
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-pressure synthesis of ε-FeOOH from β-FeOOH and its application to the water oxidation catalyst†
Kazuhiko Mukai *a,
Tomiko M. Suzukia,
Takeshi Uyamaa,
Takamasa Nonakaa,
Takeshi Morikawaa and
Ikuya Yamadab
*a,
Tomiko M. Suzukia,
Takeshi Uyamaa,
Takamasa Nonakaa,
Takeshi Morikawaa and
Ikuya Yamadab
aToyota Central Research & Development Laboratories, Yokomichi 41-1, Nagakute, Aichi 480-1192, Japan. E-mail: e1089@mosk.tytlabs.co.jp; Fax: +81-561-63-6119; Tel: +81-561-71-7698
bDepartment of Materials Science, Graduate School of Engineering, Osaka Prefecture University, 1-2 Gakuen, Sakai, Osaka 599-8570, Japan
First published on 18th December 2020
Abstract
Research on materials under extreme conditions such as high pressures provides new insights into the evolution and dynamics of the earth and space sciences, but recently, this research has focused on applications as functional materials. In this contribution, we examined high-pressure/high-temperature phases of β-FeO1−x(OH)1+xClx with x = 0.12 (β-FeOOH) and their catalytic activities of water oxidation, i.e., oxygen evolution reaction (OER). Under pressures above 6 GPa and temperatures of 100–700 °C, β-FeOOH transformed into ε-FeOOH, as in the case of α-FeOOH. However, the established pressure–temperature phase diagram of β-FeOOH differs from that of α-FeOOH, probably owing to its open framework structure and partial occupation of Cl− ions. The OER activities of ε-FeOOH strongly depended on the FeOOH sources, synthesis conditions, and composite electrodes. Nevertheless, one of the ε-FeOOH samples exhibited a low OER overpotential compared with α-FeOOH and its parent β-FeOOH, which are widely used as OER catalysts. Hence, ε-FeOOH is a potential candidate as a next-generation earth-abundant OER catalyst.
1 Introduction
One of the polymorphs of FeOOH, namely, β-FeOOH (akaganeite), has attracted interest in highly diverse fields. In the 20th century, it was examined in the context of the atmospheric rusting of iron and steel,1,2 whereas it is currently being investigated as a catalyst for water oxidation (oxygen evolution reaction; OER)3–7 or as a negative electrode for Li- and Na-ion batteries.8–11 Beside applications to functional materials, β-FeOOH has been received considerable attention in the fields of earth and space sciences and natural history because it was also discovered in the Gusev crater and the Meridiani Planum on Mars12,13 and in the cannon shot of the Mary Rose, King Henry VIII's shipwrecked flagship.14β-FeOOH was generally synthesized by hydrolyzing FeCl3 solutions at 60–100 °C,1,15–20 before it was discovered as a natural product in the Akagane metal mine, Japan.21 Fig. 1a shows the crystal structure of β-FeOOH, in which FeO6 octahedra form [2 × 2] tunnels via double corner-linked bands.22–26 A certain number of guest Cl− ions are necessary to maintain the [2 × 2] tunnels, leading to the specific chemical formula FeO1−x(OH)1+xClx.1,2,16 Its structure was initially proposed to have tetragonal symmetry with the I4/m space group,22–24 but recent X-ray diffraction (XRD) measurements revealed monoclinic symmetry with the I2/m space group.25,26 The monoclinic distortion is extremely small, as understood by its lattice parameters: am = 10.5536(7) Å, bm = 3.03449(8) Å, cm = 10.5740(4) Å, and β = 90.086(5)°.26 β-FeOOH is isostructural to α-MnO2, which contains Na+ (manjiroite), K+ (cryptomelane), Ba2+ (hollandite), or Pb2+ (coronadite) ions as the guest in the [2 × 2] tunnels.27 A series of α-MnO2 has also been employed as the catalyst for the OER28–30 and as the electrode for nonaqueous or aqueous secondary batteries.31,32
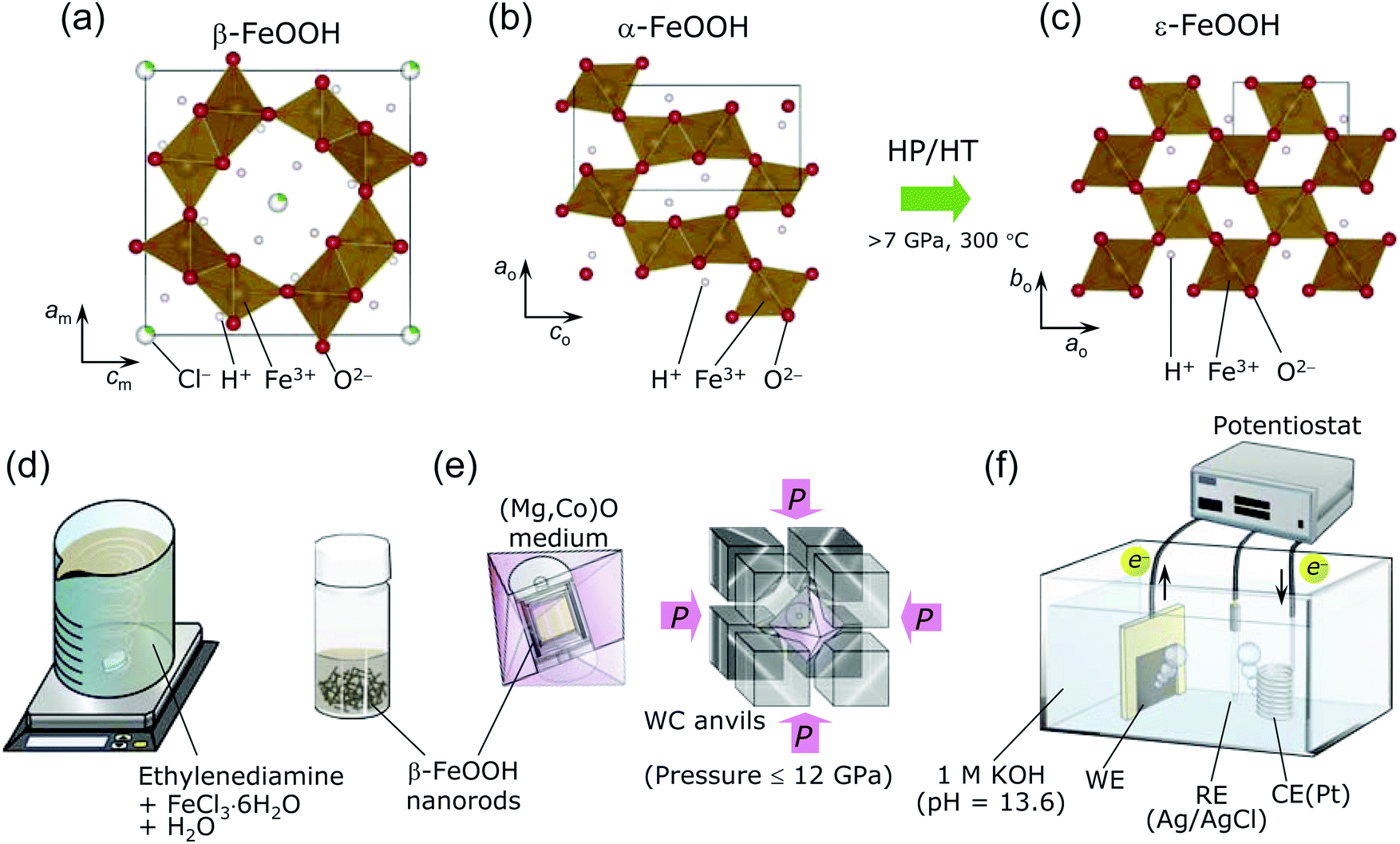 | ||
| Fig. 1 Crystal structure of (a) β-FeOOH (I2/m) with monoclinic symmetry (b) α-FeOOH (Pnma) with orthorhombic symmetry, and (c) ε-FeOOH (Pmn21) with orthorhombic symmetry. The square represents the unit cell. The Cl− ions partially occupy the β-FeOOH lattice to maintain the [2 × 2] tunnels. α-FeOOH transforms into ε-FeOOH under HP/HT conditions at above 7 GPa and 300 °C. Schematics of the procedures for (d) synthesizing β-FeOOH, (e) the HP/HT treatment, and (f) evaluating the OER activity. | ||
Many studies have been performed on β-FeOOH, but mostly under ambient pressure. However, a high-pressure/high-temperature (HP/HT) study could provide insights into its fundamental structural and electronic properties to gain an in-depth understanding of the evolution and dynamics of earth and solar system. Indeed, in the case of α-FeOOH, a pressure-induced spin state transition (t32ge2g → t52g) and hydrogen bond symmetrization occur at pressures exceeding 40 GPa.33–38 Moreover, α-FeOOH transforms into ε-FeOOH under HP/HT conditions at above 7 GPa and 300 °C.39–41 Fig. 1b and c show the crystal structures of α-FeOOH47 and ε-FeOOH,39–41 respectively, wherein the latter adopts a distorted rutile structure with the Pmn21 space group, forming [1 × 1] tunnels. Among the three FeOOH polymorphs, β-FeOOH is significantly less compressible as a function of pressure (at room temperature), owing to its high bulk modulus of 284(1) GPa.42 The HP/HT phase of β-FeOOH is currently unknown, although Xu et al.43 reported that ε-FeOOH is a HP phase of β-FeOOH without any experimental evidence.
