
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn orifice design: water insertion into C60†
Yoshifumi Hashikawa ,
Kazuro Kizaki,
Takashi Hirose and
Yasujiro Murata*
,
Kazuro Kizaki,
Takashi Hirose and
Yasujiro Murata*
Institute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan. E-mail: yasujiro@scl.kyoto-u.ac.jp
First published on 6th November 2020
Abstract
Using an open-cage C60 derivative possessing an orifice designed on the basis of computational studies, we have experimentally demonstrated the quantitative encapsulation of H2O as well as effective conversion into H2O@C60 in an overall yield remarkably higher than the previously reported methods by ca. 2–5 times.
Soon after the discovery of C60,1 the first endohedral fullerene, K+@C59B, was proven by Saunders and co-workers in 1991 under mass spectrometric conditions.2 To date, a number of endohedral fullerenes encapsulating metal ions, nitrides, and oxides, which are so-called metallofullerenes, have been produced by physical approaches, e.g., laser ablation and arc discharge methods.3 While these approaches could provide endohedral fullerenes albeit only in 0.1% yield, their properties have received growing attention from a wide research area owing to their potential applications in photovoltaic cells (Lu3N@C80 (ref. 4) and Li+@C60 (ref. 5)) and MRI (magnetic resonance imaging) contrast agents (Gd@C60 and Gd@C82).6 Additionally, DNP (dynamic nuclear polarization) was recently demonstrated by utilizing Gd2@C79N, which resulted in increased nuclear polarization of 1H and 13C spins from electron spins by ca. 50% at 1.2 K.7
In 1997, Rubin advocated an original concept to produce endohedral fullerenes via sequential steps: creation of an opening, insertion of small molecules, and closure of the opening.8 This molecular surgical synthesis based on organic reactions is conceptually different from physical approaches. Of particular importance is to chemically synthesize endohedral fullerenes encapsulating neutral species that cannot be generated by physical approaches. Since the successful synthesis of the first open-cage C60 derivative in 1995 by Wudl and co-workers,9 a number of derivatives with different structural motifs on their orifices have been reported.10 In the present time, a variety of molecules have been found to be encapsulated inside the fullerene cavities, few of which are able to be restored into the pristine caged structures such as C60,11 C70,12 and C59N13 with retaining the molecule inside. Nevertheless, organic syntheses of such endohedral fullerenes still suffer from low yielding. To overcome this problem, we focused on an orifice structure which is crucial for trapping a guest molecule as well as for controlling the reactivity of open-cage C60 derivatives. Since the orifice substructure, i.e., 1ArC–E![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(2Ar)–C(3Ar), originates from a heterocyclic azine (Fig. 1) which is used for the creation of an opening by a thermal reaction with C60,11c,14 we considered six hypothetical azines including pyridazines and triazines (1a–f) so as to design an orifice suitable both for encapsulation of guest molecules and closure of the opening. Herein, we discuss the molecular design toward efficient synthetic methodology for synthesizing endohedral fullerenes based on theoretical studies as well as experimental demonstration.
C(2Ar)–C(3Ar), originates from a heterocyclic azine (Fig. 1) which is used for the creation of an opening by a thermal reaction with C60,11c,14 we considered six hypothetical azines including pyridazines and triazines (1a–f) so as to design an orifice suitable both for encapsulation of guest molecules and closure of the opening. Herein, we discuss the molecular design toward efficient synthetic methodology for synthesizing endohedral fullerenes based on theoretical studies as well as experimental demonstration.
