
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced biomineralization and protein adsorption capacity of 3D chitosan/hydroxyapatite biomimetic scaffolds applied for bone-tissue engineering
Nguyen Kim Nga *a,
Lai Thi Thanh Tama,
Nguyen Thu Haa,
Pham Hung Vietb and
Tran Quang Huycd
*a,
Lai Thi Thanh Tama,
Nguyen Thu Haa,
Pham Hung Vietb and
Tran Quang Huycd
aSchool of Chemical Engineering, Hanoi University of Science and Technology, 1 Dai Co Viet Road, Hanoi, Vietnam. E-mail: nga.nguyenkim@hust.edu.vn; Fax: +84 24 38680 070; Tel: +84 24 38680 110
bResearch Center for Environmental Technology and Sustainable Development, Hanoi University of Science, 334 Nguyen Trai Street, Hanoi, Vietnam
cPhenikaa University Nano Institute (PHENA), Phenikaa University, Hanoi 12116, Vietnam
dFaculty of Electrical and Electronic Engineering, Phenikaa University, Hanoi 12116, Vietnam
First published on 26th November 2020
Abstract
This work presents the enhanced biomineralization and protein adsorption capacity of 3D chitosan/hydroxyapatite (CS/HAp) biomimetic scaffolds synthesized from natural sources applied for bone-tissue engineering (BTE). The scaffolds were prepared by the freeze-drying method, then characterized by X-ray diffraction, scanning electron microscopy, liquid substitution, swelling behavior, and mechanical strength. Fourier transform infrared spectroscopy was also conducted to investigate the interaction between chitosan (CS) and hydroxyapatite (HAp). The biodegradation, biomineralization and protein adsorption capabilities of the scaffolds were evaluated through tests in vitro. Results showed that the 3D CS/HAp scaffolds exhibited highly porous structures with an average pore size of 265 μm, and mean porosity of 75.01%, respectively; the tensile strength of the scaffolds was 2.45 MPa, matching well with that of cancellous bone. The addition of HAp into the CS matrix efficiently decreased the swelling percentage of the CS/HAp scaffolds and retained the suitable degradation rate of the composite scaffolds; the degradation percentage of the CS/HAp scaffolds was 46.37% after 28 days immersed in a physiological solution. The CS/HAp scaffolds demonstrated a higher biomineralization capability than that of the CS scaffolds, releasing a bone-like apatite layer on their surface after 15 days of incubation in simulated body fluids. The presence of HAp mimicking biological apatite in the composite scaffolds facilitated a higher protein adsorption capability, compared to that of the CS scaffolds. The obtained results suggest that the CS/HAp scaffolds have great potential as biocompatible materials for BTE applications.
1. Introduction
Scaffold-based tissue regeneration has become an important strategy in bone-tissue engineering (BTE) in recent years, in which a biodegradable matrix (scaffold) is combined with living cells and/or biologically active molecules to induce bone tissue repair and regeneration. This technique is a golden treatment method to the conventional grafting approaches, which relieves patients suffering from diseases and injuries.1 The biodegradable scaffold acts as a key template to support cell accommodation and direct their growth in three dimensions and degrades into biocompatible by-products over time.1,2 In this regard, the scaffolds must be biocompatible, biodegradable, and non-toxic. They must possess some characteristics, such as appropriate micro-architecture (e.g., porosity and pore size) and surface chemistry, to support cell metabolism, cell adhesion in vitro, growth, and phenotype preservation, thereby providing the necessary space for neovascularization in vivo.1–3 At present, numerous materials have been used for the preparation of scaffolds for bone tissue regeneration; three main types of materials have been classified, including polymers,4,5 bioceramics,6–8 and composites.9–12Natural bone exhibits a hierarchical structure consisting of collagen fibrils, and hydroxyapatite nanocrystallites (nHAp), which are the organic and inorganic components, respectively.13 For the past years, the development of artificial scaffolds that mimic the native bone in terms of the complex structure has attracted scientists in the field of BTE. Various kinds of synthetic and natural polymers have been combined with nHAp as biomimetic scaffolds for BTE applications.13–16 Chitosan (CS) is a natural linear polysaccharide that is composed of D-glucosamine and N-acetyl glucosamine units linked through β-(1-4) glycosidic bonds; it is derived from deacetylation of chitin found in the shells of marine crustaceans.17,18 The structural similarity of CS to glycosaminoglycans, which are the major component of the extracellular matrix (ECM) of bone, plays an important role in cell–cell adhesion by interacting with collagen fibers, thereby making CS one of the most attractive natural polymers used for BTE.19 CS scaffolds are osteoconductive, enhancing bone formation under in vitro and in vivo conditions.20 However, the lack of bone-bonding bioactivity, mechanical weakness, and incapacity to maintain a predefined shape limit their use in BTE.21–23 CS has been combined with other natural polymers, such as alginate and gelatin,22,24 as well as bioceramics, such as HAp, tricalcium phosphate, and nano SiO2, to improve the biological and mechanical properties of CS scaffolds.25–27 The incorporation of CS with nHAp as the mineral component of bone is typically based on the fabrication of biomimetic composite scaffolds, which combines the inherent characteristics of both CS and HAp for BTE.24–28 Several techniques have been used to prepare CS/HAp based scaffolds, such as electrospinning,28 freeze-drying,29 and spray drying.30 Moreover, the selection of a suitable technique plays an important role in the fabrication of biocompatible scaffolds, especially for three-dimensional (3D) structures.31 This work aims to prepare three-dimensional (3D) CS/HAp biomimetic scaffolds from natural sources using the freeze-drying technique to enhance their biomineralization and protein adsorption capacity for BTE applications. HAp nanoparticles, obtained by utilizing a bio-calcium source extracted from eggshells, following the procedure in our past work,32 were used as the inorganic component of the biomimetic scaffolds. CS flakes extracted from shrimp shells were used as the polymeric phase of the scaffolds. The obtained scaffolds were characterized by various physio-chemical methods. In contrast, their biodegradability, biomineralization capability and protein adsorption were evaluated through in vitro tests in phosphate-buffered saline (PBS), simulated body fluid (SBF), and fetal bovine serum (FBS), respectively.
2. Experimental
2.1. Materials
All reagents were of analytical grade and used without further purification. CS flakes exhibiting a degree of deacetylation of 85% and a low molecular weight were purchased from Nha Trang Aquatic Institute (Vietnam). Pre-treated chicken eggshells were used as the calcium precursor for preparing HAp nanoparticles. Sodium hydroxide (NaOH), hydrochloric acid, buffer solutions (pH 4, 7, and 9), acetic acid, and ethanol (C2H5OH) were purchased from Merck (Germany). Calcium chloride dihydrate (CaCl2·2H2O), sodium monophosphate dihydrate (Na2HPO4·2H2O), NaCl, NaHCO3, KCl, K2HPO4·3H2O, MgCl2·6H2O, Na2SO4, tris-hydroxymethyl aminomethane ((HOCH2)3CNH2), cetyltrimethylammonium bromide (CTAB), phosphate buffered saline (PBS), and sodium dodecyl sulfate (SDS) were purchased from Sigma-Aldrich. Fetal bovine serum (FBS) and minimum essential medium (MEM) were obtained from Gibco, USA. All solutions and reagents were prepared in deionized (DI) water.2.2. Methods
The X-ray diffraction (XRD) patterns of the CS and CS/HAp scaffolds were recorded on a Siemens D5005 diffractometer at the range of 2θ (20–70°) at a scan rate of 0.02° s−1 using Cu Kα radiation (λ = 0.15406 nm). Scanning electron microscopy (SEM, S4800, Hitachi, Japan) was used to observe the morphology of the scaffolds. Prior to measurement, the scaffold samples were mounted on a stub and then sputter-coated with a thin gold layer (Ion sputter E-1045, Hitachi). The average pore diameters of the scaffolds were then calculated through ImageJ software. Fourier transform infrared spectroscopy (Nicolet iS10, Thermo Scientific) with the attenuated total reflection method was conducted to investigate the expected functional groups, recorded in the region 500–4000 cm−1.