Regarding the OER catalyst, materials synthesized from β-FeOOH under HP/HT conditions could offer a wide range of material options to enhance OER performance, because current studies on the FeOOH polymorphs focus on fabricating composite electrodes or heterodoping.3–7,44–46 Furthermore, earth-abundant catalysts such as Fe-based compounds are required to realize the global scalability within the framework of sustainable energy technologies. Note that the OER activities of the HP/HT phases of β-FeOOH and ε-FeOOH have not been reported thus far, including theoretical calculations on FeOOH polymorphs.48,49 The moderate strength of metal bonds (Fe–O bonds in this case) is generally accepted to play a crucial role in the OER performance.7 Furthermore, the O–H⋯O configuration is highly asymmetric under ambient pressure; for instance, the O–H and H⋯O lengths are 0.88 and 1.94 Å, respectively, for α-FeOOH47 and 0.86–0.98 and 2.10–2.49 Å, respectively, for β-FeOOH.26 Although β-FeOOH exhibited the maximum bulk modulus,42 the strength of the Fe–O bond and the asymmetric O–H⋯O configuration should be altered under HP/HT conditions and should significantly affect the OER performance of β-FeOOH. In the case of the quadruple perovskite oxide CaCu3Fe4O12, which was synthesized under HP/HT conditions, the OER performance was enhanced when the interatomic distance between the nearest neighboring OH adsorbates was ca. 2.6 Å.50
In this contribution, we revealed HP/HT phases of β-FeOOH under pressures of 2–12 GPa and temperatures below 800 °C, as well as the OER activity of the obtained materials. Several methods such as synchrotron XRD measurements, X-ray absorption spectroscopy (XAS), and 57Fe Mössbauer spectroscopy were employed to clarify their structural and electronic properties. Consequently, β-FeOOH was found to transform into ε-FeOOH along a different pressure–temperature phase diagram from that of α-FeOOH. In addition, we found that the ε-FeOOH sample prepared at 8 GPa and 400 °C exhibited a lower OER overpotential than those of the parent β-FeOOH and α-FeOOH.
2 Experimental
2.1 Preparation of β-FeOOH
β-FeOOH is usually prepared by aging solutions at 60–100 °C for several days, offering 100–500 nm-size particles.1,15,18,20 We, however, used a recently developed method, namely, a combination of spontaneous peptization and Ostwald ripening, to obtain β-FeOOH colloidal solution at room temperature (ca. 25 °C).3–5 This method provides highly crystalline and uniform β-FeOOH nanorods with diameters of 3 nm and lengths of 15 nm,3–5 owing to the typical advantages of the Ostwald ripening method.51 As shown in Fig. 1d, a colloidal solution was prepared by mixing 500 mL of an aqueous solution consisting of 0.1 M FeCl3·6H2O and ethylenediamine (CH2)2(NH2)2 (FUJIFILM Wako Pure Chemical). The pH of this mixture was maintained at 2.0–2.4. Then the solution was continuously stirred for 30 min and finally aged overnight at ca. 25 °C.A sample of the β-FeOOH nanorods was removed from the colloidal solution before the HP/HT treatment. The remaining colloidal solution was dried in a Petri dish at ca. 25 °C and rinsed with a 0.1 M KOH (FUJIFILM Wako Pure Chemical) solution. This powder was filtered with a cellulose acetate membrane with a pore size of 0.2 μm (Toyo Roshi Kaisha) and washed three times with distilled water. The powder was finally dried at 45 °C in vacuum for 8 h. The amount of Cl− ions, x in FeO1−x(OH)1+xClx, was examined with a combination of inductively coupled plasma-atomic emission spectroscopy (ICP-AES, PS3520VDDII, Hitachi High-Technologies), and ion chromatography (IC, Dionex ICS-1500, Thermo Fisher Scientific). The ICP-AES and IC methods revealed the atomic ratios of Fe and Cl in the sample, respectively. We recorded three independent measurements to confirm the reproducibility of the x value.
2.2 HP/HT synthesis
The pristine β-FeOOH sample was heated below 800 °C and under pressures of 2–12 GPa using Walker-type equipment at Osaka Prefecture University.50,52–54 Approximately 40 mg of the sample was sealed into a Pt capsule, and then packed into an octahedral (Mg,Co)O pressure medium with one side length of 14 mm (Mino Ceramics), together with a BN sleeve (Denka) and a graphite heater (Mechanical Carbon Industry). The (Mg,Co)O octahedron was placed at the corners of eight WC anvils (TF06, Fuji Die) and compressed at ca. 25 °C and 2, 4, 6, 8, 10, or 12 GPa (see Fig. 1e). After reaching the desired pressure, each sample was heated at 100, 150, 200, 300, 400, 500, 600, 700, or 800 °C for 30 min. The time approaching each target temperature was 15 min. The WC anvils were quenched to ca. 25 °C and then slowly decompressed to ambient pressure. Moreover, we synthesized two ε-FeOOH samples using an α-FeOOH source (denoted as pristine α-FeOOH; Koujyundo Chemical Laboratory) to compare the OER activities of α- and β-FeOOH sources. The applied pressure was 10 GPa, and the heating temperature was 400 or 500 °C. Table 1 summarizes the conditions for HP/HT synthesis, such as FeOOH source, pressure, and temperature. Hereafter, the HP/HT-treated samples are represented by their synthesis conditions in the format “sample XX-YY”, where XX and YY correspond to the applied pressure and heating temperature, respectively. For example, sample 2-200 was treated with a pressure 2 GPa and 200 °C. The two samples synthesized with the α-FeOOH source are labeled with the prefix α, e.g., α-10-400 and α-10-500.| Sample no. | Sample notation | FeOOH source | Pressure/GPa | Temperature/°C | Main phase | Ej=1 mA cm−2a/V vs. RHE |
|---|---|---|---|---|---|---|
| a The potential vs. reversible hydrogen electrode (RHE) at which the anodic current density (j) was 1 mA cm−2. | ||||||
| 1 | Pristine β-FeOOH | β | — | — | β-FeOOH | 1.62 |
| 2 | 2-200 | 2 | 200 | α-Fe2O3 | ||
| 3 | 4-200 | 4 | 200 | α-FeOOH | ||
| 4 | 4-300 | 4 | 300 | α-Fe2O3 | ||
| 5 | 4-400 | 4 | 400 | α-Fe2O3 | ||
| 6 | 6-100 | 6 | 100 | ε-FeOOH | ||
| 7 | 6-200 | 6 | 200 | ε-FeOOH | ||
| 8 | 6-300 | 6 | 300 | α-FeOOH | ||
| 9 | 6-400 | 6 | 400 | α-Fe2O3 | ||
| 10 | 8-100 | 8 | 100 | β-FeOOH | ||
| 11 | 8-150 | 8 | 150 | ε-FeOOH | ||
| 12 | 8-200 | 8 | 200 | ε-FeOOH | 1.76 | |
| 13 | 8-300 | 8 | 300 | ε-FeOOH | ||
| 14 | 8-400 | 8 | 400 | ε-FeOOH | 1.56 | |
| 15 | 8-500 | 8 | 500 | α-Fe2O3 | ||
| 16 | 8-600 | 8 | 600 | α-Fe2O3 | ||
| 17 | 8-800 | 8 | 800 | Fe3O4 | ||
| 18 | 10-100 | 10 | 100 | β-FeOOH | ||
| 19 | 10-200 | 10 | 200 | ε-FeOOH | ||
| 20 | 10-300 | 10 | 300 | ε-FeOOH | ||
| 21 | 10-500 | 10 | 500 | ε-FeOOH | ||
| 22 | 10-600 | 10 | 600 | ε-FeOOH | ||
| 23 | 12-100 | 12 | 100 | β-FeOOH | ||
| 24 | 12-200 | 12 | 200 | ε-FeOOH | ||
| 25 | 12-300 | 12 | 300 | ε-FeOOH | ||
| 26 | 12-400 | 12 | 400 | ε-FeOOH | ||
| 27 | 12-500 | 12 | 500 | ε-FeOOH | 1.62 | |
| 28 | 12-600 | 12 | 600 | ε-FeOOH | ||
| 29 | 12-700 | 12 | 700 | ε-FeOOH | ||
| 30 | 12-800 | 12 | 800 | α-Fe2O3 | ||
| 31 | Pristine α-FeOOH | α | — | — | α-FeOOH | 1.82 |
| 32 | α-10-400 | 10 | 400 | ε-FeOOH | 1.64 | |
| 33 | α-10-500 | 10 | 500 | ε-FeOOH | 1.61 | |
2.3 Characterization
The crystal structures were examined with powder XRD using Fe-Kα radiation (D8 ADVANCE, Bruker AXS) and synchrotron radiation sources. The synchrotron XRD patterns were measured at the BL5S2 beamline at the Aichi Synchrotron Radiation Center using a two-dimensional (2D) detector, PILATUS 100K (Dectris). Each sample was packed into a borosilicate glass capillary with a diameter of 0.3 mm (WJM-Glas) and then exposed to X-rays for 20 min to acquire XRD data over a wide 2θ range.55 The wavelength of the X-rays was determined to be 0.799436(2) Å using standard silicon powder (NIST 640d). Rietveld analyses were performed using RIETAN-FP software,56 and crystal structures were drawn with the VESTA software.57The local structures and electronic states of pristine β-FeOOH and sample 8-400 were investigated by XAS at the Toyota beamline (BL33XU) at SPring-8. XAS was also performed on Fe metal (thickness = 10 μm, Nilaco) and α-Fe2O3 (FUJIFILM Wako Pure Chemical) as a reference. Approximately 3 mg of α-Fe2O3 and BN powder (FUJIFILM Wako Pure Chemical) were mixed with a mortar and pestle and then pressed into a pellet with a diameter of 10 mm and a thickness of ca. 0.2 mm. XAS data were recorded at ca. 25 °C in transmission mode. Fe K-edge X-ray absorption near-edge structure (XANES) and the Fourier transform of the k3-weighted Fe K-edge extended X-ray absorption fine structure (EXAFS) spectra were analyzed using ATHENA and ARTHEMIS software.58 The experimental setup for XAS and spectral analyses are described in further detail elsewhere.4,59
We performed Mössbauer spectroscopy on pristine β-FeOOH and samples 8-400 and α-10-400 to examine the valence state and local environment of their respective Fe ions. Pristine β-FeOOH and sample 8-400 were measured (without a magnetic field) at ca. 25 °C and liquid-nitrogen temperature (ca. −196 °C), whereas sample α-10-400 was examined only at ca. 25 °C. The radiation source was a 57Co/Rh matrix with a radioactivity of 370 MBq for pristine β-FeOOH and sample 8-400, while 1.85 GBq for sample α-10-400. A multi-channel-analyzer with 512 channels (CMCA-550, Wissenschaftliche Elektronik, or MCS-plus, Ortec) was employed to acquire spectral data. One channel was determined to be 0.055598 mm s−1 for pristine β-FeOOH and sample 8-400, while 0.0791871 mm s−1 for sample α-10-400, by calibrating the Doppler velocity with Fe foil. Spectral analyses were conducted using MossWinn 4.0i software.60
Thermogravimetric (TG; Thermo plus EVO2, TG8120, Rigaku) analyses were conducted for pristine β-FeOOH and samples 8-100 through 8-500, to clarify changes in x upon heating and the thermal stabilities of β-FeOOH and ε-FeOOH. TG curves were recorded with a heating rate of 20 °C min−1 up to 1000 °C under flowing air (100 mL min−1). After the TG analysis, XRD measurements were performed using an Fe-Kα radiation source (D8 ADVANCE, Bruker AXS).