 | ||
| Fig. 1 Retrosynthetic route for endohedral fullerenes (Py: 2-pyridyl group). | ||
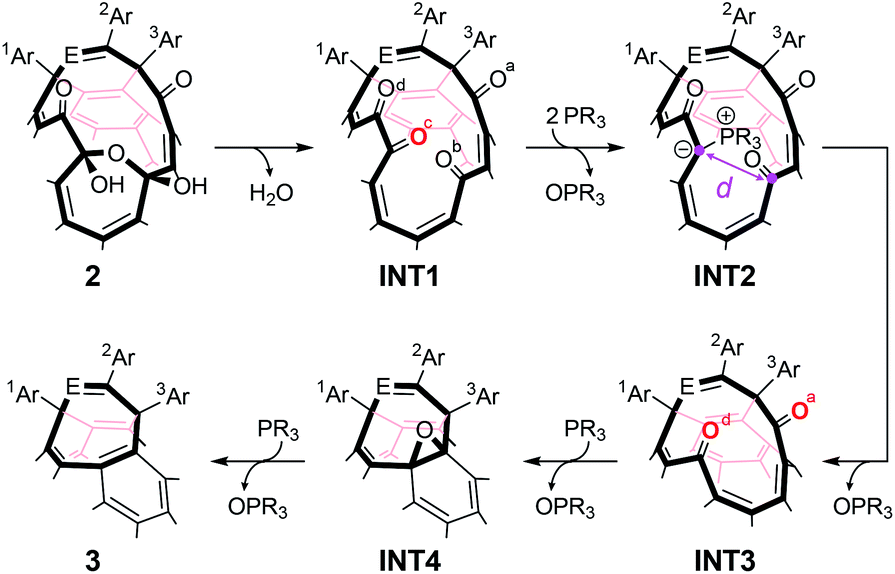
With regard to the synthesis of endohedral fullerenes, INT1 (structure in Table 1) is a key material since it can encapsulate guest molecules such as H2O,11c H2,15 and HF.16 More importantly, the orifice with a substructure of tetraketone in INT1 can be zipped up to reduce its orifice size from 16- to 8-membered ring by the reaction with phosphine and/or phosphite.11c–e The plausible mechanism on this closing process was depicted in Table 1. Firstly, a water molecule would be eliminated from the 13-membered ring of 2 to afford INT1. The ring-closing reaction is then initiated by the nucleophilic attack of PR3 to O(c) in INT1, leading to the formation of β-oxo-phosphorus ylide INT2 (ref. 17) which undergoes intramolecular Wittig reaction to give diketo derivative INT3.18 The further nucleophilic attack of another PR3 molecule on O(a) and/or O(d) in INT3 followed by the elimination of OPR3 furnishes epoxide INT4.19 Finally, deoxygenation of INT4 by PR3 provides 3 possessing an eight-membered-ring opening. Previously, we used 2a′ (which bears 6-t-butylpyridin-2-yl groups instead of 2-pyridyl groups) for preparing INT2a′. Even though INT2a′ could be formed at 100 °C for 3 h, INT2a′ was significantly decomposed during the conversion into INT3a′ at 120 °C for 8 h, resulting in 52% yield.20 Thus, open-cage C60 derivatives should have an orifice which meets criteria including (i) less polarized carbonyl groups in INT1 and INT3 to promote nucleophilic attack of PR3 to carbonyl O-atoms, (ii) formation of stable ylide INT2 to suppress undesirable decomposition pathways, and (iii) readily conversion of INT2 into INT3.