The porosity of the CS and CS/HAp scaffolds was determined by the liquid substitution method, which was described previously.11 DI water was used as the displacement liquid because it can easily move into scaffold pores without a dissolution reaction. Briefly, a dry sample of each scaffold was weighed (W1), after that it was immersed in a known volume (V1) of DI water inside a cylinder for 1 h to pervade water into the pores of the scaffold. The total volume of water and the water-impregnated scaffold was recorded as V2. The difference (V2 − V1) represents the volume of the scaffold skeleton. The water-impregnated scaffold was removed from the cylinder and weighed (W2), and the water volume remained was determined (V3). The total volume of the scaffold was given by V = (V2 − V1) + (V1 − V3) = (V2 − V3). The pore volume of the scaffold was calculated according to eqn (1), as follows:
| (W2 − W1)/ρH2O | (1) |
The porosity of the scaffold was determined using the following equation:
 | (2) |
Ten measurements were performed for each scaffold, and the results were the average of the 10 measurements.
The swelling capability of the CS and CS/HAp scaffolds was determined by immersing the samples in PBS. Before the experiments, the scaffolds were cut into small pieces of equal size (10 mm diameter). A dry sample of each scaffold with a known weight (Wd) was immersed in PBS at 37 °C for 24 h. After immersion, the sample was gently removed and blotted onto filter paper to remove the surface water. The wet weight of the swollen sample was recorded (Ww). The experiment was repeated 5 times for each scaffold. The swelling percentage of the scaffolds was calculated using the following equation:
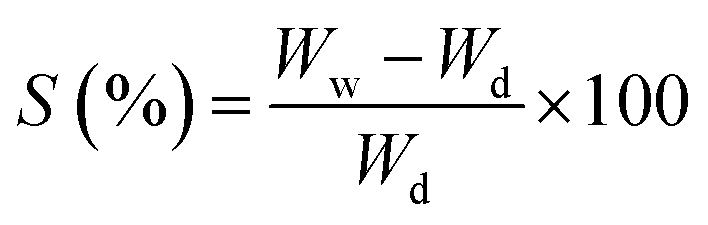 | (3) |
The mechanical properties of the scaffolds in terms of Young's modulus and tensile strength were determined using a Zwick Tensiler Z 2.5 testing machine at a crosshead speed of 1 mm min−1. Before measurement, the scaffold specimens were made in the form of thin films, which were then cut into a dumbbell shape with dimensions according to the ASTM D882 standard.
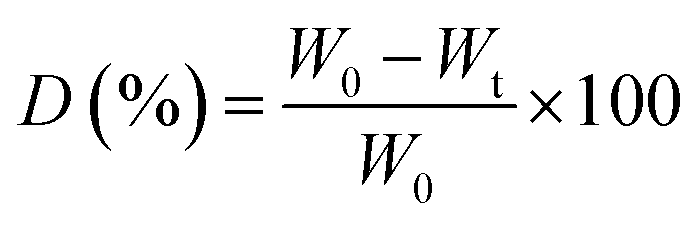 | (4) |
Changes in the pH of PBS solution were also measured over the testing time using a pH meter (HI 2211, Hanna Instruments, USA).
3. Results and discussion
3.1. 3D scaffold characterizations
3D scaffolds were synthesized using a freeze-drying method with one scaffold only containing CS and the other scaffold including CS and HAp. Some characteristics of the synthesized scaffolds are summarized in Table 1. The digital camera images of the scaffolds are shown in Fig. 1A, indicating the formation of 3D CS and CS/HAp scaffolds 40 mm in diameter and 20 mm in height. | ||
| Fig. 1 (A) Digital camera images of the 3D CS and CS/HAp scaffolds produced. (B) XRD patterns of various samples: (a) the CS scaffold, (b) the CS/HAp scaffold, and (c) HAp. | ||
The XRD patterns of the scaffolds were measured to identify their structural phase. XRD patterns of the CS and CS/HAp scaffolds and the XRD pattern of HAp nanoparticles for comparison are shown in Fig. 1B. A broad reflection around 2θ = 20° was observed in the XRD patterns of the CS [Fig. 1B(a)] and CS/HAp [Fig. 1B(b)] scaffolds, which was assigned to the presence of CS as an amorphous phase in both scaffolds. Apart from the reflection of CS, typical reflections at 2θ = 26.09°, 31.68°, 32.27°, 34.13°, 39.67°, 46.64°, 49.52°, and 53.54° could be seen in the XRD pattern of the CS/HAp scaffolds, which matched well with the JCPDS file no. 09-432 of HAp and can be indexed to the (002), (211), (112), (202), (310), (222), (213), and (004) planes, respectively. The observations confirmed the presence of the HAp crystalline phase in the composite CS/HAp scaffolds. The XRD pattern of the pure HAp nanoparticles [Fig. 1B(c)] shows typically crystalline reflections with high intensity, located at 2θ = 25.85°, 31.67°, 32.15°, 33.98°, 39.65°, 46.61°, 49.43°, and 53.24°. With regards to the typical reflections of HAp in the CS/HAp scaffolds, those in the composite scaffolds were slightly shifted to higher values than those of pure HAp. The intensity of those peaks of HAp significantly decreases in the composite scaffolds [Fig. 1B(b and c)]. The XRD results suggested that some molecular interactions between CS and HAp may have occurred, and HAp nanoparticles were integrated into the CS matrix to form composite scaffolds.
The surface morphology and pore architecture of the scaffolds were further investigated through SEM imaging. Fig. 2 represents the SEM images of the 3D CS and CS/HAp scaffolds at various magnifications. The low magnification SEM images (Fig. 2a and d) showed that both scaffolds exhibited a highly porous structure. The higher-resolution images indicated that a smooth surface morphology was observed for the CS scaffolds (Fig. 2b), whereas a rough surface morphology was produced for the CS/HAp scaffolds (Fig. 2e) because of the addition of HAp into the polymer matrix. The highest-resolution SEM image of the CS–HAp scaffolds (Fig. 2f) revealed that the HAp nanoparticles were distributed uniformly and compactly within the CS matrix, and no interface between the HAp and CS phases was observed, thereby confirming that the HAp nanoparticles exhibited a high affinity with the CS matrix. The pore walls of the CS/HAp scaffolds were observed to be thicker than those of the CS scaffolds (see Fig. 2c and f) because of the integration of HAp into the CS matrix. Further calculations based on the SEM images using ImageJ software showed that the mean pore sizes of the CS and CS/HAp scaffolds were 330 and 265 μm, which ranged from 230–457 μm to 133–420 μm (Table 1 and Fig. 3). Statistical analyses demonstrated that the pore sizes of CS/HAp scaffolds were significantly smaller than those of the CS scaffolds (p < 0.001, Fig. 3). Pores in the scaffolds play a key role in bone-tissue growth. Sufficiently large pores (greater than 100 μm) can support osteoblast cells with typical sizes of 10–30 μm to migrate and proliferate.34,35 Therefore, the scaffolds of our study contained appropriate pore sizes to support cell migration, proliferation, and further tissue growth.