Particles sizes and morphologies were examined in two different types of scanning electron microscope (SEM), that is, a conventional system with a tungsten filament (S-3600 N, Hitachi High-Technologies) and a field-emission (FE) system using an emitter tip (SU8020, Hitachi High-Technologies). For the conventional system, 1 mg of sample, which was attached to carbon tape, was coated with electrically conductive Au particles (IB-3, Eiko). Meanwhile, for the FE system, samples were coated with Os particles (HPC-1S, Vacuum Device). The accelerating voltages were 15 and 1 kV for the conventional and FE-SEM, respectively. Particle distribution and average size (dave) were determined by using the ImageJ software.61
We also investigated the distribution of Cl− ions in the particles of pristine β-FeOOH and sample 8-400 with an energy dispersive X-ray (EDX) spectrometer (Ultim Extreme, Oxford Instruments). The specimens were prepared through ion (Ar) milling under an accelerating voltage of 6 kV at −160 °C (Model685 PECS II, Gatan). We recorded FE-SEM images using Everhart–Thornley-type detector and backscattered electron (BSE) images using an in-lens detector (Ultra55, Carl Zeiss). The accelerating voltage was 5 or 7 kV.
2.4 Evaluation of OER activity
We examined the OER activity of seven samples in an Ar-purged 1 M KOH solution (pH = 13.6) with a three-electrode configuration (see Fig. 1f);3–5 i.e., pristine β-FeOOH, samples 8-200, 8-400, 12-500, pristine α-FeOOH, and samples α-10-400 and α-10-500. We chose these samples considering each crystal phase, crystallinity, particle size/morphology, and the FeOOH source employed. Approximately 0.3–0.5 mg of each sample was attached to carbon paper (CP; TGP-H-060, TORAY) with a geometrical surface area of 1 cm2 (1 cm × 1 cm) and used as a working electrode (WE). This procedure is slightly different from the method previously reported,3–5,48 where a colloidal solution of β-FeOOH nanorods was injected into CP. The total surface area of the electrode was 2 cm2 by adding both the front and back sides. The counter electrode (CE) was a Pt wire, whereas the reference electrode (RE) was Ag/AgCl. Linear sweep voltammetry (LSV; ALS612E, ALS) was performed at a scan rate of 5 mV s−1, and chronoamperometry was conducted at +0.7 V vs. Ag/AgCl for 1800 seconds to clarify the stability of the anodic currents. Electrochemical impedance spectroscopy (EIS, CHI-614C, ALS) was performed on pristine β-FeOOH and samples 8-200, 8-400, and 12-500 in the frequency range from 0.1 to 105 Hz under flowing Ar (20 mL min−1).3 Results and discussion
3.1 Crystal structure and phase diagram
Fig. 2a–i show the synchrotron XRD patterns of pristine β-FeOOH and samples 8-100 through 8-800. The XRD pattern of pristine β-FeOOH was assigned to the typical crystal structure of β-FeOOH.22–26 We first analyzed the data using the tetragonal symmetry with I4/m space group,26 providing reliability indices of Rwp = 5.99% and S = 1.09. However, employing the monoclinic symmetry with I2/m space group improved these reliability indices to Rwp = 4.11% and S = 0.74. The tetragonal symmetry only has one Fe site, whereas two Fe sites exist in monoclinic symmetry. As described later, the presence of two Fe sites was confirmed by Mössbauer spectroscopy. The lattice parameters were determined to be am = 10.4758(20) Å, bm = 3.0282(2) Å, cm = 10.5427(22) Å, and βm = 89.51(2) ° by ignoring the presence of H+ ions. The results of the Rietveld analysis are summarized in Fig. S1† and Table 2. The nearest bond distances between Fe and O ions (dFe–O) were calculated to be 1.867(13) Å × 1, 2.175(14) Å × 1, 1.948(6) Å × 2, and 2.242(7) Å × 2, whereas that between Fe and Fe ions (dFe–Fe) was 3.028(1) Å. The lattice parameters were similar to previously reported values,22–24 but two of the six dFe–O values [1.867(13) and 2.175(14) Å] slightly differ from those in the literature (1.90 and 2.05 Å).26 The relatively variable dFe–O value probably originated from the low aspect ratio of pristine β-FeOOH; i.e., the diameter and length are 3 and 15 nm, respectively.3–5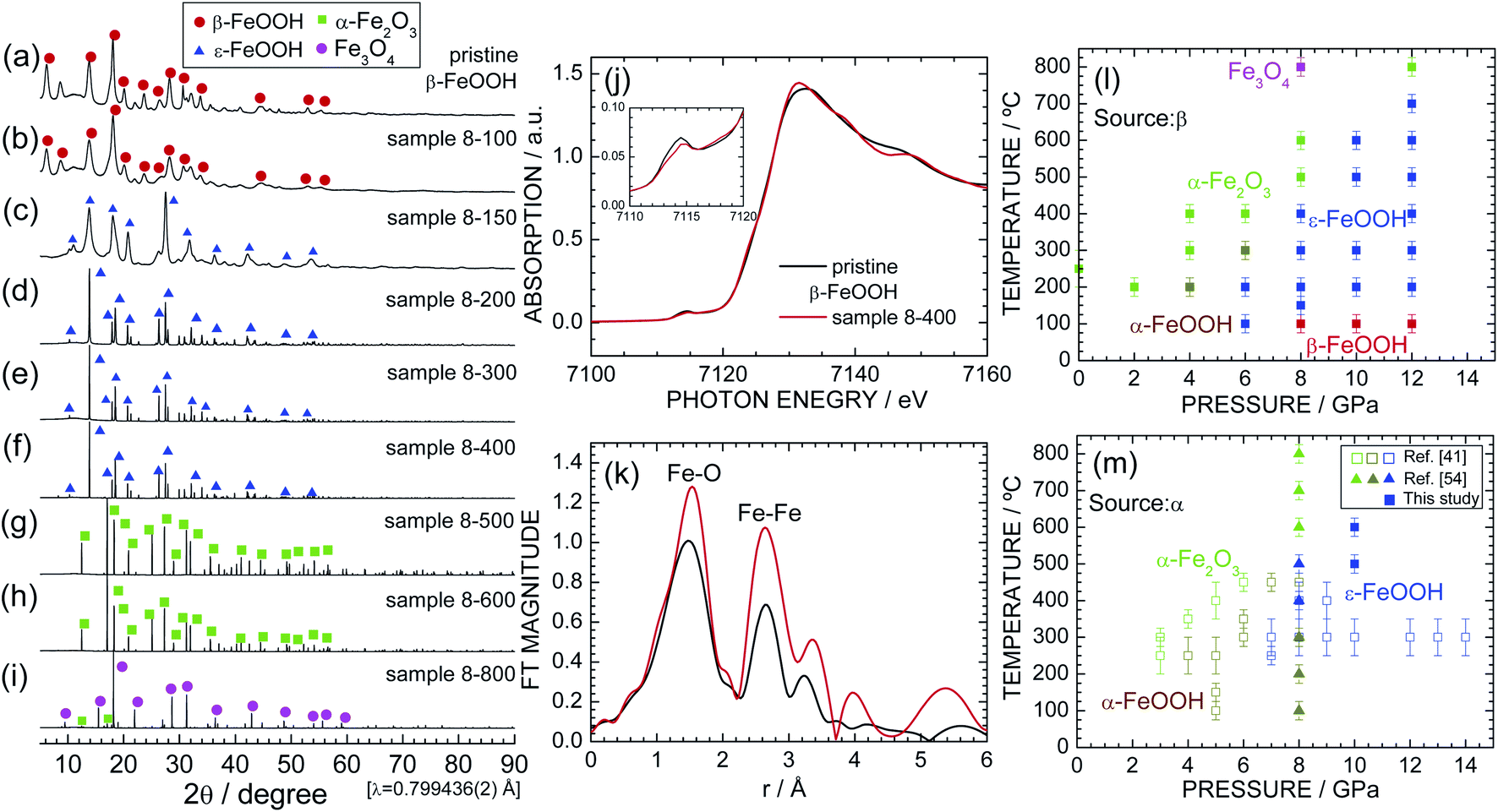 | ||
| Fig. 2 Synchrotron XRD patterns of samples (a) pristine β-FeOOH, (b) 8-100, (c) 8-150, (d) 8-200, (e) 8-300, (f) 8-400, (g) 8-500, (h) 8-600, and (i) 8-800. Diffraction lines from the β-FeOOH, ε-FeOOH, α-Fe2O3, and Fe3O4 phases are indicated by red circles, blue triangles, green squares, and purple circles, respectively. (j) Fe K-edge XANES and (k) the Fourier transforms of k3-weighted Fe K-edge EXAFS spectra of pristine β-FeOOH and sample 8-400. The pre-edge peak at ca. 7115 eV is enlarged in the inset. Pressure–temperature phase diagram for the (l) β-FeOOH and (m) α-FeOOH sources. Data from ref. 41 and 54 are also shown in (m) for comparison. | ||
| Atom | Wyckoff position | Occupancya | x | y | z | Biso (Å2) |
|---|---|---|---|---|---|---|
| a We fixed site occupancies to 1 or 0.12.b Space group: I2/m, am = 10.4758(20) Å, bm = 3.0282(2) Å, cm = 10.5427(22) Å, βm = 89.51(2) °, Rwp = 4.11%, and S = 0.74. | ||||||
| Fe1 | 4i | 1 | 0.858(1) | 0 | 0.338(1) | 0.9(1) |
| Fe2 | 4i | 1 | 0.345(1) | 0 | 0.143(1) | 0.9(1) |
| O1 | 4i | 1 | 0.661(1) | 0 | 0.276(1) | 0.4(1) |
| O2 | 4i | 1 | 0.688(1) | 0 | 0.012(1) | 0.4(1) |
| O3 | 4i | 1 | 0.303(1) | 0 | 0.377(1) | 0.4(1) |
| O4 | 4i | 1 | 0.036(1) | 0 | 0.350(1) | 1.2(1) |
| Cl1 | 2a | 0.12 | 0 | 0 | 0 | 0.3(1) |