|
|||||
|---|---|---|---|---|---|
| Addends | qNPA | ΔΔG(INT2)a (kcal mol−1) | d(INT2) (Å) | ||
| O(c)/INT1 | O(a)/INT3 | O(d)/INT3 | |||
| a ΔΔG(INT2) = ΔG(INT2) − ΔG(INT2a) where ΔG(INT2) = G(INT2) + G(OPPh3) − G(INT1) − 2G(PPh3). | |||||
| a | −0.465 | −0.510 | −0.474 | 0.0 | 3.35 |
| b | −0.464 | −0.502 | −0.469 | −1.0 | 3.36 |
| c | −0.467 | −0.511 | −0.467 | +0.1 | 3.36 |
| d | −0.461 | −0.497 | −0.469 | −1.4 | 3.34 |
| e | −0.465 | −0.501 | −0.469 | −0.4 | 3.32 |
| f | −0.464 | −0.491 | −0.469 | −3.3 | 3.32 |
To verify electronic and structural properties of INT1, INT2, and INT3, we performed theoretical calculations at the B3LYP-D3/6-31G(d) level of theory at 298 K (Table 1). From natural charges qNPA of O(c) in INT1, the substituent effect is predicted to be not so significant in the initial step. Contrastingly, the qNPA values of O(a) and O(d) in INT3 are reflected by the opening substructure. Compared with INT3a which is a model that we previously synthesized,11c both absolute values of qNPA, except for O(a) in INT3c, markedly decreased, suggestive of increased reactivity of INT3 by replacing one of Py groups with a less electron-withdrawing Ph group at the 3Ar position. Subsequently, we confirmed thermodynamic properties of β-oxo-phosphorus ylide INT2, showing considerable stabilization for INT2f (ΔΔG −3.3 kcal mol−1) which is superior to others in preventing decomposition. This is probably due to the better orientation for one of Ph groups on the P-atom in INT2f via CH/π interaction with the Ph group at the 2Ar position which is also tightly fixed via an H-bonding with the imine moiety (Fig. S8†). Furthermore, the replacement of an olefin unit with an imine moiety causes decrease in the C(b)⋯C(c) distance by 0.04 Å, implying the positive effect on the intramolecular Wittig reaction in INT2e and INT2f.
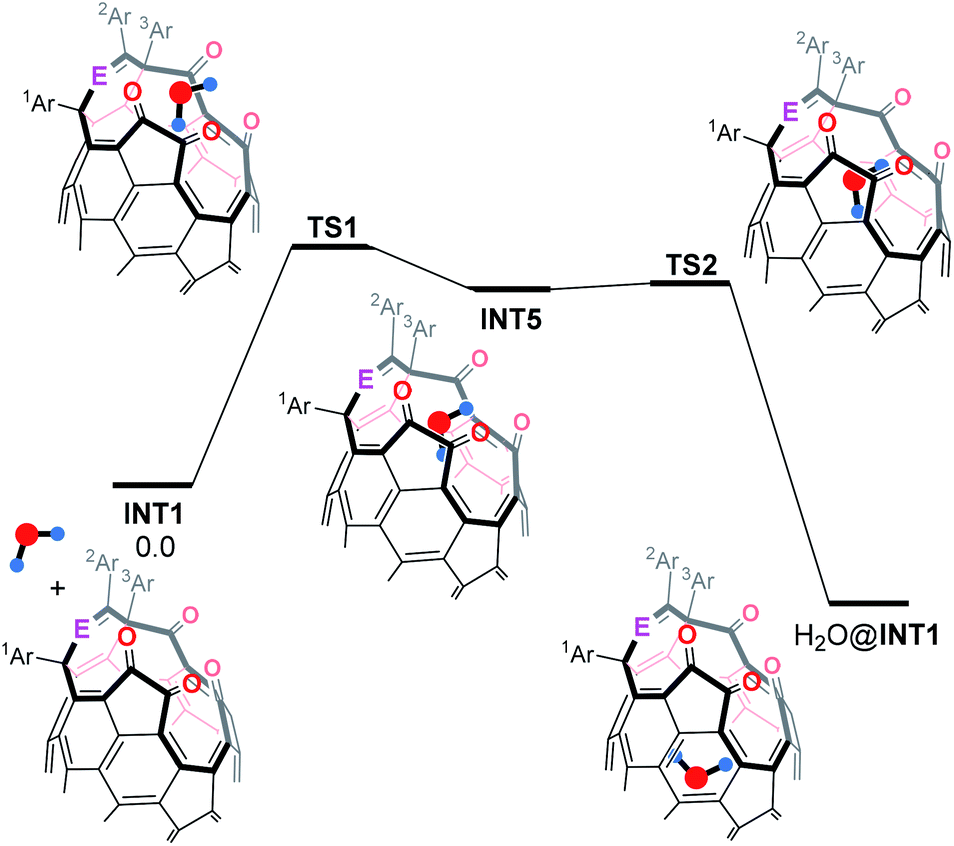
To gain insight into the orifice size of INT1, we summarized an energy profile for the H2O-insertion as shown in Table 2. Interestingly, the H2O-insertion is predicted to occur stepwisely. Firstly, the H2O molecule is trapped at a cross-section of the orifice to form INT5 via TS1 which is the rate-determining step. Subsequently, the trapped H2O molecule is encapsulated inside INT1 via TS2 to give H2O@INT1. Upon seeing activation barriers, the Ph group at the 2Ar position seems not to have considerable influence on the H2O-insertion, showing the similar ΔG‡ values of 21–22 kcal mol−1. The substantial stabilization of INT1f by the H2O encapsulation (ΔG −10.4 kcal mol−1) would be an advantage to obstruct the escape of the H2O molecule from the inside. Consequently, the orifice of 2f, which consists of one Py and two Ph groups with an imine moiety, should be preferable for the synthesis of H2O@C60 both in terms of reactivity on the closing process and capability of encapsulating a H2O molecule.