 | ||
| Fig. 2 SEM images of the CS scaffolds (a), (b), and (c) and CS/HAp scaffolds (d), (e), and (f) at different magnifications of 30, 100 and 150, respectively. | ||
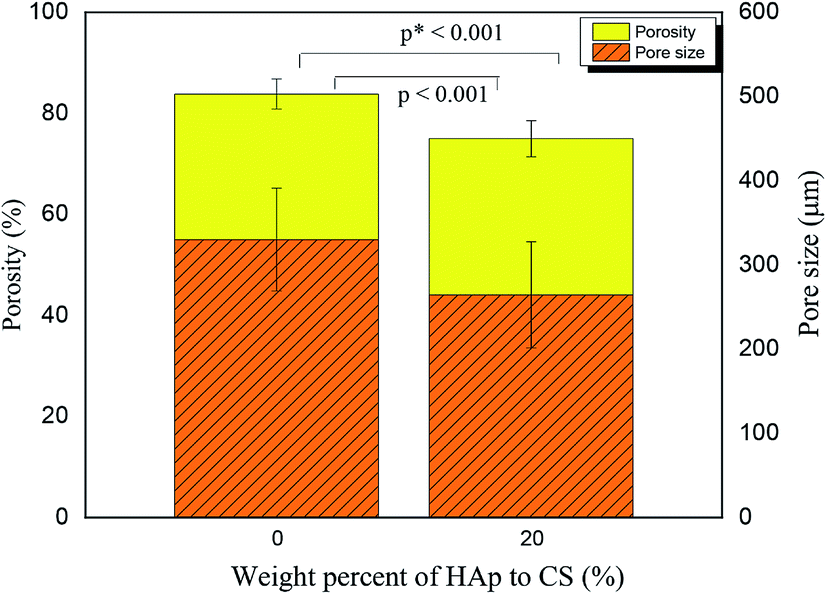 | ||
| Fig. 3 Porosity and pore sizes of CS and CS/HAp. | ||
The porosities of the CS and CS/HAp scaffolds were determined through the liquid substitution method. The porosities of the scaffolds were relatively higher than 70% with values of 83.82% ± 2.97% and 75.01% ± 3.58% for the CS and CS/HAp scaffolds, respectively. The comparison (Fig. 3) showed that the porosity of the CS/HAp scaffolds was significantly lower than that of the CS scaffolds (p* < 0.001) because of the integration of HAp nanoparticles into the CS matrix. A scaffold porosity must be at least 90% to provide a high surface area to volume ratio for cell–polymer interactions, to have sufficient space for ECM formation, and to obtain minimal diffusion constraints during cell culture.36,37 However, a compromise between porosity and scaffold mechanical properties is often made. High porosity (e.g., 90%) may decrease the mechanical properties of the scaffolds. Scaffolds with porosities at moderate levels of 60–75% were used for bone growth because of the increased mechanical properties at those porosities.38,39 The CS/HAp scaffolds exhibited optimal porosities that make them good candidates for use as bone scaffolds.
FTIR analyses were further performed to examine polymer–HAp interactions. The characteristic bands of CS and CS/HAp scaffolds are presented in Fig. 4A and summarized in Table 2. The FTIR spectrum of CS scaffolds [Fig. 4A(a)] exhibited characteristic bands at 3360 and 3289 cm−1, which were attributed to the N–H stretching vibration of NH2 group, whereas the band at 1644 and 1560 cm−1 can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of amide I and the N–H bending vibration in combination with C–N stretching (amide II), respectively. The band at 2879 cm−1 indicated the C–H stretching vibrations of –CH2, –CH3, whereas three small bands at 1416, 1373, and 1317 cm−1 showed the C–H bending vibrations of –CH2, –CH3, and the pyranose ring, respectively. The bands at 1148, 1059, and 1026 cm−1 were assigned to the asymmetric and symmetric C–O stretching vibrations of C–O–C linkage, and the small band at 893 cm−1 corresponded to the vibration of the saccharide structure of the CS scaffolds. Comparing the FTIR spectrum of the CS/HAp scaffolds [Fig. 4A(b)] with the CS scaffolds [Fig. 4A(a)], all the characteristic bands of CS are present in the composite scaffolds. Nevertheless, all the bands of the CS/HAp scaffolds were slightly shifted to higher values than those of the CS scaffolds (Table 2). Moreover, the new bands at 945, 602, 560, and 633 cm−1 were observed in the FTIR spectrum of the CS/HAp scaffolds. The band at 945 cm−1 characterized a symmetric P–O stretching vibration of PO43− in HAp. The bands at 602 and 560 cm−1 were attributed to a doubly degenerate P–O bending vibration of PO43−. The band at 633 cm−1 represented the O–H liberational vibration of HAp. However, the typical band at 3570 cm−1 corresponded to the O–H stretching vibration of HAp,32 which was not seen in the FTIR spectrum of CS/HAp, which was possibly due to the fact that this band overlapped with the N–H stretching vibration of –NH2. The observations suggested that the CS phase served as a matrix for the HAp nanoparticles and provided anchoring sites for the HAp particles, binding them together in the composite.
O stretching vibration of amide I and the N–H bending vibration in combination with C–N stretching (amide II), respectively. The band at 2879 cm−1 indicated the C–H stretching vibrations of –CH2, –CH3, whereas three small bands at 1416, 1373, and 1317 cm−1 showed the C–H bending vibrations of –CH2, –CH3, and the pyranose ring, respectively. The bands at 1148, 1059, and 1026 cm−1 were assigned to the asymmetric and symmetric C–O stretching vibrations of C–O–C linkage, and the small band at 893 cm−1 corresponded to the vibration of the saccharide structure of the CS scaffolds. Comparing the FTIR spectrum of the CS/HAp scaffolds [Fig. 4A(b)] with the CS scaffolds [Fig. 4A(a)], all the characteristic bands of CS are present in the composite scaffolds. Nevertheless, all the bands of the CS/HAp scaffolds were slightly shifted to higher values than those of the CS scaffolds (Table 2). Moreover, the new bands at 945, 602, 560, and 633 cm−1 were observed in the FTIR spectrum of the CS/HAp scaffolds. The band at 945 cm−1 characterized a symmetric P–O stretching vibration of PO43− in HAp. The bands at 602 and 560 cm−1 were attributed to a doubly degenerate P–O bending vibration of PO43−. The band at 633 cm−1 represented the O–H liberational vibration of HAp. However, the typical band at 3570 cm−1 corresponded to the O–H stretching vibration of HAp,32 which was not seen in the FTIR spectrum of CS/HAp, which was possibly due to the fact that this band overlapped with the N–H stretching vibration of –NH2. The observations suggested that the CS phase served as a matrix for the HAp nanoparticles and provided anchoring sites for the HAp particles, binding them together in the composite.
 | ||
| Fig. 4 (A) FTIR spectra of (a) the CS scaffolds and (b) CS/HAp scaffolds. (B) Swelling percentage of the CS and CS/HAp scaffolds. Values are mean ± SD (n = 5). **Significant differences compared to the CS/HAp scaffolds. | ||
| Vibration position, cm−1 | Assignments | |
|---|---|---|
| CS | CS/HAp | |
| 3360 cm−1 and 3289 cm−1 | 3361 cm−1 and 3290 cm−1 | N–H stretching vibration of –NH2 |
| 1644 cm−1 | 1645 cm−1 | CO stretching vibration of amide I |
| 1560 cm−1 | 1562 cm−1 | N–H bending vibration of –NH2 (amide II) |
| 2879 cm−1 | 2881 cm−1 | C–H stretching vibration of –CH2, –CH3 |
| 1416 cm−1, 1373 cm−1, and 1317 cm−1 | 1417 cm−1, 1375 cm−1, and 1320 cm−1 | C–H bending vibration of –CH2, –CH3, and pyranose |
| 1148 cm−1, 1059 cm−1, 1026 cm−1 | 1149 cm−1, 1024 cm−1 | Asym. and sym. C–O stretching vibration of C–O–C linkage |
| 945 cm−1 | Sym. P–O stretching vibration of PO43− in HAp | |
| 893 cm−1 | 894 cm−1 | Saccharide vibration |
| 602 cm−1, 560 cm−1 | P–O bending vibration of PO43- | |
| 633 cm−1 | O–H liberational vibration in HAp | |
Swelling percentage serves as one of the key features of biomaterials in BTE. When the scaffolds are implanted into a defective location, they are exposed to body fluids and may swell. The swelling characterizes the capability of fluid uptake by the scaffolds under physiological conditions. The swelling can affect the shape and mechanical stability of the scaffolds over time. For instance, during cell culture, the swelling results in an increase of the pore sizes, which facilitates the easy diffusion of nutrients, thus enhancing the cell infiltration and attachment. However, excessive swelling can reduce mechanical strength, thereby rendering it unsuitable in bone tissue applications.40 The swelling behavior of the CS and CS/HAp scaffolds was studied by immersing these scaffolds in PBS for 24 h, and the results obtained are shown in Fig. 4B. As shown in Fig. 4B, the swelling percentage of the CS scaffolds was 71.03% ± 6.21%, whereas that of the CS/HAp scaffolds was 55.40% ± 5.61%. Overall, the fluid uptake of the CS scaffold was significantly higher than those of the CS/HAp scaffolds (p** < 0.01). CS is a natural polymer with a high percentage of swelling, which limits its applications to the preparation of BTE scaffolds. The addition of HAp nanoparticles into the CS matrix was efficient in decreasing the swelling percentage of the composite CS/HAp scaffold.
