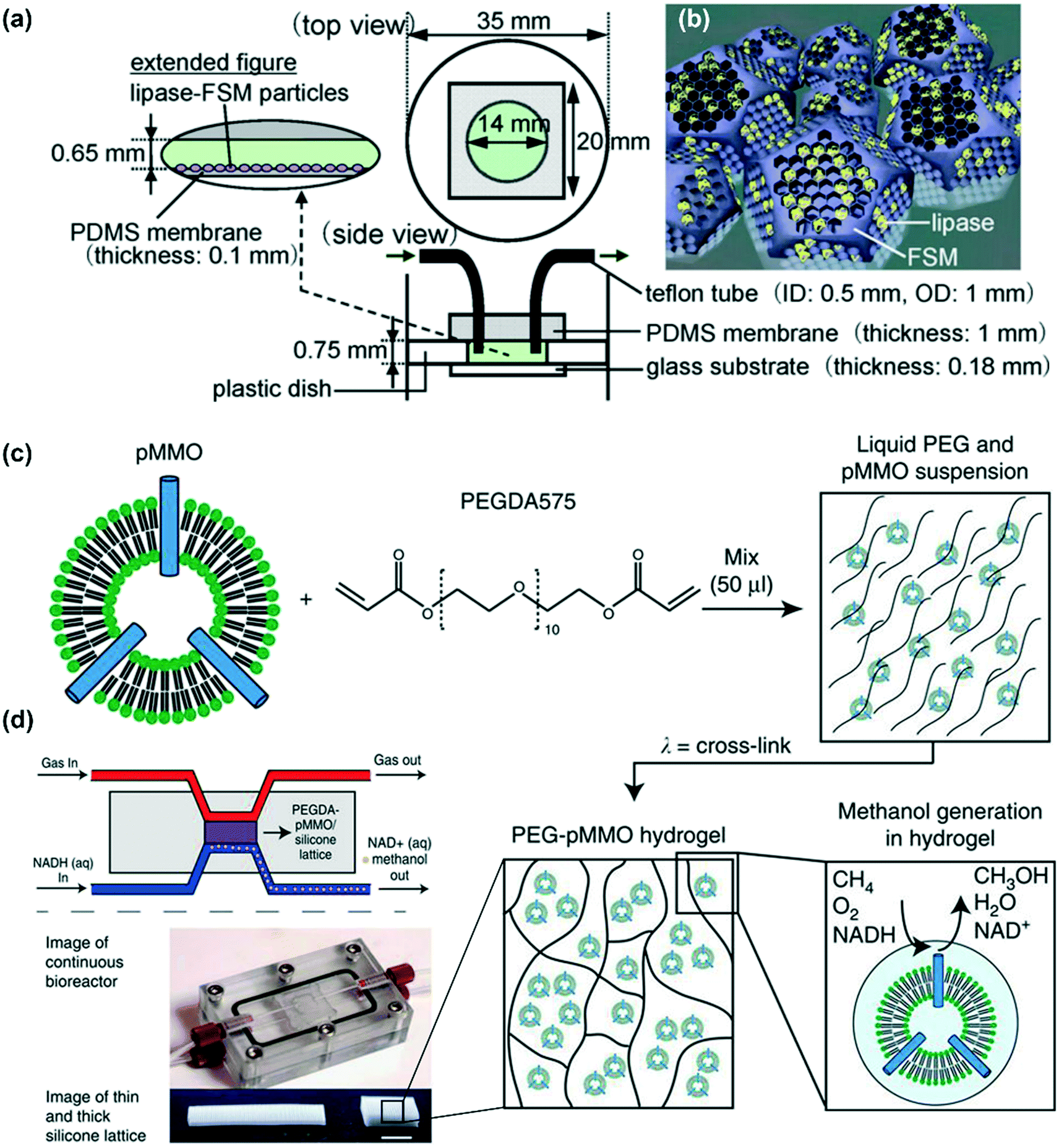 | ||
| Fig. 12 Examples of enzyme immobilization by encapsulation. (a) Schematic illustrations of the microreactor with immobilized lipase–nanoporous material (FSM) composite particles; (b) lipase molecules encapsulated in the FSM pores; these two figures were reproduced from ref. 119 with permission from Elsevier Ltd. (c) Schematic of PEG–pMMO hydrogel fabrication. Membrane-bound pMMO is mixed with PEGDA 575 and photoinitiator and exposed to ultraviolet light to crosslink the material. (d) Schematic and image of the flow-through bioreactor and two (thin and thick) silicone lattice structures used to support the PEG–pMMO hydrogel membrane (scale bar: 1 cm); these two figures were reproduced from ref. 140 with permission from Springer Nature. | ||
5. Multienzyme systems
Most natural biocatalytic reactions are catalyzed by more than one enzyme. Tremendous efforts have been devoted toward immobilizing multiple enzymes in one microfluidic reactor to artificially construct various biocatalytic reactions observed in nature.76,95,116,137,141,147–149 The methods for multienzyme immobilization are based on those involving single-enzyme immobilization. However, careful consideration should be made to maintain the activities and catalytic efficiencies of all the enzymes according to the structures and optimal environments of the different enzymes. One major issue for multienzyme immobilization is the control of their relative spatial positions in a single reactor, although it is much easier for microfluidic reactors as compared to that for batch reactors. The well-controlled spatial positions would probably promote an increase in the reaction rate and catalytic efficiency. The unwanted side reactions and accumulation of inhibitors or reactive intermediates may also be avoided.150–154Multiple enzymes can be immobilized in one pot,141,148 sequentially,115,116,137 or LBL assembly.155 Heo et al. packed GOx- and HRP-bearing microbeads into microfluidic reactors with different spatial distances, as shown in Fig. 13a.156 Reactor I packed the microbeads coimmobilizing both GOx and HRP. Reactor II had GOx-immobilized microbeads packed in front of the HRP-immobilized microbeads. In Reactor III, GOx- and HRP-immobilized microbeads were mixed and packed. It was demonstrated that better overall reaction efficiency could be obtained in Reactor I than those obtainable with the other two reactors. The increased efficiency due to the decreased enzyme distance can be attributed to the reduced diffusional loss of the intermediate product (hydrogen peroxide, H2O2) and the prevention of inhibitor accumulation. Another work presented by Wu et al. investigated the distance for multienzyme immobilization in micrometers and arrived at a similar result.116 Further, β-galactosidase and glucose oxidase were immobilized on two separate gold films patterned within a single microfluidic channel, as shown in Fig. 13b. The highest conversion efficiency of the cascaded reaction was obtained when the two enzymes were within minimum distance (50 μm). Even though the spatial positions of the enzymes are easy to control by separating the microchannels into multiple areas, they are still not very close to each other like that in the case of natural cascaded systems.157 Therefore, researchers have been paying attention to a combination of microfluidic reactors and DDI, wherein more precise spatial control can be obtained toward reaction efficiency enhancement. It has been demonstrated that when the GOx and HRP enzymes were immobilized via DNA origami tiles at a distance of 10 nm from each other, an increase of more than 15-fold in the overall cascade activity was observed as compared to that when using free enzymes, as shown in Fig. 13c.158 The reason for this activity increase was suggested to be the efficient transport of the reaction intermediate (H2O2) between the two enzymes.