|
||||
|---|---|---|---|---|
| Addends | TS1 | INT5 | TS2 | H2O@INT1 |
| a | +21.6 | +17.1 | +17.5 | −10.0 |
| b | +21.7 | +17.2 | +17.6 | −9.3 |
| c | +21.4 | +16.9 | +17.2 | −8.9 |
| d | +22.0 | +17.5 | +18.2 | −9.7 |
| e | +21.0 | +17.3 | +17.8 | −8.9 |
| f | +21.0 | +17.3 | +17.8 | −10.4 |
Thereby, we synthesized 2f by gram-scale reactions including the thermal reaction of C60 with 5,6-diphenyl-3-(pyridin-2-yl)-1,2,4-triazine (1f), photooxygenation,14 and nucleophilic oxygenation using N-methylmorpholine N-oxide.11c Recently, we found that β-oxo-phosphorus ylide INT2a′ bearing a PR3 unit with a cone angle smaller than 140° is significantly hydrolysed in the presence of water.17 As to the conversion from INT3a′ to 3a′, P(OiPr)3 was previously found to exhibit high performance.18 For the efficient synthesis of H2O@C60, we designed one-pot reaction from 2f to H2O@3f via three steps commenced with the H2O-insertion under high-pressure conditions followed by stepwise conversion of the thus formed H2O@2f into H2O@INT3f and then H2O@3f. The results were summarized in Table 3. The H2O-insertion was conducted using 20 mg of 2f in a mixed solvent system of 1-chloronaphthalene (1-ClNp) and toluene under 9000 atm for 24 h. For the closure of the opening, we used PPh3 with a cone angle of 145° (entry 1). Different from 2a′ that required totally 11 h for the conversion into INT3a′,20 2f was transformed into INT3f within 1.5 to 2 h, meeting the criteria (i)–(iii) owing to stable ylide INT2f with a shorter C(b)⋯C(c) distance. The further reaction with P(OiPr)3 at 120 °C for 0.5 h gave desired H2O@3f in 48% isolated yield with the encapsulation ratio of 98%. Whereas electron-deficient phosphine P(2-furyl)3 gave H2O@3f in lower yield (21%, entry 2), the reaction employing electron-rich P(p-tolyl)3 resulted in higher yield (69%) with keeping the 98% encapsulation ratio of H2O (entry 3). It should be noted that the decreased temperature at the first step drastically influenced on the encapsulation ratio of H2O: 98% (160 °C, entry 3), 65% (140 °C, entry 4), and 4% (120 °C, entry 5).