The mechanical properties of the CS and CS/HAp scaffolds are shown in Table 1. It was observed that the Young's modulus of the CS/HAp scaffolds (12.74 MPa) was comparable to that of the CS scaffolds (13.71 MPa). However, the tensile strength of the CS/HAp scaffolds was twice as high as that of the CS scaffolds (their tensile strengths are 2.45 MPa and 1.21 MPa, respectively). The maximum stress which the CS/HAp scaffolds can withstand before fracturing was much improved compared to that of the CS scaffolds. The results revealed that the incorporation of HAp nanoparticles in the CS matrix almost does not change the elastic modulus, but significantly increased the tensile strength of the composite scaffolds. It is reported that the tensile strength of cancellous bone ranges from 1 to 5 MPa.41 Therefore, the tensile strength of the CS/HAp scaffolds can match very well with that of cancellous bone. Thus, the CS/HAp scaffolds are appropriate biomaterials in BTE applications.
3.2. In vitro biodegradation investigation
In BTE, the scaffolding materials act as a 3D template mimicking the natural ECM for cell attachment, proliferation, and tissue growth. During the process of tissue regeneration, the matrix molecules secreted by the cells assemble to form a new ECM while the scaffold degrades.42,43 The biodegradation behavior of the scaffold plays a crucial role in the long-term performance of the tissue-engineered material construct because it provides space for tissue ingrowth and matrix deposition. An ideal scaffold must possess a controllable biodegradation rate that matches the rate of tissue regeneration to be used in BTE.44 Therefore, studies on the degradation rate of biomaterials are very important for their further applications. In this study, biodegradation tests were conducted in lysozyme containing PBS solution and the results are shown in Fig. 5a. The lysozyme concentration of 0.1 mg mL−1 was chosen to better mimic the in vivo physiological conditions because lysozyme has been found in various body fluids, such as serum and tears, with concentrations ranging from 4–13 mg L−1 and 450–1230 mg L−1, respectively.45 As shown in Fig. 5a, after only 2 days of incubation, weight loss of the scaffolds was already observed; the degradation percentages of the CS and CS/HAp scaffolds were 10.02% and 9.65%, respectively. Afterwards, the scaffolds showed continuous weight loss by increasing the incubation time up to 28 days, which indicates that the scaffolds degraded with time. Comparing the weight loss of the CS and CS/HAp scaffolds, the degradation of the CS/HAp scaffolds was slower than that of the CS scaffolds for all the tested time points. This finding may be attributed to the addition of HAp nanoparticles into the CS matrix. The addition of nano-HAp reduced the degradation rate of the composite scaffolds.46,47 In our work, the uniform dispersion of the HAp within the CS matrix led to form the compact structure of the composite scaffolds; however, the degradation rate of the composite scaffolds can be compared with that of the CS scaffolds, which is the advantage of the CS/HAp scaffolds in the present study. Our results revealed that the CS and CS/HAp scaffolds were degraded by 68.5% and 46.37% (Fig. 5a) after 28 days of degradation in vitro. The degradation profile of these scaffolds may provide a consistent time for the formation of neo-tissue and the ECM during the course of tissue regeneration.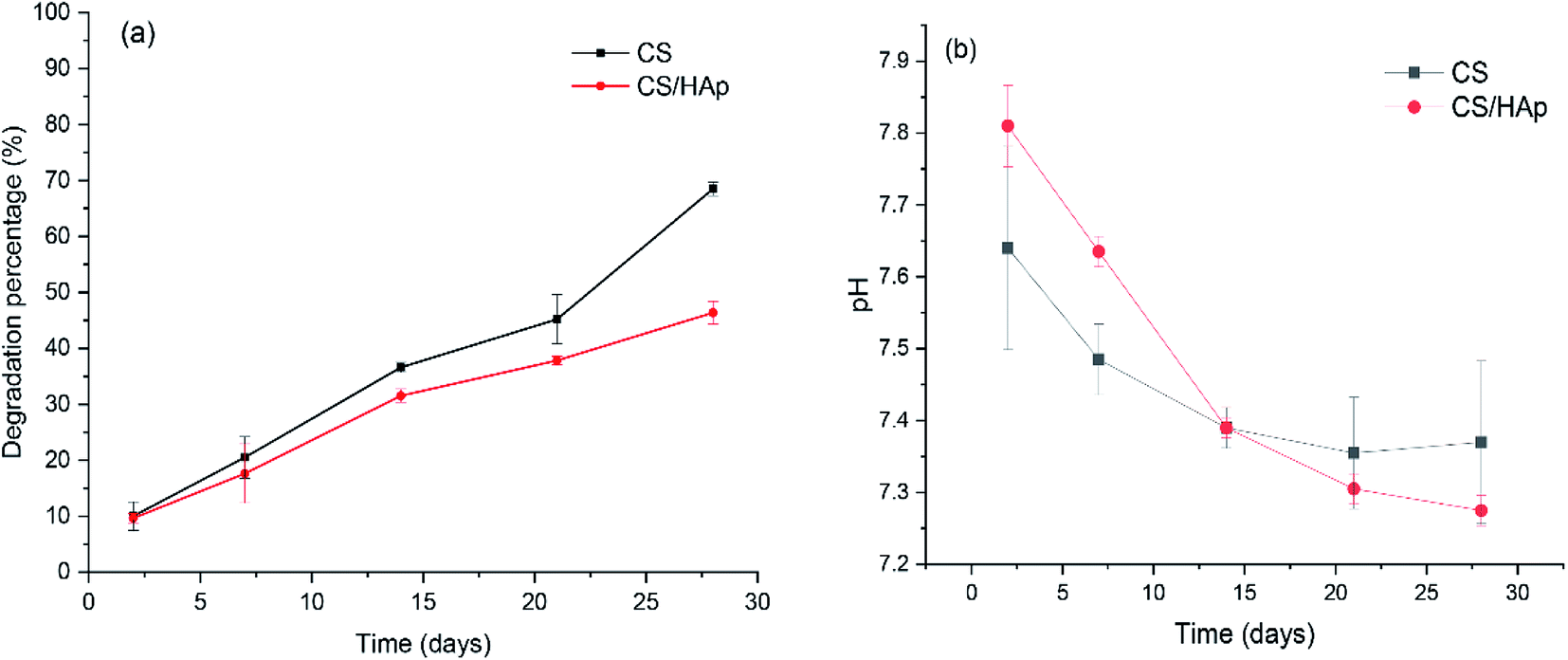 | ||
| Fig. 5 (a) In vitro biodegradation of the CS and CS/HAp scaffolds in lysozyme containing PBS solution at 37 °C for various incubation periods (2, 7, 14, 21, and 28 days). (b) pH of the PBS solution in which the scaffold samples were incubated vs. time of incubation. | ||
The solubility of the scaffolds in physiological environments can result in variation in pH and may negatively affect the cell response and its proliferation.48 The pH of the PBS solution, in which the scaffolds were incubated, was controlled over the testing time points (Fig. 5b). As shown in Fig. 5b, a slight increase in the pH of the PBS solution with respect to the initial pH 7.4 can be observed in both scaffolds in the first week of incubation. This increase in pH of the incubation medium could be attributed to the alkalescent nature of CS. During the second week of incubation, the pH of the PBS solution slowly decreased to 7.39 (Fig. 5b), which could be due to a gradual dissolution of CS in these scaffolds, indicated by the corresponding weight loss (see Fig. 5a). From the second week onward, the pH of the PBS solution decreased slowly from pH 7.39 to pH 7.37 for the CS scaffolds, but a quicker decrease in pH from 7.39 to 7.28 was observed for the composite scaffolds. This could be explained by the release of Ca2+ ions, which are acidic in nature, into the incubation medium from dissolving of the HAp phase in the composite samples, which led to the decrease in pH of the incubation medium. The results showed that the pH of the PBS solution varied little, decreasing to 7.28 from its initial value of 7.4, and remained close to that of the physiological environment, which indicated that these scaffolds maintain a highly stable biomimetic medium for the degradation process.