 | ||
| Fig. 13 Examples of enzyme immobilization in multienzyme systems. (a) Diagrams of three different configurations of microfluidic reactors. In Reactor I, GOx and HRP were coimmobilized on a single set of beads. In Reactor II, GOx- and HRP-immobilized beads were sequentially loaded. In Reactor III, the two beads were mixed in the reactor. Reprinted with permission from ref. 156. Copyright 2014 Japan Society for Analytical Chemistry. (b) Schematic of the enzyme cascade reaction confined in a microchannel. Here, β-galactosidase and glucose oxidase were assembled on gold films within controllable distances (up). Graph of the normalized response currents of H2O2 as a function of the concentration of lactose with different gap distances in the experiment (down). Reproduced from ref. 116 with permission from the PCCP Owner Societies. (c) Rectangular DNA origami tiles with assembled GOx (yellow) and HRP (purple) pairs spaced from 10 to 65 nm (up). Enhancement of the activity of the enzyme pairs on DNA nanostructures as compared to those of the free enzyme in a solution (down). The largest enhancement factor was observed when the interenzyme distance was decreased to 10 nm, as analyzed with D-glucose, ABTS2-(2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonate)), and O2 as substrates at pH 7.2. C1 and C2 refer to the tiles without nucleic acid and free enzymes, respectively. Reprinted with permission from ref. 158. Copyright 2012 American Chemical Society. (d) Schematic representation of the microfluidic setup used for performing sequential synthesis with a three-enzyme system: invertase (INV), GOX, and HRP. Enzymes were immobilized on the surface of a polymer monolith in patterned regions within a microfluidic channel. Polyethylene glycol (PEG) was grafted onto the surface of the polymer monolith to prevent nonspecific protein adsorption. Vinyl azlactone is photopatterned onto the PEG surface, which activates the surface for protein immobilization (up). Product fluorescence intensities measured with each possible arrangement of the three enzymes are shown in the figure legend (down). The substrate solution consisted of 10 mg mL−1 sucrose, 100 μmol L−1 Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine), and 1.0% (v/v) dimethyl sulfoxide (DMSO) in 50 mmol L−1 phosphate buffer, pH 7.50; pure oxygen was bubbled through this solution for 15 min prior to use. The flow rate was 0.10 μL min−1. Reprinted with permission from ref. 95. Copyright 2007 American Chemical Society. | ||
The sequential order is also very important for the overall reaction efficiency in multienzyme systems. As shown in Fig. 13d, three enzymes (INV, GOx, and HRP) were spatially immobilized by a photopatterning method on porous polymer monoliths within microfluidic devices.95 The three-enzyme system was used to perform a sequential reaction with sucrose hydrolyzed to glucose and fructose by INV in the first step. Then, GOx would oxidize glucose to gluconolactone and H2O2, which was subsequent to the oxidization by HRP to Amplex Red. Among all the six possible arrangements of the three enzymes, the correct sequential order of the catalyst (INV–GOx–HRP) resulted in the highest resorufin fluorescence by more than three folds. This demonstrates that the correct enzyme order is critical for the reaction efficiency in multienzyme-immobilized microfluidic systems.
6. Scalability of microfluidic reactors
The application of microfluidic reactors to biocatalysis exhibits several inherent benefits, such as rapid heat transfer, precise reaction control, and capability for continuous and integrated operation. Microfluidic reactors are also advantageous because of their robust structures, capacity, and ease of scalability for mass production.17,159 These are particularly attractive toward the synthesis of petrochemicals, active pharmaceutical ingredients, or value-added materials in the industry. Generally, two methods for scaling microfluidic reactors have been widely discussed: scaling up (increasing the characteristic dimensions of the channel) and scaling out (using parallel reactor systems or stacking up multiple microreactors), as shown in Fig. 14. | ||
| Fig. 14 Illustration of the concept of scaling the microreactors both upward and outward. Scaling up means increasing the characteristic dimensions of the channel. Scaling out refers to the fact that parallel microchannels are used or multiple microreactors are stacked up. | ||
Many reviews have comprehensively discussed the microreactor design and scale-up concept.20,160–162 The principle for scaling up a single-channel reactor is presented as the relationship between its characteristic dimension and the mixing and heat transfer characteristics. Some studies also use mathematical models comprising the reaction kinetics and flow dynamics to optimize the reaction conditions and reactor designs for scaling up.163–166 With regard to scaling out, it has its own advantages over scaling up in maintaining the reactions performed in each reactors to the same intensity at any level.167,168 Generally, scaling out is practicable by designing a multiple-channel reactor with only one pump and heating apparatus.169 Uniform flow distribution as well as the same residence time in all the microchannels should be ensured when designing the manifold structures. Amador studied an analytical model based on the electrical resistance networks for two manifold structures to describe the flow in each microchannel.170 Based on this model, the design and fabrication of scale-out microreactors applied to different operation conditions are feasible.
Recently, Bajić et al. conducted a scale-up study on a two-plate miniaturized packed-bed reactor (MPBR) in which LentiKats® (lens-shaped PVA particles) encapsulated with ω-transaminase (ω-TA) were uniformly or randomly packed (as shown in Fig. 15a).144 MPBRs were used for the synthesis of acetophenone (ACP) and L-alanine (L-ALA) from (S)-α-methylbenzylamine ((S)-α-MBA) and sodium pyruvate (PYR). The productivity was evaluated by increasing the capacity of the reactor from microliter to milliliter by adjusting the length, width, and depth of the channel. Fig. 15b shows the three representative reactors by scaling up in terms of width. The results revealed that increasing the length and width would increase the productivity; however, increasing the depth would reduce it. Here, the flow distribution that encourages the accessibility of the substrates to the immobilized enzyme is the key for increasing the productivity with an increase in the reactor size. This study provided a simple but efficient guide for scaling up. However, for commercial-scale production, a combination of scaling up and scaling out may be the best option.169 In addition, for a continuous reaction, the production amount can be increased by simply increasing the operation time without changing the reaction conditions. Therefore, large-scale production can be achieved in a simple, green, and cost-effective way.