|
|||
|---|---|---|---|
| Entry | Step 1 | Step 2 | Yieldb |
| a Conducted using 20 mg of 2f in a mixed solvent system of 1-chloronaphthalene (1-ClNp) and toluene.b Encapsulation ratio was determined by 1H NMR or mass spectrum. | |||
| 1 | 160 °C | PPh3 | 48% (H2O: 98%) |
| 2 | 160 °C | P(2-furyl)3 | 21% (H2O: 99%) |
| 3 | 160 °C | P(p-tolyl)3 | 69% (H2O: 98%) |
| 4 | 140 °C | P(p-tolyl)3 | 67% (H2O: 65%) |
| 5 | 120 °C | P(p-tolyl)3 | 68% (H2O: 4%) |

The structure of H2O@3f was determined spectroscopically. The molecular ion peak was observed at m/z 1020.1255 ([H2O@3f]˙−) in the negative ionization mode by the APCI (atmospheric-pressure chemical ionization) method. The 1H NMR spectrum of H2O@3f (CDCl3, 500 MHz) clearly showed a singlet signal at δ −6.11 ppm corresponding to the encapsulated H2O molecule, bearing the striking resemblance with that for H2O@3a′ (δ −6.09 ppm11c,18). Using H2O@3f with an occupation level of 60%, single crystals were grown from the benzene/hexane solution by slow evaporation at 5 °C. The solid-state structure was shown in Fig. 2. The water molecule locates at the centre of the cage and its occupancy was refined to be 0.593(8), being in accordance with the ratio determined by 1H NMR. A crystallographic disorder was seen in the orifice structure which is identical to two parts with sharing the Py group. Their occupancies were refined to be 0.854(2) for the major part and 0.146(2) for the minor part, respectively.
 | ||
| Fig. 2 Single crystal X-ray structure of H2O@3f with showing thermal ellipsoids in 50% probability (left) and disordered structures coloured with pink and light blue for major and minor parts, respectively (right). The benzene molecules co-crystallized with H2O@3f are omitted for clarity. | ||
Based on our previous approach to synthesize H2O@C60 using 2a′ in the solid state, the final step has the scale restriction up to 50 mg. Otherwise, the conversion is considerably decreased. For the scalable synthesis of H2O@C60, we examined one-pot synthesis of H2O@C60 using 160 mg of 2f. As represented in Scheme 1, 2f was firstly subjected to the optimal reaction conditions as entry 3 in Table 3. To the resultant mixture, methanol was added to obtain the crude precipitate containing H2O@3f which was further heated at 400 °C under 5 Pa for 2 h, giving H2O@C60 in 6% isolated yield. Considering the high conversion of 2f into H2O@3f (Table 3), H2O@3f should be mostly decomposed during the final step. To avoid undesirable decomposition pathways, the crude mixture was purified, prior to step 4, by silica gel column chromatography which gave H2O@3f (80% yield) together with complex mixture having higher polarity. Since this complex mixture changed into insoluble solid without formation of H2O@C60 under the same conditions as step 4, it might hamper the conversion of H2O@3f into H2O@C60. With H2O@3f as a pure form in hand, it was subjected to pyrolytic conditions (Scheme 1). As the results, H2O@C60 was obtained up to 87% yield and the overall yield from 2f reaches to 70% which is remarkably higher than that obtained by our previous approach (15% from 2a′)11c even when compared with Whitby's variant (40% from 2a′).15