3.3. Biomineralization in vitro
The essential requirement for a synthetic biomaterial to bond to living bone is the formation of bone-like apatite, so-called biomineralization on its surface when implanted in the living body. In this study, the biomineralization capabilities of the CS and CS/HAp scaffolds were examined through tests in vitro by incubating the scaffold samples in SBF solution that mimics the electrolyte composition of human blood plasma. Fig. 6 shows that the surface morphologies of the CS/HAp scaffolds were significantly changed after being incubated in SBF for 10 and 15 days. However, hardly any change was observed on the CS scaffold's surface (not shown), indicating the poor biomineralization of the CS scaffolds. The SEM image in Fig. 6a demonstrated that a mineral-like layer was already produced on the surface of the CS/HAp scaffolds after 10 days of incubation in SBF. The SEM image with a higher magnification of 10k (Fig. 6b) indicated that the mineral layer consisted of numerous flower-like particles with a mean diameter of about 1.15 μm; however, most of them did not show the entire flower-like shape, confirming that the biomineralization process was being occurred on the scaffold's surface. Next, the SEM image of a lower magnification of 2k (Fig. 6c) showed that the mineral layer was deposited all over the entire surface of the scaffold after 15 days in SBF. The SEM images with magnifications of 5k and 10k (Fig. 6d and e) further revealed that the mineral layer is composed of many flower-like mineral crystals; their mean diameter increased to 1.54 μm with a longer incubation time. The structure of each flower-like mineral crystal was clearly observed in the SEM image with 40k magnification (Fig. 6f), which was formed as the result of the association of many tiny needle-like particles. The morphology of the mineral layers produced on the scaffold's surface is typically observed for biological apatite in the bone tissue. | ||
| Fig. 6 SEM images of the CS/HAp scaffolds after incubating in SBF for 10 days (a and b), with magnifications of 5k and 10k and for 15 days (c, d, e, and f) with various magnifications of 2k, 5k, 10k and 50k, respectively. | ||
The chemical composition of the mineral layers released on the scaffolds after 10 and 15 days in SBF was analyzed through EDX analyses. The results summarized in Table 3 show that O, P, Ca, and C were the main components of both mineral layers obtained after 10 and 15 days in SBF. The presence of C can possibly be attributed to the CO32− phase and the presence of the CS matrix. Moreover, trace amounts of Na and Mg were detected as minor elements in these mineral layers, which was attributed to the trace elements in the HAp phase of the composite scaffolds, prepared from eggshells. In contrast, Cl was also detected as a minor element in the mineral layers, which could be due to a residue of the SBF and was incompletely eliminated from the scaffold samples through washing. It is known that the Ca/P ratio is a key characteristic used to identify biological minerals that can be produced under in vitro biomimetic conditions. During the incubation period, the scaffold surfaces are exposed to the biomimetic environment, which resulted in the uptake of Ca2+ and PO43− ions from the SBF solution to form biological calcium phosphate minerals. As shown in Table 3, the Ca/P ratios were 1.78 and 1.52 for the mineral layers released on the scaffold surfaces after 10 and 15 days in SBF, respectively. These values show deviations from the stoichiometric Ca/P ratio of 1.67 for stoichiometric HAp, suggesting that carbonated hydroxyapatite was released on the scaffold's surfaces after 10 and 15 days in SBF. Both SEM and EDX confirm that a bone-like apatite layer was completely formed on the surfaces of the composite CS/HAp scaffolds after 15 days of incubation in SBF.
Table 4 compares the biomineralization capability of CS/HAp scaffolds as prepared in this work with other composite scaffolds reported previously. As showed in Table 4, the bone-like apatite layer was completely produced on the surface of the CS/HAp scaffolds after 15 days of incubation in SBF. However, the previous works demonstrated that the bone-like mineral layer can be produced on the composite scaffold's surface after quite a long time of incubation in SBF. For instance, the initial formation of the mineral phase can only be observed on the surface of the CS–gelatin/HAp scaffolds after 14 days in SBF,49 while CS–alginate/SiO2 scaffolds showed the increased deposition of apatite crystals on their surface after 21 days of incubation.27 S. Srinivasan et al.50 indicated that alginate/nano-bioactive glass-ceramic composite scaffolds regenerated an apatite rich layer on their surface after 21 days of incubation in SBF. The apatite layer deposition was also prominently observed on the surfaces of chitin/nano-HAp and alginate/bioactive glass containing Zn and Mg scaffolds after 21 days of soaking in SBF.51,52 On the other hand, a mineral layer consisting of aggregated flower-like particles was formed on the surface of the β-TCP/CS–fucoidan scaffolds after 28 days of incubation.53 Comparison of the results revealed that the biomineralization greatly depends on the inorganic component of the scaffolds. In the present work, it was found that the CS/HAp scaffolds exhibited better biomineralization in vitro than the other scaffolds. This could be attributed to the presence of a biomimetic inorganic component in the scaffolds' structure. The HAp nanoparticles of the present work used as an inorganic component for fabricating CS/HAp scaffolds, produced from eggshells not only have a similar size and shape, but also have a similar composition to biological apatite.32 The trace elements such as Mg, K, and Na, which were detected in the inorganic component of the CS/HAp scaffolds play a vital role in promoting rapid formation of the bone-like apatite layer on the surface of the CS/HAp scaffolds. Although the HAp nanoparticles used for the scaffold fabrication in previous studies,49,51 synthesized from chemicals have the closest size and shape to biological apatite, they released bone-like mineral layers on the scaffold's surfaces after a longer time than the HAp nanoparticles of the present work, which was attributed to the lack of trace elements in their composition.