Sample 8-100 also maintained the β-FeOOH-type structure, whereas sample 8-150 possessed a different crystal structure from pristine β-FeOOH. After careful examination, the crystal structure of sample 8-150 was assigned to the orthorhombic symmetry with the Pmn21 space group, i.e., an ε-FeOOH-type structure. The Rietveld analysis revealed orthorhombic lattice parameters of ao = 2.9688(7) Å, bo = 4.4558(10) Å, and co = 5.0077(12) Å (not shown). The XRD pattern of 8-150 exhibits broad and weak diffraction lines, indicating low crystallinity and/or a small crystallite size. By contrast, sample 8-200 shows sharp and strong diffraction lines with the ε-FeOOH-type structure (Fig. 2d). Further, ε-FeOOH was maintained up to 400 °C, but further increasing the heating temperature caused other structural transitions. Specifically, sample 8-600 was an α-Fe2O3 (hematite) phase, and sample 8-800 consisted of α-Fe2O3 and Fe3O4 (magnetite) phases, wherein the weight fraction of the latter phase was estimated to be 92.8%. The main phase for each sample is summarized in Table 1. The formation of α-Fe2O3 is represented by
| 2FeOOH → α-Fe2O3 + H2O↑, | (1) |
| 6α-Fe2O3 → 4Fe3O4 + O2↑. | (2) |
The weight losses (Δw) were calculated to be −10.14% for eqn (1) and −3.34% for eqn (2). The formation of Fe3O4 significantly differs from the situation under ambient pressure, where α-Fe2O3 remains stable up to 1000 °C.1,16,21,22
Fig. S2† and Table 3 summarize the Rietveld analysis results of sample 8-400 as an example. The ao, bo, and co values were determined to be 3.0020(1), 4.4570(1), and 4.9527(1) Å, respectively, which slightly differ from those for sample 8-150 owing to differences in the crystallinity. The dFe–O values were calculated to be 1.942(3) Å × 1, 2.137 Å × 1, 1.970(1) Å × 2, and 2.097(1) Å × 2, whereas dFe–Fe was 3.002(1) Å. These structural parameters are comparable with those for previous ε-FeOOH compounds that were synthesized from an α-FeOOH source.40,41,54 Based on the geometry of polyhedral distortions, the distortion index in the FeO6 octahedron [DI(FeO6)] is defined as62
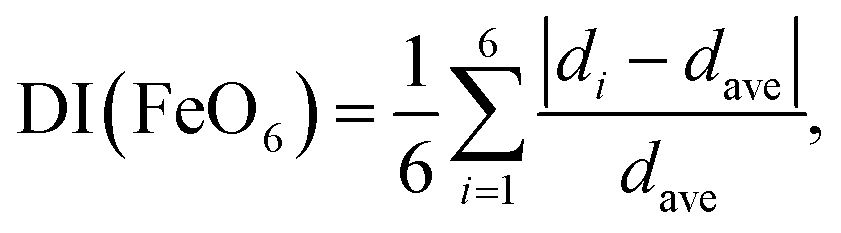 | (3) |
To clarify changes in the local structure during the β-FeOOH → ε-FeOOH transition, Fig. 2j and k show the Fe K-edge XANES and Fourier transforms of the k3-weighted Fe K-edge EXAFS spectra of pristine β-FeOOH and sample 8-400. For both samples, a pre-edge peak at ca. 7115 eV and a main edge peak at ca. 7131 eV appear in the XANES spectra. The pre-edge peak is a mixture of an electric quadrupole transition and an electric-dipole-allowed 1s → 4p transition, which is formally electric dipole forbidden.64 The presence of the pre-edge peak corresponds to a noncentrosymmetric geometry for the FeO6 octahedron, i.e., a distortion of the FeO6 octahedron, as evident from the DI(FeO6) values. As understood by the Fe K-edge XANES spectrum of α-Fe2O3 (Fig. S3†), the photon energy of the main edge peak is located at 7131 eV. Hence, Fe ions were in the trivalent state for both pristine β-FeOOH and sample 8-400.
Two major peaks appear in the EXAFS spectra centered at phase-uncorrected radial distances (r) of approximately 1.5 and 2.7 Å, which were assigned to dFe–O and dFe–Fe, respectively. More specifically, dFe–O = 1.48(1) Å and dFe–Fe = 2.66(1) Å, for pristine β-FeOOH, whereas dFe–O = 1.54(1) Å and 2.65(1) Å for sample 8-400. The EXAFS spectrum of pristine β-FeOOH is similar to the spectrum that we recently reported for β-FeOOH,3–5 although to the best of our knowledge, the EXAFS spectrum of ε-FeOOH has never been reported thus far.
To establish the pressure–temperature phase diagram for β-FeOOH, synchrotron XRD patterns of samples of 2-200–6-400, 10-100–10-600, and 12-100–12-800 are shown in Fig. S4, S5, and S6,† respectively. Based on these results, the pressure–temperature phase diagram of β-FeOOH is illustrated in Fig. 2l. The α-Fe2O3 or α-FeOOH phase exists at pressures below 4 GPa, whereas the ε-FeOOH phase forms only at pressures above 6 GPa. Surprisingly at 6 GPa, the α-FeOOH phase appears at 300 °C after the formation of ε-FeOOH below 200 °C. Thus at this pressure, the structural phase transition with temperature becomes β-FeOOH → ε-FeOOH → α-FeOOH → α-Fe2O3, although α-FeOOH was synthesized by β-FeOOH, not ε-FeOOH. At 12 GPa, ε-FeOOH appeared up to 700 °C, providing the general trend that higher pressures above 6 GPa stabilize the ε-FeOOH phase to higher temperatures above 200 °C. On the other hand at pressures above 8 GPa, the β-FeOOH phase is obtained below 100 °C. Mixtures were rarely observed, and each sample comprised an almost single phase of α-FeOOH, α-Fe2O3, Fe3O4, ε-FeOOH, or β-FeOOH.
Fig. 2m shows the pressure–temperature phase diagram for α-FeOOH for comparison, which includes data from previous HP/HT studies on α-FeOOH.41,54 Fig. S7† shows synchrotron XRD patterns of samples of pristine α-FeOOH, α-10-400 and α-10-500; among them, the latter two samples are single phases of ε-FeOOH. Obviously, the ε-FeOOH phase forms in the region of lower pressures (ca. 1 GPa) and temperatures (ca. 200 °C) in the case of β-FeOOH. As mentioned above, the bulk modulus of β-FeOOH [=284(1) GPa] at room temperature is the highest among the FeOOH polymorphs.42 Considering this result, the required pressure for the transition to ε-FeOOH would be higher in the β-FeOOH, but the obtained fact was contrary to the expectation. As illustrated in Fig. 1a, β-FeOOH possesses the open framework structure with the partial occupation of Cl− ions. The lower pressure-induced transition indicates a fragility of β-FeOOH at high temperatures above 100 °C. Indeed, β-FeOOH was reported to decompose into α-Fe2O3 and H2O at lower temperatures (under ambient pressure) compared to α-FeOOH, due to its strengthening of the hydrogen bond environments.65 Note that the structural phase transition from β-FeOOH to ε-FeOOH should be accompanied by breaking and reforming the Fe–O bonds. This is because the Pmn21 (ε-FeOOH) space group is a maximal nonisomorphic subgroup of the Pnma (α-FeOOH) space group, whereas I2/m or I4/m (ε-FeOOH) space group is not a minimal nonisomorphic supergroup of the Pmn21 space group, based on the crystallographic relationship.66 Further HP/HT studies for various amounts of Cl− ions and theoretical calculations would reveal detailed mechanisms of the structural phase transition from β-FeOOH and ε-FeOOH.