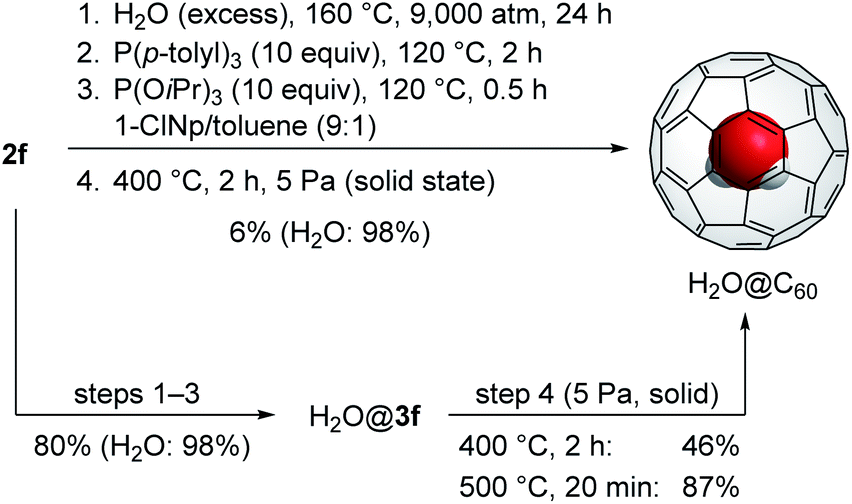 | ||
| Scheme 1 Synthesis of H2O@C60 from 2f (160 mg). | ||
In summary, we designed the orifice substructure suitable for the synthesis of endohedral fullerenes on the basis of reactivity toward closing processes and encapsulation properties. Among six candidates, the orifice possessing a phenyl imine moiety (f) was expected to have high affinity to phosphines as well as interaction with the encapsulated H2O molecule, which is strong enough to prevent its escape from the inside. The thus synthesized open-cage C60 derivative 2f demonstrated the effective conversion into H2O@3f via one-pot process, showing high yield up to 80% with nearly quantitative encapsulation of H2O. The further conversion of H2O@3f into H2O@C60 proceeded quite well when H2O@3f was used in a pure form. The method demonstrated herein could be applied for a variety of endohedral fullerenes in a practical scale.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was partially provided by the JSPS KAKENHI Grant Number JP17H06119 and JP18K14200. We thank Dr Kyusun Kim for the initial trials on the synthesis of 2f.Notes and references
- H. W. Kuroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef.
- Y. Chai, T. Guo, C. Jing, R. E. Haufler, L. P. F. Chibante, J. Fure, L. Wang, J. M. Alford and R. E. Smalley, J. Phys. Chem., 1991, 95, 7564–7568 CrossRef CAS.
- (a) A. Rpdríguez-Fortea, A. L. Balch and J. M. Poblet, Chem. Soc. Rev., 2011, 40, 3551–3563 RSC; (b) A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113(8), 5989–6113 CrossRef CAS.
- R. B. Ross, C. M. Cardona, D. M. Guldi, S. G. Sankaranarayanan, M. O. Reese, N. Kopidakis, J. Peet, B. Walker, G. C. Bazan, E. V. Keuren, B. C. Holloway and M. Drees, Nat. Mater., 2009, 8, 208–212 CrossRef CAS.
- I. Jeon, H. Ueno, S. Seo, K. Aitola, R. Nishikubo, A. Saeki, H. Okada, G. Boschloo, S. Maruyama and Y. Matsuo, Angew. Chem., Int. Ed., 2018, 57, 4607–4611 CrossRef CAS.
- K. B. Ghiassi, M. M. Olmstead and A. L. Balch, Dalton Trans., 2014, 43, 7346–7358 RSC.
- X. Wang, J. E. McKay, B. Lama, J. v. Tol, T. Li, K. Kirkpatrick, Z. Gan, S. Hill, J. R. Long and H. C. Dorn, Chem. Commun., 2018, 54, 2425–2428 RSC.
- Y. Rubin, Chem. – Eur. J., 1997, 3, 1009–1016 CrossRef CAS.
- J. C. Hummelen, M. Prato and F. Wudl, J. Am. Chem. Soc., 1995, 117, 7003–7004 CrossRef CAS.
- (a) M. Murata, Y. Murata and K. Komatsu, Chem. Commun., 2008, 6083–6094 RSC; (b) G. C. Vougioukalakis, M. M. Roubelakis and M. Orfanopoulos, Chem. Soc. Rev., 2010, 39, 817–844 RSC; (c) L. Shi and L. Gan, J. Phys. Org. Chem., 2013, 26, 766–772 CrossRef CAS.