| Scaffolds | Biomineralization capacity in terms of regenerating a bone-like apatite layer | Ref. | Scaffolds | Amount of protein adsorbed after 24 h, μg | Ref. |
|---|---|---|---|---|---|
| CS/HAp | 15 days, completed | This work | CS/HAp | 742 | This work |
| CS–gelatin/HAp | 14 days, initially formed | 49 | PLLA/HAp | 500 | 55 |
| CS/alginate/SiO2 | 21 days, completed | 27 | PLLA/CS | 150 | 56 |
| Alginate/nano-bioactive glass ceramic | 21 days, completed | 50 | CS/SiO2–ZrO2 | 100 | 57 |
| Chitin/nano-HAp | 21 days, completed | 51 | CS/alginate–fucoidan | 90 | 58 |
| Alginate/bioactive glass containing Mg–Zn | 21 days, completed | 52 | CS/nano-HAp | 16 | 59 |
| β-TCP/CS–fucoidan | 28 days, completed | 53 | CS/HAp/nCu–Zn | 20 | 59 |
3.4. Protein adsorption
The behavior of proteins on surfaces plays a vital role in determining the nature of the tissue–implant interface. Before cells adhere to the surface of biomaterials, protein is adsorbed from bodily fluids, and such protein adsorption may control subsequent cell adhesion and behavior. In this study, the protein adsorption behavior of the CS and CS/HAp scaffolds was investigated by incubating the scaffold samples in MEM supplemented with FBS containing different serum proteins. The quantity of proteins adsorbed onto the scaffold surfaces was measured for various incubation intervals of 2, 4, 6, and 24 h (Fig. 7). The relatively high amount of proteins of 128 ± 20 and 211 ± 18 μg adsorbed on the CS and CS/HAp scaffolds could be observed after the initial incubation period of 2 h, respectively. When the incubation time increased up to 4 h, the quantity of the proteins increased to the high values of 260 ± 52 and 389 ± 58 μg for the CS and CS/HAp scaffolds, respectively. By increasing the incubation time to 6 and 24 h, the quantity of proteins adsorbed onto the CS/HAp scaffolds experienced rapid growth to a significantly high value of 607 ± 47 and 742 ± 82 μg, respectively. The similar trend of increased protein adsorption was observed on CS scaffolds. The amount of protein adsorption for the CS scaffolds was determined as 338 ± 42 and 411 ± 35 μg, corresponding to 6 and 24 h of incubation, respectively. The kinetics of protein adsorption to solid surfaces includes a very rapid initial phase, followed by a slow phase upon approach to the steady-state value. Initially, proteins adsorb as quickly as they arrive at the largely empty surface. Later, in a slower phase, the arriving proteins may have difficulty finding and fitting into an empty spot on the surface.54 The protein adsorption of the scaffolds followed rapid adsorption kinetics. This feature is the reason why the amount of protein absorbed on the CS and CS/HAp scaffolds showed a slight increase beyond 6 h as the high amount of proteins became attainable. Statistical analysis in Fig. 7 indicates that the quantity of proteins adsorbed on the CS/HAp scaffolds was significantly higher than those of the CS scaffolds for all incubation periods (p*** < 0.001). The significant increase in the proteins adsorbed on the composite scaffolds was attributed to the presence of the HAp nanoparticles. The addition of the HAp may modify the characteristics of the scaffold surfaces (e.g., chemical composition, electric charge, and morphology), thereby greatly increasing the quantity of active sites on the composite scaffolds for possible interactions with proteins.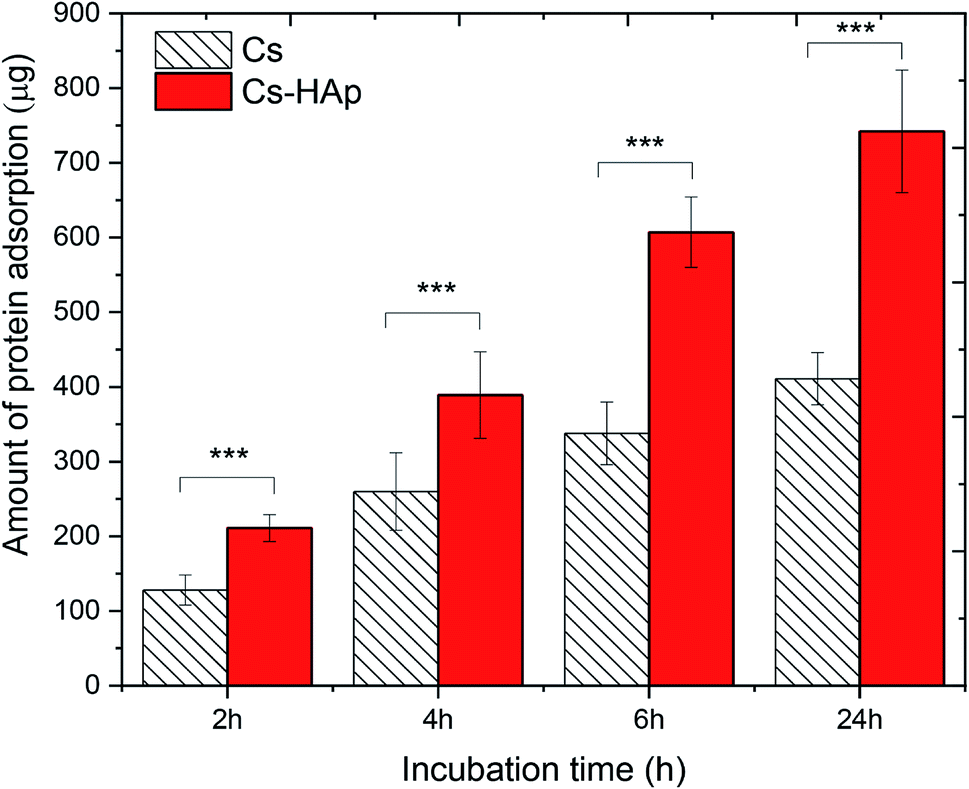 | ||
| Fig. 7 Protein adsorption studies of the CS and CS/HAp scaffolds in MEM containing 10% FBS at 37 °C for 2 h, 4 h, 6 h, and 24 h. All data expressed as mean ± SD, n = 5. ***Significant differences compared to the CS/HAp scaffolds. | ||
Table 4 compares the protein adsorption capability of the CS/HAp scaffolds of the present work with that of other scaffolds published previously. The comparison showed that the quantity of proteins adsorbed on the CS/HAp scaffolds of our work after 24 h was 742 μg, which was much higher than those of PLLA/HAp (500 μg),55 PLLA/CS (150 μg),56 CS/SiO2–ZrO2 (100 μg),57 CS/alginate–fucoidan (90 μg),58 CS/HAp/Cu–Zn (20 μg), and CS/nano-HAp.59 The much higher adsorption capability of the CS/HAp compared with the other scaffolds was possible because they possessed a more suitable pore structure (e.g., pore size and pore surface morphology). The CS/HAp scaffolds exhibited an inorganic component mimicking biological apatite, which is beneficial for increased protein adsorption. The observations suggested the higher biocompatibility of our scaffolds in terms of cell adhesion and proliferation compared with the scaffolds of the previous studies.
4. Conclusions
This work demonstrates that 3D CS/HAp biomimetic scaffolds have been successfully prepared from natural sources. The addition of nano-HAp significantly improved the characteristics, bioactivity, and biocompatibility of the 3D CS/HAp scaffolds. The CS/HAp scaffolds exhibited a suitable pore architecture, swelling ratio, tensile strength, and biodegradation capability, which are essential in bone scaffolds. Moreover, the present work revealed that the CS/HAp scaffolds showed better biomineralization in vitro and a much higher protein adsorption capability than those of the previous works. The inorganic component mimicking biological apatite plays a crucial function in the enhanced biomineralization and protein adsorption capability of the 3D CS/HAp biomimetic scaffolds. Hence, this work suggests that the CS/HAp composite scaffolds are potential biomaterials for BTE applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was funded by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.03-2019.313.References
- H. Qu, H. Fu, Z. Han and Y. Sun, Biomaterials for bone tissue engineering scaffolds, RSC Adv., 2019, 9, 26252–26262 RSC.
- J. Mitra, G. Tripathi, A. Sharma and B. Basu, Scaffolds for bone tissue engineering: role of surface patterning on osteoblast response, RSC Adv., 2013, 3, 11073–11094 RSC.
- S. Mondal and U. Pal, 3D hydroxyapatite scaffold for bone regeneration and local drug delivery applications, J. Drug Delivery Sci. Technol., 2019, 53, 101131 CrossRef CAS.