3.2 x and particle morphology
According to previous studies on β-FeOOH,16,17,19,22,24–26 x in FeO1−x(OH)1+xClx ranges from 0.003 to 0.37. Moreover, Cai et al.16 reported a decrease in x upon heating accompanying the structural phase transition to α-Fe2O3 (under ambient pressure). In this section, we discuss changes in x and the particle morphologies during the successive phase transitions of β-FeOOH → ε-FeOOH → α-Fe2O3 → Fe3O4. Only the composition of pristine β-FeOOH was analyzed, whereas TG was employed to analyze the HP/HT-treated samples because their weights were limited to ca. 40 mg.The combined ICP-AES and IC analyses confirmed x = 0.12 for pristine β-FeOOH, suggesting the chemical formula FeO0.88(OH)1.12Cl0.12. The decomposition reaction upon heating is described by26
| 2FeO0.88(OH)1.12Cl0.12 → Fe2O3 + 0.24HCl↑ + H2O↑, | (4) |
 | ||
| Fig. 3 (a) TG curves of samples of pristine β-FeOOH, 8-100, 8-150, 8-200, 8-300, 8-400, and 8-500. (b) Δw at 1000 °C as a function of the heating temperature during the HP/HT synthesis. The red and blue broken lines indicate the calculated Δw based on eqn (1) and (4), respectively. SEM images at the 50 μm-scale of samples of (c) pristine β-FeOOH, (d) 8-100, (e) 8-200, (f) 8-300, (g) 8-400, and (h) 8-800. Enlarged SEM images of pristine β-FeOOH and 8-400 are shown in (i) and (j), respectively. | ||
The TG curve of sample 8-100 is similar to that of pristine β-FeOOH except for the Δw value (=−15.9%), whereas the curves of samples 8-150, 8-200, 8-300, and 8-400 show different temperatures at which w rapidly decreases. For instance, sample 8-400 exhibits a rapid decrease in w at ca. 300 °C, which is ca. 100 °C lower than those of pristine β-FeOOH and sample 8-100. Fig. 3b reveals an almost linear relationship between Δw at 1000 °C and the heating temperature during the HP/HT synthesis. Because the Δw values of samples 8-300 and 8-400 are close to the calculated Δw value (=10.14%) based on eqn (1) (Fig. 3b), the ε-FeOOH lattice is likely to be free from Cl− ions. In other words, above 150 °C, the Cl− ions start to evaporate out of the β-FeOOH lattice, which is associated with the structural phase transition to ε-FeOOH. The disappearance of Cl− ions is strongly related to the mechanism underlying the structural phase transition; i.e., as described above, it inevitably induces the breaking and reforming of Fe–O bonds.
We confirmed the absence of Cl− ions in the ε-FeOOH lattice by comparing its TG curve with that of sample α-10-400 (Fig. S8†). Furthermore, after the TG analyses, we performed XRD and verified that pristine β-FeOOH and samples 8-100, 8-150, 8-200, 8-300, 8-400, 8-500, and α-10-400 all crystallized into the α-Fe2O3 phase (Fig. S9†). According to these results, the successive phase transitions of β-FeO0.88(OH)1.12Cl0.12 (at 8 GPa) are represented by
| 2β-FeO0.88(OH)1.12Cl0.12 → 2ε-FeOOH + 0.24HCl↑ | (5) |
| 2ε-FeOOH → α-Fe2O3 + H2O↑ | (6) |
Fig. 3c–h show SEM images of samples (c) pristine β-FeOOH, (d) 8-100, (e) 8-200, (f) 8-300, (g) 8-400, and (h) 8-800, which clarify changes in the particle morphologies and sizes during the phase transitions. Enlarged SEM images of pristine β-FeOOH and sample 8-400 are also shown in Fig. 3i and j, respectively. Particles in pristine β-FeOOH aggregate into secondary particles with an average size of 1 μm. As the heating temperature increases, secondary particles take on a scaly shape and form large secondary particles. Sample 8-400, which crystallized into the ε-FeOOH phase, shows a plate-like shape with an average size of 2 μm. Finally, sample 8-800, which crystallized into the Fe3O4 phase, exhibits a non-uniform shape with an average size of 50 μm. These changes indicate that the structural phase transitions under HP cause aggregation and change the particle size. Similar results were also observed for the phase transitions from α-FeOOH to ε-FeOOH54 and from spinel-structured Li[Li1/3Ti5/3]O4 to columbite-structured TiO2.67
We next investigated the effects of the applied pressure. Fig. S10† shows the SEM images of samples 6-200, 8-200, and 12-200, all of which were synthesized at 200 °C and crystallized into the ε-FeOOH phase. Fig. S11† shows the SEM images of samples 12-200, 12-400, 12-500, 12-700, which were synthesized at 12 GPa. The secondary particles of sample 6-200 indicate the same scaly shape as for sample 8-200, whereas those of sample 12-200 are relatively spherical. In addition, the primary particles of samples 12-400 and 12-500 are isolated from each other and are spherical with an average diameter of 3 μm. Thus, applying higher pressures gives the particles a round shape, although the samples are in the same crystalline phase. Fig. S12† shows the particle distribution of (a) β-FeOOH, (b) 8-400, and (c) 12-500 determined by the ImageJ software.61 The dave values of β-FeOOH, 8-400, and 12-500 were estimated to be 0.14, 4.7, and 2.9 μm, respectively.
3.3 OER activity
Fig. 4a shows the LSV curves of pristine β-FeOOH and samples 8-200, 8-400, and 12-500. The current density (j) was normalized by the total surface area of the electrode (2 cm2), while the potential vs. Ag/AgCl was converted into a potential vs. reversible hydrogen electrode (RHE). The anodic current of pristine β-FeOOH rapidly increases at ca. 1.60 V, which is consumed for the water oxidation accompanying with oxygen evolution.3,4 Sample 8-400 obviously exhibits a lower OER overpotential than that of pristine β-FeOOH; specifically, the anodic current rapidly increases above 1.55 V, in which threshold potential for OER is negatively shifted by ca. 60 mV compared to that of pristine β-FeOOH. For instance, the potential at which j = 1 mA cm−2 (Ej=1 mA cm−2) was 1.62 V for pristine β-FeOOH and 1.56 V for 8-400. Sample 12-500 indicates a similar threshold potential to pristine β-FeOOH, whereas sample 8-200 shows a higher threshold potential (ca. 1.75 V). The Ej=1 mA cm−2 values are summarized in Table 1. As evidenced by the XRD patterns shown in Fig. 2b, f, and S6,† samples 8-200, 8-400, and 12-500 crystallized into the ε-FeOOH phase. This suggests that OER activities of ε-FeOOH depend on synthesis conditions.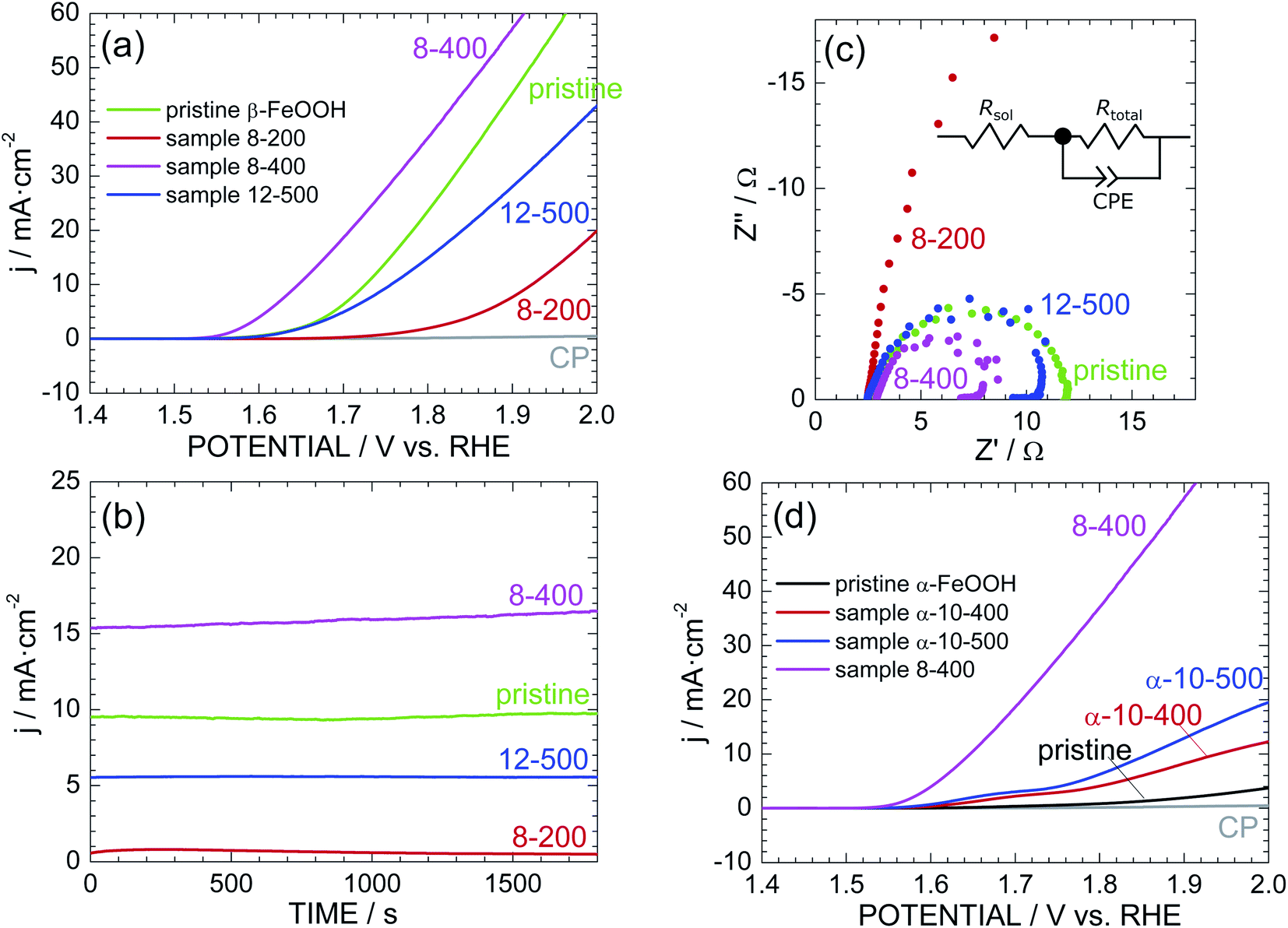 | ||
| Fig. 4 (a) LSV curves of pristine β-FeOOH and samples 8-200, 8-400, and 12-500. (b) Corresponding time courses of the anodic current measured at +1.70 V vs. RHE. (c) Cole–Cole plots of pristine β-FeOOH and samples 8-200, 8-400, and 12-500. Inset: equivalent circuit model consisting of Rsol, Rtotal, and CPE. (d) LSV curves of pristine α-FeOOH and samples α-10-400 and α-10-500 together with that of sample 8-400. | ||
Fig. 4b shows the time courses of the anodic current of pristine β-FeOOH and samples 8-200, 8-400, and 12-500, measured at 1.70 V vs. RHE. Sample 8-400 still maintains the highest anodic current density of ca. 15 mA cm−2, which is ca. 1.5 times greater than that of pristine β-FeOOH. To reveal the origins of this OER activity, Fig. 4c compares the Cole–Cole plots of pristine β-FeOOH and samples 8-200, 8-400, and 12-500. Each sample exhibits one deformed semicircle, except for sample 8-200. EIS spectra generally include several contributions originated from the bulk, grain boundary, and surface of the electrode. However, we can simply estimate the total resistance (Rtotal) of the electrode by the radius of the semicircle. The radius of sample 8-400 is apparently smaller than that of pristine β-FeOOH, indicating the decrease in Rtotal in the case for sample 8-400. More specifically, the EIS spectra were fitted to the equivalent circuit model shown in the inset of Fig. 4c, i.e., a solution resistance (Rsol), Rtotal, and a constant-phase element (CPE). Here, the CPE is represented by 1/(iwQ)α; when α = 1, Q has the dimension of capacitance (F); α < 1 indicates surface heterogeneity in the system, such as the roughness of the electrode. As summarized in Table S1,† Rtotal = 9.42 and = 5.11 Ω for pristine β-FeOOH and sample 8-400, respectively, which suggests that one cause for enhancing OER activity of sample 8-400 is due to its lower Rtotal.
As reported previously,39–41 α-FeOOH was also transformed into ε-FeOOH under HP/HT conditions. We thus examined the OER activity of pristine α-FeOOH and samples α-10-400 and α-10-500 for comparison. Here, samples α-10-400 and α-10-500, which crystallized into the ε-FeOOH phase. Fig. 4d reveals that the OER activity was enhanced by the phase transition from α-FeOOH to ε-FeOOH. However, the threshold potentials of samples α-10-400 and α-10-500 are much higher than those of samples 8-400 and 12-500, indicating the superiority of β-FeOOH over α-FeOOH. In other words, the OER activity of ε-FeOOH depends on the FeOOH source employed. The Ej=1 mA cm−2 values of pristine α-FeOOH and samples α-10-400 and α-10-500 are listed in Table 1.
If we focus on the highest OER activity, this activity was enhanced in the order by α-FeOOH → β-FeOOH → ε-FeOOH. We next investigated the reproducibility of the OER activity among different electrodes prepared with the same sample. Fig. S13† shows the additional LSV curves of pristine β-FeOOH and sample 8-400. The LSV curves of the three different electrodes fabricated using pristine β-FeOOH appear similar, whereas those of the five different electrodes from sample 8-400 vary widely. Specifically, three of the five electrodes indicate superior OER activities regarding the threshold potential and anodic current density, whereas the rest show similar or lower OER activity to pristine β-FeOOH. Each electrode was prepared by depositing the sample into CP (Fig. 1f), so the OER activity probably depends on the distribution of particles and adhesion between the particles and CP.
3.4 Further characterization of sample 8-400
As sample 8-400 exhibited the best OER activity, we further characterized sample 8-400 using Mössbauer spectroscopy and EDX analysis. Fig. 5 shows the Mössbauer spectra of (a) pristine β-FeOOH at 25 °C and sample 8-400 at (b) 25 °C and (c) − 196 °C. A doublet peak appears for pristine β-FeOOH, indicating that it is in a paramagnetic state. The Mössbauer spectrum is ideally represented by
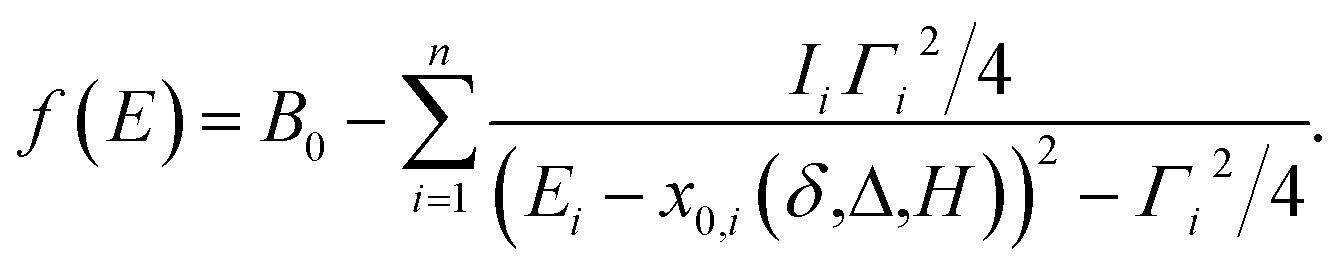 | (7) |
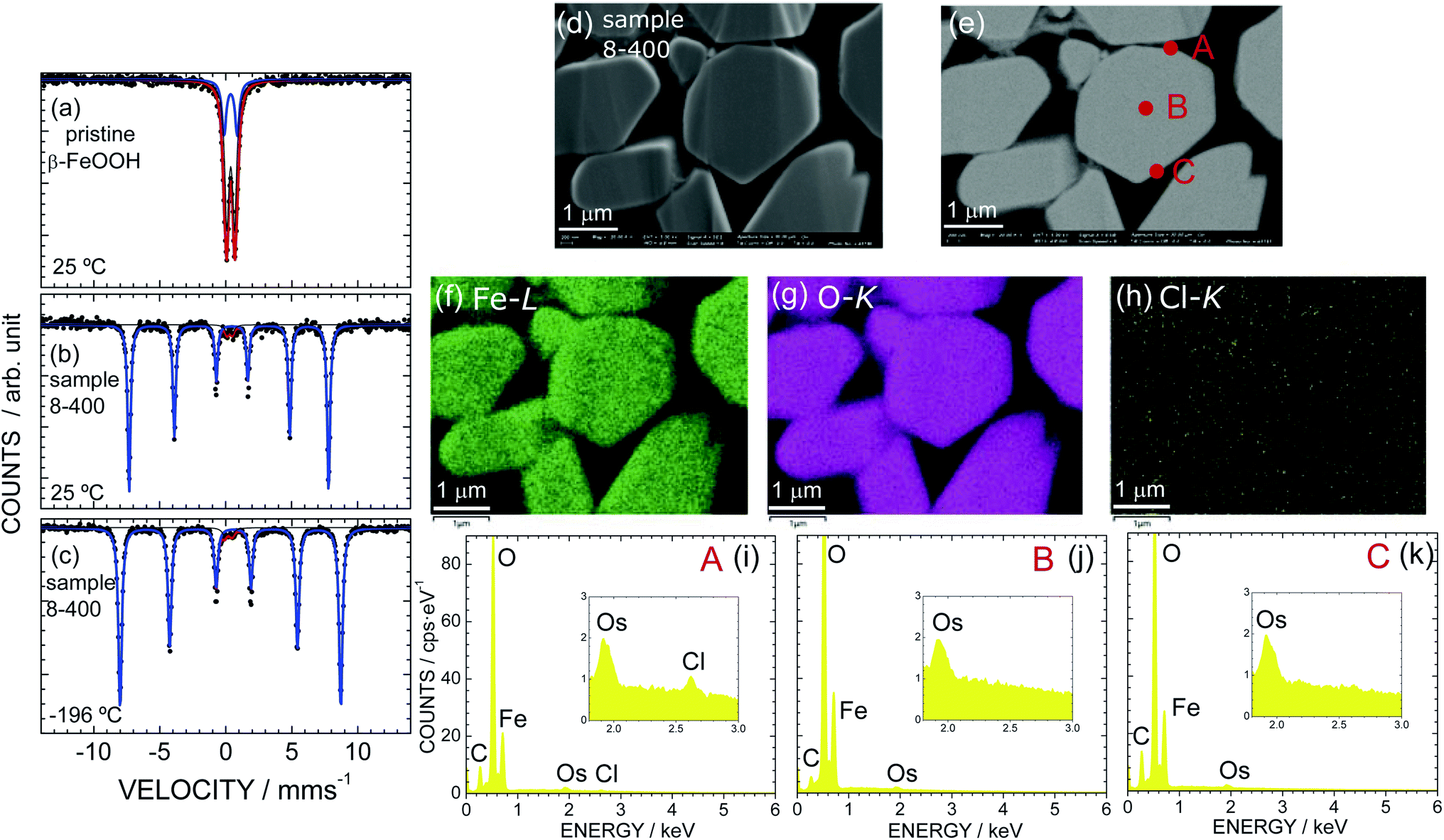 | ||
| Fig. 5 Mössbauer spectra of (a) pristine β-FeOOH at ca. 25 °C and sample 8-400 at ca. (b) 25 °C and (c) −196 °C recorded without applying a magnetic field. Red and blue lines indicate different magnetic components in the samples. (d) FE-SEM and (e) BSE images of sample 8-400. EDX mapping for (f) Fe, (g) O, and (h) Cl atoms and EDX spectra at points (i) A, (j) B, and (k) C in (e). | ||
| Sample | Component (i) | δ (mm s−1) | Δ (mm s−1) | Γ (mm s−1) | H (T) | Component ratio (%) |
|---|---|---|---|---|---|---|
| Pristine β-FeOOH (ca. 25 °C) | 1 | 0.37 | 1.00 | 0.40 | — | 28 |
| 2 | 0.37 | 0.58 | 0.40 | — | 72 | |
| 8-400 (ca. 25 °C) | 1 | 0.37 | −0.24 | 0.26 | 46.9 | 97 |
| 2 | 0.31 | 0.37 | 0.35 | — | 3 | |
| 8-400 (−196 °C) | 1 | 0.47 | −0.24 | 0.31 | 51.8 | 97 |
| 2 | 0.18 | 0.54 | 0.45 | — | 3 |
Sample 8-400 exhibits sextet peaks at both 25 and −196 °C, indicating that the sample is in an (antiferro)magnetic state (Fig. 5b and c). A careful analysis revealed that the Mössbauer spectra contain a small (3%) a paramagnetic contribution, as indicated by the red line in Fig. 5b and c. Considering each Mössbauer parameter summarized in Table 4 suggests that the paramagnetic contribution originates from the β-FeOOH phase. On the other hand, the Mössbauer parameters of the major contribution (97%) are determined to be δ = 0.37 mm s−1, Δ = −0.24 mm s−1, Γ = 0.26 mm s−1, and H = 46.9 T at ca. 25 °C. Because these Mössbauer parameters are similar to previous results for ε-FeOOH (δ = 0.15 mm s−1, Δ = −0.26 mm s−1, and H = 47.2 T at 23 °C),68 the major contribution is attributed to the ε-FeOOH phase. This mixed nature of sample 8-400 differs from the Rietveld results shown in Fig. S2† and Table 2, which suggest that the sample crystallizes into a single phase of ε-FeOOH. Note that sample α-10-400 comprises only from the ε-FeOOH phase, as understood by its Mössbauer spectrum in Fig. S14.†
According to the TG analyses (Fig. 3a and b), sample 8-400 was free from Cl− ions in the ε-FeOOH lattice. However, we further investigated the distribution of Cl− ions, particularly on the surface. Fig. 5d and e show the FE-SEM and BSE images of sample 8-400, respectively, while Fig. 5f, g, and h show the corresponding EDX mappings of Fe, O, and Cl atoms, respectively. Fe and O atoms are homogeneously distributed in the particles, whereas Cl atoms rarely appear. Based on the EDX spectra at points A, B, and C, Cl atoms are only observed at point A (Fig. 5i–k). Hence, the Cl− ions are inhomogeneously distributed on a specific particle after the phase transition from β-FeOOH to ε-FeOOH. By contrast, we confirmed that the Cl− ions are homogeneously distributed in the particles of pristine β-FeOOH, as shown in Fig. S15.† We also confirmed that there is no obvious change in the distribution of Fe and O atoms before and after the OER test (Fig. S16†).
3.5 Possible origin for high OER activity of ε-FeOOH
Considering the dave values of sample 8-400 (=4.7 μm) and pristine β-FeOOH (= 0.14 μm), the active surface area of sample 8-400 should be smaller than that of pristine β-FeOOH. This indicates that the OER activity of sample 8-400 is attributed to an intrinsic and high OER performance of ε-FeOOH. In other words, a small Rbulk contributes to the relatively low Rtotal of sample 8-400 (= 5.11 Ω).Based on studies of quadruple manganese perovskites AMn7O12 (A = La and Ca) which were synthesized by the HP/HT method,69 their OER activities relate with the nearest bond distance between O atoms (dO–O); the overpotential decreases with dO–O. Indeed, as shown in Fig. S17,† the dO–O value was decreased in the order by α-FeOOH (=3.035 Å) → β-FeOOH (=3.023 Å) → ε-FeOOH (=3.006 Å). Furthermore, it is interesting to compare the OER activities of ε-FeOOH and β-MnO2 (pyrolusite), as they are almost isostructural.27 Yan et al. clarified that the phase transition from β-MnO2 to α-MnO2 (cryptomelane) improved the OER activity,30 which is the opposite of the trend observed in this study. Note that both β-MnO2 and ε-FeOOH are classified as rutile, although ε-FeOOH is regarded as a distorted rutile structure, as in the case of columbite-type TiO2.67 Thus, there is a possibility that the superior OER activity of ε-FeOOH owes to its characteristic structural environments such as the shortest dO–O and slightly distorted FeO6 octahedra.
Another possibility would come from the mixed nature of sample 8-400. Hu et al.6 recently reported that using mixed phases of α- and β-FeOOH or β- and δ-FeOOH enhanced the OER activity owing to the formation of oxygen vacancies on the surface. As sample 8-400 contains a small amount (3%) of β-FeOOH, synergetic effects of the mixture of ε- and β-FeOOH could contribute to its OER activity.
Anyway, comparing the OER activities of samples 8-200 and 12-500 suggests that other factors besides the crystalline phase, such as the particle morphology and particle size, affect the OER activities. Further theoretical studies on FeOOH polymorphs would reveal details of the reaction mechanism underlying the OER activity of ε-FeOOH.
3.6 Perspective of ε-FeOOH as catalyst
We revealed that ε-FeOOH exhibited the best OER activity among the FeOOH polymorphs investigated. However, as summarized in Table 1, even the best OER activity of ε-FeOOH was restricted to Ej=1 mA cm−2 = 1.56 V vs. RHE. This overpotential is still higher than those of other non-noble metal-based electrocatalysts; for instance, an exfoliated NiFe layered double hydroxide indicated Ej=1 mA cm−2 ≃ 1.52 V vs. RHE.72 The relatively large overpotential of ε-FeOOH is caused by its large particle size (dave > 2 μm), namely low active site for OER, as shown in Fig. 3j and S12.† The procedure for preparing composite electrodes also results in the large overpotential, because the composite electrode with the colloridal β-FeOOH solution showed a lower (ca. 0.05 V) overpotential.3–5,48 Therefore, investigations for decreasing particle size and optimizing composite electrodes could enhance OER activity of ε-FeOOH. The pressure-phase diagram shown in Fig. 2l would be useful for these investigations. Finally, ε-FeOOH is applicable as a water oxidation photocatalyst,70,71 although the present study is focused on the electrocatalytic properties.4 Conclusion
We examined HP/HT phases of β-FeOOH nanorods, more specifically, β-FeO1−x(OH)1+xClx with x = 0.12, and their OER activities. To our best knowledge, we firstly demonstrated that β-FeOOH transformed into ε-FeOOH above 6 GPa and 100 °C. In addition, Cl− ions, which were initially hosted in the [2 × 2] tunnels, evaporated from the surface during the structural phase transition. Above 400 °C, ε-FeOOH decomposed into α-Fe2O3 or Fe3O4, while releasing oxygen from the lattice. Above 6 GPa, ε-FeOOH was stabilized at higher temperatures; for instance, at 12 GPa, ε-FeOOH decomposed into α-Fe2O3 above 700 °C. The obtained pressure–temperature phase diagram of β-FeOOH differed from that of α-FeOOH, probably owing to the open framework of β-FeOOH and partial occupation of Cl− ions. In the viewpoint from earth and space sciences, this information is crucial for in-depth understanding of evolution and dynamics of the Earth interior particularly at the upper mantle.Sample 8-400 (ε-FeOOH), which was synthesized at 8 GPa and 400 °C, exhibited superior OER activity over the parent β-FeOOH or α-FeOOH. EIS results showed that this superior OER activity is due to its low Rtotal. Moreover, Mössbauer spectroscopy revealed that sample 8-400 contains a small amount (3%) of the β-FeOOH phase, possibly leading to synergetic effects from β- and ε-FeOOH. The residual Cl− ions were inhomogeneously distributed on the surfaces of particles in sample 8-400, resulting in relatively scattered OER activities along different composite electrodes fabricated from this sample. Nonetheless, ε-FeOOH is a possible candidate as a next-generation earth-abundant OER catalyst, particularly because material options are currently limited. The established pressure–temperature phase diagram is useful for further improving the OER activity of ε-FeOOH, because particle sizes, particle morphologies, and distribution of Cl− ions strongly depend on the HP/HT conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The synchrotron XRD patterns were recorded at the BL5S2 beamline of the Aichi Synchrotron Radiation Center, Aichi Science & Technology Foundation, Japan (Proposal No. 201906021, 2020D1019, and 2020D2017). XAS was conducted at the Toyota beamline (BL33XU) of the SPring-8 with the approval of the JASRI (Proposal No. 2019A7034 and 2020A7037). We wish to thank Dr Yusaku F. Nishimura of TCRDL for help with the XAS measurements and Makoto Kondo for electrochemical measurements. We also acknowledge Dr Takashi Segi of Kobelco Research Institute, Japan, and Dr Tadahiro Nakamoto of Toray Research Center, Japan, for performing Mössbauer spectroscopy.Notes and references
- H. N. Weiser and W. O. Milligan, J. Am. Chem. Soc., 1935, 57, 238 Search PubMed.
- T. Misawa, K. Hashimoto and S. Shimodaira, Corros. Sci., 1974, 14, 131 Search PubMed.
- T. M. Suzuki, T. Nonaka, A. Suda, N. Suzuki, Y. Matsuoka, T. Arai, S. Sato and T. Morikawa, Sustainable Energy Fuels, 2017, 1, 636 Search PubMed.
- T. M. Suzuki, T. Nonaka, K. Kitazumi, N. Takahashi, S. Kosaka, Y. Matsuoka, K. Sekizawa, A. Suda and T. Morikawa, Bull. Chem. Soc. Jpn., 2018, 91, 778 Search PubMed.
- T. Morikawa, S. Gul, Y. F. Nishimura, T. M. Suzuki and J. Yano, Chem. Commun., 2020, 56, 5158 Search PubMed.
- J. Hu, S. Li, J. Chu, S. Niu, J. Wang, Y. Du, Z. Li, X. Han and P. Xu, ACS Catal., 2019, 9, 10705 Search PubMed.
- C. Feng, M. B. Faheem, J. Fu, Y. Xiao, C. Li and Y. Li, ACS Catal., 2020, 10, 4019 Search PubMed.
- K. Amine, H. Yasuda and M. Yamachi, J. Power Sources, 1999, 81–82, 221 Search PubMed.
- X. Wang, X. Chen, L. Gao, H. Zheng, M. Ji, C. Tang, T. Shen and Z. Zhang, J. Mater. Chem., 2004, 14, 905 Search PubMed.
- W. Chen, Y. Lai, Z. Zhang, Y. Gan, S. Jiang and J. Li, J. Mater. Chem. A, 2015, 3, 6447 Search PubMed.
- L. Yu, L. P. Wang, L. S. Xi, P. Yang, Y. Du, M. Srinivasan and Z. J. Xu, Chem. Mater., 2015, 27, 5340 Search PubMed.
- R. V. Morris, G. Kingelhöfer, B. Bernhardt, C. Schröder, D. S. Rodionov, P. A. de Souza Jr, A. Yen, R. Gallert, E. N. Evlanov, J. Foh, E. Kankeleit, P. Gütlich, D. W. Ming, F. Renz, T. Wdowiak, S. Squyres and R. E. Arvidson, Science, 2004, 305, 833 Search PubMed.
- T. S. Peretyazhko, M. J. Pan, D. W. Ming, E. B. Rampe, R. V. Morris and D. G. Agresti, ACS Earth Space Chem., 2019, 3, 314 Search PubMed.
- H. Simon, G. Cibin, P. Robbins, S. Day, C. Tang, I. Freestone and E. Schofield, Angew. Chem., Int. Ed., 2018, 57, 7390 Search PubMed.
- U. Schwertmann and R. M. Cornell, The Iron Oxides in the Laboratory: Preparation and Characterization, Wiley-VCH, New York, 2000 Search PubMed.
- J. Cai, J. Liu, Z. Gao, A. Navrotsky and S. L. Suib, Chem. Mater., 2001, 13, 4595 Search PubMed.
- A. E. Gash, J. H. Satcher and R. L. Simpson, Chem. Mater., 2003, 15, 3268 Search PubMed.
- L. Mazeina, S. Deore and A. Navrotsky, Chem. Mater., 2006, 18, 1830 Search PubMed.
- J. Kim and C. P. Grey, Chem. Mater., 2010, 22, 5453 Search PubMed.
- S. Jiang, S. Park, Y. Yon, J.-H. Lee, W.-H. Wu, N. P. Dan, M. Sadowsky and H.-G. Hur, Environ. Sci. Technol., 2013, 47, 10078 Search PubMed.
- M. Nambu, Ganseki Kobutsu Kosho Gakkaishi, 1968, 58, 143 Search PubMed.
- A. L. Mackay, Mineral. Mag., 1960, 32, 545 Search PubMed.
- A. L. Mackay, Mineral. Mag., 1962, 33, 270 Search PubMed.
- E. Murad, Clay Miner., 1979, 14, 273 Search PubMed.
- J. E. Post and V. F. Buchwald, Am. Mineral., 1991, 76, 272 Search PubMed.
- K. Ståhl, K. Nielsen, J. Jiang, B. Lebech, J. C. Hanson, P. Norby and J. van Lanschot, Corros. Sci., 2003, 45, 2563 Search PubMed.
- A. F. Wells, Structural Inorganic Chemistry, Oxford University Press, 5th edn, 1984 Search PubMed.
- Y. Meng, W. Song, H. Huang, Z. Ren, S.-Y. Chen and S. L. Suib, J. Am. Chem. Soc., 2014, 136, 11452 Search PubMed.
- M. Huynh, C. Shi, S. J. L. Billinge and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 14887 Search PubMed.
- G. Yan, Y. Lian, Y. Gu, C. Yang, H. Sun, Q. Mu, Q. Li, W. Zhu, X. Zheng, M. Chen, Z. Zhu and Y. Peng, ACS Catal., 2018, 8, 10137 Search PubMed.
- B. Li, G. Rong, Y. Xie, L. Huang and C. Feng, Inorg. Chem., 2006, 45, 6404 Search PubMed.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 Search PubMed.
- K. Otte, R. Pentcheva, W. W. Scmahl and J. R. Rustad, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 205116 Search PubMed.
- M. Nishi, Y. Kuwayama, J. Tuchiya and T. Tuchiya, Nature, 2017, 547, 205 Search PubMed.
- D. Tunega, J. Phys. Chem. C, 2012, 116, 6703 Search PubMed.
- C. Lu and C. Chen, J. Phys. Chem. Lett., 2018, 9, 2181 Search PubMed.
- W. Xu, E. Greenberg, G. K. Rozenberg, M. P. Pasternak, E. Bykova, T. Boffa-Ballaran, L. Dubrovinsky, V. Prakapenka, M. Hanfland, O. Y. Vekilova, S. I. Simak and I. A. Abrikosov, Phys. Rev. Lett., 2013, 111, 175501 Search PubMed.
- S.-C. Zhu, Q. Hu, W. L. Mao, H.-K. Mao and H. Sheng, J. Am. Chem. Soc., 2017, 139, 12129 Search PubMed.
- J. Chenavas, J. C. Joubert, J. J. Capponi and M. Marezio, J. Solid State Chem., 1973, 6, 1 Search PubMed.
- N. B. Bolotina, V. N. Molchanov, T. I. Dyuzheva, L. M. Lityagina and N. A. Bendeliani, Crystallogr. Rep., 2006, 51, 960 Search PubMed.
- A. E. Gleason, R. Jeanloz and M. Kunz, Am. Mineral., 2008, 93, 1882 Search PubMed.
- M. M. Reagan, A. E. Gleason, L. Daemen, Y. Xiao and W. L. Mao, Am. Mineral., 2016, 101, 1483 Search PubMed.
- C. Xu, M. Nishi and T. Inoue, Am. Mineral., 2019, 104, 1416 Search PubMed.
- J. Yan, P. Li, Y. Ji, H. Bian, Y. Li and S. Liu, J. Mater. Chem. A, 2017, 5, 21478 Search PubMed.
- W. Luo, C. Jiang, Y. Li, S. A. Shevlin, X. Han, K. Qiu, Y. Cheng, Z. Guo, W. Huang and J. Tang, J. Mater. Chem. A, 2017, 5, 2021 Search PubMed.
- J. Chi, H. Yu, G. Jiang, J. Jia, B. Qin, B. Yi and Z. Shao, J. Mater. Chem. A, 2018, 6, 3397 Search PubMed.
- A. Szytuła, A. Burewicz and Ž. Dimitrijević, Phys. Status Solidi, 1968, 26, 429 Search PubMed.
- Y. Sakamoto, Y. Noda, K. Ohno, K. Koike, K. Fujii, T. M. Suzuki, M. Morikawa and S. Nakamura, Phys. Chem. Chem. Phys., 2019, 21, 18486 Search PubMed.
- Z. Huang, F. Han, M. Li, Z. Zhou, X. Guan and L. Guo, Comput. Mater. Sci., 2019, 169, 109110 Search PubMed.
- S. Yagi, I. Yamada, H. Tsukasaki, A. Seno, M. Murakami, H. Fujii, H. Chen, N. Umezawa, H. Abe, N. Nishiyama and S. Mori, Nat. Commun., 2015, 6, 8249 Search PubMed.
- Z. Zhang, Z. Wang, S. He, C. Wang, M. Jin and Y. Yin, Chem. Sci., 2015, 6, 5197 Search PubMed.
- T. Uyama, K. Mukai and I. Yamada, Inorg. Chem., 2019, 58, 6684 Search PubMed.
- T. Uyama, K. Mukai and I. Yamada, Inorg. Chem., 2020, 59, 777 Search PubMed.
- K. Mukai and I. Yamada, ACS Omega, 2020, 5, 10115 Search PubMed.
- T. Ida, Powder Diffr., 2016, 31, 216 Search PubMed.
- F. Izumi and K. Momma, Solid State Phenom., 2007, 130, 15 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 Search PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 Search PubMed.
- Y. F. Nishimura, Y. Kondo and H. Oka, J. Power Sources, 2020, 446, 227351 Search PubMed.
- Z. Klencsör, Hyperfine Interact., 2013, 217, 117 Search PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671 Search PubMed.
- W. H. Baur, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 1195 Search PubMed.
- T. Uyama, K. Mukai and I. Yamada, RSC Adv., 2018, 8, 26325 Search PubMed.
- T. E. Westre, P. Kennepohl, J. G. Dewitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297 Search PubMed.
- X. Song and J.-F. Boily, J. Phys. Chem. A, 2016, 120, 6249 Search PubMed.
- International Tables for Crystallography, Volume A: Space-Group Symmetry, ed. T. Hahn, Kluwer Academic Publishers, Dordrecht, 5th edn, 2002 Search PubMed.
- K. Mukai and I. Yamada, Inorg. Chem. Front., 2018, 5, 1941 Search PubMed.
- M. Pernet, J. Chenavas, J. C. Joubert, C. Meyer and Y. Gros, Solid State Commun., 1973, 13, 1147 Search PubMed.
- I. Yamada, H. Fujii, A. Takamatsu, H. Ikeno, K. Wada, H. Tsukasaki, S. Kawaguchi, S. Mori and S. Yagi, Adv. Mater., 2017, 29, 1603004 Search PubMed.
- G. Ge, M. Liu, C. Liu, W. Zhou, D. Wang, L. Liu and J. Ye, J. Mater. Chem. A, 2019, 7, 9222 Search PubMed.
- J. Kim, Y. W. Lee, E.-G. Choi, P. Boonmongkolras, B. W. Jeon, H. Lee, S. T. Kim, S. K. Kuk, Y. H. Kim, B. Shin and C. B. Park, J. Mater. Chem. A, 2020, 8, 8496 Search PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Results of the Rietveld analysis of pristine β-FeOOH and 8-400; Fe K-edge XANES spectra of metallic Fe and α-Fe2O3; synchrotron XRD patterns of samples 2-200–6-400, 10-100–10-600, 12-100–12-800, pristine α-FeOOH, α-10-400, and α-10-500; TG curves of samples 2-200–6-400; XRD patterns after the TG analyses of pristine β-FeOOH, 8-100–8-500, and α-10-400; SEM images of samples 6-200, 8-200, and 12-200; SEM images of samples 12-200, 12-400, 12-500, and 12-700; particle distribution of pristine β-FeOOH, 8-400, and 12-500; fitting parameters of Cole–Cole plots; additional LSV curves; Mössbauer spectrum of sample α-10-400; SEM images and EDX mappings of pristine β-FeOOH; SEM images and EDX mappings of 8-400 before and after OER test.; crystal structure and dO–O of the three FeOOH polymorphs. See DOI: 10.1039/d0ra09895g |
| This journal is © The Royal Society of Chemistry 2020 |