 | ||
| Fig. 15 (a) Diagram of a miniaturized packed-bed reactor (MPBR) packing LentiKats® with immobilized ω-transaminase for performing continuous enzyme process. (b) MPBRs exhibiting the scale-up situation with regard to channel width. MPBRs from up to down: ∼4 mm-wide rectangular channel, hexagonal channel (rectangular part is ∼40 mm wide) with a triangular inlet and outlet parts containing pillars, and hexagonal channel (rectangular part is ∼80 mm wide) with a triangular inlet and outlet parts containing pillars. Reproduced from ref. 144 with the permission of Elsevier Ltd. | ||
As one of the earliest applications of immobilized enzyme for use in biocatalysis in a continuous flow, Liu et al. reported the continuous production of uridine diphosphate galactose (UDP-Gal) by circulating galactose (Gal), uracil monophosphate (UMP), and polyphosphate (polyP) through a column packed with seven enzyme-immobilized agarose beads, as shown in Fig. 16a.171 The enzymes were immobilized by histidine tags on nickel agarose beads. Small-scale reactions on mini Pasteur pipette columns were initially carried out to optimize the reaction conditions. Then, a packed-bed column was scaled up to the gram scale to conform to practical biosynthesis. When compared with the solution reaction, the on-column reaction results in higher product yields in a long-time reaction (50% of the UMP converted into UDP-Gal in 48 h) when compared with the solution reactions, which can be attributed to the reusability and stability of the immobilized enzyme (Fig. 16b). This continuous synthesis of UDP-Gal can help in alleviating difficulties in the production of sugar nucleotides, which is important for the synthesis of pharmaceutically valuable oligosaccharides. Orsat et al. reported a continuous acylation process to produce monoacylated Vitamin A precursors from 1,6-diol by immobilized lipase Chirazyme L2-C2 (lipase B from Candida antarctica).172 A laboratory-scale fixed-bed reactor was initially utilized to investigate the optimal reaction conditions with >99% yield and >97% selectivity at a yield of 49 g day−1. Then, a kilogram-scale reactor was accordingly prepared at a throughput of 1.6 kg day−1 over 100 days. The production of Vitamin A precursors is environment-friendly, robust, and sustainable as a result of the recyclable chemicals. Some recent studies have also demonstrated the successful scaling up of μ-IMERs for biocatalytic synthesis.173–175
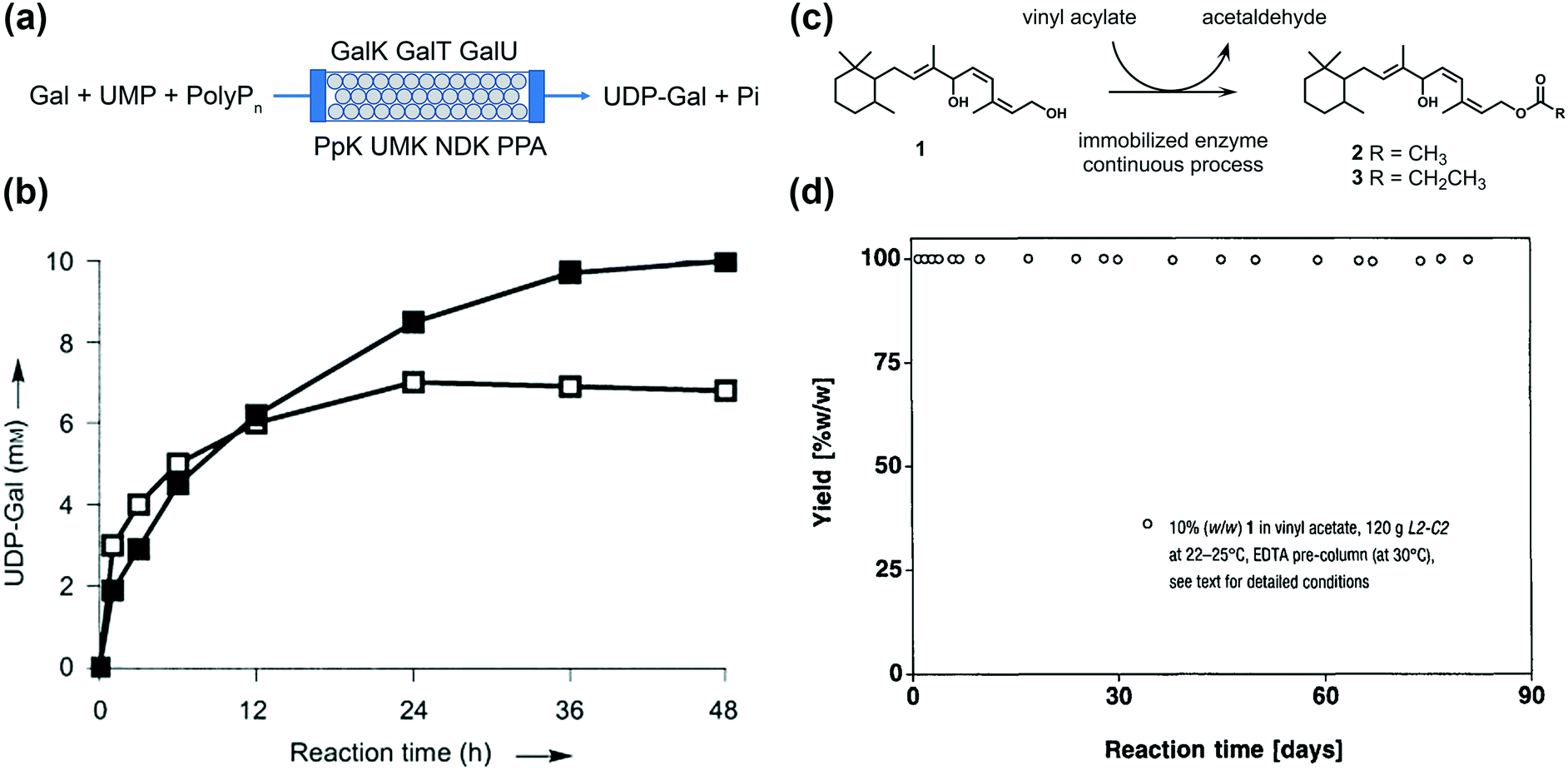 | ||
| Fig. 16 (a) Biosynthesis of UDP-Gal in a continuous packed-bed column with seven immobilized enzymes. The starting materials are Gal, UMP, and polyP. Seven enzymes were used for the catalysis: galactokinase (GalK, EC 2.7.1.6), galactose-1-phosphate uridyltransferase (GalT, EC 2.7.7.10), UDP-glucose pyrophosphorylase (GalU, EC 2.7.7.9), polyphosphate kinase (PpK, EC 2.7.4.1), uridine monophosphate kinase (UMK, EC 2.7.4.14), nucleotide diphosphate kinase (NDK, EC 2.7.4.6), and pyrophosphatase (PPA, EC 3.6.1.1). UDP-Gal and two phosphates (pi) were formed in the end. (b) Time course of UDP-Gal production. The reaction on the super-bead column (200 mL, filled squares) was performed with UMP and Gal (20 mM each), polyP (2% (w/v)), ATP, and glucose-1-phosphate (2 mM each). About 50% UMP was converted into UDP-Gal within 48 h. Reaction in the solution with the purified enzymes (50 mL, open squares) used the same reaction composition. About 35% UMP was converted in 24 h. Reproduced from ref. 171 with the permission of John Wiley and Sons. (c) Principle of the lipase Chirazyme L2-C2-catalyzed acylation of 1,6-diol (1). (d) Continuous miniplant production of 2 catalyzed by L2-C2 at 22–25 °C in the presence of 10% (w/w) of 1 in vinyl acetate. The throughput was readjusted after 74 d. Reproduced from ref. 172 with the permission of CHIMIA. | ||
7. Summary and outlook
Biocatalytic reactions play an important role in biochemistry because of their environmental-friendliness, high efficiency, and strong selectivity. However, the most popular biocatalyst—enzymes—often fail to retain the activity and stability in practical applications. Here, μ-IMERs for continuous biocatalysis have drawn on the benefits of both microfluidic reactors and enzyme immobilization techniques for effecting highly efficient, stable, reproducible, and continuous biocatalytic reactions in both laboratory and industry. In this review, different factors that affect the production efficiency, stability, and reusability in μ-IMERs have been summarized following a top-down strategy.From the macroscopic aspect, the materials used for microfluidic reactors should be temperature- and chemically stable, biocompatible with enzymes, and easy to fabricate. Among all the organic and inorganic materials (glass, silicon, PDMS, PMMA, PC, paper, etc.), PDMS is the most popular. It not only meets all the requirements mentioned above, but also has the advantages of optical transparency, promising flexibility, and easy surface modification. Once the fabrication material is chosen, the configuration of microfluidic reactors should also be effectively designed to make full use of the space for enzyme loading and substrate access. Fabrication technologies of microfluidic reactors should also be effectively chosen according to the materials and configurations.