- Y. B. Kim and G. H. Kim, Highly roughened polycaprolactone surfaces using oxygen plasma-etching and in vitro mineralization for bone tissue regeneration: fabrication, characterization, and cellular activities, Colloids Surf., B, 2015, 125, 181–189 CrossRef CAS.
- S. Shahriarpanah, J. Nourmohammadi and G. Amoabediny, Fabrication and characterization of carboxylated starch-chitosan bioactive scaffold for bone regeneration, Int. J. Biol. Macromol., 2016, 93, 1069–1078 CrossRef CAS.
- N. Ma, B. Ma, Y. Zhou, H. Zhu, Y. Zhou, Z. Huan, P. Wang and J. Chang, In vivo evaluation of the subchronic systemic toxicity of akermanite bioceramic for bone regeneration following ISO standard methods, RSC Adv., 2019, 9, 17530–17536 RSC.
- T. T. Hoai, N. K. Nga, L. T. Giang, T. Q. Huy, P. N. M. Tuan and B. T. T. Binh, Hydrothermal Synthesis of Hydroxyapatite Nanorods for Rapid Formation of Bone-Like Mineralization, J. Electron. Mater., 2017, 46(8), 5064–5072 CrossRef CAS.
- M. Sadat-Shojai, M. T. Khorasani and A. Jamshidi, Synthesis methods for nanosized hydroxyapatite with diverse structures, J. Cryst. Growth, 2012, 361, 73–84 CrossRef CAS.
- C. Sharma, A. K. Dinda, P. D. Potdar, C. F. Chou and N. C. Mishra, Fabrication and characterization of novel nano-biocomposite scaffold of chitosan–gelatin–alginate–hydroxyapatite for bone tissue engineering, Mater. Sci. Eng., C, 2016, 64, 416–427 CrossRef CAS.
- T. T. Hoai and N. K. Nga, Effect of pore architecture on osteoblast adhesion and proliferation on hydroxyapatite/poly(D,L) lactic acid-based bone scaffolds, J. Iran. Chem. Soc., 2018, 15(7), 1663–1671 CrossRef CAS.
- J. Wu, Z. Wu, Z. Xue, H. Li and J. Liu, PHBV/bioglass composite scaffolds with co-cultures of endothelial cells and bone marrow stromal cells improve vascularization and osteogenesis for bone tissue engineering, RSC Adv., 2017, 7, 22197–22207 RSC.
- A. Shavandi, A. E. D. A. Bekhit, Z. Sun, A. Ali and M. Gould, A novel squid pen chitosan/hydroxyapatite/β-tricalcium phosphate composite for bone tissue engineering, Mater. Sci. Eng., C, 2015, 55, 373–383 CrossRef CAS.
- S. Sathiyavimal, S. Vasantharaj, F. L. Oscar, R. Selvaraj, K. Brindhadevi and A. Pugazhendhi, Natural organic and inorganic–hydroxyapatite biopolymer composite for biomedical applications, Prog. Org. Coat., 2020, 147, 105858 CrossRef CAS.
- X. Cai, H. Tong, X. Shen, W. Chen, J. Yan and J. Hu, Preparation and characterization of homogeneous chitosan–polylactic acid/hydroxyapatite nanocomposite for bone tissue engineering and evaluation of its mechanical properties, Acta Biomater., 2009, 5, 2693–2703 CrossRef CAS.
- M. T. Arafat, C. X. F. Lam, A. K. Ekaputra, S. Y. Wong, X. Li and I. Gibson, Biomimetic composite coating on rapid prototyped scaffolds for bone tissue engineering, Acta Biomater., 2011, 7, 809–820 CrossRef CAS.
- S. Liu, S. Qin, M. He, D. Zhou and H. Wang, Current applications of poly(lactic acid) composites in tissue engineering and drug delivery, Composites, Part B, 2020, 199, 108238 CrossRef.
- S. Ahmed, Annu, A. Ali and J. Sheikh, A review on chitosan centred scaffolds and their applications in tissue engineering, Int. J. Biol. Macromol., 2018, 116, 849–862 CrossRef CAS.
- F. Heidari, M. E. Bahrololoom, D. Vashaee and L. Tayebi, In situ preparation of iron oxide nanoparticles in natural hydroxyapatite/chitosan matrix for bone tissue engineering application, Ceram. Int., 2015, 41(2), 3094–3100 CrossRef CAS.
- R. LogithKumar, A. KeshavNarayan, S. Dhivya, A. Chawla, S. Saravanan and N. Selvamurugan, A review of chitosan and its derivatives in bone tissue engineering, Carbohydr. Polym., 2016, 151, 172–188 CrossRef CAS.
- B. M. Chesnutt, Y. Yuan, K. Buddington, W. O. Haggard and J. D. Bumgardner, Composite chitosan/nano-hydroxyapatite scaffolds induce osteocalcin production by osteoblasts in vitro and support bone formation in vivo, Tissue Eng., Part A, 2009, 15(9), 2571–2579 CrossRef CAS.
- S. Ranganathan, K. Balagangadharan and N. Selvamurugan, Chitosan and gelatin-based electrospun fibers for bone tissue engineering, Int. J. Biol. Macromol., 2019, 133, 354–364 CrossRef CAS.
- D. Vukajlovic, J. Parker, O. Bretcanu and K. Novakovic, Chitosan based polymer/bioglass composites for tissue engineering applications, Mater. Sci. Eng., C, 2019, 96, 955–967 CrossRef CAS.
- A. Muxika, A. Etxabide, J. Uranga, P. Guerrero and K. de la Caba, Chitosan as a bioactive polymer: processing, properties and applications, Int. J. Biol. Macromol., 2018, 105(2), 1358–1368 Search PubMed.
- F. Zhao, W. L. Grayson, T. Ma, B. Bunnell and W. W. Lu, Effects of hydroxyapatite in 3-D chitosan–gelatin polymer network on human mesenchymal stem cell construct development, Biomaterials, 2006, 27, 1859–1867 CrossRef CAS.
- W. W. Thein-Han and R. D. K. Misra, Biomimetic chitosan–nanohydroxyapatite composite scaffolds for bone tissue engineering, Acta Biomater., 2009, 5, 1182–1197 CrossRef CAS.
- Y. Yin, F. Ye, J. Chu, F. Zhang, X. Li and K. Yao, Preparation characterization of macroporous chitosan-gelatin/b-tricalcium phosphate composite scaffolds for bone tissue, J. Biomed. Mater. Res., 2003, 67A, 844–855 CrossRef CAS.
- J. A. Sowjanya, J. Singh, T. Mohita, S. Sarvanan, A. Moorthi, N. Srinivasan and N. Selvamurugan, Biocomposite scaffolds containing chitosan/alginate/nano-silica for bone tissue engineering, Colloids Surf., B, 2013, 109, 294–300 CrossRef CAS.
- Y. Zhang, J. R. Venugopal, A. El-Turki, S. Ramakrishna, B. Su and C. T. Lim, Electrospun biomimetic nanocomposite nanofibers of hydroxyapatite/chitosan for bone tissue engineering, Biomaterials, 2008, 29, 4314–4322 CrossRef CAS.
- L. Kong, Y. Gao, W. Cao, Y. Gong, N. Zhao and X. Zhang, Preparation and characterization of nano-hydroxyapatite/chitosan composite scaffolds, J. Biomed. Mater. Res., 2005, 75A, 275–282 CrossRef CAS.
- G. Ruphuy, A. Saralegi, J. C. Lopes, M. M. Dias and M. F. Barreiro, Spray drying as a viable process to produce nano-hydroxyapatite/chitosan (n-HAp/CS) hybrid microparticles mimicking bone composition, Adv. Powder Technol., 2016, 27, 575–583 CrossRef CAS.