- (a) K. Komatsu, Y. Murata and Y. Murata, Science, 2005, 307, 238–240 CrossRef CAS; (b) Y. Morinaka, F. Tanabe, M. Murata, Y. Murata and K. Komatsu, Chem. Commun., 2010, 46, 4532–4534 RSC; (c) K. Kurotobi and Y. Murata, Science, 2011, 333, 613–616 CrossRef CAS; (d) A. Krachmalnicoff, R. Bounds, S. Mamone, S. Alom, M. Concistrè, B. Meier, K. Kouřil, M. E. Light, M. R. Johnson, S. Rols, A. J. Horsewill, A. Shugai, U. Nagel, T. Rõõm, M. Carravetta, M. H. Levitt and R. J. Whitby, Nat. Chem., 2016, 8, 953–957 CrossRef CAS; (e) S. Bloodworth, G. Sitinova, S. Alom, S. Vidal, G. R. Bacanu, S. J. Elliott, M. E. Light, J. M. Herniman, G. J. Langley, M. H. Levitt and R. J. Whitby, Angew. Chem., Int. Ed., 2019, 58, 5038–5043 CrossRef CAS; (f) S. Bloodworth, G. Hoffman, M. C. Walkey, G. R. Bacanu, J. M. Herniman, M. H. Levitt and R. J. Whitby, Chem. Commun., 2020, 56, 10521–10524 RSC.
- (a) M. Murata, S. Maeda, Y. Morinaka, Y. Murata and K. Komatsu, J. Am. Chem. Soc., 2008, 130, 15800–15801 CrossRef CAS; (b) R. Zhang, M. Murata, T. Aharen, A. Wakamiya, T. Shimoaka, T. Hasegawa and Y. Murata, Nat. Chem., 2016, 8, 435–441 CrossRef CAS; (c) R. Zhang, M. Murata, A. Wakamiya and Y. Murata, Sci. Adv., 2017, 3, e1602833 CrossRef; (d) Y. Morinaka, R. Zhang, S. Sato, H. Nikawa, T. Kato, K. Furukawa, M. Yamada, Y. Maeda, M. Murata, A. Wakamiya, S. Nagase, T. Akasaka and Y. Murata, Angew. Chem., Int. Ed., 2017, 56, 6488–6491 CrossRef CAS.
- (a) Y. Hashikawa, M. Murata, A. Wakamiya and Y. Murata, J. Am. Chem. Soc., 2016, 138, 4096–4104 CrossRef CAS; (b) Y. Hashikawa, M. Murata, A. Wakamiya and Y. Murata, J. Org. Chem., 2017, 82, 4465–4469 CrossRef CAS; (c) Y. Hashikawa and Y. Murata, J. Am. Chem. Soc., 2017, 139, 18468–18471 CrossRef CAS; (d) G.-Z. Zhu, Y. Liu, Y. Hashikawa, Q.-F. Zhang, Y. Murata and L.-S. Wang, Chem. Sci., 2018, 9, 5666–5671 RSC.
- Y. Murata, M. Murata and K. Komatsu, Chem. – Eur. J., 2003, 9, 1600–1609 CrossRef CAS.
- A. Krachmalnicoff, M. H. Levitt and R. J. Whitby, Chem. Commun., 2014, 50, 13037–13040 RSC.
- A. Krachmalnicoff, R. Bounds, S. Mamone, M. H. Levitt, M. Carravetta and R. J. Whitby, Chem. Commun., 2015, 51, 4993–4996 RSC.
- Y. Hashikawa, S. Okamoto and Y. Murata, Commun. Chem., 2020, 3, 90 CrossRef CAS.
- Y. Hashikawa, M. Murata, A. Wakamiya and Y. Murata, J. Am. Chem. Soc., 2017, 139, 16350–16358 CrossRef CAS.
- Y. Hashikawa, H. Yasui, K. Kurotobi and Y. Murata, Mater. Chem. Front., 2018, 2, 206–213 RSC.
- Y. Hashikawa and Y. Murata, Chem. Lett., 2020, 49, 244–247 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, spectra, and optimized geometries. CCDC 1999283. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra09067k |
| This journal is © The Royal Society of Chemistry 2020 |