From the microscopic aspect, the internal structures of microfluidic reactors should provide a large specific area for enzyme loading and a short diffusion path to facilitate the affinity of substrate to enzyme. There are three main types of microfluidic channels: wall-coated, packed-bed, and monolithic. Generally, wall-coated channels have the least impact of diffusion resistance on enzyme activity. However, they usually possess a lower specific area and longer diffusion path. Nevertheless, packed-bed channels have the shortcomings of immense pressure drops. It is also difficult to control fluids and heat transfer inside packed-bed channels. For monolithic channels, certain problems such as nonuniform permeability, poor reproducibility, and time-consuming fabrication may limit their applications. Overall, each of them has its own strengths and weaknesses, and it is difficult to say which one is the best. The design of internal structures should balance every aspect and take into account the used enzyme immobilization technique.
From the nanoscopic aspect, the choice of enzyme immobilization technique is a major factor that affects the overall biocatalytic efficiency of μ-IMERs. For most noncovalent binding methods, they have the advantages of simple fabrication, mild immobilization conditions, low chance of conformational change, and promising reversibility. However, the bonds are generally weak and dependent on pH or ionic strength. For performance improvement, covalent binding is usually employed to provide stronger and more stable interactions for the enzyme and support. However, enzyme conformation can be changed by using covalent binding, which—to some extent—may reduce the activity. In addition, these nonspecific methods cannot control the orientation of enzymes, which may cause the blockage of active sites by the support. This problem may be resolved by site-specific affinity binding, which can precisely control the orientation of enzymes and expose their active center to the substrate. Site-specific binding also enables the fine positioning of different enzymes within confined spaces, which is noted to play a key role in multienzyme systems. With regard to encapsulation, it offers a three-dimensional matrix for enzyme immobilization. Then, the enzyme-loading amount would be relatively larger than that required by surface binding methods. Nevertheless, there are certain drawbacks such as slow diffusion of the substrate to the enzyme, enzyme leakage, or enzyme contamination by encapsulation materials. On account of these, there is no perfect immobilization method. More than one strategy is often combined to optimize the activity, stability, and reusability of enzymes.
μ-IMERs for continuous biocatalysis can be expanded from laboratory to industry for large-scale production by scaling up and scaling out if the reaction kinetics and flow dynamics are carefully considered. However, difficulties persist for wider applications due to the numerous and complex issues involved. The challenges also include simplifying the fabrication process, increasing activity, and reducing cost. Some new nanomaterials or nanostructures with high SAV ratios have already been developed as enzyme immobilization carriers, such as molybdenum disulfide,176,177 halloysite nanotubes,178–180 metal–organic frameworks,181–183 and so on. However, there is still a lack of research in the integration of enzyme-loaded new nanomaterials with microfluidic reactors for use in biocatalysis. Further, it is necessary to increase the type of enzymes used for μ-IMERs and not limited to common model enzymes such as trypsin, lipase, GOx, or HRP. In the future, μ-IMERs with a new configuration design and new enzyme immobilization method could be applied to a variety of biocatalysis situations in both experimental research and industrial production.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by National Science Foundation of China (no. 61377068) and Research Grants Council (RGC) of Hong Kong (N_PolyU505/13, 152184/15E, 152127/17E, 152126/18E and 152219/19E). The authors would like to thank The Hong Kong Polytechnic University for the grants G-YBPR, 4-BCAL, 1-ZE14, 1-ZE27 and 1-ZVGH. The Science and Technology Development Fund, Macau SAR (File No. FDCT 0053/2019/A1, and AMSV SKL Fund) and University of Macau (MYRG2018-00114-AMSV) is also appreciated.References
- L. Tamborini, P. Fernandes, F. Paradisi and F. Molinari, Trends Biotechnol., 2017, 36, 73–88 CrossRef PubMed.
- A. Schmid, J. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed.
- X. Zhao, F. Qi, C. Yuan, W. Du and D. Liu, Renewable Sustainable Energy Rev., 2015, 44, 182–197 CrossRef CAS.
- D.-M. Liu, J. Chen and Y.-P. Shi, TrAC, Trends Anal. Chem., 2018, 102, 332–342 CrossRef CAS.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- S. Cantone, V. Ferrario, L. Corici, C. Ebert, D. Fattor, P. Spizzo and L. Gardossi, Chem. Soc. Rev., 2013, 42, 6262–6276 RSC.
- A. Liese and L. Hilterhaus, Chem. Soc. Rev., 2013, 42, 6236–6249 RSC.
- N. R. Mohamad, N. H. C. Marzuki, N. A. Buang, F. Huyop and R. A. Wahab, Biotechnol. Biotechnol. Equip., 2015, 29, 205–220 CrossRef CAS PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- K. Meller, M. Szumski and B. Buszewski, Sens. Actuators, B, 2016, 224, 84–106 Search PubMed.
- J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891–5918 RSC.
- M. P. van der Helm, P. Bracco, H. Busch, K. Szymańska, A. B. Jarzębski and U. Hanefeld, Catal. Sci. Technol., 2019, 9, 1189–1200 RSC.
- V. Hessel, S. Hardt and H. Löwe, A Multi-Faceted, Hierarchic Analysis of Chemical Micro Process Technology: Sections 1.1–1.5, Wiley Online Library, 2004 Search PubMed.
- E. Wolfgang, H. Volker and L. Holger, Microreactors: New Technology for Modern Chemistry, Wiley/VCH, Weinheim, 2000, pp. 41–84 Search PubMed.
- N. Wang, X. Zhang, Y. Wang, W. Yu and H. L. Chan, Lab Chip, 2014, 14, 1074–1082 RSC.
- Y. Liu and X. Jiang, Lab Chip, 2017, 17, 3960–3978 RSC.
- K. S. Elvira, X. C. I. Solvas and R. C. Wootton, Nat. Chem., 2013, 5, 905 CrossRef CAS.
- M. B. Plutschack, B. U. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS.
- F. Jia, B. Narasimhan and S. Mallapragada, Biotechnol. Bioeng., 2014, 111, 209–222 CrossRef CAS PubMed.
- J. Zhang, K. Wang, A. R. Teixeira, K. F. Jensen and G. Luo, Annu. Rev. Chem. Biomol. Eng., 2017, 8, 285–305 CrossRef.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2015, 20, 2–25 CrossRef.
- M. Planchestainer, M. L. Contente, J. Cassidy, F. Molinari, L. Tamborini and F. Paradisi, Green Chem., 2017, 19, 372–375 RSC.
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2018, 23, 9–18 CrossRef.
- E. Laurenti and A. dos Santos Vianna Jr, Biocatalysis, 2016, 1, 148–165 Search PubMed.
- S. R. Forrest, B. B. Elmore and J. D. Palmer, Catal. Today, 2007, 120, 30–34 CrossRef CAS.
- P. Abgrall and A. Gue, J. Micromech. Microeng., 2007, 17, R15 CrossRef.
- D. Kim and A. E. Herr, Biomicrofluidics, 2013, 7, 041501 CrossRef.
- K. F. Lei, in Microfluidics in Detection Science, 2014, pp. 1–28 Search PubMed.
- L. M. C. Ferreira, E. T. Da Costa, C. L. Do Lago and L. Angnes, Biosens. Bioelectron., 2013, 47, 539–544 CrossRef CAS.
- M. R. F. Cerqueira, D. Grasseschi, R. C. Matos and L. Angnes, Talanta, 2014, 126, 20–26 CrossRef CAS.
- M. R. F. Cerqueira, M. S. F. Santos, R. C. Matos, I. G. R. Gutz and L. Angnes, Microchem. J., 2015, 118, 231–237 CrossRef CAS.
- X. Hu, Y. Dong, Q. He, H. Chen and Z. Zhu, J. Chromatogr., B, 2015, 990, 96–103 CrossRef CAS.
- D. Ogończyk, P. Jankowski and P. Garstecki, Lab Chip, 2012, 12, 2743–2748 RSC.
- Y. Liu, H. Lu, W. Zhong, P. Song, J. Kong, P. Yang, H. H. Girault and B. Liu, Anal. Chem., 2006, 78, 801–808 CrossRef CAS.
- Y. Bi, H. Zhou, H. Jia and P. Wei, RSC Adv., 2017, 7, 12283–12291 RSC.
- T. Honda, M. Miyazaki, H. Nakamura and H. Maeda, Adv. Synth. Catal., 2006, 348, 2163–2171 CrossRef CAS.
- H. Becker and C. Gärtner, Electrophoresis, 2000, 21, 12–26 CrossRef CAS.
- H. Becker and L. E. Locascio, Talanta, 2002, 56, 267–287 CrossRef CAS.
- A. Alrifaiy, O. A. Lindahl and K. Ramser, Polymer, 2012, 4, 1349–1398 CAS.
- E. Roy, A. Pallandre, B. Zribi, M. C. Horny, F. D. Delapierre, A. Cattoni, J. Gamby and A. M. Haghiri-Gosnet, in Advances in Microfluidics-New Applications in Biology, Energy, and Materials Sciences, InTech, 2016 Search PubMed.
- J. P. McMullen and K. F. Jensen, Annu. Rev. Anal. Chem., 2010, 3, 19–42 CrossRef CAS.
- P. Lisowski and P. K. Zarzycki, Chromatographia, 2013, 76, 1201–1214 CrossRef CAS.
- F. Figueredo, P. T. Garcia, E. Cortón and W. K. Coltro, ACS Appl. Mater. Interfaces, 2015, 8, 11–15 CrossRef.
- K. Szymańska, M. Pietrowska, J. Kocurek, K. Maresz, A. Koreniuk, J. Mrowiec-Białoń, P. Widłak, E. Magner and A. Jarzębski, Chem. Eng. J., 2016, 287, 148–154 CrossRef.
- C. Hoffmann, I. P. R. Grundtvig, J. Thrane, N. Garg, K. V. Gernaey, M. Pinelo, J. M. Woodley, U. Krühne and A. E. Daugaard, Chem. Eng. J., 2018, 332, 16–23 CrossRef CAS.
- K. Nakagawa, A. Tamura and C. Chaiya, Chem. Eng. Sci., 2014, 119, 22–29 CrossRef CAS.
- D. B. Weibel, W. R. DiLuzio and G. M. Whitesides, Nat. Rev. Microbiol., 2007, 5, 209 CrossRef CAS.
- Y. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 550–575 CrossRef CAS.
- D. Qin, Y. Xia and G. M. Whitesides, Nat. Protoc., 2010, 5, 491 CrossRef CAS.
- M. Focke, D. Kosse, C. Müller, H. Reinecke, R. Zengerle and F. von Stetten, Lab Chip, 2010, 10, 1365–1386 RSC.
- R. Truckenmüller, S. Giselbrecht, C. van Blitterswijk, N. Dambrowsky, E. Gottwald, T. Mappes, A. Rolletschek, V. Saile, C. Trautmann and K.-F. Weibezahn, Lab Chip, 2008, 8, 1570–1579 RSC.
- S.-J. J. Lee and N. Sundararajan, Microfabrication for microfluidics, Artech house, 2010 Search PubMed.
- C.-W. Tsao and D. L. DeVoe, Microfluid. Nanofluid., 2009, 6, 1–16 CrossRef CAS.
- C. Iliescu, H. Taylor, M. Avram, J. Miao and S. Franssila, Biomicrofluidics, 2012, 6, 016505 CrossRef PubMed.
- C.-W. Tsao, Micromachines, 2016, 7, 225 CrossRef PubMed.
- P. Kim, K. W. Kwon, M. C. Park, S. H. Lee, S. M. Kim and K. Y. Suh, BioChip J., 2008, 2, 1–11 Search PubMed.
- E. Peris, O. Okafor, E. Kulcinskaja, R. Goodridge, S. V. Luis, E. Garcia-Verdugo, E. O'Reilly and V. Sans, Green Chem., 2017, 19, 5345–5349 RSC.
- S. Waheed, J. M. Cabot, N. P. Macdonald, T. Lewis, R. M. Guijt, B. Paull and M. C. Breadmore, Lab Chip, 2016, 16, 1993–2013 RSC.
- A. K. Au, W. Huynh, L. F. Horowitz and A. Folch, Angew. Chem., Int. Ed., 2016, 55, 3862–3881 CrossRef CAS PubMed.
- D. Pranzo, P. Larizza, D. Filippini and G. Percoco, Micromachines, 2018, 9, 374 CrossRef PubMed.
- J. S. Lee, S. H. Lee, J. H. Kim and C. B. Park, Lab Chip, 2011, 11, 2309–2311 RSC.
- A. Küchler, J. N. Bleich, B. Sebastian, P. S. Dittrich and P. Walde, ACS Appl. Mater. Interfaces, 2015, 7, 25970–25980 CrossRef PubMed.
- M. Jannat and K.-L. Yang, Langmuir, 2018, 34, 14226–14233 CrossRef CAS PubMed.
- Y. Zhu, Z. Huang, Q. Chen, Q. Wu, X. Huang, P.-K. So, L. Shao, Z. Yao, Y. Jia, Z. Li, W. Yu, Y. Yang, A. Jian, S. Sang, W. Zhang and X. Zhang, Nat. Commun., 2019, 10, 4049 CrossRef PubMed.
- Y. Lv, Z. Lin, T. Tan and F. Svec, Biotechnol. Bioeng., 2014, 111, 50–58 CrossRef CAS PubMed.
- F. Liu, Y. Piao, J. S. Choi and T. S. Seo, Biosens. Bioelectron., 2013, 50, 387–392 CrossRef CAS PubMed.
- A. Gong, C.-T. Zhu, Y. Xu, F.-Q. Wang, F.-A. Wu and J. Wang, Sci. Rep., 2017, 7, 4309 CrossRef PubMed.
- N. Singh, M. A. Ali, P. Rai, A. Sharma, B. D. Malhotra and R. John, ACS Appl. Mater. Interfaces, 2017, 9, 33576–33588 CrossRef CAS PubMed.
- Z. Yin, W. Zhao, M. Tian, Q. Zhang, L. Guo and L. Yang, Analyst, 2014, 139, 1973–1979 RSC.
- D. Valikhani, J. M. Bolivar, M. Viefhues, D. N. McIlroy, E. X. Vrouwe and B. Nidetzky, ACS Appl. Mater. Interfaces, 2017, 9, 34641–34649 CrossRef CAS PubMed.
- J. Liu, X. Jiang, R. Zhang, Y. Zhang, L. Wu, W. Lu, J. Li, Y. Li and H. Zhang, Adv. Funct. Mater., 2019, 29, 1807326 CrossRef.
- Y. Liu, W. Zhong, S. Meng, J. Kong, H. Lu, P. Yang, H. H. Girault and B. Liu, Chem. – Eur. J., 2006, 12, 6585–6591 CrossRef CAS PubMed.
- H. Bao, Q. Chen, L. Zhang and G. Chen, Analyst, 2011, 136, 5190–5196 RSC.
- Y. Bi, H. Zhou, H. Jia and P. Wei, Process Biochem., 2017, 54, 73–80 CrossRef CAS.
- G. H. Seong and R. M. Crooks, J. Am. Chem. Soc., 2002, 124, 13360–13361 CrossRef CAS PubMed.
- C. R. Boehm, P. S. Freemont and O. Ces, Lab Chip, 2013, 13, 3426–3432 RSC.
- S. Kundu, A. S. Bhangale, W. E. Wallace, K. M. Flynn, C. M. Guttman, R. A. Gross and K. L. Beers, J. Am. Chem. Soc., 2011, 133, 6006–6011 CrossRef CAS PubMed.
- J. Wang, X. Liu, X.-D. Wang, T. Dong, X.-Y. Zhao, D. Zhu, Y.-Y. Mei and G.-H. Wu, Bioresour. Technol., 2016, 220, 132–141 CrossRef CAS PubMed.
- G. H. Seong, J. Heo and R. M. Crooks, Anal. Chem., 2003, 75, 3161–3167 CrossRef CAS PubMed.
- L. C. Lasave, S. M. Borisov, J. Ehgartner and T. Mayr, RSC Adv., 2015, 5, 70808–70816 RSC.
- D. N. Kim, Y. Lee and W.-G. Koh, Sens. Actuators, B, 2009, 137, 305–312 CrossRef.
- A. Kecskemeti and A. Gaspar, Talanta, 2017, 166, 275–283 CrossRef CAS PubMed.
- T. Peschke, M. Skoupi, T. Burgahn, S. Gallus, I. Ahmed, K. S. Rabe and C. M. Niemeyer, ACS Catal., 2017, 7, 7866–7872 CrossRef CAS.
- R.-P. Liang, X.-N. Wang, C.-M. Liu, X.-Y. Meng and J.-D. Qiu, J. Chromatogr. A, 2013, 1315, 28–35 CrossRef CAS PubMed.
- J. Sheng, L. Zhang, J. Lei and H. Ju, Anal. Chim. Acta, 2012, 709, 41–46 CrossRef CAS PubMed.
- A. Karim, J. Bravo, D. Gorm, T. Conant and A. Datye, Catal. Today, 2005, 110, 86–91 CrossRef CAS.
- J. M. Bolivar, J. Wiesbauer and B. Nidetzky, Trends Biotechnol., 2011, 29, 333–342 CrossRef CAS PubMed.
- D. J. Strub, K. Szymańska, Z. Hrydziuszko, J. Bryjak and A. B. Jarzębski, React. Chem. Eng., 2019, 4, 587–594 RSC.
- M. Petro, F. Svec and J. M. Fréchet, Biotechnol. Bioeng., 1996, 49, 355–363 CrossRef CAS.
- T. R. Besanger, R. J. Hodgson, J. R. Green and J. D. Brennan, Anal. Chim. Acta, 2006, 564, 106–115 CrossRef CAS.
- J. Qiao, L. Qi, X. Mu and Y. Chen, Analyst, 2011, 136, 2077–2083 RSC.
- A. S. de León, N. Vargas-Alfredo, A. Gallardo, A. Fernández-Mayoralas, A. Bastida, A. Muñoz-Bonilla and J. Rodríguez-Hernández, ACS Appl. Mater. Interfaces, 2017, 9, 4184–4191 CrossRef PubMed.
- K. Nakagawa and Y. Goto, Chem. Eng. Process., 2015, 91, 35–42 CrossRef CAS.
- J. P. Lafleur, S. Senkbeil, J. Novotny, G. Nys, N. Bøgelund, K. D. Rand, F. Foret and J. P. Kutter, Lab Chip, 2015, 15, 2162–2172 RSC.
- T. C. Logan, D. S. Clark, T. B. Stachowiak, F. Svec and J. M. Frechet, Anal. Chem., 2007, 79, 6592–6598 CrossRef CAS PubMed.
- D. S. Peterson, T. Rohr, F. Svec and J. M. Fréchet, Anal. Chem., 2002, 74, 4081–4088 CrossRef CAS PubMed.
- O. Yesil-Celiktas, S. Cumana and I. Smirnova, Chem. Eng. J., 2013, 234, 166–172 CrossRef CAS.
- M. Alotaibi, J. Manayil, G. M. Greenway, S. J. Haswell, S. M. Kelly, A. F. Lee, W. Karen and G. Kyriakou, React. Chem. Eng., 2017, 3, 68–74 RSC.
- P. He, B. P. Burke, G. S. Clemente, N. Brown, N. Pamme and S. J. Archibald, React. Chem. Eng., 2016, 1, 361–365 RSC.
- Y. Yi, Y. Chen, M. A. Brook and J. D. Brennan, Chem. Mater., 2006, 18, 5336–5342 CrossRef CAS.
- J. Ma, Z. Liang, X. Qiao, Q. Deng, D. Tao, L. Zhang and Y. Zhang, Anal. Chem., 2008, 80, 2949–2956 CrossRef CAS PubMed.
- P. Liang, H. Bao, J. Yang, L. Zhang and G. Chen, Carbon, 2016, 97, 25–34 CrossRef CAS.
- D. Pirozzi, M. Abagnale, L. Minieri, P. Pernice and A. Aronne, Chem. Eng. J., 2016, 306, 1010–1016 CrossRef CAS.
- D. N. Tran and K. J. Balkus Jr, ACS Catal., 2011, 1, 956–968 CrossRef CAS.
- J. Ma, L. Zhang, Z. Liang, W. Zhang and Y. Zhang, J. Sep. Sci., 2007, 30, 3050–3059 CrossRef CAS PubMed.
- J. Nelson and E. G. Griffin, J. Am. Chem. Soc., 1916, 38, 1109–1115 CrossRef CAS.
- T. Honda, H. Yamaguchi and M. Miyazaki, Applied Bioengineering: Innovations and Future Directions, 2017, vol. 5 Search PubMed.
- L. Cao, in Carrier-bound Immobilized Enzymes, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, DOI:10.1002/3527607668.ch2, pp. 53–168.
- M. L. Shuler, F. Kargi and F. Kargi, Bioprocess engineering: basic concepts, Prentice Hall, Upper Saddle River, NJ, 2002 Search PubMed.
- Z.-m. Tang and J.-w. Kang, Anal. Chem., 2006, 78, 2514–2520 CrossRef CAS PubMed.
- A. Srinivasan, H. Bach, D. H. Sherman and J. S. Dordick, Biotechnol. Bioeng., 2004, 88, 528–535 CrossRef CAS PubMed.
- G. Kulsharova, N. Dimov, M. P. Marques, N. Szita and F. Baganz, New Biotechnol., 2017, 47, 31–38 CrossRef PubMed.
- H. Mao, T. Yang and P. S. Cremer, Anal. Chem., 2002, 74, 379–385 CrossRef CAS PubMed.
- M. A. Holden, S.-Y. Jung and P. S. Cremer, Anal. Chem., 2004, 76, 1838–1843 CrossRef CAS PubMed.
- T. Vong, S. Schoffelen, S. F. van Dongen, T. A. van Beek, H. Zuilhof and J. C. van Hest, Chem. Sci., 2011, 2, 1278–1285 RSC.
- Z.-Q. Wu, Z.-Q. Li, J.-Y. Li, J. Gu and X.-H. Xia, Phys. Chem. Chem. Phys., 2016, 18, 14460–14465 RSC.
- K. Sakai-Kato, M. Kato and T. Toyo'oka, Anal. Chem., 2003, 75, 388–393 CrossRef CAS PubMed.
- H. Wu, Y. Tian, B. Liu, H. Lu, X. Wang, J. Zhai, H. Jin, P. Yang, Y. Xu and H. Wang, J. Proteome Res., 2004, 3, 1201–1209 CrossRef CAS PubMed.
- S.-i. Matsuura, R. Ishii, T. Itoh, S. Hamakawa, T. Tsunoda, T. Hanaoka and F. Mizukami, Chem. Eng. J., 2011, 167, 744–749 CrossRef CAS.
- W.-G. Koh and M. Pishko, Sens. Actuators, B, 2005, 106, 335–342 CrossRef CAS.
- A. A. Homaei, R. Sariri, F. Vianello and R. Stevanato, J. Chem. Biol., 2013, 6, 185–205 CrossRef PubMed.
- X. Ji, F. Ye, P. Lin and S. Zhao, Talanta, 2010, 82, 1170–1174 CrossRef CAS PubMed.
- W. Min, W. Wang, J. Chen, A. Wang and Z. Hu, Anal. Bioanal. Chem., 2012, 404, 2397–2405 CrossRef CAS PubMed.
- J. J. Virgen-Ortíz, J. C. dos Santos, Á. Berenguer-Murcia, O. Barbosa, R. C. Rodrigues and R. Fernandez-Lafuente, J. Mater. Chem. B, 2017, 5, 7461–7490 RSC.
- S. Zhao, X. Ji, P. Lin and Y.-M. Liu, Anal. Biochem., 2011, 411, 88–93 CrossRef CAS PubMed.
- T.-F. Jiang, T.-T. Liang, Y.-H. Wang, W.-H. Zhang and Z.-H. Lv, J. Pharm. Biomed. Anal., 2013, 84, 36–40 CrossRef CAS PubMed.
- M. A. Camara, M. Tian, L. Guo and L. Yang, J. Chromatogr., B, 2015, 990, 174–180 CrossRef CAS PubMed.
- Z. Tang, T. Wang and J. Kang, Electrophoresis, 2007, 28, 2981–2987 CrossRef CAS PubMed.
- F. Wang, C. Guo, H.-Z. Liu and C.-Z. Liu, J. Mol. Catal. B: Enzym., 2007, 48, 1–7 CrossRef CAS.
- D. Kirstein, Adv. Mol. Cell Biol., 1996, 15, 247–256 Search PubMed.
- Q.-H. Shi, Y. Tian, X.-Y. Dong, S. Bai and Y. Sun, Biochem. Eng. J., 2003, 16, 317–322 CrossRef CAS.
- M. Miyazaki, J. Kaneno, R. Kohama, M. Uehara, K. Kanno, M. Fujii, H. Shimizu and H. Maeda, Chem. Eng. J., 2004, 101, 277–284 CrossRef CAS.
- B. Brena, P. González-Pombo and F. Batista-Viera, Immobilization of Enzymes and Cells: Third Edition, 2013, pp. 15–31 Search PubMed.
- P. C. Weber, D. Ohlendorf, J. Wendoloski and F. Salemme, Science, 1989, 243, 85–88 CrossRef CAS PubMed.
- F. Rusmini, Z. Zhong and J. Feijen, Biomacromolecules, 2007, 8, 1775–1789 CrossRef CAS PubMed.
- A. González-Campo, B. Eker, H. J. Gardeniers, J. Huskens and P. Jonkheijm, Small, 2012, 8, 3531–3537 CrossRef PubMed.
- H. Schröder, L. Hoffmann, J. Müller, P. Alhorn, M. Fleger, A. Neyer and C. M. Niemeyer, Small, 2009, 5, 1547–1552 CrossRef PubMed.
- D. Liu, R. K. Perdue, L. Sun and R. M. Crooks, Langmuir, 2004, 20, 5905–5910 CrossRef CAS PubMed.
- L. Lloret, G. Eibes, M. Moreira, G. Feijoo, J. Lema and M. Miyazaki, Chem. Eng. J., 2013, 223, 497–506 CrossRef CAS.
- C. D. Blanchette, J. M. Knipe, J. K. Stolaroff, J. R. DeOtte, J. S. Oakdale, A. Maiti, J. M. Lenhardt, S. Sirajuddin, A. C. Rosenzweig and S. E. Baker, Nat. Commun., 2016, 7, 11900 CrossRef CAS PubMed.
- D. Simon, F. Obst, S. Haefner, T. Heroldt, M. Peiter, F. Simon, A. Richter, B. Voit and D. Appelhans, React. Chem. Eng., 2019, 4, 67–77 RSC.
- S. Cumana, J. Simons, A. Liese, L. Hilterhaus and I. Smirnova, J. Mol. Catal. B: Enzym., 2013, 85, 220–228 CrossRef.
- G. Ranieri, R. Mazzei, Z. Wu, K. Li and L. Giorno, Molecules, 2016, 21, 345 CrossRef PubMed.
- M. Bajić, I. Plazl, R. Stloukal and P. Žnidaršič-Plazl, Process Biochem., 2017, 52, 63–72 CrossRef.
- H. Tan, S. Guo, N.-D. Dinh, R. Luo, L. Jin and C.-H. Chen, Nat. Commun., 2017, 8, 663 CrossRef PubMed.
- H. Wang, Z. Zhao, Y. Liu, C. Shao, F. Bian and Y. Zhao, Sci. Adv., 2018, 4, eaat2816 CrossRef PubMed.
- S. Fornera, P. Kuhn, D. Lombardi, A. D. Schlüter, P. S. Dittrich and P. Walde, ChemPlusChem, 2012, 77, 98–101 CrossRef CAS.
- F. Costantini, R. Tiggelaar, S. Sennato, F. Mura, S. Schlautmann, F. Bordi, H. Gardeniers and C. Manetti, Analyst, 2013, 138, 5019–5024 RSC.
- T. Peschke, P. Bitterwolf, S. Gallus, Y. Hu, C. Oelschlaeger, N. Willenbacher, K. S. Rabe and C. M. Niemeyer, Angew. Chem., 2018, 130, 17274–17278 CrossRef.
- R. J. Conrado, J. D. Varner and M. P. DeLisa, Curr. Opin. Biotechnol., 2008, 19, 492–499 CrossRef CAS PubMed.
- Y.-H. P. Zhang, Biotechnol. Adv., 2011, 29, 715–725 CrossRef CAS PubMed.
- A. Küchler, M. Yoshimoto, S. Luginbühl, F. Mavelli and P. Walde, Nat. Nanotechnol., 2016, 11, 409–420 CrossRef PubMed.
- T. Vong, S. Schoffelen, S. F. M. van Dongen, T. A. van Beek, H. Zuilhof and J. C. M. van Hest, Chem. Sci., 2011, 2, 1278–1285 RSC.
- J. Grant, J. A. Modica, J. Roll, P. Perkovich and M. Mrksich, Small, 2018, 14, 1800923 CrossRef PubMed.
- G. Palazzo, G. Colafemmina, C. G. Iudice and A. Mallardi, Sens. Actuators, B, 2014, 202, 217–223 CrossRef CAS.
- J. Heo, Anal. Sci., 2014, 30, 991–997 CrossRef CAS PubMed.
- S. Schoffelen and J. C. van Hest, Curr. Opin. Struct. Biol., 2013, 23, 613–621 CrossRef CAS PubMed.
- J. Fu, M. Liu, Y. Liu, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2012, 134, 5516–5519 CrossRef CAS PubMed.
- V. Hessel, C. Knobloch and H. Lowe, Recent Pat. Chem. Eng., 2008, 1, 1–16 CrossRef CAS.
- N. Kockmann, M. Gottsponer, B. Zimmermann and D. M. Roberge, Chem. – Eur. J., 2008, 14, 7470–7477 CrossRef CAS PubMed.
- N. Kockmann, M. Gottsponer and D. M. Roberge, Chem. Eng. J., 2011, 167, 718–726 CrossRef CAS.
- N. Kockmann and D. M. Roberge, Chem. Eng. Process., 2011, 50, 1017–1026 CrossRef CAS.
- P. Žnidaršič-Plazl, Biotechnol. J., 2019, 14, 1800580 CrossRef PubMed.
- N. Miložič, M. Lubej, M. Lakner, P. Žnidaršič-Plazl and I. Plazl, Chem. Eng. J., 2017, 313, 374–381 CrossRef.
- F. Strniša, M. Bajić, P. Panjan, I. Plazl, A. M. Sesay and P. Žnidaršič-Plazl, Chem. Eng. J., 2018, 350, 541–550 CrossRef.
- A. Šalić, P. Faletar and B. Zelić, Biochem. Eng. J., 2013, 77, 88–96 CrossRef.
- J.-i. Yoshida, Flash chemistry: Fast organic synthesis in microsystems, John Wiley & Sons, 2008 Search PubMed.
- S. J. Haswell and P. Watts, Green Chem., 2003, 5, 240–249 RSC.
- S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC.
- C. Amador, A. Gavriilidis and P. Angeli, Chem. Eng. J., 2004, 101, 379–390 CrossRef CAS.
- Z. Liu, J. Zhang, X. Chen and P. G. Wang, ChemBioChem, 2002, 3, 348–355 CrossRef CAS PubMed.
- B. Orsat, B. Wirz and S. Bischof, Chimia, 1999, 53, 579–584 CAS.
- G. Gasparini, I. Archer, E. Jones and R. Ashe, Org. Process Res. Dev., 2012, 16, 1013–1016 CrossRef CAS.
- U. Novak, D. Lavric and P. Žnidaršiè-Plazl, J. Flow Chem., 2016, 6, 33–38 CrossRef CAS.
- S. Pithani, S. Karlsson, H. Emtenäs and C. T. Öberg, Org. Process Res. Dev., 2019, 23, 1926–1931 CrossRef CAS.
- R. Das, H. Mishra, A. Srivastava and A. M. Kayastha, Chem. Eng. J., 2017, 328, 215–227 CrossRef CAS.
- H.-L. Shuai, K.-J. Huang, Y.-X. Chen, L.-X. Fang and M.-P. Jia, Biosens. Bioelectron., 2017, 89, 989–997 CrossRef CAS PubMed.
- J. Sun, R. Yendluri, K. Liu, Y. Guo, Y. Lvov and X. Yan, Phys. Chem. Chem. Phys., 2017, 19, 562–567 RSC.
- A. A. Kadam, J. Jang and D. S. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 15492–15501 CrossRef CAS PubMed.
- M. Z. Anwar, D. J. Kim, A. Kumar, S. K. Patel, S. Otari, P. Mardina, J.-H. Jeong, J.-H. Sohn, J. H. Kim and J. T. Park, Sci. Rep., 2017, 7, 15333 CrossRef PubMed.
- X. Lian, Y. Fang, E. Joseph, Q. Wang, J. Li, S. Banerjee, C. Lollar, X. Wang and H.-C. Zhou, Chem. Soc. Rev., 2017, 46, 3386–3401 RSC.
- P. Li, J. A. Modica, A. J. Howarth, E. Vargas, P. Z. Moghadam, R. Q. Snurr, M. Mrksich, J. T. Hupp and O. K. Farha, Chem, 2016, 1, 154–169 CAS.
- Z. Li, H. Xia, S. Li, J. Pang, W. Zhu and Y. Jiang, Nanoscale, 2017, 9, 15298–15302 RSC.
| This journal is © The Royal Society of Chemistry 2020 |