- C. Wang, W. Huang, Y. Zhou, L. He, Z. He, Z. Chen, X. He, S. Tian, J. Liao, B. Lua, Y. Wei and M. Wang, 3D printing of bone tissue engineering scaffolds, Bioact. Mater., 2020, 5, 82–91 CrossRef.
- N. K. Nga, N. T. T. Chau and P. H. Viet, Facile synthesis of hydroxyapatite nanoparticles mimicking biological apatite from eggshells for bone-tissue engineering, Colloids Surf., B, 2018, 172, 760–778 CrossRef.
- T. Kokubo and H. Takadama, How useful is SBF in predicting in vivo bone bioactivity, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS.
- M. A. Aronow, L. C. Gerstenfeld, T. A. Owen, M. S. Tassinari, G. S. Stein and J. B. Lian, Factors that promote progressive development of the osteoblast phenotype in cultured fetal rat calvaria cells, J. Cell. Physiol., 1990, 143(2), 213–221 CrossRef CAS.
- J. S. Mao, L. G. Zhao, Y. J. Yin and K. D. Yao, Structure and properties of bilayer chitosan–gelatin scaffolds, Biomaterials, 2003, 24, 1067–1074 CrossRef CAS.
- S. Ebnesajjad, Handbook of Biopolymers and Biodegradable Plastics, Elsevier, 2013 Search PubMed.
- H. E. Jazayeri, S.-M. Lee, L. Kuhn, F. Fahimipour, M. Tahriri and L. Tayebi, Polymeric scaffolds for dental pulp tissue engineering: a review, Dent. Mater., 2020, 36(2), e47–e58 CrossRef CAS.
- J. K. Sherwood, S. L. Riley, R. Palazzolo, S. C. Brown, D. C. Monkhouse, M. Coates, L. G. Griffith, L. K. Landeen and A. Ratcliffe, A three-dimensional osteochondral composite scaffold for articulate cartilage repair, Biomaterials, 2002, 23(24), 4739–4751 CrossRef CAS.
- X. X. Shao, D. W. Hutmacher, S. T. Ho, J. Goh and E. H. Lee, Evaluation of a hybrid scaffold/cell construct in repair of high-load-bearing osteochondral defects in rabbits, Biomaterials, 2006, 27(7), 1071–1080 CrossRef CAS.
- V. Vyas, T. Kaur and A. Thirugnanam, Chitosan composite three dimensional macrospheric scaffolds for bone tissue engineering, Int. J. Biol. Macromol., 2017, 104, 1946–1954 CrossRef CAS.
- Z. Wen, L. Zhang, C. Chen, Y. Liu, C. Wu and C. Dai, A construction of novel iron-foam-based calcium phosphate/chitosan coating biodegradable scaffold material, Mater. Sci. Eng., C, 2013, 33, 1022–1031 CrossRef CAS.
- Y. S. Pek, M. Spector, I. V. Yannas and L. J. Gibson, Degradation of a collagen-chondroitin-6-sulfate matrix by collagenase and by chondroitinase, Biomaterials, 2004, 25, 473–482 CrossRef CAS.
- M. Alizadeh, F. Abbasi, A. B. Khoshfetrat and H. Ghaleh, Microstructure and characteristic properties of gelatin/chitosan scaffold prepared by a combined freeze-drying/leaching method, Mater. Sci. Eng., C, 2013, 33, 3958–3967 CrossRef CAS.
- Y. M. Yang, W. Hu, X. D. Wang and X. S. Gu, The controlling biodegradation of chitosan fibers by N-acetylation in vitro and in vivo, J. Mater. Sci.: Mater. Med., 2007, 18, 2117–2121 CrossRef CAS.
- R. J. Nordtveit, K. M. Vårum and O. Smidsrød, Degradation of partially N-acetylated chitosans with hen egg white and human lysozyme, Carbohydr. Polym., 1996, 29, 163–167 CrossRef CAS.
- K. R. Mohamed and A. A. Mostafa, Preparation and bioactivity evaluation of hydroxyapatite titania/chitosan-gelatin polymeric biocomposites, Mater. Sci. Eng., C, 2008, 28, 1087–1099 CrossRef CAS.
- C. Sharma, A. K. Dinda, P. D. Potdar, C. F. Chou and N. C. Mishra, Fabrication and characterization of novel nano-biocomposite scaffold of chitosan-gelatin-alginate-hydroxyapatite for bone tissue engineering, Mater. Sci. Eng., C, 2016, 64, 416–427 CrossRef CAS.
- J. Gustavsson, M. P. Ginebra, E. Engel and J. Planell, Ion reactivity of calcium-deficient hydroxyapatite in standard cell culture media, Acta Biomater., 2011, 7(12), 4242–4252 CrossRef CAS.
- F. Farshi Azhar, A. Olad and R. Salehi, Fabrication and characterization of chitosan–gelatin/nanohydroxyapatite–polyaniline composite with potential application in tissue engineering scaffolds, Des. Monomers Polym., 2017, 17, 654–667 CrossRef.
- S. Srinivasan, R. Jayasree, K. P. Chennazhi, S. V. Nair and R. Jayakumar, Biocompatible alginate/nano bioactive glass ceramic composite scaffolds for periodontal tissue regeneration, Carbohydr, Polymers, 2012, 87, 274–283 CAS.
- P. T. Sudheesh Kumar, S. Srinivasan, V.-K. Lakshmanan, H. Tamura, S. V. Nair and R. Jayakumar, Chitin hydrogel/nano hydroxyapatite composite scaffolds for tissue engineering applications, Carbohydr. Polym., 2011, 85, 584–591 CrossRef CAS.
- D. Zamani, F. Moztarzadeh and D. Bizari, Alginate-bioactive glass containing Zn and Mg composite scaffold for bone tissue engineering, Int. J. Biol. Macromol., 2019, 137, 1256–1267 CrossRef CAS.
- S. Puvaneswary, H. B. Raghavendran, S. Talebian, M. R. Murali, S. A. Mahmod, S. Singh and T. Kamarul, Incorporation of fucoidan in β-tricalcium phosphate-chitosan scaffold prompts the differentiation of human bone marrow stromal cells into osteogenic lineage, Sci. Rep., 2016, 6 CAS.
- B. D. Ratner, A. Hoffman, F. Schoen and J. Lemons, Biomaterial Science: An introduction to Materials in Medicine, Elsevier, 2nd edn, 2004 Search PubMed.
- G. Wei and P. X. Ma, Structure and properties of nano-hydroxyapatite/polymer composite scaffolds for bone tissue engineering, Biomaterials, 2004, 25, 4749–4757 CrossRef CAS.
- T. Lou, X. Wang, X. Yan, Y. Miao, Y. Z. Long, H. L. Yin, B. Sun and G. Song, Fabrication and biocompatibility of poly(L-lactic acid) and chitosan composite scaffolds with hierarchical microstructures, Mater. Sci. Eng., C, 2016, 64, 341–345 CrossRef CAS.
- S. Pattnaik, S. Nethala, A. Tripathi, S. Saravanan, A. Moorthi and N. Selvamurugan, Chitosan scaffolds containing silicon dioxide and zirconia nano particles for bone tissue engineering, Int. J. Biol. Macromol., 2011, 49, 1167–1172 CrossRef CAS.
- J. Venkatesan, I. Bhatnagar and S. K. Kim, Chitosan-alginate biocomposite containing fucoidan for bone tissue engineering, Mar. Drugs, 2014, 12, 300–316 CrossRef CAS.
- A. Tripathi, S. Saravanana, S. Pattnaika, A. Moorthi, N. C. Partridge and N. Selvamurugana, Bio-composite scaffolds containing chitosan/nano-hydroxyapatite/nano-copper–zinc for bone tissue engineering, Int. J. Biol. Macromol., 2012, 50, 294–299 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |